

Background: Foxp3 post-translational regulation remains unclear.
Results: Foxp3 is phosphorylated by cyclin-dependent kinase 2 (CDK2) and has increased stability and function without its CDK motifs.
Conclusion: CDK2 is a negative regulator of Foxp3 function.
Significance: Understanding how Foxp3 is stabilized could lead to new therapeutic measures for autoimmunity and transplantation.
Keywords: CDK (Cyclin-dependent Kinase), Protein Phosphorylation, T Cell, T Cell Biology, Transcription Factors, Foxp3
Abstract
Foxp3 is a transcription factor required for the development of regulatory T cells (Treg). Mice and humans with a loss of Foxp3 function suffer from uncontrolled autoimmunity and inflammatory disease. Expression of Foxp3 is necessary for the anti-inflammatory capacity of Treg, but whether Foxp3 activity is further subject to regulation by extracellular signals is unclear. The primary structure of Foxp3 contains four cyclin-dependent kinase (CDK) motifs (Ser/Thr-Pro) within the N-terminal repressor domain, and we show that CDK2 can partner with cyclin E to phosphorylate Foxp3 at these sites. Consistent with our previous demonstration that CDK2 negatively regulates Treg function, we find that mutation of the serine or threonine at each CDK motif to alanine (S/T→A) results in enhanced Foxp3 protein stability in CD4+ T cells. T cells expressing the S/T→A mutant of Foxp3 showed enhanced induction (e.g. CD25) and repression (e.g. IL2) of canonical Foxp3-responsive genes, exhibited an increased capacity to suppress conventional T cell proliferation in vitro, and were highly effective at ameliorating colitis in an in vivo model of inflammatory bowel disease. These results indicate that CDK2 negatively regulates the stability and activity of Foxp3 and implicate CDK-coupled receptor signal transduction in the control of regulatory T cell function and stability.
Introduction
Foxp3, a forkhead box transcription factor, mediates the development of regulatory T cells (Treg),2 which are involved in immune regulation and tolerance (1–5). Foxp3 translocates to the nucleus to bind DNA and induce (CD25, Gitr, Ctla4) or repress (Il2, Il4, Ifnγ) the expression of various genes. Foxp3 regulates accessibility of these target genes by recruitment of chromatin remodeling factors (6, 7). The activity of many transcription factors that contribute to T cell differentiation and function is controlled by antigen, costimulatory, or growth factor receptor signaling (8); however, to what extent extracellular signals regulate Foxp3 function is largely unclear. A number of studies have recently described Foxp3 post-translational modification. Acetylated Foxp3 was first identified in response to TGFβ signaling (9). TIP60 (Tat-interactive protein, 60 kDa), p300, and sirtuin-1 were later also shown to regulate the acetylation status of Foxp3 (7, 10–12). Acetylation of Foxp3 affects its stability (11) and ability to bind to promoters (9). Other stimuli, such as hypoxia, also affect Foxp3 protein expression. The hypoxia-induced factor 1α (HIF-1α) can regulate Foxp3 ubiquitination and stability in developing Th17 (T helper 17) cells (13). Whether Foxp3 is regulated post-translationally by a specific kinase cascade, however, is not known.
The primary amino acid sequence of murine Foxp3 contains multiple putative kinase motifs, including four cyclin-dependent kinase (CDK) substrate motifs concentrated within the N-terminal repressor domain. Our previous studies showed that cyclin-dependent kinase 2 (Cdk2)-deficient Treg are more suppressive than wild type Treg, as measured by the ability to suppress the proliferation of conventional CD4+ T cells in vitro and to ameliorate colitis in an in vivo mouse model of inflammatory bowel disease (14). These findings demonstrate an important role for CDK2 in Treg biology, but did not establish the mechanism by which CDK2 functions in these cells. Considering the presence of multiple CDK motifs in the Foxp3 primary sequence, we hypothesized that CDK2 may influence Treg function through phosphorylation-dependent regulation of Foxp3.
In this study, we find that CDK2 can phosphorylate Foxp3 and that mutation of the N-terminal CDK motifs increases the half-life, steady-state level, and transcriptional activity of Foxp3. Furthermore, T cells expressing CDK mutant Foxp3 exhibit increased suppressive function as compared with cells expressing wild type Foxp3. Our results indicate that CDK2 activity controls Treg suppressive function through setting the amount of Foxp3 available in the cell.
EXPERIMENTAL PROCEDURES
Mice
Female C57BL/6 (H-2b) and Rag1-deficient mice on a C57BL/6 background were purchased from The Jackson Laboratory and maintained in our specific pathogen-free facility according to University Laboratory Animal Resources (ULAR)- and Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC)-approved institutional guidelines on animal care and usage. All mice were used at 6–14 weeks of age.
Full-length Foxp3 Purification
Murine Foxp3 cDNA was amplified from wild type C57BL/6 thymus using the following primers: Foxp3-Fwd, 5-ccggaattcatgcccaaccctaggccag-3′, Foxp3-Rev, 5-ccgctcgagtcaagggcagggattggagc-3′. The PCR-amplified DNA was then cloned into the protein expression plasmid His6-pET-28a (Novagen). Escherichia coli strain BL21 was transformed with His6-Foxp3-pET28a. A single bacterial colony was inoculated in LB containing kanamycin (50 μg/ml). The bacterial culture was grown at 37 °C, and Foxp3 expression was induced with isopropyl β-d-1-thiogalactopyranoside (0.5 mm, 12 h, 30 °C). For the purification of the His6-Foxp3 recombinant protein, cells were harvested from 500-ml culture, and the recombinant protein was purified using nickel-nitrilotriacetic acid resin by affinity chromatography under native conditions.
In Vitro Kinase Assay
0.5 μg of His6-Foxp3 or 1 μg of control histone H1 (Roche Applied Science) was incubated with CDK2-cycE (Millipore) and 5 μCi of [γ-32P]ATP (PerkinElmer) in kinase buffer for 30 min at 30 °C. The reaction was stopped with 1× SDS-PAGE lysis buffer and analyzed by SDS-PAGE. The gel was dried and exposed to x-ray film. Roscovitine (Cell Signaling) was used to inhibit CDK2 activity.
Mass Spectrometry
His6-Foxp3 was incubated with CDK2-cycE and cold ATP and analyzed by SDS-PAGE. The Foxp3 band was then excised, destained, reduced with DTT, alkylated with iodoacetamide (Sigma-Aldrich), and digested in-gel with trypsin. The extracted tryptic peptides were analyzed by reverse phase HPLC mass spectrometry using an LTQ-Orbitrap mass spectrometer. The assignment of phosphate to product ions identified was made using a Mascot search of MS2 data against the Oniprot mouse protein sequence database.
