

Background: The microtubule-associated protein p600 forms a complex with the Ca2+ sensor calmodulin.
Results: Knockdown of p600 or specific disruption of the Ca2+-dependent calmodulin/p600 binding triggers neuronal death following Ca2+ elevation.
Conclusion: p600 is required for survival of hippocampal neurons following glutamate-induced Ca2+ elevation.
Significance: This novel role for p600 in Ca2+ signaling and neuronal death has broad relevance to the study of neurodegeneration.
Keywords: Calcium, Calmodulin, Cell Biology, Cell Death, Microtubules, Neurodegeneration, Microtubule-associated Protein, UBR4, p600
Abstract
In acute and chronic neurodegeneration, Ca2+ mishandling and disruption of the cytoskeleton compromise neuronal integrity, yet abnormalities in the signaling roles of cytoskeletal proteins remain largely unexplored. We now report that the microtubule-associated protein p600 (also known as UBR4) promotes neuronal survival. Following depletion of p600, glutamate-induced Ca2+ influx through NMDA receptors, but not AMPA receptors, initiates a degenerative process characterized by endoplasmic reticulum fragmentation and endoplasmic reticulum Ca2+ release via inositol 1,4,5-trisphosphate receptors. Downstream of NMDA receptors, p600 associates with the calmodulin·calmodulin-dependent protein kinase IIα complex. A direct and atypical p600/calmodulin interaction is required for neuronal survival. Thus, p600 counteracts specific Ca2+-induced death pathways through regulation of Ca2+ homeostasis and signaling.
Introduction
Neuronal survival requires controlled Ca2+ signaling/homeostasis and the maintenance of cytoskeletal integrity. In acute (traumatic brain injury, stroke) and chronic neuronal degeneration (Alzheimer disease, Parkinson disease), perturbations in Ca2+ handling trigger neuronal death through common routes, including excitotoxicity and oxidative and endoplasmic reticulum (ER)5 stress (1–7). Genetic and biochemical alterations in cytoskeletal proteins are also observed in a wide diversity of chronic neurodegenerative disorders (Alzheimer disease, Parkinson disease, etc.) featuring deregulation of Ca2+ homeostasis and signaling (1, 5, 8). For instance, mutations at highly conserved residues within or near the microtubule-binding domain in the human microtubule-associated protein Tau gene cause inherited frontotemporal dementia and parkinsonism (9). Furthermore, the hyperphosphorylation of Tau by Ca2+-activated kinases reduces its ability to stabilize microtubules and its aggregation in toxic oligomers and/or fibrils (10). Although abnormalities in the neuronal cytoskeleton and Ca2+ mishandling both contribute to acute and chronic neuronal death, alterations in the signaling functions of cytoskeletal proteins remain largely unexplored.
Upon exposure to glutamate, the influx of Ca2+ through NMDA and AMPA receptors, and subsequently via VDCCs, activates secondary messenger systems to propagate the Ca2+ signaling cascade, leading to cytoskeletal changes, plasticity, and survival. However, excessive influx of Ca2+ via these receptors and channels triggers neuronal death via cytoskeletal disruption, whereas drugs that antagonize them prevent cell death (2, 5, 7, 11). Inside the neuron, the ER constitutes the primary intracellular source for Ca2+ mobilization and signaling. ER Ca2+ levels are controlled by Ca2+ release through ryanodine and inositol 1,4,5-triphosphate (IP3) receptors and the activity of inward pumps (5). These collectively determine the ER functioning mode and, consequently, neuronal survival. As the ER is transported on microtubules (12), collapse of microtubules is linked to ER fragmentation and dysfunction (13, 14).
We have recently discovered a novel microtubule-associated protein in CNS neurons: Protein 600 (p600, also known as UBR4) (15). p600 is required for neurite outgrowth and neuronal migration in the developing neocortex (15), two events that concomitantly require cytoskeletal dynamics and Ca2+ signaling. Thus, we hypothesized that p600 protects against Ca2+-induced neuronal dyshomeostasis and degeneration by maintaining structural stability and Ca2+ homoeostasis.
MATERIALS AND METHODS
Culture of Dissociated Primary Hippocampal Neurons
Hippocampi were dissected from perinatal rat pups of either sex. Tissues were dissociated and plated on 24-well plates in basal medium Eagle (Invitrogen) supplemented with penicillin/streptomycin (Invitrogen), 2 mm l-glutamine (Invitrogen), 10% FBS (Invitrogen), and 10 mm HEPES (pH 7.3) to a final density of 200,000 cells/well. Cultures were incubated until DIV10 with two partial medium changes.
Generation and Characterization of RNAi and mCherry-ER Vectors
RNAi sequences were selected based on the criteria of Ambion, Inc. Complementary hairpin sequences were commercially synthesized and cloned into pSilencer 2.0 under the U6 promoter (Ambion). Sequences for the rat p600 RNAi are base pairs AAGCAGTACGAGCCATTCTAC and AAGCCTGTCAAGTACGATGAA. A random sequence without homology to any known mRNA was used for control RNAi. All RNAi constructs were tested in primary neuronal cultures by both Western blot and immunofluorescent staining. In order to achieve the higher transfection efficiency required for Western blotting (Fig. 1C), the Amaxa nucleofector system was used as described previously (16). The mCherry vector was a gift from Dr. R. Y. Tsien. The mCherry cDNA was subcloned into pcDNA 3.1+ flanked by an N-terminal calreticulin ER-targeting sequence and a C-terminal ER retention KDEL tag as described previously (17). ER-specific expression was confirmed by BiP co-immunofluorescence in transfected HeLa cells.
FIGURE 1.

p600 is enriched in the brain and required for neuronal survival. A, p600 is enriched in the brain, as detected by Western blots. B, in situ hybridization of coronal mouse adult brain sections shows the mRNA expression pattern of p600. Pyr, pyramidal layer of the hippocampus; Dg, dentate gyrus; Cx, cortex; Pir, piriform corex; Tha, thalamus; Pvn, paraventricular nucleus; Sch, suprachiasmatic nucleus; Son, supraoptic nucleus; Dm, dorsomedial nucleus; Ag, amygdala; Hyp, hypothalamus; Hb, habenula. The sense probe did not produce any signal, providing specificity to the observed pattern of expression. C, two short hairpin RNA (shRNA) constructs targeting mouse p600 mRNA (15) tested by electroporation in primary hippocampal neurons effectively knock down p600 expression. Note the absence of difference in levels of CaMKIIα, activated CaMKIIα (phospho-Thr286-CaMKIIα; p-CaMKIIα), and the microtubule-associated proteins Lis1 and Ndel1 between untreated control RNAi and untreated p600 RNAi-treated neurons, indicating specific loss of p600. Actin was used as a control for protein loading. Values are shown ± S.D. (error bars); **, p (T < t) two-tailed < 0.01). D, at the magnification used for scoring neuronal survival and GFP-CaMKIIα aggregation, neurons (top) were readily distinguished from glia (bottom). Morphological differences in transfected cells (green) were confirmed by the presence or absence (arrowheads) of NeuN staining (red). Scale bar, 10 μm (E). When depleted of p600 by RNAi, 42% of neurons die over the time frame indicated in the experimental timeline below. There were no significant differences in the number of neurons with cleaved caspase-3, an indicator of apoptosis, or which were permeable to propidium iodide (P.I.), an indicator of necrosis (see values and statistics under “Results”). Shown are means ± S.E. (error bars) of six repeats. ∼200 neurons were analyzed per condition.
Transfection, Glutamate Treatment, and Assessment of GFP-CaMKIIα Aggregation
Hippocampal cultures were transfected with vectors encoding one of two RNAi sequences directed against p600 or with a control RNAi along with rat GFP-CaMKIIα in a 4:1 ratio. 16 h post-transfection, cultures were treated with bath solution (135 mm NaCl, 5 mm KCl, 2 mm MgCl2, 3 mm CaCl2, 10 mm d-glucose, 10 mm HEPES, pH 7.3) with or without 30 μm Glu, 3 μm Gly for 3 min, washed for 2 min with PBS, fixed, and scored with a fluorescent microscope. Aggregation was quantified as a percentage of neurons showing any form of non-synaptic CaMKIIα aggregation relative to total GFP-positive neurons. All neurons were scored with a Nikon T2000E fluorescent microscope using a ×40 or ×60 objective such that there was no ambiguity as to whether the transfected cells were neurons or glia (confirmed by NeuN staining; Fig. 1D). For better comparison between culture sets, data on GFP-CaMKIIα aggregation are mostly presented as normalized values.
Quantification of Neuronal Degeneration
In order to assess neuronal survival, cultures were transfected as described above, except GFP-CaMKIIα was replaced with pEGFP N2. After the prescribed incubation time, cultures were fixed in 4% paraformaldehyde in PBS, and GFP-positive neurons were counted as a measure of neuronal survival. In the experiment reported in Fig. 1E, wherein neurons were depleted of p600 by RNAi, neurons were transfected for 16 h and counted 6 h thereafter. The assessment of survival with p600P peptide (Fig. 5B) required a longer incubation of 24 h due to the higher expression requirement for competitive inhibition by the peptide.
