

Background: The peripheral stator stalk of Escherichia coli ATP synthase contains two b subunits.
Results: Using disulfide bond formation, one b subunit was cross-linked to a, α, and δ and the other to β.
Conclusion: The b subunits adopt distinct positions within the stator to generate stability.
Significance: The different positions imply different roles in counteracting the torque generated by the rotor.
Keywords: ATP Synthase, Disulfide, Escherichia coli, Membrane Proteins, Protein Cross-linking, F-ATPase, Peripheral Stator Stalk
Abstract
FOF1 ATP synthases are rotary nanomotors that couple proton translocation across biological membranes to the synthesis/hydrolysis of ATP. During catalysis, the peripheral stalk, composed of two b subunits and subunit δ in Escherichia coli, counteracts the torque generated by the rotation of the central stalk. Here we characterize individual interactions of the b subunits within the stator by use of monoclonal antibodies and nearest neighbor analyses via intersubunit disulfide bond formation. Antibody binding studies revealed that the C-terminal region of one of the two b subunits is principally involved in the binding of subunit δ, whereas the other one is accessible to antibody binding without impact on the function of FOF1. Individually substituted cysteine pairs suitable for disulfide cross-linking between the b subunits and the other stator subunits (b-α, b-β, b-δ, and b-a) were screened and combined with each other to discriminate between the two b subunits (i.e. bI and bII). The results show the b dimer to be located at a non-catalytic α/β cleft, with bI close to subunit α, whereas bII is proximal to subunit β. Furthermore, bI can be linked to subunit δ as well as to subunit a. Among the subcomplexes formed were a-bI-α, bII-β, α-bI-bII-β, and a-bI-δ. Taken together, the data obtained define the different positions of the two b subunits at a non-catalytic interface and imply that each b subunit has a different role in generating stability within the stator. We suggest that bI is functionally related to the single b subunit present in mitochondrial ATP synthase.
Introduction
FOF1 ATP synthases utilize the energy of an electrochemical ion gradient (ΔμH+ or ΔμNa+) across biological membranes to catalyze the synthesis of ATP from ADP and inorganic phosphate. In many bacteria, the enzyme can also work in the reverse direction, generating a proton or Na+ motive force by hydrolysis of ATP. ATP synthases are rotary nanomachines that couple the translocation of ions in FO to ATP synthesis/hydrolysis within the catalytic F1 part. The flow of H+ or Na+ through two half channels within subunit a drives the rotation of the subunit c ring in FO as well as of the elongated γϵ central stalk in F1. Subunit γ rotates inside a molecular bearing composed of the alternately arranged α3β3 hexamer and generates cyclic conformational changes within the three catalytic nucleotide binding sites because of its eccentric rotation, thereby promoting ATP synthesis and its release. To counteract the tendency of the α3β3 hexamer to follow the rotation of the rotor, a peripheral stalk, composed of a b dimer and subunit δ in most bacterial enzymes, is essential to hold the α3β3 hexamer in position (1–3).
Peripheral stalks are present in all three evolutionarily related types of rotary ATPases. Interestingly, FOF1 ATP synthases (F-type ATPases) contain only one peripheral stalk. A-type ATPases, which function primarily as ATP synthases but are evolutionarily more closely related to V-type ATPases, have two peripheral stalks, whereas eukaryotic vacuolar V-type ATPases working as ion pumps contain three peripheral stalks per enzyme complex (4, 5). Furthermore, although each peripheral stalk of A-type as well as V-type ATPases studied so far is composed of a 1:1 heterodimer of nonhomologous subunits E and G (6–11), the single peripheral stalk of FOF1 ATP synthases shows variations in subunit composition dependent on the organism studied (12–18).
In mitochondrial FOF1, the peripheral stalk contains a single b subunit with two transmembrane helices together with the additional subunits d, F6, and OSCP5, the latter being homologous to bacterial subunit δ. Structures of bovine mitochondrial stalk subcomplexes revealed that the hydrophilic region of subunit b forms a continuous, slightly curved α-helix that is stiffened by surrounding shorter helical stretches of subunits d and F6. The binding between OSCP and the C-terminal region of subunit b is strengthened by extensive α-helical interactions (17, 18). In contrast, in chloroplasts and some bacteria, two different b-like proteins, named subunits I/II and subunits b/b′, respectively, are encoded, and both have been shown to be present in ATP synthase complexes purified, e.g., from spinach chloroplasts, Rhodobacter capsulatus, and the hyperthermophilic bacterium Aquifex aeolicus (12, 14, 16). Furthermore, heterodimer formation has been demonstrated for the soluble domains of subunits b and b′ of the cyanobacterium Synechocystis PCC6803 (13) and for the chimeric b and b′ constructs of Escherichia coli and Thermosynechococcus elongatus functionally assembled into E. coli ATP synthase (19, 20).
In E. coli as well as most bacteria, the peripheral stalk contains a dimer of identical b subunits, each with a single transmembrane helix and a soluble domain extending from the membrane to the top of F1 (15, 21, 22). Four distinct functional domains have been defined for the b dimer (23), starting at the N terminus with a mainly α-helical transmembrane domain (b1–24) anchoring the protein in the membrane by strong direct interactions with subunit a (24, 25). The tether domain (b25–52) spanning the surface of the membrane and the region beneath the F1 part appears to be a highly flexible region. Deletions of up to 11 amino acids or insertions of up to 14 amino acids can be positioned in this domain without loss of function (26, 27). Furthermore, functional FOF1 complexes can contain dimers of b subunits differing in the length of this region (28). Cross-linking data suggest that the dimerization domain (b53–122) extends along the F1 part at one of the non-catalytic (α/β) clefts (29, 22). The C-terminal δ-binding domain (b123–156) is essential for the binding of F1. A disulfide cross-link can be formed between b158C and δM158C (30, 31). Information about the structure of E. coli subunit b has been obtained from the analyses of peptides representing segments of subunit b, in particular of the membrane domain (fragment b1–34), the tether domain (fragment b30–82), the dimerization domain (fragment b62–122), and a part of the δ-binding domain (b140–156). In each case, only monomeric subunit b segments yielded to structural analysis, revealing an extended, highly α-helical configuration (24, 32–34). A low-resolution structure of the dimeric, soluble b22–156 domain exhibits a 160-Å-long, slightly curved shape (33) that resembles the density of the peripheral stalk observed in electron microscopy projections of FOF1 (21). Whether the two b subunits are present in a staggered or in an in-register conformation and whether in a right-handed coiled coil, like in A-type ATPases (9, 10), or in a left-handed coiled coil, which is observed in a great number of proteins (35), is still under discussion (20, 36–38).
A number of studies have shown that in the E. coli enzyme, the two b subunits interact with, or are proximal to, subunits a, c, α, β, and δ (25, 29–31, 39, 40). Because ATP synthase complexes do not have 2-fold symmetry, the two b subunits must occupy dissimilar positions within the enzyme and have different subunit-subunit interactions, but identifying features that distinguish the two positions has been difficult because the subunits themselves are identical, encoded by the same gene. However, genetic complementation between two differently defective b subunits supports the view that each b subunit contributes uniquely to the function of the peripheral stalk (41). In addition, the use of two differently shortened C-terminal hydrophilic forms of subunit b (bsyn) suggested that the two b subunits have different roles in binding of F1 (37).
In this study, we determined individual interactions of both b subunits within the stator of assembled FOF1 by use of antibody binding studies as well as disulfide cross-linking, applying two or three different cysteine pairs at a time. Within region b92–118, a part of the b dimer known to be associated with the side of F1 (29, 42), one b subunit, which we designated bI, is proximal to subunit α, whereas the other one (bII) can be linked to subunit β. They can also be distinguished by tagging the bI subunit with subunit a via the cross-linking pair bL16C/aV239C. Furthermore, the C-terminal region of only bI formed disulfides with subunit δ, whereas the corresponding region of bII is accessible for antibody binding. The results clearly demonstrate that, although identical in sequence, the two b subunits have different positions and competences within the stalk. We suggest that subunit bI is functionally related to the single subunit b present in mitochondrial ATP synthase, whereas subunit bII may have a more stabilizing function like mitochondrial subunits d and F6.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Growth Conditions
E. coli strains ML308-225 (lacIZ), CM1470 (F+ asnB32 thi1 relA1 spoT1 atp706 (ΔatpIBEFHA)), and KY7485 (F+ asn-31 thi rif λasn5), which overproduces the ATP synthase after heat induction, were grown as described (43).
For epitope mapping and cross-linking experiments, strain DK8 (HfrPO1, bglR, thi1, relA1, ilv::Tn10 (Tetr), ΔatpBEFHAGDC) (44) was used as host for plasmids carrying the wild-type or mutated atpBEFHAGDC genes. For plasmids coding only for mutated atpF (and wild-type atpH) genes (provided by R. D. Simoni and M. Futai), strains 1100ΔuncFH (HfrPO1, bglR, thi1, relA1, ΔatpFH) (45) and KF92rA (atpF92, thi, recA1) (46), respectively, were used. Cells were grown in Luria-Bertani medium in the presence of the appropriate antibiotic and, if necessary, 0.1 mm isopropyl-β-d-thiogalactopyranoside or in minimal medium with glycerol or succinate as a carbon source (25).
Construction of Mutants
All plasmids were derived from plasmid pBWU13 (atpBEFHAGDC) (47). The endogenous cysteine residues within the subunits under investigation were changed to alanine using plasmid pFV2 as template or source for cloning (48). Cysteine substitutions were introduced by a two-step PCR overlap extension method using two mutagenic oligonucleotides containing the codon change necessary and two wild-type primers with the corresponding restriction sites (49). PCR products were directly transferred into the suitable pBWU13 derivative using unique sites for cloning (49). All mutations were verified by DNA sequencing of the cloned fragment through the ligation sites.
