

Background: Dystrophic skeletal muscles overexpress ankyrin repeat domain protein 2 (ANKRD2), which inhibits myoblast differentiation.
Results: Skeletal muscles of the mdm mouse overexpress ANKRD2 and inhibitor of DNA binding 3 (ID3) proteins, which cooperatively inhibit myoblast differentiation by physical interaction.
Conclusion: Activation of SREBP-1/ANKRD2/ID3 pathway impairs, at least in part, skeletal muscle development in mdm mice.
Significance: We provide evidence revealing a novel mechanism by which expression of ANKRD2 inhibits myoblast differentiation.
Keywords: Molecular Cell Biology, mRNA, Muscular Dystrophy, Myogenesis, Skeletal Muscle, Ankrd2, SREBP-1(Sterol Regulatory Element Binding Protein-1), Helix-Loop-Helix Protein Id3, Mdm Mouse
Abstract
Ankyrin repeat domain protein 2 (ANKRD2) translocates from the nucleus to the cytoplasm upon myogenic induction. Overexpression of ANKRD2 inhibits C2C12 myoblast differentiation. However, the mechanism by which ANKRD2 inhibits myoblast differentiation is unknown. We demonstrate that the primary myoblasts of mdm (muscular dystrophy with myositis) mice (pMBmdm) overexpress ANKRD2 and ID3 (inhibitor of DNA binding 3) proteins and are unable to differentiate into myotubes upon myogenic induction. Although suppression of either ANKRD2 or ID3 induces myoblast differentiation in mdm mice, overexpression of ANKRD2 and inhibition of ID3 or vice versa is insufficient to inhibit myoblast differentiation in WT mice. We identified that ANKRD2 and ID3 cooperatively inhibit myoblast differentiation by physical interaction. Interestingly, although MyoD activates the Ankrd2 promoter in the skeletal muscles of wild-type mice, SREBP-1 (sterol regulatory element binding protein-1) activates the same promoter in the skeletal muscles of mdm mice, suggesting the differential regulation of Ankrd2. Overall, we uncovered a novel pathway in which SREBP-1/ANKRD2/ID3 activation inhibits myoblast differentiation, and we propose that this pathway acts as a critical determinant of the skeletal muscle developmental program.
Introduction
In general, the regeneration ability of normal postnatal skeletal muscle fibers relies on the successful activation of satellite cells into proliferating myoblasts, differentiation of myoblasts into myotubes, and fusion of myotubes with the adjacent muscle fibers. Occurrence of defects in any of these processes severely affects the formation of new muscle fibers. In line with this, studies have shown that impairment in the myogenic program hinders skeletal muscle development in many physiological and experimental conditions (1–4). Although protein catabolism can accelerate skeletal muscle wasting in cancer patients, an impaired myogenic program can also play an important role in muscle wasting in cancer cachexia (5, 6). However, the mechanism that inhibits the myogenic ability of muscle precursor cells is incomplete.
ANKRD2 (ankyrin repeat domain protein 2) is a member of the family of muscle ankyrin repeat proteins, expressed mostly in skeletal muscles. In proliferating myoblasts, ANKRD2 generally resides in the nucleus, whereas in differentiating myoblasts, ANKRD2 begins to accumulate in the cytoplasm (7). Enforced expression of ANKRD2 in C2C12 myoblasts inhibits differentiation (7, 8). However, the mechanism by which ANKRD2 inhibits myoblast differentiation is unknown. Moreover, data from microarray analyses have shown that inhibition of MyoD in myogenic C2C12 cells down-regulates ANKRD2 expression (9, 10). We have previously shown NF-κB-mediated regulation of ANKRD2 in mouse diaphragm muscle after a mechanical stimulus (11). These results suggest that both basal expression and differential regulation of ANKRD2 in skeletal muscles are a complex process with many unknown details.
ID3 (inhibitor of DNA binding 3) belongs to the family of ID proteins containing four distinct genes, Id1 through Id4, in humans and mice. These proteins contain a helix-loop-helix (HLH)2 structural motif that facilitates formation of heterodimers with the ubiquitous basic HLH transcription factors known as E-proteins (12, 13). Sequestration of E-proteins prevents them from forming transcriptionally active dimers with tissue-specific basic HLH proteins. ID proteins are transcriptional regulators that influence the proliferation and differentiation of many cell types including skeletal muscle cells (14, 15). Developing somites, activated satellite cells, and proliferating and terminally differentiated myoblasts express ID3 (16–19). These studies suggest that ID3 plays a key role in the skeletal muscle development especially in the activation and proliferation of muscle precursor cells. However, little is known about the role of ID3 and its interaction with other proteins in the skeletal muscle myogenic program.
SREBPs (sterol regulatory element binding proteins) belong to the basic helix-loop-helix leucine zipper family of DNA-binding proteins such as MyoD. In mammals, two distinct genes encode SREBP isoforms, SREBP-1 and SREBP-2, each with distinct structural, regulatory, and functional features (20). The SREBP-1 gene encodes SREBP-1a and SREBP-1c proteins via promoters. Like liver and adipose tissues (21, 22), skeletal muscles express high levels of SREBP-1 proteins (23–25). Whereas SREBP-1 regulates genes that are associated with lipid metabolism, recent studies have shown the role of SREBP-1 in skeletal muscle development. For example, microarray analysis of human myotubes over expressing SREBP-1a or SREBP-1c identified many potential targets of SREBP-1 proteins, including a number of muscle-specific genes and markers of muscle differentiation (26). Whereas SREBP-1 regulates genes that are associated with lipid metabolism, recent studies have shown a role for SREBP-1 in skeletal muscle development. Interestingly, enforced expression of SREBP-1 proteins in human myoblasts inhibit their differentiation, and expression of SREBP-1 proteins in mature myotubes in vitro and in mouse skeletal muscle in vivo can induce muscle atrophy (27). These studies suggest the negative role of SREBP-1 in skeletal muscle development. However, how SREBP-1 proteins inhibit skeletal muscle development is yet to be uncovered.
To explore the mechanism of ANKRD2 regulation and its role in skeletal muscle pathogenicity, we have used the skeletal muscles of mdm (muscular dystrophy with myositis) mice. We show that the skeletal muscles of mdm mice overexpress ANKRD2 and ID3 proteins, which cooperatively inhibit myoblast differentiation by physical interaction. Although MyoD regulates ANKRD2 in the skeletal muscle of WT mice, we show that SREBP-1 regulates ANKRD2 in mdm skeletal muscle, suggesting that differential regulation of ANKRD2 significantly affects muscle regeneration in this model of muscular dystrophy.