FIGURE 5.

Disruption of the p600/CaM interaction causes aggregation of GFP-CaMKII followed by neuronal degeneration. A, ∼30% increase in the number of neurons with GFP-CaMKIIα aggregation following transfection of the peptide and treatment with 30 μm Glu, 3 μm Gly for 3 min (the molar ratio of GFP-CaMKIIα/peptide was 1:2.4). Three experiments were performed for each condition. *, p < 0.05; **, p < 0.01; N.S., p > 0.05 (see values and statistics under “Results”). Representative confocal pictures show the effect of the p600P on CaMKIIα distribution. B, when exposed to exogenous glutamate, neurons transfected with p600P show a ∼40% decrease in survival relative to empty vector-transfected control cultures. A similar effect is seen when cultures are sham-treated with a glutamate-free wash. However, when endogenous NMDA receptor activation is blocked, supplementing the growth medium with 100 μm 2-APV, no decrease in survival is seen in neurons transfected with p600P. *, p < 0.05; **, p < 0.01; N.S., p > 0.05 (see values and statistics under “Results”). Error bars, S.E.
For analysis of necrosis, neurons were transfected for 16 h, incubated in growth medium substituted with the vital dye propidium iodide (1 μg ml−1; Molecular Probes) for 15 min, washed twice with PBS, and then fixed. Propidium iodide-positive cells were all rounded but still GFP-positive. For analysis of apoptosis, neurons were transfected for 16 h and then returned to growth medium for 6 h, at which point caspase-3 activity is maximal in similar culture systems (18). Neurons were then fixed and immunostained with an antibody directed against cleaved caspase-3 (9661, Cell Signaling).
Quantification of ER Fragmentation
In order to quantify the relationship between CaMKIIα aggregation and ER fragmentation, cells were co-transfected with GFP-CaMKIIα and an ER-targeted fluorophore, mCherry-ER, and treated as described above. The measure of bimodal bias was used to quantify CaMKIIα aggregation and ER fragmentation. For calculation of the bimodal bias, the pixel density outputs of the GFP-CaMKIIα fluorophore were analyzed using NOCOM (19) to assess bimodality of pixel distribution, a measure of aggregation. The difference between the likelihood of a single-mode fit versus that of a bimodal fit of the pixel density, the so-called bimodal bias, was used as a measure of pixel/protein aggregation.
Pharmacological Treatments
Dissociated primary hippocampal neuronal cultures (DIV10) were transfected with p600 RNAi and GFP-CaMKIIα in a 4:1 ratio. 16 h post-transfection, cultures were incubated with a bath solution containing a compound of interest or a vehicle control for 15 min (except EGTA, for which a 2-min preincubation was used). Cultures were then treated with a bath solution containing the compound of interest/vehicle control and 30 μm Glu, 3 μm Gly for 3 min. Cultures were then washed for 2 min with PBS and fixed, and GFP-CaMKIIα aggregation was scored as described above. All compounds were from Sigma except nimodipine and ryanodine (Enzo).
p600 Blocking Peptide
Primary hippocampal neuronal cultures (DIV10) were co-transfected with GFP-CaMKIIα vector and an empty vector (pcDNA3.1+), a pcDNA3.1+ vector encoding the p600/calmodulin (CaM) interaction sequence (residues 4086–4113 on human p600 (MAPSKSELRHLYLTEKYVWRWKQFLSRRG)), or the same vector containing an additional negative control, the blocking peptide with a W4103E mutation on a conserved residue on the CaM-binding motif (MAPSKSELRHLYLTEKYVERWKQFLSRRG) in a 1:2 ratio. 16 h post-transfection, cultures were treated with bath solution with or without 30 μm Glu, 3 μm Gly for 3 min, washed for 2 min with PBS, fixed, and scored for prevalence of CaMKIIα aggregation with a fluorescent microscope. In order to assess the effect of the p600/CaM blocking peptide on survival, cultures were transfected as above, with pEGFP N2 substituted for GFP-CaMKIIα. 16 h post-transfection, they were treated with bath solution with or without 30 μm Glu, 3 μm Gly for 3 min and returned to growth medium for 24 h. Where indicated, this growth medium was supplemented with 100 μm 2-APV (Sigma) or 20 μm benzyloxycarbonyl-VAD-fluoromethyl ketone (Millipore). Neuronal survival counts are normalized within each experiment and expressed as a percentage in order to compare values between experiments.
Statistical Analyses
The correlation between CaMKIIα aggregation and ER fragmentation was analyzed with two-tailed Pearson's correlation. Data sets acquired by comparison of neuronal cultures were checked for normality by the Anderson-Darling test. Data sets that were not normally distributed were compared by the Mann-Whitney U test. Data generated by scoring CaMKIIα aggregation were found by Bartlett's test to have unequal variance between test groups and hence were compared by two-tailed Welch's t test. All other normally distributed data were compared by two-tailed Student's t test. Curve fitting was done in MATLAB (MathWorks).
Western Blots and in Situ Hybridization
The mice were sacrificed by intraperitoneal injection of chloral hydrate. Protein extracts of the animals or primary hippocampal neurons were obtained by homogenization in SDS-urea (0.5% SDS, 8 m urea in phosphate buffer, pH 7.4) or radioimmunoprecipitation assay lysis buffer with a mixture of protease inhibitors (PMSF, leupeptin, pepstatin, aprotinin) and 1 mm sodium orthovanadate for phosphatase inhibition. The protein concentration in tissue and cell homogenates was estimated by the Bradford procedure (Bio-Rad). Proteins (20–50 μg) were fractionated on 6, 8, or 10% SDS-PAGE and blotted on a nitrocellulose or PVDF membrane for Western blot analysis. Membranes were incubated with antibodies against p600 (F4, F7) (15), actin (C4, MAB1501, Chemicon), synaptophysin (MAB329, Chemicon), CaMKIIα (MAB8699, Chemicon), PSD-95 (75-028, NeuroMab), and calmodulin (05-173, Millipore). Quantifications were corrected with levels of actin and performed with the Labscan program (Image Master 2D software version 3.10, Amersham Biosciences). The in situ hybridization was performed as described previously (20) with both sense and antisense probes.
Subcellular Fractionation
Mouse brain was homogenized in 5 ml of 10 mm HEPES, 0.32 m sucrose (with the addition of 0.2 mm PMSF, 1 mm EGTA, and a complete protease inhibitor tablet (Roche Applied Science)) at 1000 × g at 4 °C for 10 min. Separated from the pellet (P1), the supernatant (S1) was then centrifuged again for 15 min at 12,000 × g. The resulting pellet (P2) corresponds to the crude synaptosome preparation, whereas the supernatant (S2) corresponds to the cleared soluble cytosolic fraction.
Gel Filtration Assay
Recombinant Xenopus laevis calmodulin was prepared as described previously (21). A peptide derived from p600 encompassing the sequence identified by the CTDB search program (SKSELRHLYLTEKYVWRWKQFLSRRG) was synthesized by the Queen's University Peptide Synthesis Laboratory (Kingston, Canada). For non-denaturing urea PAGE analysis, calmodulin and p600 peptide at several molar ratios were incubated for 1 h at room temperature in 1 mm Tris, pH 7.5, 50 mm CaCl2, and 10 m urea. The latter was included to disrupt nonspecific interactions. Electrophoresis was performed as described previously (22).
Immunoprecipitations
For the experiments testing the effect of glutamate pretreatment on the interaction between p600 and CaMKIIα, the medium was replaced with KRH buffer (containing 128 mm NaCl, 5 mm KCl, 1 mm NaH2PO4, 2.7 mm CaCl2, 1.2 mm MgSO4, 10 mm glucose, and 20 mm HEPES (pH 7.4)) for 30 min at 37 °C. Neurons were then treated with 100 μm/10 μm glycine in Mg2+-free KRH buffer for 5 min and then were lysed in radioimmune precipitation assay buffer (10 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm EDTA, and 1% Triton X-100) containing protease and phosphatase inhibitors. The homogenized cells were centrifuged at 13,000 × g for 5 min at 4 °C. The resulting supernatant was collected for co-immunoprecipitation. 1 mg of protein was used per immunoprecipitation. Immunoprecipitations were performed as described (16) and according to standard protocols.
RESULTS
p600 Is Enriched in the Brain and Required for Neuronal Survival
In mice, the protein levels of p600 are highest in the brain (Fig. 1A). In the adult cortex and hippocampus, p600 is predominantly expressed in neurons (15) (Fig. 1B). The embryonic lethality of two different p600 null mouse strains (24, 25), however, precluded an investigation of the mechanistic roles of p600 in adult brain neurons but hints at the requirement of p600 for cell survival in vivo. Because of this lethal phenotype and expression of p600 in the adult hippocampus (15) (Fig. 1B), we sought to use primary cultured hippocampal neurons to study the roles of p600 in neuronal survival. In our experimental settings, DIV10 neurons had differentiated, elaborated synapses and responded to glutamate treatment, indicating functional maturity (see “Materials and Methods”).