Preparative Procedures
FO subunits a, b, and c (43), F1 (50), FO (43), and FOF1 (51) were purified as described. mAbs against subunit b were prepared as described using purified subunit b as antigen (43). Antibodies from culture supernatants were purified by caprylic acid and ammonium sulfate precipitation according to the method described by McKinney and Parkinson (52) and stored as lyophilized powder. Inverted membrane vesicles with and without F1 were prepared from E. coli strains ML308-225 and CM1470 (53). Binding of F1 to F1-stripped inverted membrane vesicles of strain ML308-225 and the influence of the anti-subunit b mAbs on the function of the ATP synthase was studied as described by Deckers-Hebestreit et al. (53).
Binding of mAbs to purified FOF1 was performed as follows. First, FOF1 and mAbs were independently purified on a Superdex SPX-200 gel filtration column to remove aggregates and, in the case of FOF1, incomplete protein complexes using 20 mm Tris-HCl (pH 7.8), 1 mm MgCl2, 0.2 mm phenylmethyl-sulfonyl fluoride, and 0.02% (w/v) n-dodecyl-β-d-maltoside as buffer system. Homogeneous fractions were pooled and concentrated by ultrafiltration (exclusion size, 100 kDa; Millipore) to a final concentration of 30 μm. Equimolar amounts of FOF1 and mAbs were mixed and incubated for 1 h at 37 °C. Subsequently, the mixture was separated by gel filtration using the same buffer system and analyzed by SDS-PAGE as well as immunoblotting for antibody binding.
Cross-linking with Copper-1,10-phenanthroline
Inverted membrane vesicles were prepared according to Krebstakies et al. (54) using TMG buffer (50 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10% (v/v) glycerol). In each case, the mutant strains used for membrane preparation were named according to the cysteine substitutions present within the FOF1 subunits. For cross-linking experiments, membranes were adjusted to a final protein concentration of 5 mg/ml with TMG (pH 8.2) (or TMG (pH 7.5), as stated in the corresponding figure legends). The CuP stock solution was prepared in TMG (pH 8.2 or pH 7.5) and added to the samples to provide 1.5 mm CuSO4 and 4.5 mm o-phenanthroline as the final concentrations. The cross-linking reaction was incubated for 1 h at 23 °C and stopped by addition of 50 mm EDTA-KOH (pH 8.0) (500 mm stock solution) and 50 mm N-ethylmaleimide (500 mm stock solution in dimethyl sulfoxide), while incubating the samples for another 15 min at 23 °C. An equal volume of double-concentrated sample loading buffer without 2-mercaptoethanol was added to solubilize the membranes and incubated for 15 min at 23 °C (24, 49).
Assays
Protein concentrations were determined by the method of Dulley and Grieve (55) or with the bicinchoninic acid protein assay as recommended by the supplier (Pierce). Proteins were separated by SDS-PAGE using 10% separating gels (56) with a Page-RulerTM prestained protein ladder (Fermentas) as the standard.
Immunoblotting was performed according to Birkenhäger et al. (43) using 10 mm NaHCO3, 3 mm Na2CO3 (pH 9.9), 20% (v/v) methanol as transfer buffer and the ECL Western blotting detection system (Amersham Biosciences) for visualization of immunolabeling. For cross-linking experiments, blot membranes were incubated with the corresponding primary antibodies raised in mice or rabbits (see figure legends), with IRDyeTM 800DX-labeled goat-anti-mouse IgG (H+L), or IRDyeTM 700DX-labeled goat-anti rabbit IgG (H+L) (Rockland or LI-COR) as secondary antibodies and finally detected with the two-channel infrared detection system Odyssey (LI-COR). Both secondary antibodies were affinity-purified for low cross-reactivities with serum proteins of other species, and, therefore, allowed the simultaneous detection of fluorescence (shown in red for IRDyeTM 700DX and in green for IRDyeTM 800DX) after immunolabeling of two proteins on one blot membrane. As a consequence, an overlay of both labels because of cross-linking of proteins appears as a yellow band. However, a clear yellow band can only be obtained in the presence of a 1:1 balance of the two fluorescent dyes. Because of the varying affinities and titers of the antibodies used (primary as well as secondary), the color of the yellow bands may shift to orange or lime green (chartreuse).
DCCD-sensitive ATPase activity of membrane vesicles was determined using a continuous flow system as described by Jäger et al. (57). ATP- and NADH-driven proton translocation was performed through ACMA (Sigma, catalog no. A-5806) fluorescence quenching as described by Birkenhäger et al. (43).
RESULTS
Epitope Mapping of mAbs Raised against Subunit b of the FO Complex
mAbs were prepared against subunit b of the E. coli ATP synthase using subunit b purified under non-denaturing conditions dissolved in a cholate-containing buffer as antigen for immunization (43). For epitope mapping, the binding of the individual mAbs to N- or C-terminally truncated b subunits (Fig. 1) as well as to b or bsyn subunits (59) carrying single amino acid substitutions (Fig. 2 and supplemental Table S1) was determined by immunoblot analyses using cell lysates. The results of the binding analyses for the different mAbs, summarized in Fig. 2, reveal that the epitopes of the different mAbs are spread over the hydrophilic part of subunit b. GDH 10-1A4 recognizes an epitope located at the C-terminal end of subunit b comprising amino acids b150–156. Even the removal of only the last (two) C-terminal amino acids resulted in a complete loss of mAb binding (Fig. 1). In addition, an elongation of subunit b by two residues (bsyn158) reduced mAb recognition significantly, clearly indicating that the free C terminus is critical in mAb binding (Fig. 2). GDH 1-5A2 and GDH 10-1B1, which also bind within the δ-binding domain of subunit b, cannot be distinguished by the use of truncated b subunits (Fig. 1). However, the analyses of substituted b subunits clearly demonstrate a slight shift between the binding sites of the two mAbs, b131–138 versus b128–138 (Fig. 2). GDH 10-6D1 binds within region b77–85 of the dimerization domain, as observed with N- and C-terminally truncated proteins as well as substituted b subunits, whereas region b32–41 in the tether domain is recognized by mAb GDH 1-4D3.
FIGURE 1.

Epitope mapping by binding of mAbs to N- or C-terminally truncated b subunits. Cells synthesizing plasmid-encoded N- or C-terminally truncated b subunits (1100ΔuncFH/pRPG derivatives (45), KF92rA/pBMF derivatives (46), and DK8/pSD or pDM derivatives (23, 29, 30, 32)) were separated on SDS-PAGE (20 μg of cell protein/lane) and analyzed by immunoblotting with different mAbs using the ECL detection system for immunodecoration. The amount of protein was determined on the basis of the calculation that 1 ml of cells with an optical density (578 nm) of 1.0 contains ∼160 μg of protein (58). The differences observed for b and bsyn truncated for the C-terminal amino acid bLeu-156 obviously depend on the varying expression level of the corresponding proteins because of different plasmid backgrounds. +, mAb binding; ±, weak mAb binding; -, no binding; n.d., not determined.
FIGURE 2.

Epitope mapping by binding of mAbs to mutated subunit b or bsyn carrying single amino acid substitutions. Right panel, sections of immunoblot analyses comparing the binding of mAbs to cells synthesizing plasmid-encoded mutated b or bsyn subunits. The position of the amino acid substitution in each mutant is indicated. In most cases, the addressed amino acid residue has been substituted with cysteine. For exceptions, see supplemental Table S1 summarizing the complete analysis. The immunoblot analyses were performed as described in legend to Fig. 1. To verify that residue bAla-68 is not a part of the epitope of mAb GDH 10-6D1, GDH 1-5A2 has been shown as a control, revealing that, in general, the expression of the bsynA68C variant is lower. The differences observed for b and bsyn depend on the varying expression level of the corresponding proteins because of different plasmid backgrounds. Left panel, structural model of FOF1 with the b dimer in blue and the other subunits in light gray as drawn by RasMol 2.7.2.1.1 to mark the positions of the epitopes recognized by mAbs in subunit b. The structural homology model of E. coli FOF1 is a composite of several partial structures combined with biochemical data, as described in detail by Junge et al. (1).
Specificity of Anti-b mAb GDH 10-1A4 Binding
GDH 10-1A4 binds to the C-terminal region of subunit b, which is also known to be directly involved in binding of subunit δ. Thus, we further investigated its binding characteristics. A direct binding of GDH 10-1A4 to subunit b in assembled FOF1 was shown by purification of an FOF1-mAb complex by size exclusion chromatography (Fig. 3A). After incubation of purified FOF1 with mAbs, the mixture was separated by gel filtration, showing a slight shift of the FOF1 peak to a higher molecular mass (Fig. 3A, peak A), with a shoulder representing residual free FOF1 (peak B). A further peak represents free mAb molecules (Fig. 3A, peak C) and is identical to that obtained in mAb purification (not shown). A silver-stained SDS gel showed that peak A separated under non-reducing conditions contained, in addition to the subunits of the FOF1 complex, a molecular mass of approximately 150 kDa that can be detected in a corresponding immunoblot analysis exclusively by mouse IgG-specific antibodies conjugated with horseradish peroxidase (Fig. 3A, inset). The presence of only one single sharp FOF1-mAb peak as well as its elution volume indicates that only one mAb molecule was bound per FOF1 complex. Corresponding results were obtained by application of the other mAb molecules (data not shown).
FIGURE 3.