EXPERIMENTAL PROCEDURES
Primary Myoblast Isolation
The mdm (B6.B6C3Fe-Ttnmdm-J/Cx) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). The animal protocol for our experiments was approved by the Animal Care and Use Committee of the Baylor College of Medicine. Heterozygous (Ttnmdm/+) breeder pairs were used to generate homozygous WT (Ttn+/+) and mdm (Ttnmdm/mdm) mice (28). Gastrocnemius, tibialis anterior, and soleus muscles were excised from the hind limb muscle of 4-week-old wild-type and mdm mice. The excised skeletal muscles were weighed and enzymatically dissociated by incubating 30 min in a solution containing 1.5 units/ml collagenase D, 2.4 units/ml dispase II, and 2.5 mm CaCl2 per gram tissue at 37 °C with 75 rpm in a rotation incubator. The resulting suspension was filtered through 50-μm nylon mesh, centrifuged for 5 min at 350 × g, and resuspended in F-12-based primary myoblast growth medium (GM; 80% Ham's F-12, 20% fetal calf serum, 0.025% basic fibroblast growth factor in 0.5% BSA, 100 units/ml penicillin, and 100 μg/ml streptomycin). After cell counting, 4000–10,000 cells were cultured in a 60-mm collagen-coated culture dish containing GM at 37 °C and 5% CO2. At 70–80% confluents, cells were dislodged in PBS with no trypsin or EDTA by rocking the dish firmly. (This treatment facilitates the myoblasts to come off freely and leave most of the fibroblasts in the dish.) The myoblasts were plated in a new dish and cultured in F-12 GM. This step was repeated for the first week of culture expansion or until most of the fibroblasts were gone from the culture. After the fibroblasts were no longer visible in the culture, the medium was changed to F-12/DMEM-based growth medium (50% F-12 growth medium and 50% DMEM). The existence of myoblasts was confirmed by immunofluorescent staining for desmin.
Construction of Expression Plasmids
Sense and antisense Ankrd2 (1100 bp) and sense Id3 (960 bp) cDNAs were synthesized and cloned into pcDNA 3.1D/V5-His-TOPO vector (Invitrogen) according to the manufacturer's instructions. PCRs were performed to synthesis inserts with AccuPrime Pfx DNA polymerase according to the manufacturer's instructions (Invitrogen). Primer information is detailed in Table 1. Constructs were sequenced by the DNA Sequence Core Facility of Baylor College of Medicine to verify insert identities.
TABLE 1.
Primers used in PCR
| Primer name | Sequence (5′–3′) | Underlined residues | Purpose |
|---|---|---|---|
| ANKRD2 sense (forward) | CCGGAATTCATGGAGGGTACCATGGAGGGG | EcoRI | Cloning |
| ANKNRD2 sense (reverse) | AACCCGGCTCGAGCGGCCGCGAGCAAAGCCAGCACTTTATTG | NotI | Cloning |
| ANKRD2 anti-sense (forward) | AACCCGGCTCGAGCGGCCGCATGGAGGGTACCATGGAGGG′ | EcoRI | Cloning |
| ANKNRD2 anti-sense (reverse) | CCGGAATTCAGCAAAGCCAGCACTTTATTG′ | NotI | Cloning |
| ID3 sense (forward) | CCGGAATTCTGTTTGCTGCTTTAGGTGTCTCTTTT | EcoRI | Cloning |
| ID3 sense (reverse) | CCCGGCTCGAGCGGCCGCATAAACCATTTTTGAACACTTTG | NotI | Cloning |
| ANKRD2 (forward) | GCCTCGGGGTTCAGAGTCCT | qPCR | |
| ANKRD2 (reverse) | GATCTCACGTCGCAGGTCCA | qPCR | |
| ID3 (forward) | ACCTGGAGCCCGAGAGAAGG | qPCR | |
| ID3 (reverse) | AGGGTGGGGACAGAGTGACG | qPCR |
ChIP Assays
ChIP assays were performed in the skeletal muscles of WT and mdm mice as described previously (11).
Transfection and Luciferase Assay
Cells were grown in Opti-MEM I medium (Invitrogen) for 24 h before transfection. Cells were transfected with 2.2 μg of pcDNA expression vector bearing Ankrd2 or Id3 using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. For siRNA-mediated knockdown studies, cells were transfected with 500 pmol of siRNA specific for mouse ID3, MyoD, SREBP-1, or nonspecific siRNA (Santa Cruz Biotechnology). RNA transfection studies were performed with Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. After 8 h, the transfection medium was replaced with the growth medium. Subsequent assays were made after 24 to 48 h of transfection.
Immunocytochemistry
Primary myoblasts were cultured on sterile glass coverslips. At 60–70% confluence, the cells were gently washed twice in PBS, fixed, and permeabilized with methanol-acetone (1:1) at −20 °C for 5 min. After washing twice in PBS, the cells were blocked in 1% normal goat serum for 1 h at room temperature. The cells were incubated in primary antibody (rabbit anti-mouse tubulin) for 2 h at room temperature. After washing in PBS, the cells were incubated in secondary antibody (goat anti-rabbit) conjugated with Alexa Fluor 547 for 2 h at room temperature followed by washing in PBS. The cells then were mounted with DAPI (nuclear stain) based mounting media for fluorescent microscope analysis.
RT-PCR
Real-time RT-PCRs were performed as described previously (11). The amount of amplified transcripts (2−ΔCT) was estimated by the comparative CT (ΔCT) method and normalized to an endogenous reference (GAPDH) relative to a calibrator. All PCR products were verified on agarose gel stained with ethidium bromide to discriminate between the correct amplification products and the potential primer dimers.
Western Blot
Western blots were performed as described previously (11). Anti-ANKRD2 (sc-138111), anti-MyoD (sc-377186), anti-Myf5 (sc-302), anti-myogenin (sc-52903), anti-ID3 (sc-490), anti-SREBP-1 (sc-8984), or anti-tubulin (sc-53646) antibodies were used to detect respective proteins. All of the antibodies were purchased from Santa Cruz Biotechnology.
In Vitro Pulldown Assay
After transfection, cells were collected by gentle scraping and lysed in radioimmune precipitation assay buffer containing PMSF and protease inhibitor mixture. After centrifugation, supernatants were precleared with blocked protein G beads (Sigma) for 1 h at 4 °C. Target protein-protein complexes were immunoprecipitated using either 10 μg of anti-ANKRD2, anti-His, or anti-ID3 antibody (Santa Cruz Biotechnology) overnight at 4 °C with rotation. Immunocomplexes were captured by incubating with blocked protein G-agarose/Sepharose bead (Sigma) and enhanced by adding a bridging antibody to the other samples (Pierce Biotechnology). Agarose/Sepharose beads were collected by centrifugation and immunoprecipitated antibody-protein-protein complexes eluted with sample buffer and dissociated by boiling. Supernatants were then transferred to fresh micro centrifuge tubes and used in appropriate Western blotting.
Statistical Analysis
All experiments were repeated at least three times. In addition, assays producing quantitative data were run in triplicate. Statistical significance was determined by one-way analysis of variance followed by Bonferroni test or the unpaired Student's t test, as appropriate. The criterion for significance (α) was set at 0.05.
RESULTS
Myoblasts from the Skeletal Muscles of mdm Mice Display Impaired Differentiation Program
Studies have shown that the myogenic ability of muscle precursor cells becomes progressively reduced in muscular dystrophies (1, 2). Because mdm mice have severe muscular dystrophy, we sought to determine whether the primary myoblasts of mdm mice (pMBmdm) exhibit a normal differentiation program. Surprisingly, culture of pMBmdm in differentiation media (DM) were unable to generate large multinucleated myotubes (Fig. 1A) and showed a significant decrease in total myotube numbers (Fig. 1B) compared with similar culture of wild-type (pMBWT) myoblasts. These results suggest that the severe skeletal muscle wasting and muscular dystrophy in mdm mice could be due to, at least in part, the impaired differentiation program of the myoblasts. The coordinated expressions and functions of the myogenic regulators MyoD, myogenin, and Myf5 are crucial for the normal differentiation program (29–32). We therefore examined the expression levels of myogenic regulators in myogenic-induced pMBWT and pMBmdm. Although there were no apparent changes in the expression levels of myogenin and Myf5 between pMBWT and pMBmdm, the MyoD expression levels were markedly altered in pMBmdm (Fig. 1C). However, the MyoD levels were still detectable in pMBmdm. These results indicate that the impaired differentiation program in the pMBmdm may be due to dysregulation of proteins other than the myogenic regulators.