We first sought to establish a loss-of-function methodology by which we could study the role of p600 in a culture system. First, we established the efficacy of an RNAi knockdown strategy. In primary hippocampal neurons electroporated with a vector encoding one of the two p600-specific small interference RNA oligonucleotides directed against p600 mRNA (15) (RNAi 1 or 2), endogenous p600 protein was diminished by 60–70%, as revealed by Western blots (Fig. 1C) (15). The strong decrease in p600 was further confirmed at the single cell level by immunofluorescent staining followed by confocal microscopy (15) (data not shown). No decrease in p600 was observed in cells treated with a random oligonucleotide sequence without homology to any known mRNA (control RNAi).
In order to test the hypothesis that p600 is required for neuronal survival, we transfected DIV10 primary hippocampal neurons with a control or a p600 RNAi vector together with pEGFP N2 and scored cultures for number of viable neurons 22 h after transfection. Under normal culture conditions, primary mature hippocampal neurons depleted of p600 degenerate more than control RNAi-treated neurons (where control RNAi = 100 ± 13% survival, p600 RNAi 1 = 58.1 ± 7.5% survival, t10 = 2.87, p = 0.017; Fig. 1E). This result was confirmed with p600 RNAi 2 (where control RNAi = 100 ± 10% survival, p600 RNAi 2 = 65.9 ± 10% survival, t10 = 2.39, p = 0.038).
We next explored the manner of neuronal demise. By microscopy, we found that depletion of p600 by RNAi did not increase the number of cells permeable to propidium iodide (where control RNAi = 100 ± 24%, p600 RNAi = 96.5 ± 13%, t10 = 0.41, p = 0.69) or increase the number of cells showing cleaved caspase-3 immunoreactivity (where control RNAi = 100 ± 31%, p600 RNAi = 74.6 ± 51%, t10 = 0.14, p = 0.89; Fig. 1E). We thus conclude that neurons depleted of p600 undergo a non-apoptotic and non-necrotic demise (see “Discussion”).
Glutamate-treated Hippocampal Neurons Exhibit Ca2+-induced Dyshomeostasis When Depleted of p600
Having found that p600 is required for neuronal survival, we determined to look at the events preceding this demise. Because the survival of hippocampal neurons is so highly susceptible to perturbations in Ca2+ homeostasis, we hypothesized that p600 was regulating Ca2+ homeostasis in these cells. For this purpose, we used GFP-CaMKIIα as a marker for neuronal dyshomeostasis. In whole ischemic brains, CaMKIIα forms high molecular weight aggregates (26). CaMKIIα aggregation can also be induced within minutes by toxic insults, specifically by increasing intracellular [Ca2+], energy depletion (of ATP) or by lowering intracellular pH (26–32). This somatic/dendritic aggregation is distinct from the synaptic clustering of CaMKIIα during heterosynaptic plasticity, as evidenced by a decrease in CaMKIIα activity (33). Under the microscope, these non-synaptic aggregates appear as puncta distributed throughout the cell soma and processes and are therefore easily detected in fixed or live cells. The aggregation of GFP-CaMKIIα is therefore a convenient biomarker for neuronal dyshomeostasis and an early correlate of neuronal demise.
In order to induce a rise in intracellular calcium by physiologically relevant means, we used glutamate treatment. We first constructed a dose-response curve in which we determined that neurons depleted of p600 by RNAi are more susceptible to undergoing GFP-CaMKIIα aggregation (Fig. 2A). Based on this finding, we selected a dose of 30 μm Glu, 3 μm Gly (3 min), to which control RNAi-transfected neurons were generally unresponsive by the metric of GFP-CaMKIIα aggregation. Neurons depleted of p600 by RNAi are, however, ∼2.5-fold more likely to exhibit CaMKIIα aggregation than neurons transfected with control RNAi following treatment with 30 μm Glu, 3 μm Gly (control RNAi versus p600 RNAi 1: 100 ± 24 and 239 ± 65%, t8 = 5.04, p = 0.0010; control RNAi versus p600 RNAi 2: 100 ± 16 and 330 ± 67%, t10 = 3.34, p = 0.020; Fig. 2, B and C). Two p600 RNAs targeting different sequences of p600 mRNA generated the same result, thereby providing specificity to the observed phenotype.
FIGURE 2.

Neurons depleted of p600 by RNAi undergo a rapid dyshomeostasis characterized by the aggregation of CaMKIIα and fragmentation of the ER. A, in control neurons, treatment with glutamate (supplemented with glycine, Glu/Gly, 10:1) for 3 min generates a sigmoid dose-response curve with a EC50 of 50–60 μm. Depletion of p600 by RNAi shifts this dose-response curve leftward, indicating greater response for the same dose of glutamate. An average of 500 neurons were scored in total for each condition (control RNAi, n = 18; p600 RNAi, n = 9). B, neurons transfected with either one of the two shRNA constructs targeting p600 were found to be more susceptible to Glu/Gly-induced dyshomeostasis, as indicated by the 2–3-fold increase in CaMKIIα aggregation. n = 5 for control/p600 RNAi 1; n = 6 for control/p600 RNAi 2; more than 500 neurons were analyzed per condition. C, representative confocal pictures of control RNAi-treated and p600 RNAi-treated primary hippocampal neurons (DIV10) transfected with GFP-CaMKIIα following treatment with 30 μm Glu, 3 μm Gly for 3 min. Bar, 20 μm. D, representative confocal pictures of ER morphology visualized with an ER-targeted mCherry protein in control RNAi/GFP-CaMKIIα/mCherry-ER and p600 RNAi/GFP-CaMKIIα/mCherry-ER transfected neurons following treatment with 50 μm Glu, 5 μm Gly for 3 min. The RNAi/GFP-CaMKIIα/mCherry-ER ratio for transfection is 4:1:1. Bar, 10 μm. E, within single neurons, the degree of CaMKIIα aggregation correlates strongly with the degree of fragmentation of the ER (r(17) = 0.89, p < 0.0001). b.b, bimodal bias (see “Materials and Methods”). Error bars, S.E.
Based on the role of p600 in microtubule stabilization (15), we first tested whether stabilization of microtubules with 100 nm paclitaxel could rescue p600 depletion-induced GFP-CaMKIIα aggregation. Intriguingly, paclitaxel was found to have no effect (Table 1). This result indicates that upon glutamate treatment, p600 does not maintain neuronal homeostasis by stabilizing microtubules. Based on our previous finding that p600 associates with the ER (15), we hypothesized that p600 maintains ER structure in response to Ca2+ influx. To test this, we co-transfected neurons depleted of p600 by RNAi with GFP-CaMKIIα and an mCherry-tagged ER (mCherry-ER) construct. This allowed us to simultaneously visualize the aggregation of the kinase and the fragmentation of the ER reported in degenerating neurons (4, 5, 34, 35). Using a quantification of bimodal bias (see “Materials and Methods”) to quantify kinase aggregation and ER fragmentation, we found a strong positive correlation between these two pathocellular processes in glutamate-treated p600-depleted neurons (Fig. 2, D and E); cells with high levels of kinase aggregation also exhibit a high degree of ER fragmentation. This constitutes one of the early Ca2+-induced events observed in p600-depleted neurons and presages their demise.
TABLE 1.
GFP-CaMKIIα aggregation is mediated by a rise in intracellular Ca2+, primarily through NMDAR-mediated Ca2+ entry and via metabotropic ER Ca2+ release
| Compound + 30 μm Glu, 3 μm Glya | Percentage of neurons with aggregation ± S.E. (n) | t | p | Significanceb |
|---|---|---|---|---|
| Taxol (100 nm) | 97.9 ± 2.5 (6) | 0.692 | 0.50 | N.S. |
| Vehicle (0.1% DMSO) | 100 ± 1.7 (6) | |||
| EGTA (2 mm) | 46.0 ± 7.9 (5) | 5.37 | 0.0007 | *** |
| Vehicle (ddH2O) | 100 ± 6.2 (5) | |||
| 2-APV (100 μm) | 23.5 ± 5.5 (6) | 7.84 | <0.0001 | *** |
| Vehicle (ddH20) | 100 ± 8.0 (6) | |||
| MK801 (1 μm) | 4.61 ± 2.7 (6) | 7.05 | 0.0009 | *** |
| Vehicle (0.1% MeOH) | 100 ± 13 (6) | |||
| Nimodipine (50 nm) | 86.7 ± 5.6 (6) | 2.26 | 0.047 | * |
| Vehicle (10−2% DMSO) | 100 ± 1.7 (6) | |||
| CNQX (10 μm) | 106 ± 24 (6) | 0.230 | 0.82 | N.S. |
| Vehicle (10−4 % DMSO) | 100 ± 18 (6) | |||
| Dantrolene (10 μm) | 98.7 ± 2.9 (6) | 0.317 | 0.70 | N.S. |
| Vehicle (0.1% MeOH) | 100 ± 2.1 (6) | |||
| Ryanodine (50 μm) | 97.1 ± 2.6 (6) | 0.621 | 0.55 | N.S. |
| Vehicle (0.05% DMSO) | 100 ± 3.9 (6) | |||
| 2-APB (50 μm) | 43.8 ± 9.1 (6) | 4.18 | 0.0019 | ** |
| Vehicle (0.05% DMSO) | 100 ± 9.9 (6) | |||
| KN62 (1 μm) | 101 ± 6.0 (6) | 0.148 | 0.89 | N.S. |
| Vehicle (0.1% DMSO) | 100 ± 1.7 (6) | |||
| KN93 (1 μm) | 98.7 ± 3.1 (6) | 0.890 | 0.40 | N.S. |
| Vehicle (0.1% MeOH) | 100 ± 2.1 (6) |
a Compounds that were effective in preventing GFP-CaMKIIα aggregation are shown in boldface type.
b *, p < 0.05; **, p < 0.01; ***, p < 0.001; N.S., not significant (p > 0.05).