Biochemical characteristics of anti-b mAbs. A, binding of mAb GDH 10-1A4 to purified ATP synthase. Equimolar amounts of mAb GDH 10-1A4 and purified FOF1 complexes were incubated for 1 h at 37 °C prior to separation of the protein complexes by size exclusion chromatography on a Superdex SPX-200, as described under “Experimental Procedures.” Peak fraction A was separated in comparison to purified FOF1 under non-reducing conditions on SDS-PAGE, silver-stained (61) (lanes 1 and 2), and analyzed by immunoblotting (lanes 3 and 4) using exclusively goat anti-mouse IgG antibodies conjugated with horseradish peroxidase for ECL detection. Lanes 1 and 3, purified FOF1; lanes 2 and 4, peak fraction A. mAU, milliabsorption unit. B, competition between bound subunit b and free membrane vesicles. Microtiter plates were coated with purified subunit b (0.1 μg/ml) and subsequently incubated with a mixture of different amounts of free membrane vesicles and culture supernatant of mAb GDH 10-1A4. The competitive inhibition ELISA was performed as described by Jäger et al. (57). ■ (dark gray), F1-containing inverted membrane vesicles of the atp wild-type strain ML308-225; ♦ (black), F1-stripped inverted membrane vesicles of strain ML308-225; ▴ (light gray), inverted membrane vesicles of the atp mutant strain CM1470. The data represent average values of three independent measurements. C, influence of subunit b-specific mAb GDH 10-1A4 on the function of FOF1. Membrane vesicles (MV; 2 mg/ml) were incubated in the absence or presence of mAb GDH 10-1A4 (50 μg/ml) as described under “Experimental Procedures.” NADH-driven proton translocation by respiration and ATP-driven proton translocation by FOF1 were measured via ACMA fluorescence quenching. DCCD-sensitive ATPase activities were determined as described. The presence of mAb GDH 10-1A4 on the membrane vesicles after the washing steps was determined by competitive inhibition ELISA before measuring ATPase and proton translocation activities. a, NADH-dependent ACMA fluorescence quenching after incubation of F1-stripped membrane vesicles with DCCD (40 μm, 20 min at room temperature) for inhibition of FO. b, rebinding of F1 to F1-stripped inverted membrane vesicles was performed after incubation with mAb, as described by Deckers-Hebestreit et al. (53).
Binding of mAb molecules to membrane vesicles of inside-out orientation in the presence and absence of F1 was tested via a competitive inhibition ELISA using subunit b as a fixed antigen. GDH 10-1A4 showed competition between subunit b and F1-stripped inverted membrane vesicles as well as F1-containing inverted membrane vesicles of the atp wild-type strain ML308-225 (Fig. 3B). This indicates the accessibility of the antigenic determinants of subunit b even in the presence of the membrane. However, the greater amount of F1-containing membranes, compared with F1-stripped membranes, which is required to achieve competition, suggests that the presence of F1 may hinder recognition of the epitope in some way. A comparable picture was obtained for the other mAbs, except GDH 1-4D3, which showed no binding to inverted membrane vesicles, indicating that region b32–41 is at least partly shielded by the phospholipid bilayer. Taken together, although mAb GDH 10-1A4 recognizes the C-terminal part of the δ-binding domain of subunit b, it is still able to bind to its epitope, even in the presence of F1 and, therefore, subunit δ.
Influence of Subunit b-specific mAb GDH 10-1A4 on the Function of FO
Independent of the presence or absence of F1, the incubation of inverted membrane vesicles with anti-b mAbs had no influence on the DCCD-sensitive ATPase activity or on the proton-translocating activity of the FO complex (Fig. 3C). Although, in the presence of F1, the generation of a pH gradient by energization with ATP as well as NADH could be followed by ACMA quenching, in the absence of F1, energization of the membranes even by use of NADH was not possible because of the passive proton translocation through the open FO complex. After preincubation with DCCD, however, a pH gradient could be generated by addition of NADH independent of added mAbs.
In contrast, the binding of F1 to F1-stripped inverted membrane vesicles was affected after preincubation with mAbs, probably because of steric hindrance (Fig. 3C). With increasing amounts of mAb molecules up to saturation (not shown), the energization of the membranes with either NADH or ATP was reduced, as was the DCCD-sensitive ATPase activity, revealing that the antibodies interfered with the binding of F1 to FO.
In summary, binding of mAb GDH 10-1A4 had no influence on the function of assembled FOF1, although its antigenic binding site overlaps with the region essential for binding of subunit δ. This result can only be explained by a simultaneous binding of both molecules, subunit δ and mAb GDH 10-1A4, to the b dimer and demonstrates that one b subunit is far more important than the other for binding of subunit δ, as has also been deduced from a completely different set of experiments by Wood and Dunn (37).
Effect of Cysteine Substitutions on the Function of FOF1
As a prerequisite for cross-linking experiments, the native cysteine residues present in the corresponding subunits were exchanged to alanine with no loss of function (supplemental Table S2). Indeed, a substitution of all 10 native cysteine residues present in FOF1 to alanine has no effect on the ATPase activity of the enzyme (62). Furthermore, each of the mutants used for cross-linking, containing up to five individually introduced cysteine residues, grew similar to the wild type on succinate as the sole carbon and energy source, indicating a functional oxidative phosphorylation system (supplemental Table S2). To quantify the influence of the cysteine substitutions on the activity of ATP synthase, DCCD-sensitive ATPase activities of membrane vesicles were measured (supplemental Table S2). For most mutants, the maximal reduction in ATPase activity was by a factor of 2 in the presence of two cysteine substitutions or by a factor of 3 in the presence of four or five cysteine substitutions. Combinations of mutations causing a greater reduction in activity were not used for cross-linking experiments. However, although reductions in ATPase activity were observed, DCCD sensitivity, which indicates a precise coupling between FO and F1, was maintained in each case.
Identification of Amino Acid Pairs for Cross-linking of Subunit b to Subunits α or β
To characterize the interactions of the b dimer in FOF1 complexes in more detail, nearest neighbor analyses via disulfide-based intermolecular cross-linking are an ideal tool. The proximity of bA92C to region α464–483 shown via benzophenone-4-maleimide cross-linking (29) was used as starting point to determine neighboring amino acid pairs between subunits b and α. Region α464–483 has an α-helical conformation (1, 63) and, therefore, residues αSer-466, αAla-469, αAla-473, αArg-477, and αAla-480, which are all present at the surface of the α3β3 hexamer, were exchanged to cysteine. Membrane vesicles from mutants with each of these substitutions, along with bA92C, were analyzed for disulfide-linked b-α heterodimer formation mediated by copper-1,10-phenanthroline (CuP) using non-reducing SDS-PAGE and immunoblotting with subunit-specific antibodies (Fig. 4A). A direct detection of cross-linking products was carried out in one immunoblot analysis through the use of primary antibodies from different sources and detection by corresponding secondary antibodies labeled with different fluorophores. Heterodimers of b-α were observed in a high yield for residues αA473C and αR477C with bA92C, whereas αA480C showed only a weak signal. No cross-linking could be detected for αS466C and αA469C. When possible, cross-linking yields were estimated from both the appearance/increase of the cross-linked product and the reduction of the band intensities of the corresponding monomeric subunits. However, because of the presence of two b and three α subunits within FOF1, but cross-linking of only one of each, and because of the signal saturation obtained by the conditions used for the immunoblot analyses, the removal of single polypeptides could hardly be determined in many cases. Nevertheless, even an increase of pH to 8.2, which lies close to the pK value of sulfhydryl groups under standard conditions, during the oxidation reaction with CuP had no influence, suggesting a high cross-linking yield for these interaction partners. In addition, membrane vesicles containing no cysteines within the two subunits studied and those carrying only the substitution bA92C were used as controls. As side reactions, the formation of b dimers and of a band with an apparent molecular weight of ∼120 kDa could be observed in small amounts, which could both be completely abolished by reduction of the disulfide bonds with 2-mercaptoethanol (data not shown). The b dimer formation was not unexpected because the two b subunits are known to closely interact with each other (compare Refs. 22, 23, 28). To analyze the proteins present within the 120-kDa band, immunoblotting using antibodies raised against the individual eight FOF1 subunits was performed, but the band could only be detected with antibodies against subunit α (data not shown). Furthermore, using membranes with otherwise cysteine-free FOF1 complexes, in which only αR477C is present, the cross-linking band could still be observed (data not shown), indicating the probability that an α dimer had been formed, likely through an interenzyme reaction. Substitution of a cysteine residue in α (or β) will result in incorporation of three thiol groups into FOF1. Although one may be proximal to the b dimer, the other two located at the surface of the hexamer will be freely available for other reactions. Because membrane vesicles are rich in FOF1, this intermolecular cross-linking was not unexpected.
FIGURE 4.

Cu2+-catalyzed cross-linking between cysteine substitutions of subunit b at position 92 and subunit α or β. Inverted membrane vesicles of different mutant strains (termed by the cysteine substitutions present within FOF1 subunits) were incubated in the absence (−) or presence (+) of CuP, separated by SDS-PAGE (20 μg/lane) under non-reducing conditions, and analyzed by immunoblotting using two antibodies simultaneously, as described under “Experimental Procedures.” A, cross-linking between bA92C and residues of subunit α of region α464–483 exposed at the surface of the α3β3 hexamer. Cross-linking yields obtained after incubation with CuP at pH 7.5 and 8.2, respectively, were compared. For immunolabeling, mouse anti-b antibodies (GDH 10-1B1, green) and rabbit anti-α antibodies (red) were applied. In each mutant, endogenous cysteine residues in subunits b (bCys-21) and α (αCys-47, αCys-90, αCys-193, and αCys-243) were substituted by alanine. St, molecular mass standard. B, cross-linking of bA92C with residues of subunit β located at a non-catalytic cleft of F1. Rabbit anti-b (red) and mouse anti-β antibodies (green) were applied for detection. In each mutant, endogenous cysteine residues in subunits b (bCys-21) and β (βCys-137) were changed to alanine. St, molecular mass standard; unspec., unspecific immunolabeling. C, structure of the E. coli α3β3 hexamer (63) drawn by RasMol 2.7.2.1.1 with marked amino acid residues of subunits α and β, which were cross-linked to subunit b at position bA92C. Red, high cross-linking yield; orange, intermediate cross-linking yield; yellow, low cross-linking yield; white, no cross-linking.