FIGURE 1.
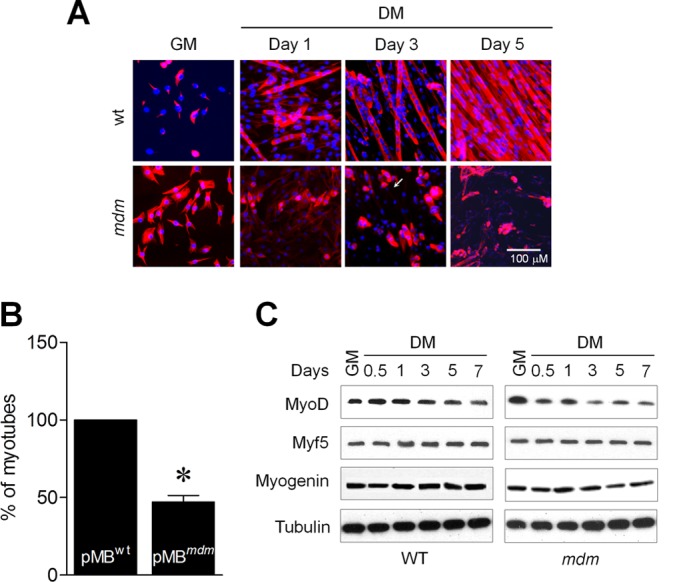
Myoblasts of mdm mice show impaired-differentiation program. A, pMBWT and pMBmdm were cultured in DM for 5 days. Formation of myotubes was determined by immunocytochemistry. B, the number of myotube formations were counted on day 3 in pMBWT and pMBmdm cultured in DM. C, Western blot analyses showed MyoD, myogenin, and Myf5 expressions between pMBWT and pMBmdm during differentiation. Gel and immunocytochemistry pictures are representative of three separate experiments. Each error bar indicates mean ± S.E. (n = 3). *, p < 0.05.
Overexpression of ANKRD2 Inhibits Differentiation of pMBmdm
Our earlier study has shown up-regulation of ANKRD2 in the diaphragm muscle of mdm mice (11). ANKRD2 negatively regulates myoblast differentiation (7, 8) and is under the control of MyoD (9). These results suggest the possibility that the impaired differentiation program in pMBmdm may be due to the overexpression of ANKRD2. To explore this, we first determined the kinetics of ANKRD2 expression in the skeletal muscles of WT and mdm mice at different ages. We excised different hind limb skeletal muscles from mdm mice and their WT littermates at 1–8 weeks of age. The data show that ANKRD2 mRNA and protein levels in skeletal muscles were not significantly different at one week of age, but levels increased to 3-fold higher at 2 weeks and 4.5-fold higher at 4 weeks in mdm mice compared with their WT littermates (Fig. 2A). Interestingly, the elevated ANKRD2 levels in mdm mice were similar in all skeletal muscles that were analyzed (Fig. 2B) and were also elevated in cultured pMBmdm compared with pMBWT (Fig. 2C). These results indicate that the skeletal muscles of mdm mice begin to overexpress ANKRD2 from 2 weeks of age and that overexpression is consistent in all skeletal muscles.
FIGURE 2.
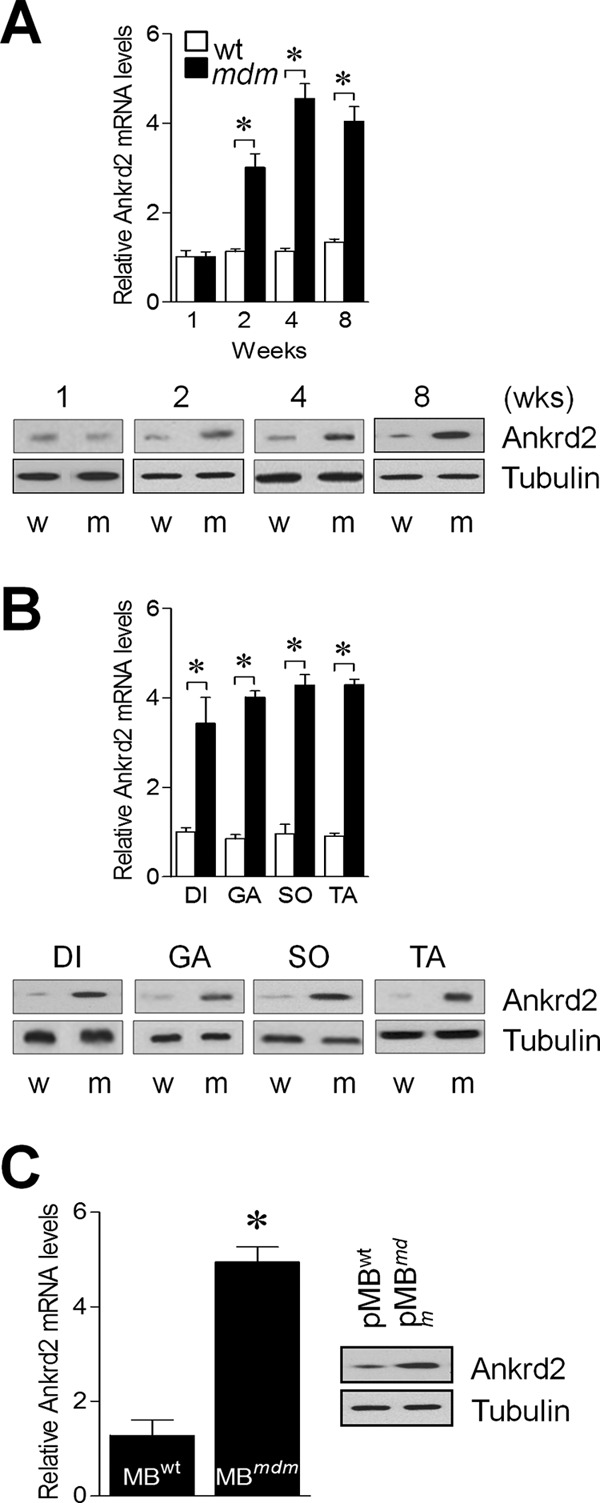
Skeletal muscles of mdm mice over express ANKRD2. Hind limb skeletal muscles were excised from WT and mdm mice, and ANKRD2 expression was determined by real-time RT-PCR and Western blot methods (A). diaphragm (DI), gastrocnemius (GA), soleus (SO), tibialis anterior (TA), and primary myoblasts (pMB) were isolated from the hind limb skeletal muscles of WT and mdm mice, and ANKRD2 expression was determined by real-time RT-PCR and Western blot (B and C). w and m indicate wild-type and mdm, respectively. Gel images are representative of three separate experiments. Each error bar indicates mean ± S.E. (n = 3). *, p < 0.05. Wks., weeks.