Analysis of GFP-CaMKIIα aggregation also provided a means of rapidly assessing the manner of involvement of Ca2+ entry while avoiding the long term effects of pharmacological treatments. To this end, primary hippocampal neurons transfected with control or p600 RNAi 1 were treated with the Ca2+ chelator EGTA or a vehicle prior to treatment with Glu/Gly for 3 min and scored for CaMKIIα aggregation after a 2-min rinse. Chelation of extracellular Ca2+ with EGTA significantly rescued CaMKIIα aggregation in these cells (Table 1). These results revealed that entry of extracellular Ca2+ renders p600-depleted cells vulnerable to Ca2+-induced dyshomeostasis.
Glu/Gly treatment triggers Ca2+ influx through NMDA receptors and Ca2+-permeable AMPA receptors. To determine which of these receptors are involved in the Ca2+-induced dyshomeostasis phenotype, we treated p600-depleted cells with the NMDAR inhibitors 2-APV and MK-801 or the AMPA/kainate inhibitor CNQX prior to 3-min Glu/Gly treatment and then assessed CaMKIIα aggregation after a 2-min rinse. We found that 100 μm 2-APV rescued kinase aggregation by 80%, whereas 1 μm MK-801 almost completely abolished the formation of the aggregates (Table 1). Blockade of L-type VDCCs with 50 nm nimodipine also had a minimal yet significant effect in blocking the formation of the aggregates. In contrast, blockade of the AMPA and kainate receptors with 10 μm CNQX had no effect (Table 1). These results revealed that NMDA receptors are the primary source of Ca2+ influx in p600-depleted neurons.
The fragmentation of the ER in glutamate-treated and p600-depleted cells (Fig. 2, D and E) suggests that Ca2+ release from the ER contributes to the observed response. ER Ca2+ levels are controlled by Ca2+ release channels, such as ryanodine receptors and IP3 receptors, that have been shown to protect against excitotoxicity when blocked (4). No difference in CaMKIIα aggregation was observed between p600-depleted neurons treated with vehicles or the ryanodine receptor inhibitors dantrolene or ryanodine (10 and 50 μm, respectively; Table 1). However, inhibition of IP3 receptor with 50 μm 2-APV reduces the number of neurons with GFP-CaMKIIα aggregation by over 50%. These results indicate that IP3 receptors, but not ryanodine receptors, contribute to neuronal CaMKIIα aggregation caused by p600 depletion. Finally, to test whether CaM-dependent activation of CaMKIIα is critical for p600 RNAi-induced dyshomeostasis, we treated the neurons with 1 μm KN-62 or 1 μm KN-93, two CaMKIIα inhibitors that prevent the binding of CaM to CaMKIIα. Interestingly, these drugs had no effect on CaMKIIα aggregation (Table 1). These results show that dyshomeostasis caused by p600 depletion does not involve CaM-dependent activation of CaMKIIα. In sum, these experiments using GFP-CaMKIIα aggregation as an early marker for Ca2+-induced dyshomeostasis highlighted the involvement of Ca2+, NMDA receptors, and IP3 receptors in a pathway initiated by the loss of p600 (Table 1).
p600 and CaM-activated CaMKIIα Associate in a Ca2+-dependent Manner
Degeneration in the absence of p600 requires Ca2+ influx through NMDA receptors, which signal through CaM and CaMKIIα. We have also previously published in vitro data showing that p600 has a Ca2+-dependent association with the Ca2+ sensor CaM (36), which in turn has been shown to associate with CaMKIIα in a Ca2+-dependent manner. We thus hypothesized that p600 physiologically associates with CaM and CaMKIIα in the presence of intracellular Ca2+. First, the distribution of p600, CaM, and CaMKIIα was assessed in mouse brains and primary mature hippocampal neurons. In a subcellular fractionation of adult brain, p600 predominantly co-purified with cytosolic pools of CaM and CaMKIIα (S2) (Fig. 3A). The synaptic marker PSD-95 is used as a control, enriched in the crude synaptosomal fraction (P2) but not present in the cytosolic fraction. By immunofluorescent confocal microscopy, p600 co-localized with CaM and activated phosphothreonine 286-CaMKIIα in cell bodies and neuronal processes of primary hippocampal neurons (Fig. 3B). These first two panels confirm that p600, CaM, and CaMKIIα co-localize in neurons, a prerequisite for a direct and physiologically relevant interaction. Next, we found that p600 co-immunoprecipitated with CaM and CaMKIIα from adult brain lysates (Fig. 3C). The interactions between p600 and CaM and between p600 and CaMKIIα in immunoprecipitates from brain tissues were further strengthened by the exogenous application of 1 mm CaCl2 (data not shown). These results suggested that the increase in intracellular [Ca2+] triggered by neuronal activity may enhance the interaction between p600 and activated CaMKIIα. To test this hypothesis, hippocampal neurons were treated with 100 μm Glu, 10 μm Gly, and co-immunoprecipitations using p600 antibodies were performed on soluble lysates derived from these cells. As shown in Fig. 3D, a 2-fold increase in association between p600 and CaMKIIα was detected in glutamate-challenged neurons, whereas total levels of p600 and CaMKIIα in the soluble fraction remained unaffected (−Glu/Gly: 7.9% ± 0.4%; +Glu/Gly: 16.9% ± 2.2%; n = 6; **, p < 0.01 by two-tailed Student's t test). Taken together, these results indicate that the association between p600 and Ca2+/CaM-activated CaMKIIα is regulated physiologically by neuronal activity.
FIGURE 3.

p600, CaMKII, and CaM form a glutamate-potentiated complex in hippocampal neurons. A, p600 co-fractionates with CaM and CaMKIIα in significant amounts in the cytosolic fraction (S2) but to a far lesser extent in the synaptic compartment (P2) of adult brain. Input, unprocessed homogenate. The molecular masses (kDa) of the ladder are indicated on the left. B, confocal images depicting the co-localization of p600 and CaM and phospho-Thr286 active CaMKIIα in neuronal processes and the cell body of primary rat hippocampal neurons (DIV10). Bar, 20 μm. C, CaM and CaMKIIα co-immunoprecipitate with p600 in the adult mouse brain. CaM and CaMKIIα antibodies co-immunoprecipitate their cognate ligands (CaM, CaMKIIα, and the Cdk5 co-activator p35, which has been shown to associate with CaMKIIα (23)). Conditions with beads only (No Ab) or with species-isotypic GST antibodies were used as negative controls. D, primary hippocampal neurons at a density of 10 million/10-cm dish were treated with 100 μm Glu, 10 μm Gly, and co-immunoprecipitations using p600 antibodies were performed. A 2-fold increase in association between p600 and CaMKIIα was detected in neurons pretreated with Glu/Gly, whereas total levels of p600 and CaMKIIα remained unaffected. **, p (T < t) two-tailed < 0.01. Ab, antibody; IP, immunoprecipitation. Error bars, S.E.
p600 Interacts Directly with CaM
Previously, we found that p600 interacts directly with CaM protein in a Ca2+-dependent manner (36). These data are consistent with the report of isolation of CALO/pushover, a Drosophila counterpart of p600, in a screen for CaM-binding proteins (37). Having established a physiological Ca2+-dependent interaction between p600 and CaM, we hypothesized that disruption of the p600/CaM interaction would lead to neuronal dyshomeostasis and death. First, to map the CaM-interacting domain of p600, we generated various p600 fragments and found that the fragment containing residues 4293–4534 interacts with CaM (data not shown). In light of the CaM binding sequence prediction algorithm available at the CaM Target Database (38), various fragments covering the C-terminal region of p600 (residues 3029–4073, 3362–3761, 3754–4158, 4048–4367, 4152–4548, 4545–4548, 4545–4868, and 4861–5183) were expressed as FLAG-tagged proteins in SF9 cells using a baculovirus expression system. In far-Western blotting, p600 fragments containing residues 3754–4158 and 4048–4367 directly bound to CaM. Two constructs expressing either the region of overlap defined by these experiments (residues 4058–4158) or a subset of this region (residues 4076–4112) were then expressed as glutathione S-transferase (GST) fusions in Escherichia coli. Far-Western blotting showed that both of the p600 fusion proteins interact with CaM, whereas GST alone does not (Fig. 4A).