Site-directed Mutagenesis
Ser/Thr→Ala-Foxp3 mutant was generated using the Stratagene QuikChange II site-directed mutagenesis kit. Mutagenesis primers used were: S19A-Fwd, 5′-cttggcccttggcccagccccaggag-3′, S19A-Rev, 5′-ctcctggggctgggccaagggccaag-3′, S88A-Fwd, 5′-ccgactaggtcccgcaccccacctaca-3′, S88A-Rev, 5′-tgtaggtggggtgcgggacctagtcgg-3, T114A-Fwd, 5′-gcccatgcccaggcccctgtgctcc-3′, T114A-Rev, 5′-ggagcacaggggcctgggcatgggc-3′, T175A-Fwd, 5′-cccacgctcgggtgcacccaggaaaga-3′, T175A-Rev, 5′-tctttcctgggtgcacccgagcgtggg-3′.
In Vivo Phosphorylation
HEK293 cells were transfected (Lipofectamine 2000, Invitrogen). 48 h after transfection, cells were harvested, lysed, and subjected to FLAG immunoprecipitation according to the manufacturer's protocol (Sigma-Aldrich). Lysis buffer was supplemented with protease and phosphatase inhibitor mixture (Sigma-Aldrich) and β-glycerophosphate (Sigma-Aldrich). Where indicated, cells were treated with the proteasome inhibitor MG132 (Sigma-Aldrich) and the CDK2 inhibitor roscovitine (Sigma-Aldrich). Affinity-purified p+Ser-19-Foxp3 rabbit polyclonal antibody (YenZym) was used to detect phosphorylated species of Foxp3.
Peptide Dot Blot
Lyophilized Foxp3 peptide (aa 12–24), either unmodified or phosphorylated at serine 19, was resuspended at 10 mg/ml and serially diluted with PBS. Amounts of peptide from 500 μg to 0.05 ng were placed on nitrocellulose in log10 dilutions. Western blot was then performed using p+Ser-19-Foxp3 (YenZym).
Retroviral Transduction
Murine Foxp3 cDNA was amplified from wild type C57BL/6 thymus and cloned into the murine stem cell virus-based retroviral vector (6) expressing the reporter gene GFP (MIGR) or human NGFR (MINR1) and containing an in-frame, N-terminal FLAG epitope. For generation of retrovirus, constructs were cotransfected (Lipofectamine 2000, Invitrogen) with the pCLeco (Invitrogen) helper plasmid into the 293T-based Phoenix ecotropic packaging cell line (provided by G. Nolan, Stanford University). Wild type C57BL/6 CD4+CD25− naive T cells were activated with phorbol 12-myristate 13-acetate (3 ng/ml, Sigma-Aldrich), ionomycin (1 μm, Sigma-Aldrich), and IL-2 (10 units/ml, Roche Applied Science) for 24 h, washed, and transduced by spinfection (15) with supernatants from 48 h transfected Phoenix cells. Transduced cells were expanded in IL-2 for 3–4 days. CD4+ T cell transduction efficiencies were ≥90%. Where necessary, transduced T cells were purified to near 100% purity using vector reporter markers.
FACS, ELISA, Quantitative PCR, and Western Blot Analysis of Transduced Cells
Before analysis, transduced cells were restimulated 4–6 h with 1 μg/ml CD3 (2C11, Bio X Cell), 1 μg/ml CD28 (37.51, Bio X Cell), and IL-2 (10 units/ml, Roche Applied Science). FACS analysis was performed on a Beckman Coulter CyAn ADP. Cells were stained for FACS analysis with conjugated mouse anti-Foxp3-APC (FJK-16s, eBioscience), mouse anti-CD25-APCcy7 (PC61, BioLegend), anti-NGFR-PE (CD271, BD Pharmingen), and anti-CD4-PacificBlue (GK1.5, BioLegend). Supernatants from restimulated cells were harvested and probed for levels of IL-2 protein by ELISA according to the manufacturer's protocol (eBioscience kit). Manufacturers' protocols were followed for kits to extract RNA from transduced cells (Qiagen) and convert it into cDNA (Bio-Rad). Relative Foxp3 and IL-2 message levels were determined following quantitative PCR according to the 2ΔΔCt method after normalization to actin. Whole cell extracts were generated using the radioimmune precipitation buffer lysis kit (Sigma-Aldrich) and run on precast 10% SDS-PAGE Criterion gels (Bio-Rad). Western blots were performed using mouse/rat anti-Foxp3 (FJK-16s, eBioscience) and rabbit anti-p+Ser-19-Foxp3 (YenZym).
Stability Assay
Following restimulation of transduced cells with CD3, CD28, and IL-2, cells were harvested, washed, and replated in the presence of cycloheximide (25 μg/ml, Sigma). Cells were harvested at the indicated time points, and whole cell extracts were prepared and analyzed for expression of Foxp3 by Western blot as described above.
In Vitro Treg Suppression
CD4+CD25− (Tconv) and CD90-negative APC were isolated from splenocytes of wild type C57BL/6 mice using magnetic bead-conjugated mAbs (Miltenyi). APC (1 × 105) were irradiated (1000 radians, 3 min) and plated onto 96-well round-bottomed plates along with Tconv effector cells (5 × 104/well) labeled with CellTrace (Invitrogen Molecular Probes, violet carboxyfluorescein succinimidyl ester equivalent) and 4 μg/ml soluble anti-CD3 mAb. T cells transduced with empty vector control, wild type Foxp3, or Ser/Thr→Ala-Foxp3 (serving as suppressor Treg) cells were added to each well in varying ratios to Tconv cells and cultured for 72 h. After 3 days, suppression of responder cell proliferation was measured by flow cytometry to assess the degree of inhibition of CellTrace (Invitrogen, violet carboxyfluorescein succinimidyl ester equivalent) dilution as previously published (16).
Adoptive Transfer Colitis
To induce experimental colitis, conventional CD4+25− T cells were purified from naive, wild type C57BL/6 mice and adoptively transferred (1 × 106, intravenously) into Rag1−/− B6 recipients (17, 18). 21 days from the initial transfer, groups of 3–7 mice then received either PBS or T cells transduced with empty vector control, wild type Foxp3, or Ser/Thr→Ala-Foxp3 mutant (2 × 106, intraperitoneally). Recipients were weighed and observed for symptoms of diarrhea approximately every 2 days. At the end of the experiment, spleens, mesenteric lymph nodes, and intestines were harvested for examination of gross pathology, and histology.
Gross Pathology
Colitis-induced animals were sacrificed and analyzed for signs of gross pathology using a modified version of established methods (19). Scores of 0 (no colitis) to 4 (worst disease) were assigned according to colon rigidity, visible inflammation, and presence of blood in intestines, as well as diarrhea and presence of fat tissue.