Second, we generated transgenic myoblasts stably overexpressing an antisense ANKRD2 mRNA (pMBmdm/ANKRD2↓) that effectively decreased ANKRD2 protein levels (Fig. 3A). Interestingly, inhibition of ANKRD2 by antisense ANKRD2 induced differentiation of pMBmdm/Ankrd2↓, and co-transfection of ANKRD2-sense vector in pMBmdm/Ankrd2↓ voided the antisense Ankrd2-induced differentiation program (Fig. 3B). To further study whether ANKRD2 indeed inhibits myoblast differentiation, we generated ANKRD2 overexpressing WT transgenic myoblasts (pMBWT/ANKRD2↑). Culture of pMBWT/ANKRD2↑ in DM was unable to differentiate into myotubes and inhibition of ANKRD2 in these myoblasts by antisense ANKRD2 reinstated the differentiation program (Fig. 3C). The levels of ANKRD2 expression were confirmed in pMBWT/ANKRD2↑ (Fig. 3D). These results suggest that ANKRD2 is a negative regulator of the myoblast differentiation program and that ANKRD2 up-regulation in the skeletal muscles of mdm mice could be a critical factor causing skeletal muscle growth deficiency through inhibition of myoblast differentiation.
FIGURE 3.
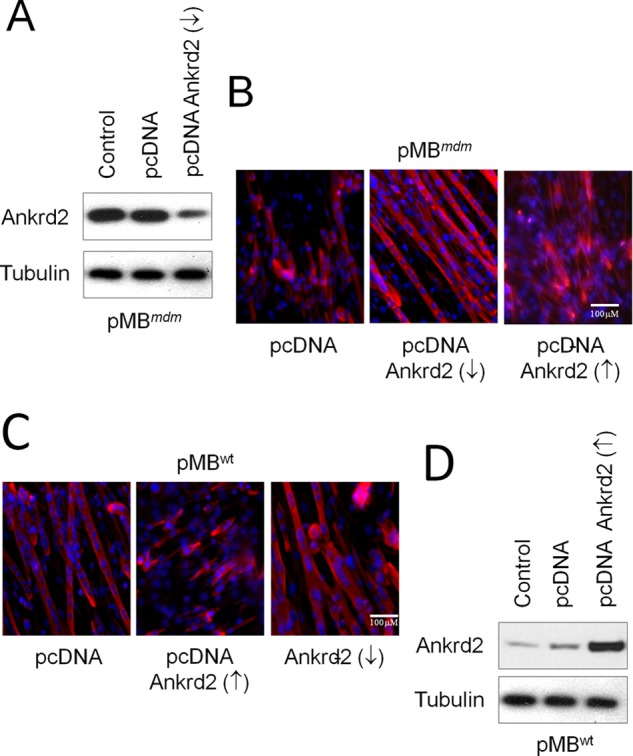
ANKRD2 negatively regulates myoblast differentiation program. pMBWT and pMBmdm cultured in GM were transfected with either pcDNA, pcDNA-ANKRD2-sense or pcDNA-ANKRD2-antisense vector for 48 h. A, knockdown of ANKRD2 protein was confirmed by Western blot in pMBmdm/Ankrd2↓. B, knockdown of ANKRD2 promotes differentiation of pMBmdm/Ankrd2↓, and co-transfection of ANKRD2-sense vector in pMBmdm/Ankrd2↓ voids the antisense ANKRD2-induced differentiation program. C, overexpression of ANKRD2 inhibits differentiation of pMBWT/Ankrd2, and co-transfection of ANKRD2-antisense vector in pMBWT/Ankrd2 voids the sense Ankrd2-induced differentiation program. D, overexpression of ANKRD2 protein was confirmed by Western blot in pMBmdm/Ankrd2. Gel and immunocytochemistry pictures are representative of three separate experiments. ↑, overexpression; ↓, down-regulation.
Overexpression of ID3 in pMBmdm Inhibits Differentiation Program
Studies have shown that overexpression of ID3 in C2C12 myoblasts inhibits the differentiation program (12, 33). Thus, we tested whether ID3, similar to ANKRD2, inhibits the differentiation of pMBmdm. We show that the skeletal muscles of mdm mice overexpressed Id3 mRNA and protein (Fig. 4A). The level of Id3 in the skeletal muscles of mdm mice was ∼3.5-fold higher than that in the skeletal muscles of WT mice as determined by quantitative PCR (qPCR) (Fig. 4B). Furthermore, the myogenic pMBWT showed a rapid decline in the levels of ID3 at 12 h, which were barely detectable after 24 h. In contrast, pMBmdm in DM showed no significant decline in the levels of ID3. To study whether knockdown of ID3 in pMBmdm could induce differentiation, we applied an siRNA-based strategy to inhibit ID3 protein expression. Interestingly, inhibition of ID3 by Id3 siRNA was capable of inducing pMBmdm differentiation (Fig. 4C), and overexpression of ID3 in these myoblasts inhibited differentiation. To determine whether this was a unique feature caused by the mdm mutation, we overexpressed ID3 in pMBWT. Enforced expression of ID3 in pMBWT hindered the serum withdrawal-induced myoblast differentiation (Fig. 4D). These results indicate that similar to ANKRD2, overexpression of ID3 in pMBWT and pMBmdm inhibits differentiation program. We also determined ID3 levels in pMBmdm after Id3 siRNA transfection and confirmed that it significantly reduced the endogenous ID3 levels. Similarly, the enforced-expression of ID3 in pMBWT significantly increased the endogenous ID3 levels, suggesting the specificity of the siRNA and overexpression constructs (Fig. 4E).
FIGURE 4.
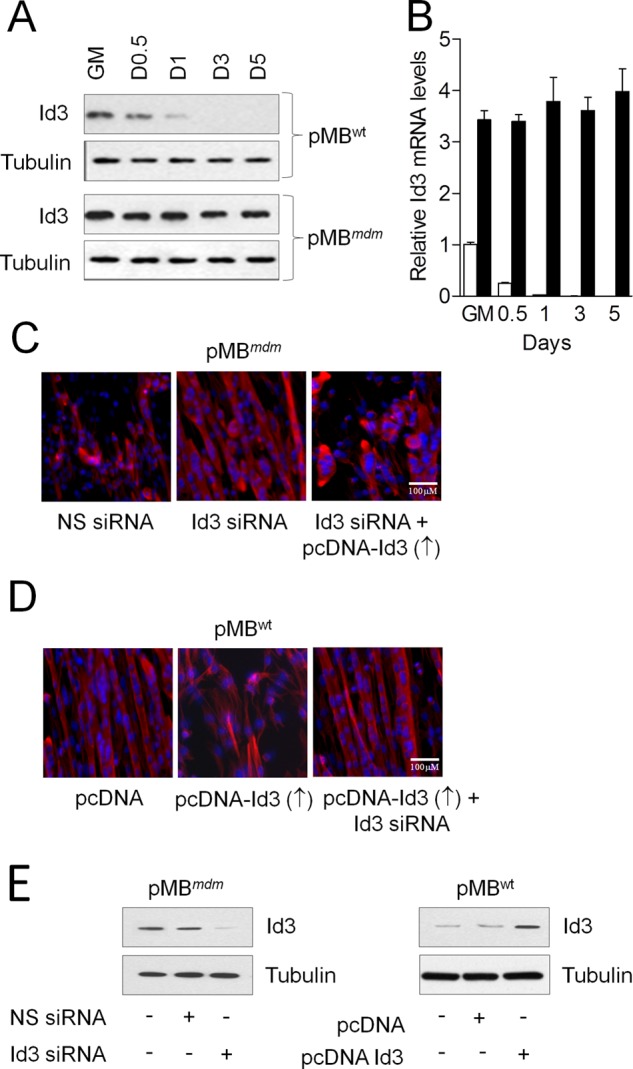
Up-regulation of ID3 in the skeletal muscle of mdm mice inhibits myoblast differentiation program. A and B, total RNA and protein were isolated from proliferating and myogenic pMBWT and pMBmdm to determine ID3 mRNA and protein expressions. C, pMBmdm were transfected with nonspecific (NS) or ID3 siRNA with or without pcDNA-ID3 construct. D, pMBWT were transfected with either pcDNA or pcDNA-ID3 with or without ID3 siRNA. After 24 h, the GM was replaced with DM, and the myogenic program was determined by ICH. Overexpression or knockdown of ID3 was determined by Western blot (E). Gel and immunocytochemistry images are representative of three separate experiments. Each error bar indicates mean ± S.E. (n = 3). *, p < 0.05. ↑, overexpression; ↓, down-regulation.