The same approach was used to identify amino acid residues of subunit β that are in close proximity to bA92C (Fig. 4B). Amino acid residues βLeu-347, βGln-351, βGlu-352, βArg-425, βLys-428, and βGlu432 were substituted by cysteine. These residues are all located within a comparable distance to the non-catalytic cleft as those residues of subunit α that were able to form an S-S bridge with residue bA92C. Intense cross-link formation of bA92C to βL347C and βQ351C was observed, whereas the reaction with βE352C was much weaker (Fig. 4B). Because of the localization of the interacting amino acid residues within an αβ pair, it can be concluded that the b dimer is located at a non-catalytic α/β interface, as suggested previously (22, 29).
In summary, Fig. 4C shows the localization of the amino acid residues of subunits α and β interacting with residue bAla-92. At this point, however, it is not apparent if both b subunits are involved in interaction with the αβ pair (one b interacting with α and the other with β) or whether only one of the two b subunits can interact at its position 92 with either subunit α or β depending on the cysteine residue provided in the experiment.
To further define the relationship between the αβ pair and the b dimer, residues bAla-103, bGln-106, bLys-110, bArg-113, bArg-117, and bGlu-118, all with side chains located on one side of the α-helix (32), were exchanged to cysteine, combined with several cysteine substitutions in subunits α or β, and analyzed for disulfide bond formation. To summarize those results, the pairs identified and used for further experiments were: b92-α477, b92-β351, b92-β122, b103-α215, b103-β122, b103-β286, b106-β287, b118-α118, b118-α120, and b118-β117 (see Fig. 6, lanes b-α and b-β, respectively).
FIGURE 6.

Cu2+-catalyzed cross-linking between cysteine substitutions of subunits b, α, and β combined with a b-a (bL16C/aV239C) cross-linking pair to distinguish both b subunits. A, immunoblot analysis of the a-bI-α and bII-β complexes, respectively, generated by combination of the following cysteine pairs: b-a, bL16C/aV239C; b-α, bE118C/αK118C; b-β, bE118C/βE117C; and b-α,β, bE118C/αK118C/βE117C. Inverted membrane vesicles of the corresponding mutant strains were incubated with CuP as described in the legend to Fig. 4. Immunodecoration: left panel, rabbit anti-b (red) and mouse anti-a (green) antibodies. Right panel, mouse anti-α (green) and rabbit anti-β antibodies (red). In each mutant, the corresponding endogenous cysteine residues in subunits b, α, and β (compare legend to Fig. 4) were substituted by alanine. Because of the use of 10% N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine-SDS-polyacrylamide gels (56), a high resolution of protein bands is observed within the range up to 50 kDa, whereas bands with higher molecular weights are observed as dense, highly focused bands (compare also the molecular mass standard). The yellow intensities observed in the blot analysis at the right side arise from a partial overlay because of the close proximity of the bands representing α and β as well as to a slight degradation of subunit α. St, molecular mass standard. B, sections of immunoblot analyses comparable with A showing the cross-linking products obtained by several combinations of different cysteine-substituted subunits b, α, and β as announced in the corresponding boxes at the left side. a-b always represents the presence of cross-linking pair bL16C-aV239C. For clarity, during the simultaneous presence of cysteine substitutions in subunits α and β for interaction with subunit b (last lane of the blot membranes), the complexes formed are marked in green for complexes involving subunit α and in red for those containing subunit β.
Identification of a Cross-linking Pair for Tagging One Subunit b with Subunit a
Subunit a is known to stably interact with the b dimer (25), and the cysteine pairs bN2C-aG227C and bN2C-aL228C allow cross-link formation (39). However, hydrophilic loops as well as N-terminal regions are in many cases highly flexible elements, and, therefore, we set out to identify a cross-linking pair between the more tightly packed transmembrane helices to increase the probability that a defined cysteine in the single subunit a might only react with the cysteine of one of the two b subunits. bLeu-16, located outside the known b-b interaction area (24), was exchanged to cysteine and combined with individual cysteine substitutions within region a237–250, which is proposed to be part of the C-terminal transmembrane helix of subunit a (64, 65). The region was chosen on the basis of the finding that mutation bG9D can be partially suppressed by mutation aP240A/L in subunit a (66). The experiments shown in Fig. 5 demonstrate that only cysteine pair b16-a239 formed a cross-link with a high yield in the presence of CuP (note the conversion of virtually all subunits a to a-b). Therefore, the cross-linking pair b16-a239 was used in further experiments where several cross-linking pairs were combined to determine the interacting partners for both b subunits.
FIGURE 5.
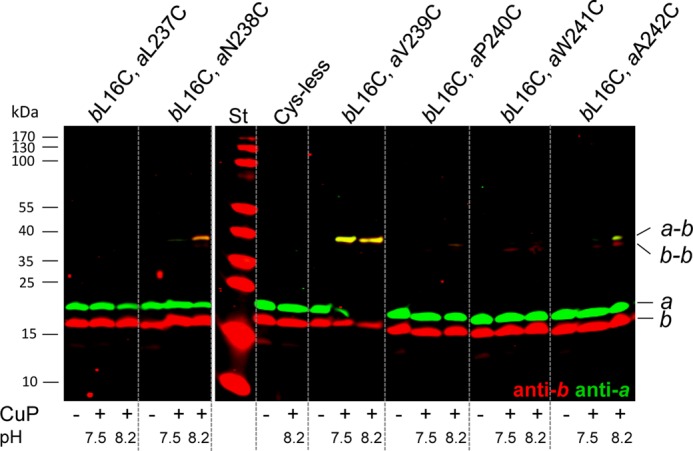
Cu2+-catalyzed cross-linking between cysteine substitutions of subunit b at position 16 in the N-terminal transmembrane helix and subunit a. Inverted membrane vesicles of different mutant strains (termed by the cysteine substitutions present within FOF1 subunits) were incubated in the absence (-) or presence (+) of CuP, separated by SDS-PAGE (20 μg/lane) under non-reducing conditions, and analyzed by immunoblotting using two antibodies simultaneously as described under “Experimental Procedures.” St, molecular mass standard. Cross-linking yields obtained after incubation with CuP at pH 7.5 and 8.2, respectively, were compared. For immunolabeling rabbit anti-b antibodies (red) and mouse anti-a antibodies (green) were applied. In each mutant, the endogenous cysteine residue in subunit b (bCys-21) was substituted by alanine.
Generation of a-b-α and b-β Complexes by Combining Cysteine Pairs for Cross-linking
In the next step, the cross-linking pair b16-a239 was combined with a second cysteine on b that would allow disulfide linkage to identified sites on either subunit α or β. Furthermore, FOF1 variants containing single cysteine residues in both α and β as well as the corresponding cysteine residue in the b subunit were used to identify preferred linkages between subunit α or β with one of the two b subunits. Note that in Fig. 6, lanes are identified by the pairs of subunits containing cross-linkable cysteines identified previously, with a-b always being b16-a239, whereas the varying positions of cysteines for b-α and b-β cross-links are indicated within the corresponding boxes. Fig. 6A shows a typical experiment with pairs b16-a239, b118-α118, and b118-β117, appropriate controls, and immunolabeling of all FOF1 subunits involved in cross-linking, i.e. subunits a, b, α, and β. When only individual cross-linking pairs were present, strong, specific disulfide linkage was observed, although, in each case, a b-b band could also be detected. The heterodimers b-α and b-β were also observed in the absence of CuP, probably produced by oxygen present during incubation (Fig. 6A, lanes b-α and b-β). In each case, disulfide bonds could be reduced by addition of 2-mercaptoethanol (data not shown). When the b-a pair was also present, we observed a shift of b-α as well as of b-β to a molecular mass corresponding to a-b-α or a-b-β, and, indeed, the immunolabeling revealed the presence of all three subunits (Fig. 6A, lanes b-a/b-α and b-a/b-β). In addition, small amounts of an a-b-b product could be observed, because of the presence of two cysteine residues within subunit b, namely bL16C for linkage to subunit a and bE118C for linkage to subunit α or β, whereas the amount of b dimer was strongly reduced. Surprisingly, when CuP was provided in the simultaneous presence of single cysteine residues in both subunits α and β, only an a-b-α cross-linked product was formed, indicating that the b subunit bridged with subunit a is preferentially cross-linked to subunit α, whereas no b-β or a-b-β was observed (Fig. 6A, lane b-a/b-α,β). Thus, it appears from the results obtained with the set of cysteine substitutions used in Fig. 6A, that one of the b subunits, which we designated as bI, can be tagged with the a subunit through the b16-a239 pair and can also be linked to either subunit α or subunit β, but preferentially to α when the choice is available. In contrast, it appears that the second b subunit, bII, which is not tagged with subunit a, can be linked to neither α nor β in this case.
Comparable experiments performed for six other combinations of cross-linking pairs yielded a range of results as shown in Fig. 6B and summarized in Fig. 7. The cysteine residues present in FOF1 in addition to b16-a239 are noted at the left side of the figure in the corresponding boxes. In each case, only the upper part of the immunoblot analysis is presented, showing the formation of b-α, b-β, a-b-α, and a-b-β, respectively. Bands with molecular masses corresponding to α-α or β-β were also detected after immunodecoration with subunit-specific antibodies, depending on the cysteine substitution present.