FIGURE 4.
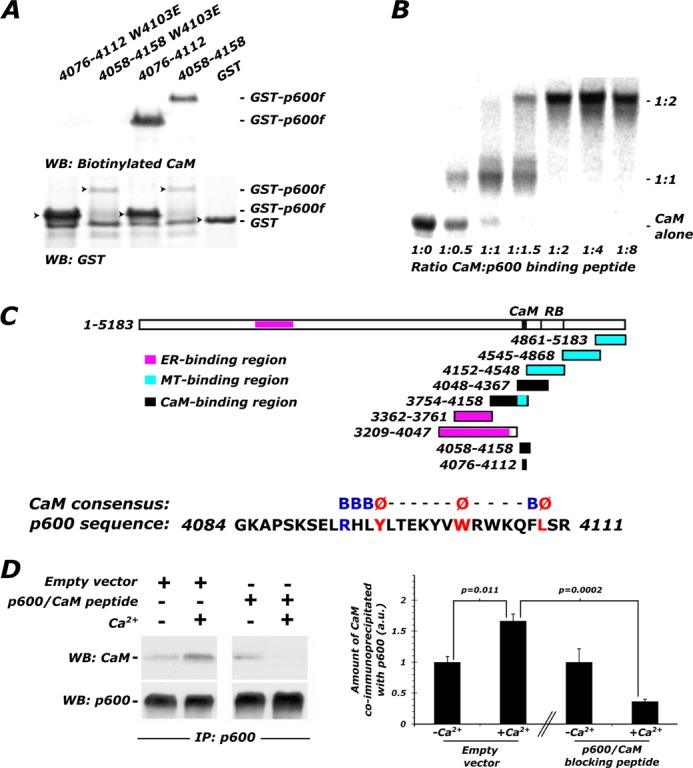
p600 directly binds to CaM with atypical stoichiometry via a C-terminal binding motif. A, overlay assays for mapping of the CaM-binding domain on p600. Biotinylated CaM bound directly to GST-tagged p600 fragments (GST-p600f) containing residues 4058–4158 and 4076–4112 but not to GST alone. To test if residues 4089–4111 define the p600 CaM-binding motif, the tryptophan residue at 4103 was mutated to glutamic acid in GST constructs containing residues 4058–4158 and 4076–4112 (W4103E). Overlay assays showed that the W4103E mutation in both constructs completely abolishes the CaM binding activity. The ER and microtubule-binding fragments of p600 are depicted for reference. The consensus sequence and conserved residues of the CaM-binding motif are illustrated on the atypical CaM-binding motif found in p600 (B). The native gel shift titration assay method was used to define the stoichiometric ratio between CaM and p600. The p600 peptide SKSELRHLYLTEKYVWRWKQFLSRRG (p600P (residues 4088–4113)) was generated for the gel shift titration assay. The CaM-p600P complex was saturated at an atypical stoichiometric ratio of 1:2. An additional intermediate complex formed at a ratio of 1:1 was also detected. C, residues 4086–4113 of p600 (APSKSELRHLYLTEKYVWRWKQFLSRRG), containing the CaM-binding consensus sequence, p600P, is localized to the C terminus of p600. Regions containing previously defined functions are shown. D, p600P was transfected into CAD cells in order to confirm expression. Immunoprecipitation from CAD extracts using p600 antibodies effectively co-immunoprecipitates CaM, more so when 2 mm Ca2+ (Ca2+ +; lane 2) is included in the lysis buffer as opposed to 1 mm EDTA (Ca2+ −; lane 2). In the presence of the p600P motif and Ca2+, the interaction between endogenous p600 and CaM is reduced by 78% (lane 4). WB, Western blot; IP, immunoprecipitation. Error bars, S.E.
Within the p600 segment of 4076–4112, the CaM Target Database (38) identified a strong hit at residues 4089–4111 with moderate to high scores. This region does not encode any of the canonical CaM binding motifs that were well characterized previously (39) but contains conserved hydrophobic residues, such as Trp-4103. To test whether the segment comprising residues 4089–4111 constitutes a functional p600 CaM-binding site, Trp-4103 was mutated to glutamate in GST constructs containing residues 4058–4158 and 4076–4112. Far-Western blotting showed that the mutation of W4103E in both constructs almost completely abolished the CaM binding activity (Fig. 4A), demonstrating that Trp-4103 is a key hydrophobic anchoring residue for CaM binding.
To further understand the nature of the interaction between p600 and CaM, we generated the peptide SKSELRHLYLTEKYVWRWKQFLSRRG (p600P) that encompasses residues 4089–4111 of p600. Using the native gel shift titration assay method, we found that the complex was saturated at an atypical stoichiometric ratio of 1:2 CaM/p600P and that an intermediate 1:1 complex was also observed (Fig. 4B). These data suggest that CaM could recruit two p600 molecules by virtue of two target binding sites in CaM. Taken together, our in vitro results revealed a direct interaction between p600 and CaM through residues 4088–4111 on p600 and indicate an atypical p600/CaM interaction.
A Direct p600/CaM Interaction Promotes Neuronal Survival
Finally, we tested whether the interaction between p600 and CaM is required for neuronal survival. In order to verify the expression of p600P, the peptide was transfected into CAD cells, a CNS-derived cell line that expresses neuronal markers (40) and shows robust expression of CaM and p600. Most importantly, these cells have sufficient transfection efficiency to assess the inhibition of the p600/CaM interaction in a lysate. They therefore served as a useful proxy for sparsely transfected hippocampal neurons. Immunoprecipitation of endogenous p600 co-precipitated CaM in the absence of Ca2+ (lysis buffer supplemented with 1 mm EDTA). The addition of 2 mm Ca2+ enhanced the amount of immunoprecipitated CaM by 66% (Fig. 4D; n = 3, t4 = 4.52, p = 0.011 by two-tailed Student's t test), in keeping with our previous results (Fig. 3). By contrast, in the presence of p600P, there was a marked 78% reduction in the amount of immunoprecipitated CaM (Fig. 4D; n = 3, t4 = 13.9, p = 0.0002 by two-tailed Student's t test). This finding confirmed p600P expression and validated its use as an inhibitor of the p600/CaM interaction.
We then co-transfected DIV10 neurons with p600P, an empty vector, or the mutant p600 peptide W4103E (p600P W4103E, which does not bind CaM) and GFP-CaMKIIα. Upon treatment with exogenous glutamate for 3 min (30 μm Glu, 3 μm Gly), neurons in which the p600/CaM interaction is disrupted with p600P were 35 and 54% more likely to display GFP-CaMKIIα aggregation (Fig. 5A) when compared with the empty vector and p600P W4103E controls, respectively (U = 27, p = 0.016 and U = 17, p = 0.0015 by two-tailed Mann-Whitney U test). Predictably, transfection with the mutant p600P W4103E had no effect when compared with the empty vector (U = 48, p = 0.17). In accordance with the Ca2+ dependence of the p600/CaM interaction, in the absence of exogenous glutamate, there were no significant differences in the number of neurons in which CaMKIIα is aggregated (Fig. 5A; for all comparisons, p ≥ 0.37). 12 coverslips totaling an average of 593 cells were analyzed for each condition. From these results, we conclude that disruption of the p600/CaM interaction increases the susceptibility of neurons to undergo a Ca2+-induced dyshomeostasis response symptomized by aggregation of GFP-CaMKIIα.
In order to determine if the p600/CaM interaction is required for cell survival, we transfected DIV10 neurons with p600P, an empty vector, or p600P W4103E and an enhanced GFP construct; treated with 30 μm Glu, 3 μm Gly for 3 min; and scored for cell death 24 h after treatment. The expression of p600P, but not p600P W4103E, reduced cell survival by 43%, as indicated by the significantly lower number of remaining GFP-positive cells (p600P versus empty vector control: t14 = 3.39, p = 0.0044; p600P versus p600P W4103E: t14 = 2.88, p = 0.012; Fig. 5B). The same experiment was repeated using a sham 0 μm Glu/Gly wash but yielded a similar 44% decrease in p600P-transfected neuronal survival (p600P versus empty vector control: t14 = 2.28, p = 0.039; p600P versus p600P W4103E: t14 = 2.21, p = 0.048; Fig. 5B). This suggested that over the course of 24 h, the cumulative dose of endogenous glutamate is vastly in excess of the dose of exogenous glutamate, rendering it insignificant. Indeed, regardless of transfection group, our mild glutamate treatment did not create a significant difference in neuronal survival within transfection groups after 24 h (p > 0.15 for all comparisons). This finding was confirmed by repeating the 0 μm Glu/Gly treatment for 3 min and then supplementing culture medium with 100 μm 2-APV, previously shown to block GFP-CaMKIIα aggregation caused by p600 loss of function (Table 1). Predictably, in the presence of 2-APV, the differences in percentage of survival between cultures transfected with p600P, p600P W4103E, and an empty vector control were no longer statistically significant (p600P versus empty vector control: t14 = 0.627, p = 0.54; p600P versus p600P W4103E: t14 = 1.27, p = 0.23; Fig. 5B).