ANKRD2 and ID3 Cooperatively Inhibit pMB differentiation by Physical Interaction
Because ANKRD2 and ID3 play similar roles in the myoblast differentiation program, we sought to determine whether these two proteins act cooperatively or independently. We used transgenic myoblasts expressing either ANKRD2 (pMBWT/ANKRD2↑) or ID3 (pMBWT/ID3↑). Although pMBWT/ANKRD2↑ or pMBWT/ID3↑ were unable to differentiate into myotubes, knockdown of ID3 in pMBWT/ANKRD2↑ using ID3 siRNA or ANKRD2 in pMBWT/ID3↑ using the ANKRD2-antisense construct effectively induced myoblast differentiation (Fig. 5A). These results suggest that both ANKRD2 and ID3 inhibit myoblast differentiation in a cooperative manner. To determine whether the inhibition of myoblast differentiation by ANKRD2 and ID3 could be caused by a physical interaction between these proteins, we conducted a series of co-immunoprecipitation experiments. First, we isolated cell lysate from pMBWT/ANKRD2↑ or pMBWT/pcDNA and immunoprecipitated with either anti-His or anti-ID3. Cell lysates from pMBWT/pcDNA showed no co-precipitation of ANKRD2 with ID3, whereas cell lysate from pMBWT/ANKRD2↑ immunoprecipitated with anti-His showed co-precipitation of ID3. Similarly, myoblasts immunoprecipitated with anti-ID3 showed co-precipitation of ANKRD2 (Fig. 5B). These results indicate that there is likely a physical interaction between exogenously expressed ANKRD2 and endogenous ID3. To further support this result and rule out artifacts due to overexpression, we attempted to detect interactions between endogenous ANKRD2 and ID3. We used pMBmdm, which overexpresses both ANKRD2 and ID3 proteins. Immunoprecipitation of cell lysate of pMBmdm with anti-ANKRD2 antibody showed co-precipitation of ID3. Similarly, immunoprecipitation of cell lysate of pMBmdm with anti-ID3 antibody showed co-precipitation of ANKRD2. These results corroborate the existence of a physical interaction between the endogenous ANKRD2 and ID3 proteins and further support the hypothesis that their combined actions cooperatively inhibit myoblast differentiation.
FIGURE 5.
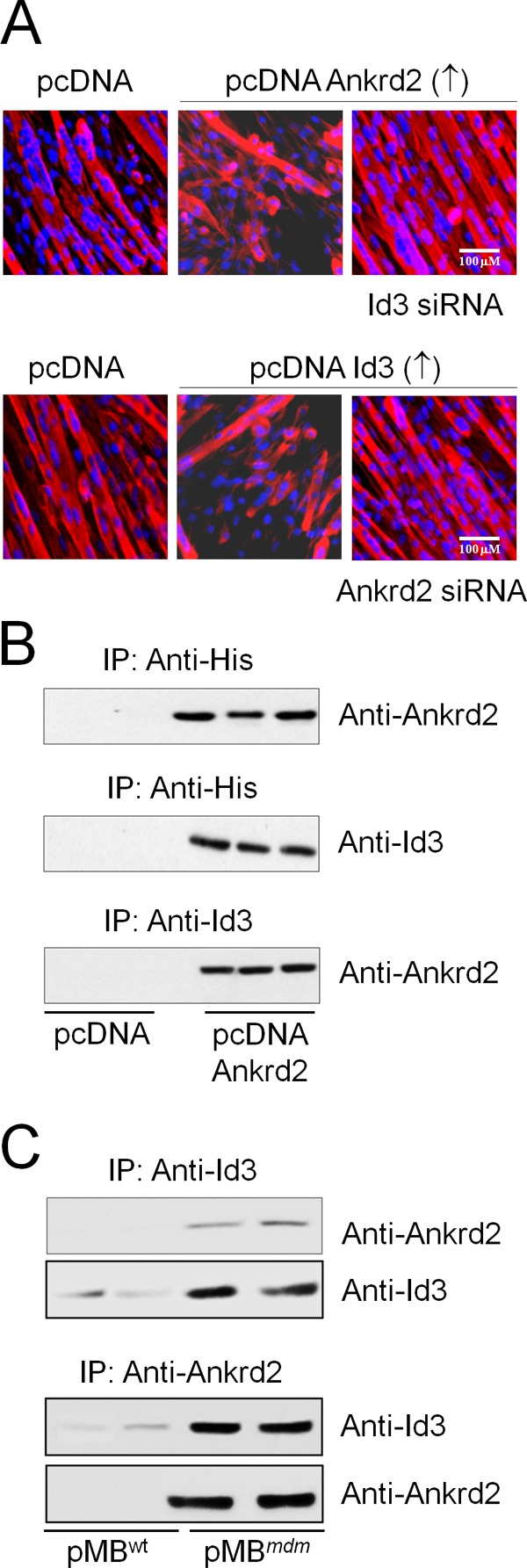
ANKRD2 and ID3 physically interact with each other and cooperatively inhibit myoblast differentiation program. A, pMBWT/ANKRD2↑ were transfected with or without ID3 siRNA. After 24 h, the GM was replaced with DM and differentiation was determined by ICH. B, total cell lysate was isolated from pMBWT/ANKRD2 and immunoprecipitated with anti-His antibody, and the pellets were resolved on SDS-PAGE and subjected to immunoblot analysis with anti-ANKRD2 or anti-ID3 antibody. The cell lysate was also immunoprecipitated with anti-ID3 antibody followed by resolving on SDS-PAGE and subjected to immunoblot analysis with anti-ANKRD2 antibody. C, total cell lysate was isolated from pMBmdm and immunoprecipitated with either anti-ANKRD2 or anti-ID3 antibody, and the pellets were resolved on SDS-PAGE and subjected to immunoblot analysis with anti-ANKRD2 or anti-ID3 antibody. Gel and immunocytochemistry images are representative of three separate experiments. ↑, overexpression; ↓, down-regulation.