Histopathology
Intestines from colitis-induced animals were fixed in formaldehyde, imbedded in paraffin, sliced, stained with hematoxylin and eosin, and mounted onto glass slides for histological analysis. Blinded evaluation of H&E-stained paraffin sections was performed by a pathologist and scored using established criteria (20) to reach a maximum colitis score of 23. The criteria and scores were: inflammation, 0–3; mucin depletion, 0–2; reactive epithelial changes, 0–3; number of intraepithelial lymphocytes, 0–3; crypt architectural distortion, 0–3; number of inflammatory foci per 10 high power field, 1–3; inflammatory activity, 0–2; transmural inflammation, 0–2; mucosal ulceration, 0–2.
Statistical Analysis
All p values were calculated by Student's paired t test using Prism software (GraphPad).
RESULTS
Foxp3 Is Phosphorylated by CDK2
We analyzed the Foxp3 amino acid sequence as a primary approach to identify potential sites of post-translational modification. We found many putative kinase binding elements, including a cluster of four cyclin-dependent kinase (CDK) motifs, Ser/Thr-Pro (Fig. 1). We recently demonstrated that cyclin-dependent kinase 2 is a negative regulator of Foxp3+ Treg function, but a biochemical basis was not established (14). The presence of multiple CDK motifs in Foxp3 suggests that CDK2 might control Treg function through a phosphorylation-dependent mechanism. The Foxp3 CDK motifs are concentrated in the N-terminal half of the protein. This segment of Foxp3 (aa 1–198) is necessary and sufficient for repression of the Il2 gene and is therefore referred to as a repressor domain (21, 22). This domain contains no defined structural motifs, so how it contributes to Foxp3 function is not understood.
FIGURE 1.
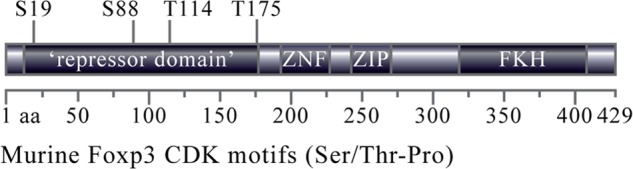
Murine Foxp3 contains four putative CDK consensus binding sites. Analysis of the murine Foxp3 sequence revealed the presence of four putative cyclin-dependent kinase motifs (Ser/Thr-Pro (S/T-P)) in the N-terminal repressor domain. Also shown are the Foxp3 zinc finger (ZNF), leucine zipper (ZIP), and forkhead domains (FKH).
To test whether CDK2 can phosphorylate Foxp3, we purified full-length recombinant Foxp3 (Fig. 2A) and performed standard in vitro kinase assays. Recombinant CDK2 together with its G1/S phase binding partner cyclin E was able to phosphorylate Foxp3, and the reaction could be abrogated by the CDK inhibitor roscovitine (Fig. 2B). To map the residues phosphorylated by CDK2-cyclin E, we performed the kinase reaction with cold ATP and subjected the purified Foxp3 substrate to RP-HPLC mass spectrometry (MS). Product ions b14 and y9 from the peptide PAKPMAPSLALGPSPGVLPSWK (Fig. 2C) contain an 80-dalton increase in their expected mass-to-charge (m/z) ratio, indicative of a phosphate group at the serine followed by the proline (aa 19). Product ions containing only the other serine residues in the peptide do not contain an 80-dalton increase in m/z and are therefore not phosphorylated (Fig. 2C, supplemental Fig. S1a). An 80-dalton increase is also found in the fragment ion b5 from the peptide SGTPRKDSNLLAAPQGSYPLLANGVCK (Fig. 2D, supplemental Fig. S1b). The absence of earlier b-series ions or later y-series ions for this peptide precludes specific assignment of the phosphate group to the first serine (aa 173) or threonine (aa 175) residue in the peptide. However, CDK2 is an obligate proline-directed kinase, and therefore must be acting on threonine 175. The phosphorylation of the other two CDK target residues, serine 88 and threonine 114, was not determined as peptides containing these residues could not be isolated during enzymatic digestion preceding RP-HPLC. None of the other serine or threonine residues in Foxp3 outside these motifs were phosphorylated by CDK2 (data not shown). These data suggest that Foxp3 is phosphorylated by CDK2 on at least two of its CDK motifs, Ser-19 and Thr-175.
FIGURE 2.

Foxp3 is phosphorylated by CDK2 in vitro. A, recombinant His6-Foxp3 was purified as described under “Experimental Procedures.” IPTG, isopropyl β-d-1-thiogalactopyranoside; Ni-affinity, nickel affinity. B, a standard in vitro kinase assay was performed using His6-Foxp3 (500 ng) or control substrate histone H1 (1 μg) and recombinant CDK2-cycE. Roscovitine was used to inhibit CDK2 activity. The autoradiograms shown are representative of three separate experiments. C and D, purified His6-Foxp3 was digested with trypsin, and the resulting peptides were analyzed by RP-HPLC mass spectrometry. Tandem MS spectra for Foxp3 phospho-peptides shown. b- and y-series ions are shown in red and blue, respectively, and ions involving neutral loss of the elements of phosphoric acid, water, or ammonia are shown in green. Phosphorylation site assignments are made for serine 19 (C) and threonine 175 (D). The data shown are from two separate mass spectrometry runs with parent error <5 ppm. AMU, atomic mass units.
We next generated an antibody that recognizes an N-terminal Foxp3 peptide, aa 12–24, PSLALGPpSPGVLP, containing phosphorylated serine 19. To confirm that the antibody detects only phosphorylated serine 19, we performed a dot blot using serial dilutions of unphosphorylated and phosphorylated peptide (Fig. 3A). Our results demonstrate that our antibody recognizes only p+Ser-19, with the limit of detection in the nanogram range. To determine whether Foxp3 can be phosphorylated on Ser-19 in an in vivo, cellular environment, we transfected HEK293 cells with a control FLAG-tagged vector, wild type Foxp3, or a mutant of Foxp3 in which the serine or threonine of all four CDK motifs was mutated to alanine (Ser/Thr→Ala). After 48 h, transfected cells were harvested and lysed in the presence of both phosphatase and proteasome inhibitors. FLAG immunoprecipitation was performed on all lysates. Whole cell extracts and FLAG immunoprecipitation eluate were analyzed by Western blot for phosphorylated and total Foxp3 (Fig. 3B). Wild type Foxp3 transfected cell lysates and FLAG immunoprecipitation eluates contain a species of Foxp3 phosphorylated on Ser-19, as detected using the p+Ser-19-Foxp3 antibody. No phosphorylated Foxp3 was detected in extracts from Ser/Thr→Ala mutant Foxp3. These data demonstrate that Foxp3 is phosphorylated at Ser-19 CDK motif by endogenous cellular machinery.
FIGURE 3.