Based on the association of p600 with the ER and its role in maintaining ER integrity, we then speculated that p600 may maintain Ca2+ homeostasis and neuronal survival by preventing ER stress. However, we found that in neurons in which the p600/CaM interaction is blocked with p600P, 24-h treatment with the chemical chaperone and ER stress inhibitor 4-phenylbutyrate (4-PBA) does not improve neuronal survival rates (experimental protocol as in Fig. 5, where p600P = 100 ± 26%, p600P + 4-PBA = 87.5 ± 13%; t10 = 0.39, p = 0.72). We then questioned whether the glutamate-dependent aggregation of GFP-CaMKIIα upon depletion of p600 by RNAi was a result of activation of ER stress pathways. However, 4-PBA was again ineffective at preventing the aggregation of GFP-CaMKIIα (experimental protocol as in Table 1, where p600 RNAi + 0.002% DMSO = 100 ± 10%, p600 RNAi + 1 mm 4-PBA = 101 ± 17%; t8 = 0.03, p = 0.97).
Finally, although we did not find evidence of apoptosis following depletion of p600 by RNAi, we tested the possibility that over the course of 24 h, neurons were dying of delayed apoptosis. We therefore tried to rescue neuronal death in neurons transfected with p600/CaM peptide with a 24-h treatment with the pancaspase inhibitor benzyloxycarbonyl-VAD-fluoromethyl ketone (20 μm). This inhibitor, however, had no significant effect (experimental protocol as in Fig. 5, where 0.05% DMSO vehicle = 100 ± 17%, benzyloxycarbonyl-VAD-fluoromethyl ketone = 80.7 ± 8.5%; t10 = 0.88, p = 0.40), confirming our earlier finding that neurons wherein p600 function is disrupted do not die by apoptotic pathways. In summary, these results indicate that physiological and non-pathological levels of glutamate-induced Ca2+ influx cause a direct interaction between p600 and CaM that is important to maintain neuronal homeostasis and to promote neuronal survival.
DISCUSSION
Although chronic neurodegenerative diseases are thought of as slow progressive disorders, the final steps resulting in cell death are remarkably similar to those described in acute neuronal degeneration (1–7). In both cases, influx of Ca2+ and disruption of the cytoskeleton compromise neuronal integrity, yet the Ca2+ signaling roles of cytoskeletal proteins remain largely unexplored. On the basis of the mechanistic similarities between chronic and acute neurodegeneration and the dual CaM-binding and microtubule-stabilizing roles of p600, we explored p600 as an interface for these roles in neuronal pathology. However, p600 does not promote neuronal survival in response to glutamate-induced Ca2+ influx through its microtubule-stabilizing function, as evidenced by the ineffectiveness of paclitaxel in preventing Ca2+ dyshomeostasis after p600 depletion (Table 1). The neuronal death observed upon p600 depletion (Fig. 1E) is preceded, within 5 min of glutamate treatment, by a rapid Ca2+-induced dyshomeostasis characterized by rapid aggregation of GFP-CaMKIIα and fragmentation of the ER. Interestingly, the close temporal link between Ca2+-induced CaMKIIα aggregation and fragmentation of the ER is also seen in smooth muscle cells with the δ isoform of CaMKII, where the aggregation is also thought to be indicative of Ca2+ overload (41). Of note, the observed aggregation of CaMKIIα is distinct from the localization of CaMKII to microtubules upon local NMDA stimulation, which can be blocked by KN-93 (42). It is also distinct from the other pathological localization of CaMKII to the postsynaptic density, which can be blocked by KN-62 (43) (reviewed in Ref. 44). This finding, following previous reports of CaMKII aggregation in pathological circumstances (26–32), lends support to our analysis of CaMKIIα aggregation as a biomarker for early Ca2+-induced neuronal dyshomeostasis.
Using pharmacological and molecular approaches, we delineate a path whereby glutamate stimulation of NMDARs and activation of ER IP3 receptors (via presumptive metabotropic glutamate receptor activity) allow Ca2+ entry, which causes p600, CaM, and CaMKIIα to form a complex mediated by a direct p600/CaM interaction (summarized in the left panel of Fig. 6). The p600/CaM interaction was found to be atypical in its stoichiometry, and p600P has no primary sequence homology elsewhere in the proteome. Hence, p600P is probably extremely specific to the p600/CaM interaction and thereby useful as a tool to specifically disrupt the p600/CaM interaction. It is worthwhile to note that Fig. 4D shows a basal interaction (lane 1) between p600 and CaM, which is enhanced in the presence of Ca2+ (lane 2) and unaffected by the blocking peptide (lane 3) but disrupted by both together (lane 4). This observation presaged our conclusion that the effects deriving from the p600/CaM interaction are Ca2+-dependent.
FIGURE 6.
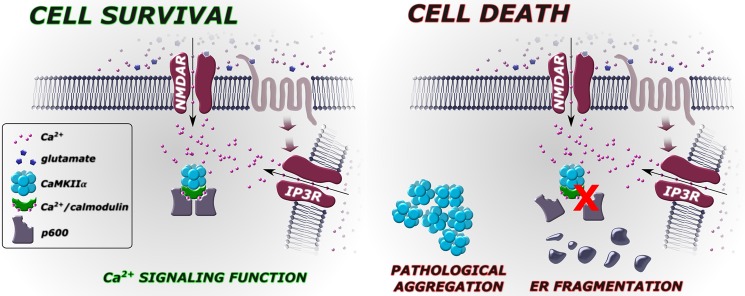
p600 prevents neuronal death in response to glutamate-induced cytosolic Ca2+ elevation. Left, Ca2+ influx via NMDARs or presumptive mGluR-activated endoplasmic reticulum Ca2+ efflux via IP3 receptors causes p600 to complex with CaM and CaMKIIα, promoting neuronal survival. Right, disruption of the p600/CaM interaction or depletion of p600 by RNAi causes a rapid aggregation of CaMKIIα, fragmentation of the endoplasmic reticulum, and ultimately neuronal death.
Fig. 5 shows that the direct p600/CaM binding is required to prevent the GFP-CaMKIIα aggregation (A) for survival (B), indicating that in the normal neuron, p600 promotes survival by a Ca2+-dependent binding to calmodulin. NMDAR blockade prevented both GFP-CaMKIIα (Table 1) and cell demise under ambient activity (Fig. 5B), further indicating mutual cause. Importantly, the unexpected finding that the brief 3-min dose of glutamate used in the analyses of rapid perturbations to neuronal homeostasis (Figs. 2 and 5A) experiments had no detectable effect 24 h later in cell death experiments (Fig. 5B, left and center) was an important validation of the experimental design; a 30 μm dose of glutamate, although required to show a difference in CaMKIIα aggregation minutes later, was insignificant compared with 24 h of ambient activity in these cultures. The rescue of cell death by blockade of ambient NMDAR Ca2+ influx (Fig. 5B, right) further proves that p600 promotes survival at ambient (subpathological) levels of neuronal activity. The lack of observed differences in CaMKIIα aggregation (Fig. 5A) or base-line CaMKIIα phosphorylation (Fig. 1C) without 24-h incubation probably reflects a less complete knockdown and a lower summation of spontaneous activity.
This study opens a line of questioning pertaining to the short term significance of the p600/CaM interaction; although this interaction is certainly required for survival, it is unknown what events transpire to promote survival or to cause death in the absence of the p600/CaM interaction. p600 appears not to counteract the microtubule-destabilizing effects of CaM because paclitaxel did not prevent CaMKIIα aggregation after glutamate treatment. That said, our data reported here suggest that p600 promotes survival entirely by transducing Ca2+ signals. Importantly, the base line of CaM·CaMKIIα signaling is not altered in the absence of p600 because no difference was observed in the levels of phosphothreonine 286-CaMKIIα (activated CaMKIIα) between control RNAi and p600-depleted RNAi neurons (Fig. 1C). The significance of the p600/CaMKIIα interaction (Fig. 3) remains to be determined, although p600 is one of many cytoskeletal proteins with which CaMKII associates (see Ref. 45 for a review). The inability of treatment with KN-62 and KN-93, inhibitors of the CaM/CaMKII interaction, to rescue neurons from Ca2+-induced dyshomeostasis also demonstrates that p600 promotes survival by pathways that do not involve classical CaM-dependent CaMKIIα activation. Neuronal survival seems to rather be dependent on the Ca2+-dependent p600/CaM interaction. A probable explanation is that the p600/CaM interaction forms a feedback mechanism that inhibits further Ca2+ influx following abnormally high bouts of glutamatergic stimulation, although this possibility remains untested. The relationship between the findings that p600 is an ER- and microtubule-associated protein (15) and the Ca2+-dependent function reported here is similarly unclear, and it remains a possibility that there are multiple subcellular pools of p600 with distinct functions.