WT and mdm Mice Skeletal Muscles Differentially Regulate ANKRD2
Data from microarray analyses show that MyoD-silenced C2C12 myoblasts down-regulate Ankrd2 gene expression (9). We have shown that mechanical stretch can up-regulate ANKRD2 expression through NF-κB in WT and mdm mouse diaphragm muscles (11). These results suggest a differential regulation of Ankrd2 in skeletal muscle. It has been shown that SREBP-1 overexpression can inhibit myoblast differentiation and promotes skeletal muscle atrophy (19). However, the mechanism of regulation of the basal level ANKRD2 expression in skeletal muscle is unknown. To end this, we analyzed the Ankrd2 promoter region bioinformatically, focusing on MyoD, NF-κB, and SREBP-1 transcription factors. A scan of the 1.7-kb genomic sequence located upstream of the ATG of the Ankrd2 gene identified two putative MyoD (−865 and −1449; green), five NF-κB (−271, −1036, −1207, −1531, and −1656; red), and two SREBP-1 (−929 and −1451; blue) binding sites (Fig. 6A), which led us to consider whether ANKRD2 is a transcriptional target of any of these regulators. Using ChIP assays, we identified that MyoD but not NF-κB and SREBP-1 bound the Ankrd2 promoter in skeletal muscles of WT mice as evidenced by qPCR and the visualization of PCR products on 1% agarose gel (Fig. 6B). In contrast, SREBP-1 but not MyoD and NF-κB bound the Ankrd2 promoter in skeletal muscles of mdm mice. To further confirm this, we generated a 1500-bp (−200 to −1700) Ankrd2 promoter construct, encompassing MyoD, NF-κB, and SREBP-1 binding sites, named pGL-WT (Fig. 6C) and a mutant Ankrd2 promoter construct with mutated-MyoD or mutated-SREBP-1 binding site, named pGL-mtMyoD or pGL-mtSREBP-1 (Fig. 6C). Transfection of pGL-WT construct into pMBWT and pMBmdm showed higher luciferase activities than those cells transfected with pGL vector alone (Fig. 6D). In contrast, transfection of pMBWT with pGL-mtMyoD but not pGL-mtSREBP-1 or transfection of pMBmdm with pGL-mtSREBP-1 but not pGL-mtMyoD showed less luciferase activity. Finally, we tested whether the endogenous MyoD and SREBP-1 is able to influence the promoter activity of Ankrd2 in pMBWT and pMBmdm, respectively. To achieve this, we knocked down MyoD in pMBWT and SREBP-1 in pMBmdm using siRNA-based strategy, which decreased the endogenous levels of MyoD in pMBWT and SREBP-1 in pMBmdm (Fig. 6E). Transfection of pMBWT with pGL-WT construct increased the luciferase activity, but knockdown of MyoD by siRNA abolished the luciferase activity (Fig. 6F). Similarly, transfection of pMBmdm with pGL-WT construct increased the luciferase activity, but knockdown of SREBP-1 by siRNA abolished the luciferase activity (Fig. 6F). Taken together, these data indicate that the Ankrd2 promoter is a direct transcriptional target of both MyoD and SREBP-1 in the skeletal muscles of WT and mdm mice, respectively.
FIGURE 6.

MyoD and SREBP-1 regulate ANKRD2 expression. A, schematic representation of Ankrd2 promoter. The region between −1700 and +1 bp contains putative binding elements for MyoD (green), NF-κB (red), and SREBP-1 (pink). B, chromatin was isolated from the skeletal muscles of WT and mdm mice and precipitated with anti-c-MyoD, anti-NF-κB, anti-SREBP-1, anti-RNA poly II, or nonspecific IgG. qPCRs were performed with three sets of primers, specific for ANKRD2 promoter to identify the specific transcription factor and its region of binding to the ANKRD2 promoter and resolved in 1% agarose gel. C, 1500-bp pGL-WT, 700-bp (pGL-mtMyoD), or 600-bp (pGL-SREBP-1) promoter regions were synthesized and linked to luciferase (Luc) reporter gene. D, pMBWT and pMBmdm were transfected with empty vector, pGL-WT, pGL-mtMyoD, or pGL-SREBP-1 vector. Forty-eight h after transfection, firefly luciferase activities were estimated and normalized to Renilla luciferase activities. E and F, MBWT were transfected with MyoD siRNA or nonspecific siRNA (NS siRNA) followed by transfection of either pGL or pGL-WT. MBmdm were transfected with SREBP-1 siRNA or nonspecific siRNA followed by transfection of either pGL or pGL-WT. After 48 h, total protein was isolated, and MyoD and SREBP-1 protein expression was determined by Western blot (E). Forty-eight h after transfection, firefly luciferase activities were estimated and normalized to Renilla luciferase activities (F). Gel pictures are representative of three separate experiments. Each error bar indicates mean ± S.E. (n = 3). *, p < 0.05.
DISCUSSION
In the present study, we demonstrated that primary myoblasts and skeletal muscles of mdm mice overexpress ANKRD2 and ID3 proteins. The myoblasts of mdm mice were incompetent to differentiate into myotubes upon myogenic induction. We found that an ANKRD2-ID3 complex inhibits myoblast differentiation by physical interaction. Although MyoD activates the Ankrd2 promoter in skeletal muscles of WT mice, SREBP-1 activates this promoter in the skeletal muscles of mdm mice. Overall, we provide the first experimental evidence demonstrating that the ANKRD2/ID3 pathway is critical for the developmental program of skeletal muscles.
Impaired muscle development in skeletal muscle diseases such as muscular dystrophies is associated with an impaired myogenic program (1–4). Studies have shown that many different genetic muscular dystrophies display overexpression of ANKRD2 (34), which is known to negatively regulate the differentiation program in C2C12 myoblasts (7, 8). However, it is not known whether overexpression of ANKRD2 in dystrophic skeletal muscles directly leads to disease pathology disruption of the myogenic program or is a consequence of the disease process. To address this issue, we used primary myoblasts and skeletal muscles of mdm mice, a genetic model of muscular dystrophy caused by a small in-frame deletion within the titin gene. Skeletal muscles of mdm mice began to uniformly overexpress ANKRD2 from 2 weeks of age uniformly in all skeletal muscles analyzed, suggesting that ANKRD2 is an early player in mdm skeletal muscle pathology. This observation is in agreement with our previous study showing that the mdm mouse diaphragm begins to display muscle weakness and histopathological signs of muscular dystrophy, including centrally located nuclei, increased variation in myofiber diameter, hypertrophy, and fatty or connective tissue infiltration at 2 weeks of age (28). To explore the role of ANKRD2 more precisely in mdm skeletal muscle, we isolated primary myoblasts from 2-week-old mdm mice (pMBmdm) and their wild-type littermates (pMBWT). Down-regulation of ANKRD2 or ID3 in mdm myoblasts restored the ability to differentiate. Overexpressing either ANKRD2 or ID3 similarly disrupted differentiation of WT myoblasts. This result suggests that both proteins are critical to the disease process and that they function independently of the titin mutation. In the present study, we did not notice any impairment in cell proliferation between pMBmdm and pMBWT/ANKRD2↑, suggesting that ANKRD2 does not perturb cell proliferation. This result is in agreement with an earlier study showing that proliferating myoblasts and injured skeletal muscles accumulate ANKRD2 in the nucleus (35).