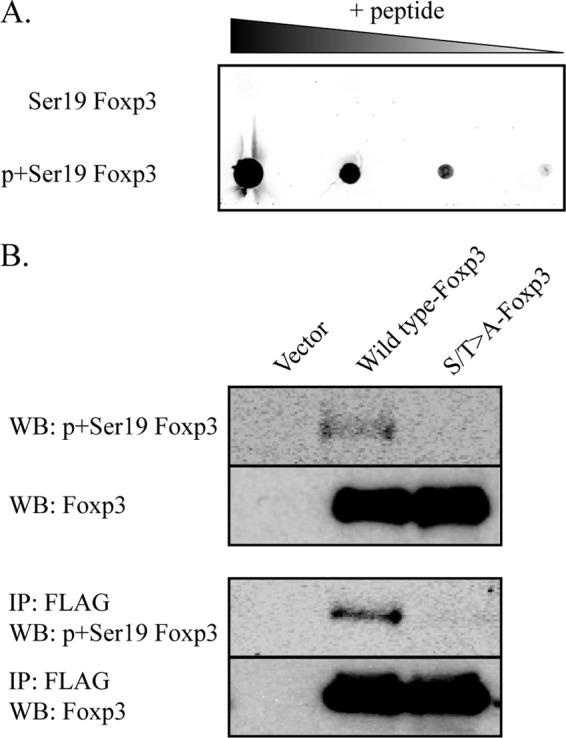
Foxp3 is phosphorylated at CDK motifs in vivo. A, Foxp3 peptide (aa 12–24) containing either unmodified or phosphorylated serine 19 was serially diluted and used for a dot blot assay. Membranes containing diluted peptides were probed with p+Ser-19-Foxp3 antibody. B, HEK293 cells were transfected with vector control, wild type Foxp3, or S/T→A-Foxp3 mutant. Whole cell extracts were prepared at 48 h after transfection. FLAG immunoprecipitation (IP) and Western blotting (WB) were performed as described under “Experimental Procedures.” Phosphorylated and total Foxp3 is shown. Data are representative of three separate experiments.
CDK Motifs Contribute to Foxp3 Protein Stability
Kinase activity and phosphorylation can be coupled to protein stability (23). To test whether Foxp3 N-terminal CDK motifs contribute to protein stability, conventional CD4+CD25− T lymphocytes were transduced with murine stem cell virus-based retroviral vectors encoding wild type Foxp3 or the Ser/Thr→Ala-Foxp3 mutant (Fig. 4A). We treated empty vector-, wild type Foxp3-, or Ser/Thr→Ala-Foxp3-transduced CD4+ T cells with cycloheximide (CHX) and measured Foxp3 protein levels over time (Fig. 4B). Wild type Foxp3 was expressed at high levels following transduction; however, inhibition of protein synthesis with CHX revealed rapid turnover of Foxp3 within 5 h with a half-life of 2 h. Ser/Thr→Ala-Foxp3 exhibited increased protein stability; following treatment with CHX, the S/T→A(ble)-Foxp3 mutant showed a significantly increased half-life of 7–9 h as compared with wild type Foxp3 (Fig. 4C). Importantly, the transcription of S/T→A(ble)-Foxp3 is not elevated over that of wild type Foxp3 (Fig. 4D). These data indicate that CDK phosphorylation motifs are involved in the regulation of Foxp3 protein stability.
FIGURE 4.
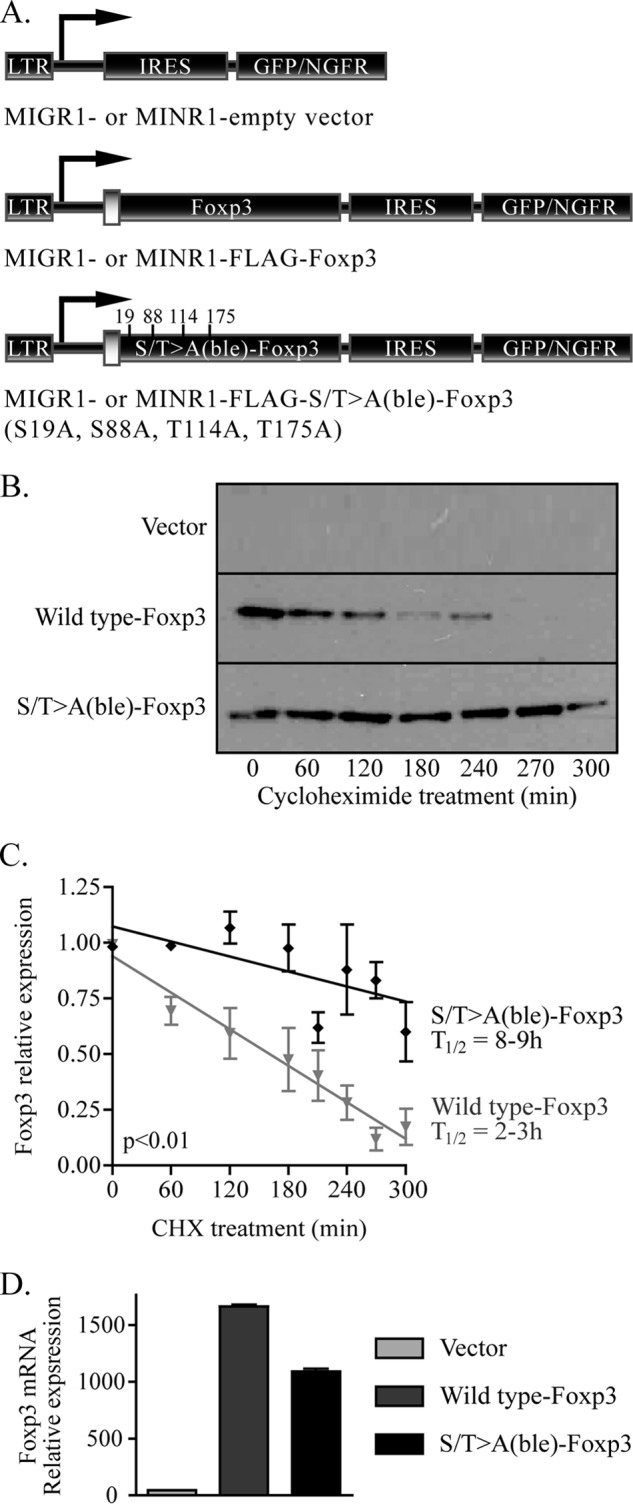
Foxp3 lacking CDK consensus elements has increased protein stability. A, CD4+25− T lymphocytes were transduced with vector control, wild type Foxp3, or the S/T→A-Foxp3 mutant. LTR, long terminal repeat; IRES, internal ribosomal entry site. B, transduced cells were rested overnight and restimulated for 4 h with plate-bound CD3 and CD28 antibodies, incubated with 25 μg/ml CHX, and harvested at the indicated times. Whole cell extracts from treated cells were analyzed by SDS-PAGE, blotted to membranes, and probed with Foxp3 antibody. One representative image of three experiments is shown. C, average stability of wild type and S/T→A-Foxp3 over three experiments is shown. Half-life was calculated based on these values. Error bars are S.E. for biological replicate cultures. D, Foxp3 transcription normalized to actin is shown. Message level is represented in arbitrary units, before the addition of CHX to transduced cells.