Perhaps the most conspicuous question remaining is the precise relationship between the rapid indicators of Ca2+ dyshomeostasis, namely the fragmentation of the ER and aggregation of CaMKIIα, and neuronal demise hours later. Although these analyses provide enticing detail of mechanisms of Ca2+ dyshomeostasis, we cannot conclude that such mechanisms are more than mere correlates of the neuronal demise we separately quantify. Accordingly, the conclusions drawn by this study are conservative and derived entirely from direct assessment of neuronal demise (especially in Figs. 1E and 5B), whereas our analysis of Ca2+-induced neuronal dyshomeostasis (Fig. 2 and Table 1) is included in order to provide the reader with mechanistic insight. We must also make clear that the aggregation of CaMKIIα and the fragmentation of the ER are used here as markers of cellular dyshomeostasis rather than markers of cell death.
The question of the precise manner of neuronal demise is likewise still open. We used only mild concentrations of exogenous glutamate and even observed NMDAR-dependent death under ambient culture treatment, making necrotic neuronal death unlikely (46). Thus, although we observed necrotic cells in our cultures, depletion of p600 by RNAi did not increase their frequency (Fig. 1E). We also found no apoptotic indicators following depletion of p600 by RNAi (Fig. 1E) and similarly no delayed apoptosis over the 24 h after which we saw neuronal death when we blocked the p600/CaM interaction. Because p600 is an ER-associated protein (15), ER stress pathways may be activated (47, 48). We can, however, rule out ER stress-induced apoptosis (49) based on the failure of the pancaspase inhibitor benzyloxycarbonyl-VAD-fluoromethyl ketone to rescue neuronal demise (see “Results”). Direct inhibition of ER stress with the chemical chaperone and ER stress inhibitor 4-PBA was similarly ineffective at preventing both neuronal demise and the aggregation of CaMKIIα. These data together absolve ER stress as a causal factor in the neuronal demise following disruption of p600 function. However, a closely related possibility is that p600 is required for autophagy and/or protein degradation-dependent survival. Broadly speaking, neurons can die by necrosis, apoptosis, or autophagy. Because we were unable to detect signs of the former two mechanisms, we are bound to suspect the latter. This suspicion is well grounded in the published role of p600 in the N-end rule pathway (50) and especially in new work (24) showing that p600 knock-out mice die during embryogenesis, with yolk sac development defects characterized by aberrant autophagy (elaborated in Ref. 51). Another recent analysis of a second distinct p600 knock-out mouse strain found severe defects in the embryonic heart but also found no indications of apoptosis (25). Unfortunately, analysis of the autophagic process or defects therein is complex and well beyond the scope of this study. In fact, the role of calcium in the induction of autophagy, or even the role of autophagy in cell death, are not completely understood in any system (52, 53). However due to its established role in protein degradation pathways and its role in Ca2+ signaling presented in this study, p600 is an excellent candidate molecule for a link between Ca2+ signaling, autophagy, and neurodegeneration.
Although excessive cytosolic Ca2+ normally bodes ill for hippocampal neurons, p600 is in a unique position to promote survival via a Ca2+/CaM-dependent interaction. The prevalence of p600, CaM, and CaMKIIα in neurons susceptible to excitotoxicity further suggests a role for p600 in the array of acute and chronic degenerative neuropathologies in which Ca2+ dysregulation in neurons plays a central role.
Acknowledgment
We are grateful to Dr. H. Le Thi for technical help.
This work was authored, in whole or in part, by National Institutes of Health staff. This work was supported by an operating grant from the Canadian Institutes of Health Research (CIHR), the Human Frontier Science Program Organization (HFSPO), and the Brenda Strafford Chair Foundation in Alzheimer's disease (to M. D. N.).
- ER
- endoplasmic reticulum
- IP3
- inositol 1,4,5-trisphosphate
- DIV10
- day in vitro 10
- CaM
- calmodulin
- CaMKIIα
- calmodulin-dependent protein kinase IIα
- 4-PBA
- 4-phenylbutyrate
- NMDAR
- NMDA receptor
- 2-APV
- DL-2-amino-5-phosphonovaleric acid
- CNQX
- 6-cyano-7-nitroquinoxaline-2,3-dione
- 2-APB
- 2-aminoethyl diphenylborinate.
REFERENCES
- 1. LaFerla F. M. (2002) Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat. Rev. Neurosci. 3, 862–872 [DOI] [PubMed] [Google Scholar]
- 2. Mattson M. P. (2003) Excitotoxic and excitoprotective mechanisms. Abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromol. Med. 3, 65–94 [DOI] [PubMed] [Google Scholar]
- 3. Mattson M. P., Chan S. L. (2003) Neuronal and glial calcium signaling in Alzheimer's disease. Cell Calcium 34, 385–397 [DOI] [PubMed] [Google Scholar]
- 4. Verkhratsky A. (2004) Endoplasmic reticulum calcium signaling in nerve cells. Biol. Res. 37, 693–699 [DOI] [PubMed] [Google Scholar]
- 5. Verkhratsky A. (2005) Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol. Rev. 85, 201–279 [DOI] [PubMed] [Google Scholar]
- 6. Mattson M. P. (2007) Calcium and neurodegeneration. Aging Cell 6, 337–350 [DOI] [PubMed] [Google Scholar]
- 7. Wojda U., Salinska E., Kuznicki J. (2008) Calcium ions in neuronal degeneration. IUBMB Life 60, 575–590 [DOI] [PubMed] [Google Scholar]
- 8. Trojanowski J. Q., Schmidt M. L., Shin R. W., Bramblett G. T., Rao D., Lee V. M. (1993) Altered Tau and neurofilament proteins in neuro-degenerative diseases. Diagnostic implications for Alzheimer's disease and Lewy body dementias. Brain Pathol. 3, 45–54 [DOI] [PubMed] [Google Scholar]
- 9. Ballatore C., Lee V. M., Trojanowski J. Q. (2007) Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 8, 663–672 [DOI] [PubMed] [Google Scholar]
- 10. Lee V., Goedert M., Trojanowski J. (2001) Neurodegenerative Tauopathies. Annu. Rev. Neurosci. 1121–1159 [DOI] [PubMed] [Google Scholar]
- 11. Weiss J. H., Hartley D. M., Koh J., Choi D. W. (1990) The calcium channel blocker nifedipine attenuates slow excitatory amino acid neurotoxicity. Science 247, 1474–1477 [DOI] [PubMed] [Google Scholar]
- 12. Waterman-Storer C. M., Salmon E. D. (1998) Endoplasmic reticulum membrane tubules are distributed by microtubules in living cells using three distinct mechanisms. Curr. Biol. 8, 798–806 [DOI] [PubMed] [Google Scholar]
- 13. Xu C., Bailly-Maitre B., Reed J. C. (2005) Endoplasmic reticulum stress. Cell life and death decisions. J. Clin. Invest. 115, 2656–2664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ogburn K. D., Figueiredo-Pereira M. E. (2006) Cytoskeleton/endoplasmic reticulum collapse induced by prostaglandin J2 parallels centrosomal deposition of ubiquitinated protein aggregates. J. Biol. Chem. 281, 23274–23284 [DOI] [PubMed] [Google Scholar]
- 15. Shim S. Y., Wang J., Asada N., Neumayer G., Tran H. C., Ishiguro K., Sanada K., Nakatani Y., Nguyen M. D. (2008) Protein 600 Is a microtubule/endoplasmic reticulum-associated protein in CNS neurons. J. Neurosci. 28, 3604–3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nguyen M. D., Shu T., Sanada K., Larivière R. C., Tseng H. C., Park S. K., Julien J. P., Tsai L. H. (2004) A NUDEL-dependent mechanism of neurofilament assembly regulates the integrity of CNS neurons. Nat. Cell Biol. 6, 595–608 [DOI] [PubMed] [Google Scholar]
- 17. Ribeiro C. M., McKay R. R., Hosoki E., Bird G. S., Putney J. W. (2000) Effects of elevated cytoplasmic calcium and protein kinase C on endoplasmic reticulum structure and function in HEK293 cells. Cell Calcium 27, 175–185 [DOI] [PubMed] [Google Scholar]
- 18. Tenneti L., Lipton S. A. (2000) Involvement of activated caspase-3-like proteases in N-methyl-d-aspartate-induced apoptosis in cerebrocortical neurons. J. Neurochem. 74, 134–142 [DOI] [PubMed] [Google Scholar]
- 19. Ott J. (1979) Detection of rare major genes in lipid levels. Hum. Genet. 51, 79–91 [DOI] [PubMed] [Google Scholar]