We also found that the primary myoblasts and the skeletal muscle of mdm mice overexpressed ID3 protein (DNA-binding protein inhibitor 3). ID3 belongs to the ID family of proteins, encoded by four distinct genes, Id1 through Id4, which are transcriptional regulators that influence the proliferation and differentiation of many cell types, including skeletal muscle cells (14, 15). Satellite cells express ID3 under the direct transcriptional control of Pax7 (36) and skeletal muscle markedly up-regulates ID3 within 24 h of injury (37). Although the proliferating C2C12 myoblasts actively express ID3, differentiating myoblasts show reduced levels of ID3 (38). Enforced expression of ID3 in C2C12 myoblasts effectively inhibits the myogenic program (12, 33). These studies suggest that ANKRD2 and ID3 may have similar roles in skeletal muscle development. In agreement with this hypothesis, we found that ID3 expression levels were detectable in proliferating pMBWT and drastically reduced during differentiation. In contrast, ID3 overexpression in pMBmdm did not change upon myogenic induction. Similar to ANKRD2, inhibition of ID3 in pMBmdm induced differentiation, whereas enforced expression of ID3 in pMBWT inhibited differentiation, suggesting that ANKRD2 and ID3 negatively regulate myoblast differentiation. We further expanded our study to identify the potential mechanism by which ANKRD2 and ID3 impair myoblast differentiation. ID proteins form heterodimers with the ubiquitous transcription factors known as E-proteins and that interaction prevents them from forming transcriptionally active dimers (12, 13). It has been shown that ID3 inhibits the myogenic program through physical interaction with E class basic HLH proteins to form inactive heterodimers (39, 40). In addition, ANKRD2 physically interacts with the structural proteins titin (41), myopalladin, and telethonin (42), and with the regulatory nuclear proteins promyelocytic leukemia protein, p53, and YB-1 (42). These studies provide a strong rationale to explore whether ANKRD2 and ID3 cooperatively inhibit myoblast differentiation by physical interaction. Although knockdown of either ANKRD2 or ID3 in pMBmdm was adequate to induce differentiation program, ANKRD2 overexpression combined with ID3 knockdown or vice versa in pMBWT does not inhibit myoblast differentiation. Furthermore, our data from co-immunoprecipitation assays revealed the existence of a strong physical interaction between ANKRD2 and ID3 proteins. These data indicate that ANKRD2 and ID3 cooperatively inhibit myoblast differentiation by physical interaction. We have previously shown the Akt-mediated up-regulation of ANKRD2 in diaphragm muscle (11). In agreement with this, other studies have shown that phosphorylation of ANKRD2 by Akt2 inhibited C2C12 myoblast differentiation (8), and myoblasts lacking Akt2 have a normal differentiation program (43, 44), suggesting that ANKRD2 is a downstream substrate of Akt2. In the present study, we have shown ID3 as a new substrate of ANKRD2 in the differentiation program.
Our earlier study shows that NF-κB binds the Ankrd2 promoter and up-regulates Aknrd2 expression in diaphragm muscle after mechanical stimuli (11). A microarray study shows that MyoD-silenced myogenic C2C12 myoblasts down-regulate ANKRD2 expression (9). These studies suggest that there are multiple key regulators of ANKRD2 expression. In this study, we have provided new insight to the regulation of ANKRD2 in skeletal muscles in WT and mdm mice. We show that MyoD bound to and transactivated the Ankrd2 promoter in skeletal muscles of WT mice, as demonstrated by ChIP and luciferase assays. In contrast, activation of the Ankrd2 promoter in the skeletal muscle of mdm mice was completely dependent on SREBP-1-binding sites and independent of MyoD and NF-κB binding sites, suggesting that SREBP-1 inhibits the differentiation of pMBmdm through ANKRD2 up-regulation. The SREBP-1 gene produces SREBP-1a and SREBP-1c proteins through alternate promoters. Like liver and adipose tissues, which highly express SREBP-1 proteins (21, 22), skeletal muscles express significant levels of SREBP-1 proteins (23–25). For example, microarray analysis of human myotubes over expressing SREBP-1a or SREBP-1c identified many potential targets of SREBP-1 proteins, including a number of muscle-specific genes and markers of muscle differentiation (26). In line with this result, a previous study demonstrated that enforced-expression of SREBP-1 proteins inhibit differentiation in human myoblasts and induce atrophy in mouse myotubes in vitro and skeletal muscle in vivo (27). Moreover, our site-directed promoter mutagenesis and knockdown of MyoD and SREBP-1 confirmed the differential regulation of ANKRD2 between the skeletal muscles of WT and mdm mice. These data provide novel evidence demonstrating that aberrant regulation of SREBP-1 in mdm skeletal muscle inhibits myoblast differentiation through ANKRD2 up-regulation.
In summary, we have shown that ANKRD2 up-regulation by SREBP-1 negatively regulates myoblast differentiation by interacting with ID3 in mdm skeletal muscle. A schematic summarizing our data is depicted in Fig. 7. These results prompt the speculation that ANKRD2 inhibition could be utilized in the context of skeletal muscle wasting to enhance new muscle fiber formation.
FIGURE 7.
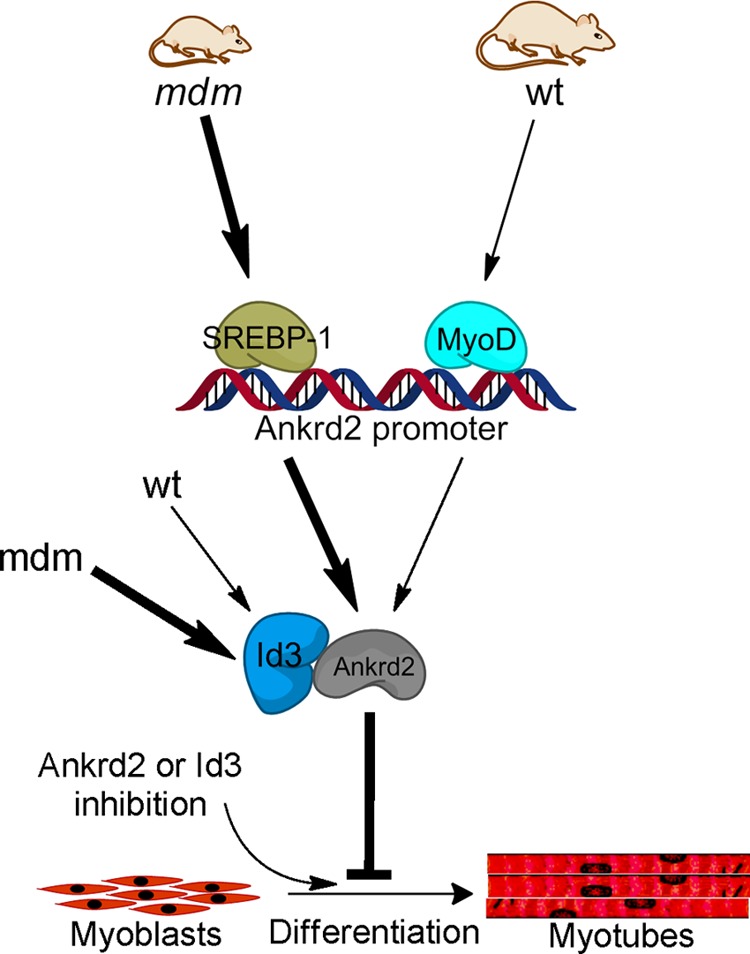
Proposed pathway illustrating the mechanism by which ANKRD2 inhibits skeletal differentiation program. The skeletal muscle of mdm mice up-regulates ANKRD2 and ID3 proteins, which cooperatively inhibit pMBmdm differentiation program through physical interaction. Although MyoD regulates the Ankrd2 gene in the skeletal muscle of WT mice, SREBP-1 regulates Ankrd2 in the skeletal muscle of mdm mice. Inhibition of ANKRD2 or ID3 is sufficient to recover the myogenic program. Arrows in lightface and boldface type indicate up-regulation and basal-level regulation, respectively.
This work was supported by the National Science Foundation.
- HLH
- helix-loop-helix
- GM
- growth medium
- SREBP
- sterol regulatory element binding protein
- MB
- myoblast
- mdm
- muscular dystrophy with myositis
- qPCR
- quantitative PCR.