CDK Motifs Are Important for Foxp3-dependent Genetic Events
Foxp3 drives a specific transcriptional program including the induction of genes such as the α chain of the IL-2 receptor, CD25, and the repression of cytokine genes such as Il2. To determine whether these CDK motifs affect the transcriptional activity of Foxp3, we compared CD4+25− naive T cells transduced with either wild type or S/T→A(ble)-Foxp3 mutant. Wild type Foxp3 induced an expected Treg-like signature (1) including high Foxp3 expression, elevated CD25 levels, and decreased IL-2 production as compared with cells transduced with empty vector. Mutation of all four CDK motifs resulted in significant elevation of Foxp3 and CD25 protein levels (Fig. 5A), as well as enhanced repression of IL-2 message (Fig. 5B) and protein (Fig. 5C) as compared with wild type. These data suggest that the CDK motifs negatively regulate Foxp3 function.
FIGURE 5.
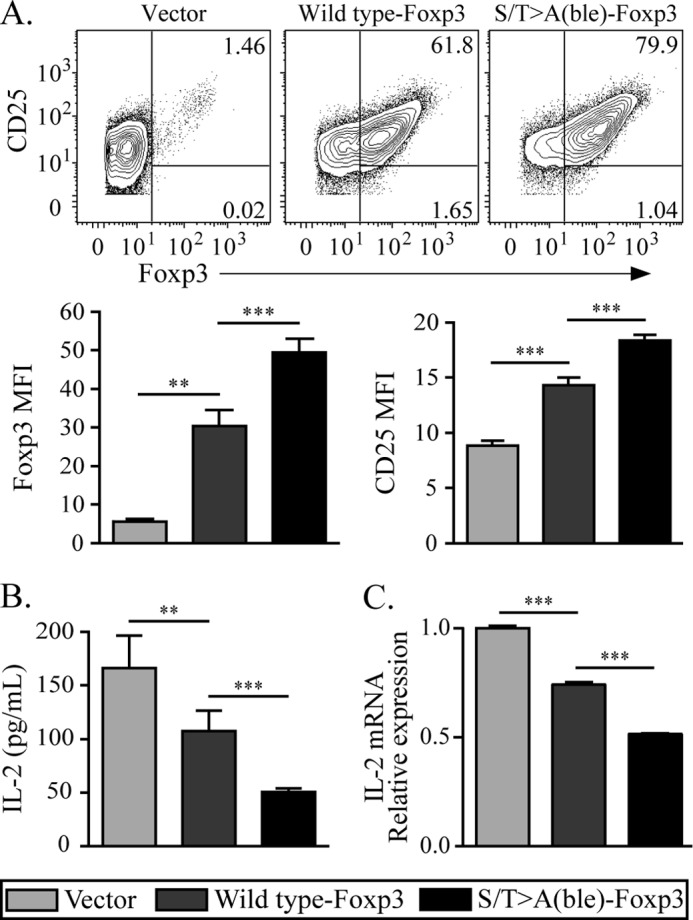
Mutation of the N-terminal CDK motifs results in enhanced Foxp3 expression and transcriptional function. A, CD4+25− T lymphocytes were transduced with wild type or S/T→A(ble) Foxp3, restimulated as in Fig. 4, and Foxp3 and CD25 expression was assessed by flow cytometry. The mean fluorescence intensity (MFI) of each parameter was calculated and graphed. B, supernatants from restimulated cells in A were assessed for IL-2 production by ELISA. C, Il2 transcription normalized to actin is shown. Message level is represented in arbitrary units. The data shown are representative of at least two experiments. Error bars are S.E. for biological replicate cultures. **, p < 0.01, ***, p < 0.001.
S/T→A(ble)-Foxp3-expressing Cells Have Increased Suppressive Function
Mutation of the N-terminal CDK motifs in Foxp3 resulted in elevated protein stability and increased Foxp3 transcriptional activity. Therefore, we next asked whether T cells expressing the S/T→A(ble) mutant of Foxp3 exhibited a gain of suppressive function. To test this, we used a standard, anti-CD3-driven in vitro Treg suppression assay (24). CD4+ T cells expressing wild type Foxp3 were able to suppress the proliferation of conventional T cells as compared with empty vector-transduced control cells. However, cells expressing the S/T→A(ble) mutant showed significantly elevated suppressive capacity as compared with cells expressing wild type (Fig. 6). From these results, we conclude that the N-terminal CDK motifs restrain Foxp3 activity and suppressive function.
FIGURE 6.
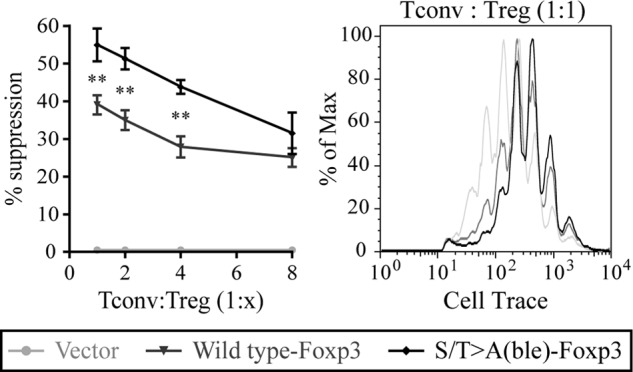
S/T→A(ble)-Foxp3 exhibits enhanced suppressive function in vitro. CD4+25− T lymphocytes transduced with wild type or S/T→A(ble) Foxp3 were used as suppressors in a standard in vitro Treg suppression assay against conventional CD4+25− T cells labeled with CellTrace to track cell division. Suppressors and effectors were cultured with irradiated wild type APCs and soluble CD3 for 72 h. Proliferation data shown are gated on CD4+ lymphocytes. Percentage of suppression was calculated as described previously (16). Data are representative of two separate experiments. Error bars are S.E. for biological replicate cultures. **, p < 0.01, ***, p < 0.001.