- 20. Nguyen M. D., D'Aigle T., Gowing G., Julien J. P., Rivest S. (2004) Exacerbation of motor neuron disease by chronic stimulation of innate immunity in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 24, 1340–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ikura M., Kay L. E., Bax A. (1990) A novel approach for sequential assignment of 1H, 13C, and 15N spectra of proteins. Heteronuclear triple-resonance three-dimensional NMR spectroscopy. Application to calmodulin. Biochemistry 29, 4659–4667 [DOI] [PubMed] [Google Scholar]
- 22. Yuan T., Vogel H. J. (1998) Calcium-calmodulin-induced dimerization of the carboxyl-terminal domain from petunia glutamate decarboxylase. A novel calmodulin-peptide interaction motif. J. Biol. Chem. 273, 30328–30335 [DOI] [PubMed] [Google Scholar]
- 23. Dhavan R., Greer P. L., Morabito M. A., Orlando L. R., Tsai L. H. (2002) The cyclin-dependent kinase 5 activators p35 and p39 interact with the α-subunit of Ca2+/calmodulin-dependent protein kinase II and α-actinin-1 in a calcium-dependent manner. J. Neurosci. 22, 7879–7891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tasaki T., Kim S. T., Zakrzewska A., Lee B. E., Kang M. J., Yoo Y. D., Cha-Molstad H. J., Hwang J., Soung N. K., Sung K. S., Kim S.-H., Nguyen M. D., Sun M., Yi E. C., Kim B. Y., Kwon Y. T. (2013) UBR box N-recognin-4 (UBR4), an N-recognin of the N-end rule pathway, and its role in yolk sac vascular development and autophagy. Proc. Natl. Acad. Sci. U.S.A. 110, 3800–3805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakaya T., Ishiguro K. I., Belzil C., Rietsch A. M., Yu Q., Mizuno S. I., Bronson R. T., Geng Y., Nguyen M. D., Akashi K., Sicinski P., Nakatani Y. (2013) p600 plays essential roles in fetal development. PLoS ONE 8, e66269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aronowski J., Grotta J. C., Waxham M. N. (1992) Ischemia-induced translocation of Ca2+/calmodulin-dependent protein kinase II. Potential role in neuronal damage. J. Neurochem. 58, 1743–1753 [DOI] [PubMed] [Google Scholar]
- 27. Taft W. C., Tennes-Rees K. A., Blair R. E., Clifton G. L., DeLorenzo R. J. (1988) Cerebral ischemia decreases endogenous calcium-dependent protein phosphorylation in gerbil brain. Brain Res. 447, 159–163 [DOI] [PubMed] [Google Scholar]
- 28. Kochhar A., Saitoh T., Zivin J. A. (1991) Spinal cord ischemia reduces calcium/calmodulin-dependent protein kinase activity. Brain Res. 542, 141–146 [DOI] [PubMed] [Google Scholar]
- 29. Dosemeci A., Reese T. S., Petersen J., Tao-Cheng J. H. (2000) A novel particulate form of Ca2+/calmodulin-dependent [correction of Ca2+/CaMKII-dependent] protein kinase II in neurons. J. Neurosci. 20, 3076–3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tao-Cheng J. H., Vinade L., Smith C., Winters C. A., Ward R., Brightman M. W., Reese T. S., Dosemeci A. (2001) Sustained elevation of calcium induces Ca2+/calmodulin-dependent protein kinase II clusters in hippocampal neurons. Neuroscience 106, 69–78 [DOI] [PubMed] [Google Scholar]
- 31. Tao-Cheng J. H., Vinade L., Pozzo-Miller L. D., Reese T. S., Dosemeci A. (2002) Calcium/calmodulin-dependent protein kinase II clusters in adult rat hippocampal slices. Neuroscience 115, 435–440 [DOI] [PubMed] [Google Scholar]
- 32. Hudmon A., Lebel E., Roy H., Sik A., Schulman H., Waxham M. N., De Koninck P. (2005) A mechanism for Ca2+/calmodulin-dependent protein kinase II clustering at synaptic and nonsynaptic sites based on self-association. J. Neurosci. 25, 6971–6983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rose J., Jin S. X., Craig A. M. (2009) Heterosynaptic molecular dynamics. Locally induced propagating synaptic accumulation of CaM kinase II. Neuron 61, 351–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mattson M. P., Gary D. S., Chan S. L., Duan W. (2001) Perturbed endoplasmic reticulum function, synaptic apoptosis and the pathogenesis of Alzheimer's disease. Biochem. Soc. Symp., 151–162 [DOI] [PubMed] [Google Scholar]
- 35. Verkhratsky A., Toescu E. C. (2003) Endoplasmic reticulum Ca2+ homeostasis and neuronal death. J. Cell Mol. Med. 7, 351–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nakatani Y., Konishi H., Vassilev A., Kurooka H., Ishiguro K., Sawada J., Ikura T., Korsmeyer S. J., Qin J., Herlitz A. M. (2005) p600, a unique protein required for membrane morphogenesis and cell survival. Proc. Natl. Acad. Sci. U.S.A. 102, 15093–15098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu X.-Z., Wes P. D., Chen H., Li H. S., Yu M., Morgan S., Liu Y., Montell C. (1998) Retinal targets for calmodulin include proteins implicated in synaptic transmission. J. Biol. Chem. 273, 31297–31307 [DOI] [PubMed] [Google Scholar]
- 38. Yap K. L., Kim J., Truong K., Sherman M., Yuan T., Ikura M. (2000) Calmodulin target database. J. Struct. Funct. Genom. 1, 8–14 [DOI] [PubMed] [Google Scholar]
- 39. Hoeflich K. P., Ikura M. (2002) Calmodulin in action. Diversity in target recognition and activation mechanisms. Cell 108, 739–742 [DOI] [PubMed] [Google Scholar]
- 40. Qi Y., Wang J. K., McMillian M., Chikaraishi D. M. (1997) Characterization of a CNS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. J. Neurosci. 17, 1217–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Van Riper D. A., Schworer C. M., Singer H. A. (2000) Ca2+-induced redistribution of Ca2+/calmodulin-dependent protein kinase II associated with an endoplasmic reticulum stress response in vascular smooth muscle. Mol. Cell. Biochem. 213, 83–92 [DOI] [PubMed] [Google Scholar]
- 42. Lemieux M., Labrecque S., Tardif C., Labrie-Dion É., Lebel É., De Koninck P. (2012) Translocation of CaMKII to dendritic microtubules supports the plasticity of local synapses. J. Cell Biol. 198, 1055–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Meng F., Guo J., Zhang Q., Song B., Zhang G. (2003) Autophosphorylated calcium/calmodulin-dependent protein kinase II α (CaMKIIα) reversibly targets to and phosphorylates N-methyl-d-aspartate receptor subunit 2B (NR2B) in cerebral ischemia and reperfusion in hippocampus of rats. Brain Res. 967, 161–169 [DOI] [PubMed] [Google Scholar]
- 44. Colbran R. J. (2004) Targeting of calcium/calmodulin-dependent protein kinase II. Biochem. J. 378, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Colbran R. J. (1992) Regulation and role of brain calcium/calmodulin-dependent protein kinase II. Neurochem. Int. 2 [DOI] [PubMed] [Google Scholar]
- 46. Bonfoco E., Krainc D., Ankarcrona M., Nicotera P., Lipton S. A. (1995) Apoptosis and necrosis. Two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc. Natl. Acad. Sci. U.S.A. 92, 7162–7166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang K., Kaufman R. J. (2006) The unfolded protein response. A stress signaling pathway critical for health and disease. Neurology 66, S102–S109 [DOI] [PubMed] [Google Scholar]
- 48. Shibata N., Kobayashi M. (2008) [The role for oxidative stress in neurodegenerative diseases]. Brain Nerve 60, 157–170 [PubMed] [Google Scholar]
- 49. Smith M. I., Deshmukh M. (2007) Endoplasmic reticulum stress-induced apoptosis requires bax for commitment and Apaf-1 for execution in primary neurons. Cell Death Differ. 14, 1011–1019 [DOI] [PubMed] [Google Scholar]
- 50. Tasaki T., Mulder L. C., Iwamatsu A., Lee M. J., Davydov I. V., Varshavsky A., Muesing M., Kwon Y. T. (2005) A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol. Cell. Biol. 25, 7120–7136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim S. T., Tasaki T., Zakrzewska A., Yoo Y. D., Sa Sung K., Kim B. Y., Cha-Molstad H., Hwang J., Kim K. A., Kwon Y. T. (2013) The N-end rule proteolytic system in autophagy. Autophagy [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rami A. (2009) Autophagy in Neurodegeneration. Firefighter and/or incendiarist? Neuropathol. Appl. Neurobiol. 35, 449–461 [DOI] [PubMed] [Google Scholar]
- 53. Decuypere J.-P., Bultynck G., Parys J. B. (2011) A dual role for Ca2+ in autophagy regulation. Cell Calcium 50, 242–250 [DOI] [PubMed] [Google Scholar]