FIGURE 7.

Schematic showing zero-length cross-links of b-α and b-β with single cysteine residues present simultaneously in each subunit and with one of the b subunits tagged with subunit a. The schematic summarizes the data obtained for lanes b-a/b-α and b-a/b-β (A) and for lanes b-a/b-α,β (B) in Fig. 6. The numbering indicates the amino acid residues substituted by cysteine within the corresponding subunit. Solid bars, high cross-linking yields; dashed bars, low cross-linking yields.
In each case, b-α and b-β cross-links could be observed, although the cross-linking yield of b106-α118 was rather low. Upon combination with b16-a239, a shift of the b-α bands corresponding to a-b-α could be detected in each case, again, with only a weak band for b16-a239/b106-α118. However, for the b-β cross-linking pairs, a different picture emerged after combination with a-b. A complete shift could only be observed for b118-β117. Otherwise, depending on the b-β pair present, a moderate change (b103-β122), weak change (b103-β286, b92-β351, b92-β122), or no change (b106-β287) of the b-β band to an a-b-β band was obtained (Fig. 6B and summarized in Fig. 7A). Remarkably, in the simultaneous presence of cysteine residues in subunits α and β, an a-b-α complex was formed in each case, whereas subunit β preferentially formed a b-β cross-linking product. Although there are two exceptions (no cross-linking between b118-β117 and a weak a-b-β product for b16-a239/b103-β122), the results obtained clearly demonstrate that it is possible to distinguish between the two b subunits by their different interaction partners, as summarized in Fig. 7B. These results strongly support the idea that the sulfhydryl group of aV239C forms a specific disulfide bond with the sulfhydryl group (bL16C) of only one of the two b subunits present (bI).
In summary, for most positions within the region tested, cysteines in subunit bI (marked by subunit a) cross-linked strongly to subunit α. If α contained no suitably positioned cysteine residue, most bI positions cross-linked also to β, as seen by the a-b-β band. These results imply that bI is located at an α/β interface, positioned most closely to α. In contrast, cysteines in subunit bII were usually linked to subunit β, as seen by the formation of b-β in the presence of the bI-a cross-link, except for residue bE118C. We saw no evidence that cysteines in bII would cross-link to sites on α. This would have been indicated by formation of b-α in the presence of the b-a, b-α, and b-β cross-linking pairs. These results imply that subunit bII is positioned largely along the surface of the β subunit.
Formation of an α-bI-bII-β Complex
To further test the observation that the two b subunits show a different interaction pattern toward subunits α and β, cross-linking pairs b118-α118 and b106-β287 were combined with an additional cysteine residue at position b68, which allows b dimer formation in a high yield (59). If the hypothesis is correct, a covalently linked quaternary subcomplex composed of subunits α and β connected through the b dimer should be generated, as shown in Fig. 8B. Experimentally, each of the individual cross-linking pairs generated the expected homo- or heterodimer in addition to b-b, α-α, or β-β as side products (Fig. 8A). By combining all three pairs, the formation of α-b-b-β as well as of α-b-b can be detected by immunolabeling as two newly formed bands with corresponding molecular masses (Fig. 8A). Although the bands were densely focused because of the behavior of proteins with high molecular masses in 10% N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine-SDS gels (56), the product yields appeared to be unusually high given the number of cross-links required. In the presence of CuP, free subunit b as well as b-α and b-β were completely absent, and b-b was drastically reduced compared with the samples containing individual cross-linking pairs, indicating that a large majority of the b subunit is involved in cross-linking. These results confirm that subunits bI and bII of the peripheral stalk can be clearly distinguished by their individual cross-linking properties, as described above.
FIGURE 8.

Cu2+-catalyzed cross-linking between cysteine substitutions of subunits b, α, and β generating an α-b-b-β complex. A, immunoblot analysis of an α-b-b-β complex generated by combination of the following cysteine pairs: b-b, bA68C; b-α, bE118C/αK118C; and b-β, bQ106C/βT287C. Inverted membrane vesicles of the corresponding mutant strains were incubated with CuP as described in the legend to Fig. 4. For immunolabeling, rabbit anti-b antibodies (red) and mouse anti-α (green, left panel) or mouse anti-β antibodies (green, right panel) were applied. In each mutant, the corresponding endogenous cysteine residues in subunits b, α, and β (compare legend to Fig. 4) were substituted by alanine. St, molecular mass standard. B, schematic to illustrate the disulfide bonds formed between the different stator subunits allowing a clear discrimination between both b subunits present in FOF1. Black bars, zero-length cross-links present to obtain the quaternary α-bI-bII-β complex; gray bar, zero-length cross-link used to mark subunit bI with subunit a.; unspec., unspecific reaction of the secondary antibody IRDyeTM 700DX-labeled goat-anti rabbit IgG (H+L).
Identification of the b Subunit Preferentially Involved in Binding of Subunit δ
Subunit δ is known to interact through its N-terminal domain with the N-terminal region of subunit α (67, 68), whereas the C-terminal region of δ is in contact with the C-terminal region of b, and one of the b subunits is much more critical for binding of δ than the other (30, 31, 37). To identify this b subunit, the same design as described above was applied, using the a-b cross-linking pair to discriminate between the two b subunits. In a first step, bE155C (30) was combined with subunit δ derivatives carrying cysteine substitutions within region δ149–172 to screen for a suitable b-δ cross-linking pair. Residues δA149C, δI160C, and δG162C were identified for disulfide bond formation (data not shown). However, a detection of the b-δ cross-linked product was only possible with anti-δ-specific antibodies, whereas, surprisingly, the polyclonal anti-b antiserum, which exhibits an extremely high avidity to subunit b, cannot bind to the cross-link product formed. The dominant epitopes recognized by the antibodies present are apparently shielded by interacting subunit δ (Fig. 9C). Applying the different mAbs for detection revealed that the mAbs binding within region b123–156, namely the δ-binding domain (23), cannot recognize subunit b within b-δ (Fig. 9D). However, with mAbs GDH 1-4D3 and GDH 10-6D1 with their epitopes located within the tether and dimerization domains, respectively, subunit b could be identified within b-δ, although the binding was comparatively weak (Fig. 9D).
FIGURE 9.

Cu2+-catalyzed cross-linking between cysteine substitutions of subunits b, a, and δ generating an a-b-δ complex. A, immunoblot analysis of an a-b-δ complex generated by combination of the following cysteine pairs: a-b, bL16C/aV239C and b-δ, bE155C/δI160C. Inverted membrane vesicles of the corresponding mutant strains were incubated with CuP as described in the legend to Fig. 4. For immunolabeling, mouse anti-δ (top panel), anti-b (center panel; GDH 1-4D3), and anti-a antibodies (bottom panel) were applied. In each mutant, the corresponding endogenous cysteine residues in subunits b (bCys-21) and δ (δCys-64 and δCys-140) were substituted by alanine. unspec., unspecific immunolabeling. B, schematic to illustrate the disulfide bonds formed between the different stator subunits. Black bars, zero-length cross-links to obtain the a-bI-δ complex; gray bar, zero-length cross-links used for the α-bI-bII-β complex. C, immunoblot analysis corresponding to the top panel in A using two antibodies simultaneously: mouse anti-δ (green) and rabbit anti-b (red) antibodies. The prominent yellow band observed in each lane is due to an unspecific binding of antibodies (compare with A), which has not been marked for clarity. St, molecular mass standard. D, detection of the b-δ cross-linking product (bE155C/δI160C or bE155C/δG162C) by different anti-b mAbs. bdegrad, degradation product of subunit b detected by mAb.
Next, the b155-δ160 cross-linking pair was combined with b16-a239, and a cross-linking experiment with CuP was performed (Fig. 9A). As expected, no b-δ product could be observed in the presence of only δI160C or bE155C, although bE155C showed strong b-b formation. b155-δ160 showed disulfide bond formation even in the absence of oxidizing agent, reaching a high cross-link yield in the presence of CuP. In each case, cross-links were abolished by the addition of 2-mercaptoethanol (data not shown). In the presence of both cross-linking pairs b155-δ160 and b16-a239, a complete shift of the band representing b-δ toward a band with a molecular weight corresponding to an a-b-δ product was observed after treatment with CuP using anti-δ antiserum for detection (Fig. 9A, top panel). However, with anti-b mAb GDH 1-4D3, three new bands were detected, whereas the b-δ product was eliminated (Fig. 9A, center panel). Comparing the same immunoblot analysis as in the top panel decorated with anti-δ (green) and anti-b antisera (red) (Fig. 9C), it can be seen that only the band with the highest molecular mass is decorated by anti-δ antibodies, whereas the other two products were only detected by anti-b antiserum, indicating that the double band observed represents two different cross-linked products. From examination of the control lanes containing samples with cysteines at b16-a239 and b16-a239/b155, but lacking the substitution at δ160, it is evident that, in addition to an a-b-δ product, an a-b as well as an a-b-b cross-link was formed during treatment with CuP (Fig. 9A, center and bottom panels). Comparable results were obtained when combining b16-a239 with b155-δ149 and b155-δ162 (data not shown). Taken together, these results demonstrate that subunit bI is the direct interaction partner of subunit δ within the peripheral stalk, as summarized in Fig. 9B (black bars).