To test whether the N-terminal CDK motifs contribute to the anti-inflammatory capacity of Foxp3-expressing cells in vivo, we used an adoptive transfer model of inflammatory bowel disease (17, 18). Immunodeficient Rag1−/− mice were injected with congenically marked (CD45.1+) naive CD4+CD25− T cells and allowed to develop colitis highlighted by diarrhea and weight loss (Fig. 7A). 3 weeks after the initial injection, animals were adoptively transferred with congenically marked (CD45.2+) CD4+ T cells transduced with wild type or S/T→A(ble) mutant Foxp3. Control mice receiving either PBS or empty vector-transduced cells developed a severe wasting disease including significant and rapid weight loss (Fig. 7A). These animals had Foxp3+ cells in the mesenteric lymph nodes (Fig. 7B), but the expression was restricted to a CD45.1+NGFR− pool (data not shown), representing a population of in vivo induced Foxp3+ cells, which were not sufficient to ameliorate disease. Control mice also developed diarrhea, intestinal wall thickening, and intestinal inflammation and ulceration, including destruction of intestinal architecture as measured by gross and histopathological analysis (Fig. 7, C and D). As shown previously (1), mice treated with transduced CD4+ T cells expressing wild type Foxp3 regained weight, no longer exhibited diarrhea, showed decreased intestinal thickness and rigidity, and showed decreased inflammation and restored intestinal architecture. Mice that received S/T→A(ble) mutant-expressing cells experienced a similar degree of weight gain and restored intestinal architecture as compared with wild type treated mice. However, animals that received S/T→A mutant-expressing cells exhibited significantly less colon rigidity and hemorrhaging and increased mesenteric fat tissue mass as compared with mice receiving wild type Foxp3 transduced cells (Fig. 7D). This decrease in gross pathology was accompanied by an elevated frequency of Foxp3+ donor cells in the mesenteric lymph nodes of S/T→A(ble)-Foxp3 treated animals (Fig. 7B). In addition, mice receiving mutant-expressing cells exhibited fewer outward signs of morbidity; they were more ambulant and had normal grooming behaviors (data not shown), whereas animals that received wild type Foxp3-expressing cells were more hunched, less active, and did not exhibit normal grooming behavior. These data demonstrate that Foxp3 CDK motifs are not required for the suppressive activity of Foxp3-expressing cells in vivo, but rather, function to temper Treg anti-inflammatory activity.
FIGURE 7.

Anti-inflammatory activity of S/T→A(ble) mutant Foxp3 in vivo. Rag1−/− mice were injected intravenously with 1 × 106 conventional CD4+25− T lymphocytes to induce colitis. After 21 days, mice were administered a therapeutic injection intraperitoneally with 2 × 106 transduced cells (vector, wild type Foxp3, or S/T→A(ble)-Foxp3) or a PBS control. A, mice were weighed every 2–3 days until day 70 after initial injection. B, mice were sacrificed, and mesenteric lymph node cells were harvested and stained for CD4 and Foxp3. Shown is the percentage of Foxp3+ gated on CD4+ lymphocytes. *, p < 0.05, ***, p < 0.001. C, large intestines were harvested and prepared for histological analysis, and histopathological scoring was performed blinded. D, gross pathological analysis of sacrificed mice was performed and scored as described under “Experimental Procedures.” Each group contained 3–7 mice.
DISCUSSION
Foxp3 is a transcription factor necessary for the development of regulatory T cell suppressive function (1, 3–5). It recruits chromatin remodeling factors to the nucleus to induce or repress the expression of multiple genes (6), but to what extent Foxp3 is subject to regulation by extracellular signals remains unclear. Foxp3 can be acetylated by Tip60 and p300, increasing its stability (7, 10, 11). This effect is countered by the histone deacetylase, sirtuin-1 (10, 12), which negatively affects Foxp3+ Treg function (25). The histone acetyl transferase p300 competes for lysine residues with E3 ubiquitin ligase activity; acetylation or mutation of target lysine residues to arginine increases Foxp3 stability by inhibiting ubiquitination (10). Foxp3 degradation can also be directed by hypoxia-induced factor 1α through a ubiquitin-mediated mechanism. This occurs under hypoxic conditions during Th17 development (13). Phosphorylation of Foxp3 was recently shown to occur at serine 418, affecting Foxp3 transcriptional activity (26). However, the kinase responsible for this modification was not identified. We previously showed that CDK2 opposes Treg suppressive function both in vitro and in vivo, but did not determine a mechanism by which CDK2 acts in Treg (14). We hypothesize that CDK2 modifies Foxp3 and controls Treg function by a phosphorylation-dependent mechanism. Our analysis reveals four putative CDK motifs in the Foxp3 amino acid sequence. We demonstrate that CDK2 phosphorylates Foxp3 and that the CDK motifs oppose Foxp3 protein stability and activity. This suggests that Foxp3 is subject to regulation by a kinase cascade and that the effects of this phosphorylation oppose its function.
Cyclin-dependent kinases are ubiquitously expressed, growth factor receptor-coupled enzymes primarily thought to drive cell cycle progression (27, 28). CDK2 and its binding partner cyclin E were initially described to be indispensable for progression past the G1/S phase checkpoint of cell cycle (29). CDK1, however, is now known to be sufficient for cell cycle progression in most cell types and can compensate for the absence of other interphase CDKs, (30–32). For example, Cdk2−/− mice are viable and present virtually no defect in mouse embryonic fibroblast cell cycle progression (33, 34). T lymphocyte development (35) and proliferation (14) are also normal in the absence of CDK2. These findings suggest that CDK2 could have cell cycle-independent roles in T lymphocytes. We have previously identified such a role, showing that CDK2 regulates Treg suppressive function (14). Considering our current data, we predict that CDK2 affects Treg function through phosphorylation and destabilization of Foxp3. However, our studies do not rule out whether other CDKs are acting on Foxp3 as they all recognize the same motif. More work will need to be performed to dissect the effect of each CDK on Foxp3.
Our data suggest that Foxp3 is a CDK2 substrate in T lymphocytes, but when is CDK2 active in the cell? Thymus-derived regulatory T cells have stable long term expression of Foxp3 (36) and may not be subject to regulation by CDK. Interestingly, a thymic Treg does repress Cdk1, Cdk2, and Cdk6 expression (37). This could be an adapted mechanism to regulate these kinase cascades and maintain Foxp3 stability. Foxp3 can also be induced peripherally in conventional T cells (38), providing an influx of suppressive cells. CDK2 is active in conventional T cells (14), driving differentiation and cytokine production, and our current data suggest that CDK2 negatively affects Foxp3 stability. Therefore, we predict that to generate an extrathymic Treg, the cell must not only induce Foxp3, but must also oppose CDK2 activity. Consistent with this idea, TGFβ signaling not only induces Foxp3 expression (38), but is known to induce p27Kip1, an inhibitor of CDK2 (39, 40). In murine B cells, TGFβ induces p27Kip1, which results in increased association with CDK2 and decreased CDK2 kinase activity (40). We hypothesize that TGFβ-induced p27kip1 reinforces the extrathymic Treg cell fate by opposing CDK2-mediated destabilization of Foxp3.
How are CDK motifs involved in regulating Foxp3 stability and function? One possibility is through phosphorylation-dependent ubiquitination. There are numerous examples of this type of cross-talk, in which phosphorylation at one residue can prime ubiquitination at a nearby lysine. Such peptides are referred to as phospho-degrons (23). CDK2 phosphorylation has already been linked to this process, pairing if with the E3 ligase SCF/Fwb7 to target cyclin E for degradation (41). We propose that the CDK motifs and nearby lysine residues cooperate to form a phospho-degron that regulates phosphorylation-dependent ubiquitination and degradation of Foxp3. This hypothesis is consistent with our data showing that S/T→A(ble)-Foxp3 has increased stability as compared with wild type Foxp3. It is also possible that the Foxp3 CDK motifs are involved in subnuclear localization or nuclear export, leading to subsequent cytoplasmic degradation. The CDK2 inhibitor, p27, is degraded in this manner (42, 43). Either of these possibilities is consistent with our data.
Our data show that CDK2 negatively regulates Treg suppressive function (14), likely through phosphorylation-dependent destabilization of Foxp3. Furthermore, we propose that Foxp3 CDK motifs act to moderate the cell-extrinsic anti-inflammatory activity of Foxp3. Modulating CDK2 activity could therefore be used to affect the balance between immunity and tolerance. Roscovitine (seliciclib), a pharmacologic inhibitor that targets CDK2, CDK7, and CDK9 (44), is in clinical trials to treat cancer (45) and was shown to ameliorate graft-versus-host disease in mice (46). CDK2 may therefore be a relevant target in the treatment of autoimmunity or during transplantation.
Acknowledgment
We thank Steven Seeholzer for helpful discussion of mass spectrometry data.
This work was supported, in whole or in part, by National Institutes of Health Grant R01-AI054643-06 (to A. D. W.). This work was also supported by the 2011 Goldie Simon Preceptorship Award sponsored by the Lupus Foundation of America Philadelphia Tri-State Chapter and the Pennsylvania Department of Health (to P. A. M.).

This article contains supplemental Fig. S1.
- Treg
- regulatory T cell(s)
- CDK
- cyclin-dependent kinase
- HIF-1α
- hypoxia-induced factor 1α
- cycE
- cyclin E
- CHX
- cycloheximide
- NGFR
- nerve growth factor receptor
- p+Ser-19-Foxp3
- phosphorylated Ser-19-Foxp3.
REFERENCES
- 1. Hori S., Nomura T., Sakaguchi S. (2003) Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061 [DOI] [PubMed] [Google Scholar]
- 2. Taylor P. A., Noelle R. J., Blazar B. R. (2001) CD4+CD25+ immune regulatory cells are required for induction of tolerance to alloantigen via costimulatory blockade. J. Exp. Med. 193, 1311–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fontenot J. D., Gavin M. A., Rudensky A. Y. (2003) Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4, 330–336 [DOI] [PubMed] [Google Scholar]
- 4. Khattri R., Cox T., Yasayko S. A., Ramsdell F. (2003) An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 4, 337–342 [DOI] [PubMed] [Google Scholar]
- 5. Kasprowicz D. J., Smallwood P. S., Tyznik A. J., Ziegler S. F. (2003) Scurfin (FoxP3) controls T-dependent immune responses in vivo through regulation of CD4+ T cell effector function. J. Immunol. 171, 1216–1223 [DOI] [PubMed] [Google Scholar]
- 6. Chen C., Rowell E. A., Thomas R. M., Hancock W. W., Wells A. D. (2006) Transcriptional regulation by Foxp3 is associated with direct promoter occupancy and modulation of histone acetylation. J. Biol. Chem. 281, 36828–36834 [DOI] [PubMed] [Google Scholar]
- 7. Li B., Samanta A., Song X., Iacono K. T., Bembas K., Tao R., Basu S., Riley J. L., Hancock W. W., Shen Y., Saouaf S. J., Greene M. I. (2007) FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc. Natl. Acad. Sci. U.S.A. 104, 4571–4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhu J., Paul W. E. (2010) Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol. Rev. 238, 247–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Samanta A., Li B., Song X., Bembas K., Zhang G., Katsumata M., Saouaf S. J., Wang Q., Hancock W. W., Shen Y., Greene M. I. (2008) TGF-β and IL-6 signals modulate chromatin binding and promoter occupancy by acetylated FOXP3. Proc. Natl. Acad. Sci. U.S.A. 105, 14023–14027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kwon H.-S., Lim H. W., Wu J., Schnölzer M., Verdin E., Ott M. (2012) Three novel acetylation sites in the Foxp3 transcription factor regulate the suppressive activity of regulatory T cells. J. Immunol. 188, 2712–2721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van Loosdregt J., Vercoulen Y., Guichelaar T., Gent Y. Y., Beekman J. M., van Beekum O., Brenkman A. B., Hijnen D.-J., Mutis T., Kalkhoven E., Prakken B. J., Coffer P. J. (2010) Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood 115, 965–974 [DOI] [PubMed] [Google Scholar]
- 12. van Loosdregt J., Brunen D., Fleskens V., Pals C. E. G. M., Lam E. W. F., Coffer P. J. (2011) Rapid temporal control of Foxp3 protein degradation by sirtuin-1. PLoS ONE 6, e19047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dang E. V., Barbi J., Yang H.-Y., Jinasena D., Yu H., Zheng Y., Bordman Z., Fu J., Kim Y., Yen H.-R., Luo W., Zeller K., Shimoda L., Topalian S. L., Semenza G. L., Dang C. V., Pardoll D. M., Pan F. (2011) Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell 146, 772–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chunder N., Wang L., Chen C., Hancock W. W., Wells A. D. (2012) Cyclin-dependent kinase 2 controls peripheral immune tolerance. J. Immunol. 189, 5659–5666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pear W. S., Nolan G. P., Scott M. L., Baltimore D. (1993) Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. U.S.A. 90, 8392–8396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wells A. D., Gudmundsdottir H., Turka L. A. (1997) Following the fate of individual T cells throughout activation and clonal expansion. Signals from T cell receptor and CD28 differentially regulate the induction and duration of a proliferative response. J. Clin. Invest. 100, 3173–3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Powrie F., Leach M. W., Mauze S., Caddle L. B., Coffman R. L. (1993) Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 5, 1461–1471 [DOI] [PubMed] [Google Scholar]