DISCUSSION
Despite many studies of the stator stalk of bacterial F-ATPases, in particular the b homodimer of E. coli, its relationship to the rest of the enzyme has remained ill-defined in many ways. In particular, the asymmetry of the enzyme requires that the two b subunits interact with other subunits in different ways. The b dimer itself appears to be intrinsically asymmetric because the helices of the dimerization domain form an unusual coiled coil in which they are offset rather than in-register (36). However, features distinguishing the different positions of the b subunits within FOF1 have not been identified previously. At this point, single particle electron microscopic analyses have provided the most detailed images of the holoenzyme (69). No high-resolution structure of the holoenzyme is yet available, and it cannot be anticipated when one will be obtained. In this work, we have used two tools, mAb binding analyses and disulfide-based cross-linking of subunits in membrane-bound FOF1, to provide a more discriminating view of the relationships of the two b subunits to the remainder of the enzyme.
Binding between Subunits b and δ
A shortening of the C-terminal region of both b subunits by four amino acids (bV153end) prevents the assembly of functional FOF1 (46, 41), whereas a heterodimer, in which only one of the two b subunits was shortened by molecular genetics, allows the formation of functional FOF1 (41). Studies on the binding of F1 to purified, soluble heterodimeric bsyn subunits indicated that although one b subunit was most critical, the second b subunit played a lesser role (37). Here, the interaction of this region of one b subunit, designated bI, to the highly conserved C-terminal region of subunit δ could be observed, because bE155C was cross-linked to residues δA149C, δI160C, or δG162C, consistent with the cross-link pair described previously, b158C-δM158C (30). Immunoblot analyses showed that cross-linking of b-δ prevented not only the immunodecoration by mAb GDH 10-1A4 but also by mAbs GDH 1-5A2 and GDH 10-1B1 (Fig. 9D), revealing that the δ-binding domain is mostly shielded by subunit δ in the presence of the covalent S-S bond, even under denaturing conditions. This finding fits well with the extensive helix-helix interaction observed for the C-terminal regions of subunit b and OSCP (homologous to bacterial subunit δ) of bovine mitochondrial ATP synthase (18).
We observed that antibody GDH 10-1A4 was stably bound to its epitope in FOF1 but less strongly than in FO, and that this binding had no influence on the functional activities of the wild-type FOF1 complex. Together, these results imply that the C-terminal region of the second b subunit, here designated bII, which is less critical to the interaction with δ, is accessible to the antibody, but to a more limited extent than when F1 has been removed. In this context, it is worthwhile mentioning that the 10-Å cryoelectron microscopic structure of the A-type ATP synthase of Thermus thermophilus suggests that subunits E and G of the two peripheral stator stalks are differently associated with the A3B3 hexamer, with only subunit E being closely apposed to the non-catalytic subunit B (11). Assuming a similar arrangement of the E. coli b dimer with the α3β3 hexamer, it becomes feasible that the epitope of at least one of the two b subunits is accessible for mAb binding.
Tagging of One of the b Subunits by Subunit a
Several systems have been developed to allow for discrimination between the two b subunits within the E. coli homodimer. Using a genetic approach, two different b-encoding sequences were labeled with different tags (NH2-His tag and COOH-V5 tag) (28) or with diverse mutations (bR36I/bV153end) (41). In addition, chimeric b proteins were constructed by combination of b and b′ DNA sequences from T. elongatus with the E. coli atpF gene (19, 20). An in vitro approach utilized specific disulfide bond formation between purified bsyn derivatives of different sizes (b34–156 and b53–156) (37). Although valuable tools, these approaches have been limited by unequal expression, less-than-specific heterodimer incorporation into ATP synthase, or limited applicability. Furthermore, the individual locations of the two subunits have not been distinguished.
Here, we have identified pairs of cysteine-substituted positions between subunit b and subunits a, α, β, and δ that can be cross-linked through disulfide bond formation, and we then used these pairs in combinations to form multiple cross-links within ATP synthase solely by engineering the plasmid-encoded atp operon. This has allowed us to discriminate between the positions occupied by the two identical b subunits in the enzyme complex. Subunit a, known to tightly interact with the b dimer (25), was chosen to mark one of the b subunits by S-S bond formation between positions b16 and a239. The contact areas of the single, asymmetric subunit a must differ for the two b subunits. Using bL16C, which has little or no tendency to form b-b cross-links (24), a disulfide could be formed with aV239C in a high yield. A number of observations indicate an exclusive linkage of aV239C to bL16C of just one b subunit. First, in the presence of the b16-a239 cross-link, a complete shift of b-δ to a-b-δ was observed (Fig. 9, A and C, lane b-δ/a-b), and only one b subunit is involved in binding of subunit δ (see above). If cross-linking between subunits a and b was a statistical process, one would expect two bands corresponding to b-δ and a-b-δ in comparable amounts. Second, the b subunit tagged by subunit a (bI) showed a preferred cross-linking to subunit α, whereas the unmodified subunit b (bII) was exclusively linked to subunit β. The formation of an α-b-b-β cross-linked product in a high yield further supports our conclusion.
Individual Cross-links of the Two b Subunits to α3β3
The data show that, in general, the b subunit positions can be distinguished by their specific sites of disulfide cross-linking to either α or β (Fig. 9B, black and gray bars). The proximity of bA92C and region α464–483 was inferred previously through cross-linking with benzophenone-4-maleimide, which provides a 10-Å spacer between the sites (29), and disulfide formation between bA92C and αR477C confirmed this. The sites of β that supported disulfide formation to bA92C were located slightly above the region proposed after benzophenone-4-maleimide cross-linking (22, 29). Using bA92C as starting point, several additional cross-linkable pairs between the b dimer and the α3β3 hexamer were identified. However, before tagging subunit bI with subunit a, it was not evident that subunit α exclusively interacts with subunit bI, whereas subunit β predominantly makes contact to subunit bII.
The b cross-link sites in subunits α and β are located along a non-catalytic α/β interface, as proposed previously (22, 29) and as shown by a structural analysis for the peripheral stalk of bovine mitochondrial ATP synthase (18). Our confirmation of this placement is significant because, unlike the experimental approaches used previously, it is done on the basis of work with the holoenzyme in its native membrane-bound state. Random movements of b2 are restricted by specific interactions at both ends. We found that the S-S bond between b92-α477 had no influence on the ATP-hydrolyzing activity (data not shown). Previous work (70) has shown that only small differences exist in the dwell times of the three catalytic sites, despite the asymmetry provided by the stator stalk. Evidently, the non-catalytic α/β interface, falling between two catalytic αβ pairs, provides an unobtrusive placement for this asymmetric feature. However, in this context it is important to note that, although the α subunit cross-links specifically to bI, the β subunit may cross-link to either bI or bII, depending on the presence of an additional cysteine residue in subunit α (Figs. 6 and 7). These observations argue in favor of bI being located at the α/β subunit interface, whereas bII may be along the adjacent surface of β. It should be noted that the recent 10-Å cryoelectron microscopic structure of the T. thermophilus A-type ATPase (11) showed each of the two heterodimeric EG stator stalks to be positioned along the outer surfaces of non-catalytic B subunits, with interactions only between subunits B and E. It seems unlikely to us that a similar arrangement of the α and b subunits of E. coli FOF1 would yield the results we obtained, in particular the b-β disulfide cross-links, suggesting that some variation in stator placement may exist within the family of rotary ATPases. Subunit bII may be less closely associated with α3β3 than bI, given the lower levels of cross-linking to β that was observed for most positions, but one cannot draw firm conclusions from weak or negative cross-linking results.
Subunit bI has also been shown to be proximal to subunit δ. Comparing the peripheral stalks in bacterial FOF1 complexes to that of mitochondria, it can be inferred that subunit bI is functionally related to the single subunit b present in mitochondrial ATP synthase, which makes the major contact to OSCP (17, 18). Subunit bII might have a more stabilizing effect, like mitochondrial subunits d and F6, which appear to stiffen the continuous α helix of subunit b through their shorter structural elements (18).
Staggered versus Non-staggered, In-register Conformation of the b Dimer
A staggered assembly within the dimerization domain of the b dimer with an offset of ∼5.5 amino acid residues has been shown by different experimental approaches (20, 36, 37). The more N-terminally shifted b subunit (bN) was shown to be more important for binding of subunit δ or the F1 complex than the b subunit in the bC position (37). Therefore, it is reasonable to propose that the N-terminally shifted bN position corresponds to the bI subunit described in this study. If one compares the positions on α and β that formed disulfides with particular positions of b, there is in general a trend that the positions on β are located farther up the surface of α3β3 than those on α, consistent with bII being C-shifted and bI being N-shifted.
The bL16C-aV239C Cross-link
Identification of a high-yield disulfide formation between aV239C and bL16C was quite unexpected. It is generally accepted that bLeu-16 is located within the lipid core of the membrane (24), with bTyr-24 and bTrp-26 at the cytoplasmic membrane/water interface (71). In addition, the binding characteristics of mAb GDH 1-4D3 revealed that at least a part of region b32–41 is shielded by the membrane, most probably by the polar head groups of the phospholipids. Although a structure for subunit a is still missing, Fillingame and co-workers (Ref. 64 and references therein) have analyzed the four C-terminal transmembrane helices of subunit a in great detail. In their topology model, residue aVal-239 is located close to the periplasmic surface of the membrane so that, even assuming the two helices to be adjacent, bL16C and aV239C would still be several angstroms apart. At this point we can only speculate on the explanation for such efficient cross-linking. Possibly, the membrane-spanning b helices may also be present in a staggered conformation, as proposed for the dimerization region. This would place bL16C of one subunit closer to the periplasmic surface. Alternatively, thermal fluctuations in the membrane domains may be greater than ordinarily expected. Finally, some modification of the topological models may be in order. In each case, additional studies examining these possibilities are required. In this context, it is worthwhile mentioning that a recent study of DeLeon-Rangel et al. (60) suggests that the two b subunits show individual interactions with different transmembrane helices of the four helix bundles of subunit a.
Acknowledgments
We thank Patrick Bischoff, Julika Pulst, Bettina Rieger, Stefanie Treidel, and Stephanie Konrad for generating some of the plasmids used for cross-linking experiments and Dr. Siegfried Engelbrecht for providing the structural homology model of the E. coli FOF1 ATP synthase. We also thank Drs. Robert D. Simoni, Masamitsu Futai, Brian D. Cain, and Steven B. Vik for providing plasmids and strains used for cloning or determination of the antigenic epitopes within subunit b.
This work was supported by Deutsche Forschungsgemeinschaft Grant DE482/1-1 (to G. D.-H.) and by Canadian Institutes of Health Research Grant MT-10237 (to S. D. D.).

This article contains supplemental Tables S1 and S2 and references.
- OSCP
- oligomycin-sensitivity conferring protein
- DCCD
- N,N′-dicyclohexylcarbodiimide
- CuP
- copper-1,10-phenanthroline
- ACMA
- 9-amino-6-chloro-2-methoxyacridine.
REFERENCES
- 1. Junge W., Sielaff H., Engelbrecht S. (2009) Torque generation and elastic power transmission in the rotary FOF1-ATPase. Nature 459, 364–370 [DOI] [PubMed] [Google Scholar]
- 2. von Ballmoos C., Wiedenmann A., Dimroth P. (2009) Essentials for ATP synthesis by F1FO ATP synthases. Annu. Rev. Biochem. 78, 649–672 [DOI] [PubMed] [Google Scholar]
- 3. von Ballmoos C., Cook G. M., Dimroth P. (2008) Unique rotor ATP synthase and its biological diversity. Annu. Rev. Biophys. 37, 43–64 [DOI] [PubMed] [Google Scholar]
- 4. Cross R. L., Müller V. (2004) The evolution of A-, F-, and V-type ATP synthases and ATPases. Reversals in function and changes in the H+/ATP coupling ratio. FEBS Lett. 576, 1–4 [DOI] [PubMed] [Google Scholar]
- 5. Muench S. P., Trinick J., Harrison M. A. (2011) Structural divergence of the rotary ATPases. Q. Rev. Biophys. 44, 311–356 [DOI] [PubMed] [Google Scholar]
- 6. Diepholz M., Venzke D., Prinz S., Batisse C., Flörchinger B., Rössle M., Svergun D. I., Böttcher B., Féthière J. (2008) A different conformation of EGC stator subcomplex in solution and in the assembled yeast V-ATPase: Possible implications for regulatory disassembly. Structure 16, 1789–1798 [DOI] [PubMed] [Google Scholar]
- 7. Oot R. A., Huang L.-S., Berry E. A., Wilkens S. (2012) Crystal structure of the yeast vacuolar ATPase heterodimeric EGChead peripheral stalk complex. Structure 20, 1881–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kish-Trier E., Wilkens S. (2009) Domain architecture of the stator complex of the A1AO-ATP synthase from Thermoplasma acidophilum. J. Biol. Chem. 284, 12031–12040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee L. K., Stewart A. G., Donohoe M., Bernal R. A., Stock D. (2010) The structure of the peripheral stalk of Thermus thermophilus H+-ATPase/synthase. Nat. Struct. Mol. Biol. 17, 373–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stewart A. G., Lee L. K., Donohoe M., Chaston J. J., Stock D. (2012) The dynamic stator stalk of rotary ATPases. Nat. Commun. 3, 687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lau W. C., Rubinstein J. L. (2012) Subnanometre-resolution structure of the intact Thermus thermophilus H+-driven ATP synthase. Nature 481, 214–218 [DOI] [PubMed] [Google Scholar]
- 12. Herrmann R. G., Steppuhn J., Herrmann G. S., Nelson N. (1993) The nuclear-encoded polypeptide Cfo-II from spinach is a real, ninth subunit of chloroplast ATP synthase. FEBS Lett. 326, 192–198 [DOI] [PubMed] [Google Scholar]
- 13. Dunn S. D., Kellner E., Lill H. (2001) Specific heterodimer formation by the cytoplasmic domains of the b and b′ subunits of cyanobacterial ATP synthase. Biochemistry 40, 187–192 [DOI] [PubMed] [Google Scholar]
- 14. Gabellini N., Gao Z., Eckerskorn C., Lottspeich F., Oesterhelt D. (1988) Purification of the H+-ATPase from Rhodobacter capsulatus, identification of the F1FO components and reconstitution of the active enzyme. Biochim. Biophys. Acta 934, 227–234 [Google Scholar]
- 15. Dunn S. D., Cipriano D. J., Del Rizzo P. A. (2004) in Handbook of ATPases. Biochemistry, Cell Biology, Pathophysiology (Futai M., Wada Y., Kaplan J. H., eds), pp. 311–338, Wiley-VCH, Weinheim, Germany [Google Scholar]
- 16. Peng G., Bostina M., Radermacher M., Rais I., Karas M., Michel H. (2006) Biochemical and electron microscopic characterization of the F1FO ATP synthase from the hyperthermophilic eubacterium. Aquifex aeolicus. FEBS Lett. 580, 5934–5940 [DOI] [PubMed] [Google Scholar]
- 17. Dickson V. K., Silvester J. A., Fearnley I. M., Leslie A. G., Walker J. E. (2006) On the structure of the stator of the mitochondrial ATP synthase. EMBO J. 25, 2911–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rees D. M., Leslie A. G., Walker J. E. (2009) The structure of the membrane extrinsic region of bovine ATP synthase. Proc. Natl. Acad. Sci. U.S.A. 106, 21597–21601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Claggett S. B., Grabar T. B., Dunn S. D., Cain B. D. (2007) Functional incorporation of chimeric b subunits into F1FO ATP synthase. J. Bacteriol. 189, 5463–5471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Claggett S. B., O'Neil Plancher M., Dunn S. D., Cain B. D. (2009) The b subunits in the peripheral stalk of F1FO ATP synthase preferentially adopt an offset relationship. J. Biol. Chem. 284, 16531–16540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wilkens S., Capaldi R. A. (1998) ATP synthase's second stalk comes into focus. Nature 393, 29. [DOI] [PubMed] [Google Scholar]
- 22. Weber J. (2006) ATP synthase. Subunit-subunit interaction in the stator stalk. Biochim. Biophys. Acta 1757, 1162–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Revington M., McLachlin D. T., Shaw G. S., Dunn S. D. (1999) The dimerization domain of the b subunit of the Escherichia coli F1FO-ATPase. J. Biol. Chem. 274, 31094–31101 [DOI] [PubMed] [Google Scholar]
- 24. Dmitriev O., Jones P. C., Jiang W., Fillingame R. H. (1999) Structure of the membrane domain of subunit b of the Escherichia coli FOF1 ATP synthase. J. Biol. Chem. 274, 15598–15604 [DOI] [PubMed] [Google Scholar]
- 25. Stalz W.-D., Greie J.-C., Deckers-Hebestreit G., Altendorf K. (2003) Direct interaction of subunits a and b of the FO complex of Escherichia coli ATP synthase by forming an ab2 subcomplex. J. Biol. Chem. 278, 27068–27071 [DOI] [PubMed] [Google Scholar]
- 26. Sorgen P. L., Caviston T. L., Perry R. C., Cain B. D. (1998) Deletions in the second stalk of F1FO-ATP synthase in Escherichia coli. J. Biol. Chem. 273, 27873–27878 [DOI] [PubMed] [Google Scholar]
- 27. Sorgen P. L., Bubb M. R., Cain B. D. (1999) Lengthening the second stalk of F1FO ATP synthase in Escherichia coli. J. Biol. Chem. 274, 36261–36266 [DOI] [PubMed] [Google Scholar]
- 28. Grabar T. B., Cain B. D. (2003) Integration of b subunits of unequal lengths into F1FO-ATP synthase. J. Biol. Chem. 278, 34751–34756 [DOI] [PubMed] [Google Scholar]
- 29. McLachlin D. T., Coveny A. M., Clark S. M., Dunn S. D. (2000) Site-directed cross-linking of b to the α, β, and a subunits of the Escherichia coli ATP synthase. J. Biol. Chem. 275, 17571–17577 [DOI] [PubMed] [Google Scholar]
- 30. McLachlin D. T., Bestard J. A., Dunn S. D. (1998) The b and δ subunits of the Escherichia coli ATP synthase interact via residues in their C-terminal regions. J. Biol. Chem. 273, 15162–15168 [DOI] [PubMed] [Google Scholar]
- 31. McLachlin D. T., Dunn S. D. (2000) Disulfide linkage of the b and δ subunits does not affect the function of the Escherichia coli ATP synthase. Biochemistry 39, 3486–3490 [DOI] [PubMed] [Google Scholar]
- 32. Del Rizzo P. A., Bi Y., Dunn S. D., Shilton B. H. (2002) The “second stalk” of Escherichia coli ATP synthase. Structure of the isolated dimerization domain. Biochemistry 41, 6875–6884 [DOI] [PubMed] [Google Scholar]
- 33. Priya R., Tadwal V. S., Roessle M. W., Gayen S., Hunke C., Peng W. C., Torres J., Grüber G. (2008) Low resolution structure of subunit b (b22–156) of Escherichia coli F1FO ATP synthase in solution and the b-δ assembly. J. Bioenerg. Biomembr. 40, 245–255 [DOI] [PubMed] [Google Scholar]