- 18. Powrie F., Leach M. W., Mauze S., Menon S., Caddle L. B., Coffman R. L. (1994) Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1, 553–562 [DOI] [PubMed] [Google Scholar]
- 19. Conner E. M., Brand S., Davis J. M., Laroux F. S., Palombella V. J., Fuseler J. W., Kang D. Y., Wolf R. E., Grisham M. B. (1997) Proteasome inhibition attenuates nitric oxide synthase expression, VCAM-1 transcription, and the development of chronic colitis. J. Pharmacol. Exp. Ther. 282, 1615–1622 [PubMed] [Google Scholar]
- 20. Aranda R., Sydora B. C., McAllister P. L., Binder S. W., Yang H. Y., Targan S. R., Kronenberg M. (1997) Analysis of intestinal lymphocytes in mouse colitis mediated by transfer of CD4+, CD45RBhigh T cells to SCID recipients. J. Immunol. 158, 3464–3473 [PubMed] [Google Scholar]
- 21. Bettelli E., Dastrange M., Oukka M. (2005) Foxp3 interacts with nuclear factor of activated T cells and NF-κB to repress cytokine gene expression and effector functions of T helper cells. Proc. Natl. Acad. Sci. U.S.A. 102, 5138–5143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lopes J. E., Torgerson T. R., Schubert L. A., Anover S. D., Ocheltree E. L., Ochs H. D., Ziegler S. F. (2006) Analysis of FOXP3 reveals multiple domains required for its function as a transcriptional repressor. J. Immunol. 177, 3133–3142 [DOI] [PubMed] [Google Scholar]
- 23. Hunter T. (2007) The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol. Cell 28, 730–738 [DOI] [PubMed] [Google Scholar]
- 24. Thornton A. M., Donovan E. E., Piccirillo C. A., Shevach E. M. (2004) Cutting edge: IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J. Immunol. 172, 6519–6523 [DOI] [PubMed] [Google Scholar]
- 25. Beier U. H., Wang L., Bhatti T. R., Liu Y., Han R., Ge G., Hancock W. W. (2011) Sirtuin-1 targeting promotes Foxp3+ T-regulatory cell function and prolongs allograft survival. Mol. Cell Biol. 31, 1022–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nie H., Zheng Y., Li R., Guo T. B., He D., Fang L., Liu X., Xiao L., Chen X., Wan B., Chin Y. E., Zhang J. Z. (2013) Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-α in rheumatoid arthritis. Nat. Med. 19, 322–328 [DOI] [PubMed] [Google Scholar]
- 27. Kim Y. H., Proust J. J., Buchholz M. J., Chrest F. J., Nordin A. A. (1992) Expression of the murine homologue of the cell cycle control protein p34cdc2 in T lymphocytes. J. Immunol. 149, 17–23 [PubMed] [Google Scholar]
- 28. Kim Y. H., Buchholz M. A., Chrest F. J., Nordin A. A. (1994) Up-regulation of c-myc induces the gene expression of the murine homologues of p34cdc2 and cyclin-dependent kinase-2 in T lymphocytes. J. Immunol. 152, 4328–4335 [PubMed] [Google Scholar]
- 29. Coppock D. L., Buffolino P., Kopman C., Nathanson L. (1995) Inhibition of the melanoma cell cycle and regulation at the G1/S transition by 12-O-tetradecanoylphorbol-13-acetate (TPA) by modulation of CDK2 activity. Exp. Cell Res. 221, 92–102 [DOI] [PubMed] [Google Scholar]
- 30. Aleem E., Kiyokawa H., Kaldis P. (2005) Cdc2-cyclin E complexes regulate the G1/S phase transition. Nat. Cell Biol. 7, 831–836 [DOI] [PubMed] [Google Scholar]
- 31. Bashir T., Pagano M. (2005) Cdk1: the dominant sibling of Cdk2. Nat. Cell Biol. 7, 779–781 [DOI] [PubMed] [Google Scholar]
- 32. Satyanarayana A., Kaldis P. (2009) Mammalian cell-cycle regulation: several Cdks, numerous cyclins, and diverse compensatory mechanisms. Oncogene 28, 2925–2939 [DOI] [PubMed] [Google Scholar]
- 33. Berthet C., Aleem E., Coppola V., Tessarollo L., Kaldis P. (2003) Cdk2 knockout mice are viable. Curr. Biol. 13, 1775–1785 [DOI] [PubMed] [Google Scholar]
- 34. Ortega S., Prieto I., Odajima J., Martín A., Dubus P., Sotillo R., Barbero J. L., Malumbres M., Barbacid M. (2003) Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat. Genet. 35, 25–31 [DOI] [PubMed] [Google Scholar]
- 35. Berthet C., Rodriguez-Galan M. C., Hodge D. L., Gooya J., Pascal V., Young H. A., Keller J., Bosselut R., Kaldis P. (2007) Hematopoiesis and thymic apoptosis are not affected by the loss of Cdk2. Mol. Cell Biol. 27, 5079–5089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rubtsov Y. P., Niec R. E., Josefowicz S., Li L., Darce J., Mathis D., Benoist C., Rudensky A. Y. (2010) Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. König S., Probst-Kepper M., Reinl T., Jeron A., Huehn J., Schraven B., Jänsch L. (2012) First insight into the kinome of human regulatory T cells. PLoS ONE 7, e40896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen W., Jin W., Hardegen N., Lei K. J., Li L., Marinos N., McGrady G., Wahl S. M. (2003) Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med. 198, 1875–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Toyoshima H., Hunter T. (1994) p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 78, 67–74 [DOI] [PubMed] [Google Scholar]
- 40. Kamesaki H., Nishizawa K., Michaud G. Y., Cossman J., Kiyono T. (1998) TGF-β1 induces the cyclin-dependent kinase inhibitor p27Kip1 mRNA and protein in murine B cells. J. Immunol. 160, 770–777 [PubMed] [Google Scholar]
- 41. Koepp D. M., Schaefer L. K., Ye X., Keyomarsi K., Chu C., Harper J. W., Elledge S. J. (2001) Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 294, 173–177 [DOI] [PubMed] [Google Scholar]
- 42. Besson A., Gurian-West M., Chen X., Kelly-Spratt K. S., Kemp C. J., Roberts J. M. (2006) A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev. 20, 47–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nakayama K. I., Nakayama K. (2006) Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer 6, 369–381 [DOI] [PubMed] [Google Scholar]
- 44. De Azevedo W. F., Leclerc S., Meijer L., Havlicek L., Strnad M., Kim S. H. (1997) Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. Eur. J. Biochem. 243, 518–526 [DOI] [PubMed] [Google Scholar]
- 45. Gallorini M., Cataldi A., di Giacomo V. (2012) Cyclin-dependent kinase modulators and cancer therapy. BioDrugs 26, 377–391 [DOI] [PubMed] [Google Scholar]
- 46. Li L., Wang H., Kim J.s., Pihan G., Boussiotis V. (2009) The cyclin dependent kinase inhibitor (R)-roscovitine prevents alloreactive T cell clonal expansion and protects against acute GvHD. Cell Cycle 8, 1794–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]