- 34. Priya R., Biukovic G., Gayen S., Vivekanandan S., Grüber G. (2009) Solution structure, determined by nuclear magnetic resonance, of the b30–82 domain of subunit b of Escherichia coli F1FO ATP synthase. J. Bacteriol. 191, 7538–7544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Walshaw J., Woolfson D. N. (2001) Open-and-shut cases in coiled coil assembly. α-sheets and α-cylinders. Protein Sci. 10, 668–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Del Rizzo P. A., Bi Y., Dunn S. D. (2006) ATP synthase b subunit dimerization domain. A right-handed coiled coil with offset helices. J. Mol. Biol. 364, 735–746 [DOI] [PubMed] [Google Scholar]
- 37. Wood K. S., Dunn S. D. (2007) Role of the asymmetry of the homodimeric b2 stator stalk in the interaction with the F1 sector of Escherichia coli ATP synthase. J. Biol. Chem. 282, 31920–31927 [DOI] [PubMed] [Google Scholar]
- 38. Wise J. G., Vogel P. D. (2009) Accommodating discontinuities in dimeric left-handed coiled coils in ATP synthase external stalks. Biophys. J. 96, 2823–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fillingame R. H., Jiang W., Dmitriev O. Y. (2000) Coupling H+ transport to rotary catalysis in F-type ATP synthases. Structure and organization of the transmembrane rotary motor. J. Exp. Biol. 203, 9–17 [DOI] [PubMed] [Google Scholar]
- 40. Jones P. C., Hermolin J., Jiang W., Fillingame R. H. (2000) Insights into the rotary catalytic mechanism of FOF1 ATP synthase from the cross-linking of subunits b and c in the Escherichia coli enzyme. J. Biol. Chem. 275, 31340–31346 [DOI] [PubMed] [Google Scholar]
- 41. Grabar T. B., Cain B. D. (2004) Genetic complementation between mutant b subunits in F1FO ATP synthase. J. Biol. Chem. 279, 31205–31211 [DOI] [PubMed] [Google Scholar]
- 42. Motz C., Hornung T., Kersten M., McLachlin D. T., Dunn S. D., Wise J. G., Vogel P. D. (2004) The subunit b dimer of the FOF1-ATP synthase. Interaction with F1-ATPase as deduced by site-specific spin-labeling. J. Biol. Chem. 279, 49074–49081 [DOI] [PubMed] [Google Scholar]
- 43. Birkenhäger R., Greie J.-C., Altendorf K., Deckers-Hebestreit G. (1999) FO complex of the Escherichia coli ATP synthase. Not all monomers of the subunit c oligomer are involved in F1 interaction. Eur. J. Biochem. 264, 385–396 [DOI] [PubMed] [Google Scholar]
- 44. Klionsky D. J., Brusilow W. S., Simoni R. D. (1984) In vivo evidence for the role of the ϵ subunit as an inhibitor of the proton-translocating ATPase of Escherichia coli. J. Bacteriol. 160, 1055–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Porter A. C., Kumamoto C., Aldape K., Simoni R. D. (1985) Role of the b subunit of the Escherichia coli proton-translocating ATPase. A mutagenic analysis. J. Biol. Chem. 260, 8182–8187 [PubMed] [Google Scholar]
- 46. Takeyama M., Noumi T., Maeda M., Futai M. (1988) FO portion of Escherichia coli H+-ATPase. Carboxyl-terminal region of the b subunit is essential for assembly of functional FO. J. Biol. Chem. 263, 16106–16112 [PubMed] [Google Scholar]
- 47. Moriyama Y., Iwamoto A., Hanada H., Maeda M., Futai M. (1991) One-step purification of Escherichia coli H+-ATPase (FOF1) and its reconstitution into liposomes with neurotransmitter transporters. J. Biol. Chem. 266, 22141–22146 [PubMed] [Google Scholar]
- 48. Ishmukhametov R. R., Galkin M. A., Vik S. B. (2005) Ultrafast purification and reconstitution of His-tagged cysteine-less Escherichia coli F1FO ATP synthase. Biochim. Biophys. Acta 1706, 110–116 [DOI] [PubMed] [Google Scholar]
- 49. Ballhausen B., Altendorf K., Deckers-Hebestreit G. (2009) Constant c10 ring stoichiometry in the Escherichia coli ATP synthase analyzed by cross-linking. J. Bacteriol. 191, 2400–2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Steffens K., Schneider E., Deckers-Hebestreit G., Altendorf K. (1987) FO portion of Escherichia coli ATP synthase. Further resolution of trypsin-generated fragments from subunit b. J. Biol. Chem. 262, 5866–5869 [PubMed] [Google Scholar]
- 51. Friedl P., Friedl C., Schairer H. U. (1979) The ATP synthetase of Escherichia coli K12. Purification of the enzyme and reconstitution of energy-transducing activities. Eur. J. Biochem. 100, 175–180 [DOI] [PubMed] [Google Scholar]
- 52. McKinney M. M., Parkinson A. (1987) A simple, non-chromatographic procedure to purify immunoglobulins from serum and ascites fluid. J. Immunol. Methods 96, 271–278 [DOI] [PubMed] [Google Scholar]
- 53. Deckers-Hebestreit G., Simoni R. D., Altendorf K. (1992) Influence of subunit-specific antibodies on the activity of the FO complex of the ATP synthase of Escherichia coli. I. Effects of subunit b-specific polyclonal antibodies. J. Biol. Chem. 267, 12364–12369 [PubMed] [Google Scholar]
- 54. Krebstakies T., Aldag I., Altendorf K., Greie J.-C., Deckers-Hebestreit G. (2008) The stoichiometry of subunit c of Escherichia coli ATP synthase is independent of its rate of synthesis. Biochemistry 47, 6907–6916 [DOI] [PubMed] [Google Scholar]
- 55. Dulley J. R., Grieve P. A. (1975) A simple technique for eliminating interference by detergents in the Lowry method of protein determination. Anal. Biochem. 64, 136–141 [DOI] [PubMed] [Google Scholar]
- 56. Schägger H., von Jagow G. (1987) Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166, 368–379 [DOI] [PubMed] [Google Scholar]
- 57. Jäger H., Birkenhäger R., Stalz W.-D., Altendorf K., Deckers-Hebestreit G. (1998) Topology of subunit a of the Escherichia coli ATP synthase. Eur. J. Biochem. 251, 122–132 [DOI] [PubMed] [Google Scholar]
- 58. Neidhardt F. C., Ingraham J. L., Schaechter M. (1990) Physiology of the bacterial cell. A molecular approach. Sinauer Associates, Sunderland, MA [Google Scholar]
- 59. McLachlin D. T., Dunn S. D. (1997) Dimerization interactions of the b subunit of the Escherichia coli F1FO-ATPase. J. Biol. Chem. 272, 21233–21239 [DOI] [PubMed] [Google Scholar]
- 60. DeLeon-Rangel J., Ishmukhametov R. R., Jiang W., Fillingame R. H., Vik S. B. (2013) Interactions between subunits a and b in the rotary ATP synthase as determined by cross-linking. FEBS Lett. 587, 892–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Heukeshoven J., Dernick R. (1988) Improved silver staining procedure for fast staining in PhastSystem development unit. I. Staining of sodium dodecyl sulfate gels. Electrophoresis 9, 28–32 [DOI] [PubMed] [Google Scholar]
- 62. Kuo P. H., Ketchum C. J., Nakamoto R. K. (1998) Stability and functionality of cysteine-less FOF1-ATP synthase from Escherichia coli. FEBS Lett. 426, 217–220 [DOI] [PubMed] [Google Scholar]
- 63. Cingolani G., Duncan T. M. (2011) Structure of the ATP synthase catalytic complex (F1) from Escherichia coli in an auto-inhibited conformation. Nat. Struct. Mol. Biol. 18, 701–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dong H., Fillingame R. H. (2010) Chemical reactivities of cysteine substitutions in subunit a of ATP synthase define residues gating H+ transport from each side of the membrane. J. Biol. Chem. 285, 39811–39818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang D., Vik S. B. (2003) Close proximity of a cytoplasmic loop of subunit a with c subunits of the ATP synthase from. Escherichia coli. J. Biol. Chem. 278, 12319–12324 [DOI] [PubMed] [Google Scholar]
- 66. Kumamoto C. A., Simoni R. D. (1986) Genetic evidence for interaction between the a and b subunits of the FO portion of the Escherichia coli proton translocating ATPase. J. Biol. Chem. 261, 10037–10042 [PubMed] [Google Scholar]
- 67. Dunn S. D., Heppel L. A., Fullmer C. S. (1980) The NH2-terminal portion of the α subunit of Escherichia coli F1 ATPase is required for binding the δ subunit. J. Biol. Chem. 255, 6891–6896 [PubMed] [Google Scholar]
- 68. Weber J., Muharemagic A., Wilke-Mounts S., Senior A. E. (2004) Analysis of sequence determinants of F1FO-ATP synthase in the N-terminal region of α subunit for binding of δ subunit. J. Biol. Chem. 279, 25673–25679 [DOI] [PubMed] [Google Scholar]
- 69. Wilkens S. (2000) F1FO-ATP synthase. Stalking mind and imagination. J. Bioenerg. Biomembr. 32, 333–339 [DOI] [PubMed] [Google Scholar]
- 70. Zimmermann B., Diez M., Zarrabi N., Gräber P., Börsch M. (2005) Movements of the ϵ-subunit during catalysis and activation in single membrane-bound H+-ATP synthase. EMBO J. 24, 2053–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Killian J. A., von Heijne G. (2000) How proteins adapt to a membrane-water interface. Trends Biochem. Sci. 25, 429–434 [DOI] [PubMed] [Google Scholar]