REFERENCES
- 1. Webster C., Blau H. M. (1990) Accelerated age-related decline in replicative life-span of Duchenne muscular dystrophy myoblasts: implications for cell and gene therapy. Somat. Cell Mol. Genet. 16, 557–565 [DOI] [PubMed] [Google Scholar]
- 2. Bulfield G., Siller W. G., Wight P. A., Moore K. J. (1984) X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. U.S.A. 81, 1189–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mitchell P. O., Pavlath G. K. (2004) Skeletal muscle atrophy leads to loss and dysfunction of muscle precursor cells. Am. J. Physiol. Cell Physiol. 287, C1753–C1762 [DOI] [PubMed] [Google Scholar]
- 4. Peterson J. M., Bryner R. W., Alway S. E. (2008) Satellite cell proliferation is reduced in muscles of obese Zucker rats but restored with loading. Am. J. Physiol. Cell Physiol. 295, C521–C528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guttridge D. C., Mayo M. W., Madrid L. V., Wang C. Y., Baldwin A. S., Jr. (2000) NF-κB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 289, 2363–2366 [DOI] [PubMed] [Google Scholar]
- 6. Penna F., Costamagna D., Fanzani A., Bonelli G., Baccino F. M., Costelli P. (2010) Muscle wasting and impaired myogenesis in tumor bearing mice are prevented by ERK inhibition. PLoS One 5, e13604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bean C., Facchinello N., Faulkner G., Lanfranchi G. (2008) The effects of Ankrd2 alteration indicate its involvement in cell cycle regulation during muscle differentiation. Biochim. Biophys. Acta 1783, 1023–1035 [DOI] [PubMed] [Google Scholar]
- 8. Cenni V., Bavelloni A., Beretti F., Tagliavini F., Manzoli L., Lattanzi G., Maraldi N. M., Cocco L., Marmiroli S. (2011) Ankrd2/ARPP is a novel Akt2 specific substrate and regulates myogenic differentiation upon cellular exposure to H2O2. Mol. Biol. Cell 22, 2946–2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bean C., Salamon M., Raffaello A., Campanaro S., Pallavicini A., Lanfranchi G. (2005) The Ankrd2, Cdkn1c and calcyclin genes are under the control of MyoD during myogenic differentiation. J. Mol. Biol. 349, 349–366 [DOI] [PubMed] [Google Scholar]
- 10. Lassar A. B., Skapek S. X., Novitch B. (1994) Regulatory mechanisms that coordinate skeletal muscle differentiation and cell cycle withdrawal. Curr. Opin. Cell Biol. 6, 788–794 [DOI] [PubMed] [Google Scholar]
- 11. Mohamed J. S., Lopez M. A., Cox G. A., Boriek A. M. (2010) Anisotropic regulation of Ankrd2 gene expression in skeletal muscle by mechanical stretch. FASEB J. 24, 3330–3340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jen Y., Weintraub H., Benezra R. (1992) Overexpression of Id protein inhibits the muscle differentiation program: in vivo association of Id with E2A proteins. Genes Dev. 6, 1466–1479 [DOI] [PubMed] [Google Scholar]
- 13. Sun X. H., Copeland N. G., Jenkins N. A., Baltimore D. (1991) Id proteins Id1 and Id2 selectively inhibit DNA binding by one class of helix-loop-helix proteins. Mol. Cell Biol. 11, 5603–5611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zebedee Z., Hara E. (2001) Id proteins in cell cycle control and cellular senescence. Oncogene 20, 8317–8325 [DOI] [PubMed] [Google Scholar]
- 15. Lasorella A., Uo T., Iavarone A. (2001) Id proteins at the cross-road of development and cancer. Oncogene 20, 8326–8333 [DOI] [PubMed] [Google Scholar]
- 16. Hara E., Yamaguchi T., Nojima H., Ide T., Campisi J., Okayama H., Oda K. (1994) Id-related genes encoding helix-loop-helix proteins are required for G1 progression and are repressed in senescent human fibroblasts. J. Biol. Chem. 269, 2139–2145 [PubMed] [Google Scholar]
- 17. Ellmeier W., Weith A. (1995) Expression of the helix-loop-helix gene Id3 during murine embryonic development. Dev. Dyn. 203, 163–173 [DOI] [PubMed] [Google Scholar]
- 18. Melnikova I. N., Christy B. A. (1996) Muscle cell differentiation is inhibited by the helix-loop-helix protein Id3. Cell Growth Differ. 7, 1067–1079 [PubMed] [Google Scholar]
- 19. Cornelison D. D., Olwin B. B., Rudnicki M. A., Wold B. J. (2000) MyoD(-/-) satellite cells in single-fiber culture are differentiation defective and MRF4 deficient. Dev. Biol. 224, 122–137 [DOI] [PubMed] [Google Scholar]
- 20. Eberlé D., Hegarty B., Bossard P., Ferré P., Foufelle F. (2004) SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86, 839–848 [DOI] [PubMed] [Google Scholar]
- 21. Horton J. D., Goldstein J. L., Brown M. S. (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yokoyama C., Wang X., Briggs M. R., Admon A., Wu J., Hua X., Goldstein J. L., Brown M. S. (1993) SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 75, 187–197 [PubMed] [Google Scholar]
- 23. Dif N., Euthine V., Gonnet E., Laville M., Vidal H., Lefai E. (2006) Insulin activates human sterol-regulatory-element-binding protein-1c (SREBP-1c) promoter through SRE motifs. Biochem. J. 400, 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ducluzeau P. H., Perretti N., Laville M., Andreelli F., Vega N., Riou J. P., Vidal H. (2001) Regulation by insulin of gene expression in human skeletal muscle and adipose tissue. Evidence for specific defects in type 2 diabetes. Diabetes 50, 1134–1142 [DOI] [PubMed] [Google Scholar]
- 25. Guillet-Deniau I., Mieulet V., Le Lay S., Achouri Y., Carré D., Girard J., Foufelle F., Ferré P. (2002) Sterol regulatory element binding protein-1c expression and action in rat muscles: insulin-like effects on the control of glycolytic and lipogenic enzymes and UCP3 gene expression. Diabetes 51, 1722–1728 [DOI] [PubMed] [Google Scholar]
- 26. Rome S., Lecomte V., Meugnier E., Rieusset J., Debard C., Euthine V., Vidal H., Lefai E. (2008) Microarray analyses of SREBP-1a and SREBP-1c target genes identify new regulatory pathways in muscle. Physiol Genomics 34, 327–337 [DOI] [PubMed] [Google Scholar]
- 27. Lecomte V., Meugnier E., Euthine V., Durand C., Freyssenet D., Nemoz G., Rome S., Vidal H., Lefai E. (2010) A new role for sterol regulatory element binding protein 1 transcription factors in the regulation of muscle mass and muscle cell differentiation. Mol. Cell Biol. 30, 1182–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lopez M. A., Pardo P. S., Cox G. A., Boriek A. M. (2008) Early mechanical dysfunction of the diaphragm in the muscular dystrophy with myositis (Ttnmdm) model. Am. J. Physiol. Cell Physiol 295, C1092–C1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Edmondson D. G., Olson E. N. (1993) Helix-loop-helix proteins as regulators of muscle-specific transcription. J. Biol. Chem. 268, 755–758 [PubMed] [Google Scholar]
- 30. Megeney L. A., Rudnicki M. A. (1995) Determination versus differentiation and the MyoD family of transcription factors. Biochem. Cell Biol. 73, 723–732 [DOI] [PubMed] [Google Scholar]
- 31. Olson E. N., Klein W. H. (1994) bHLH factors in muscle development: dead lines and commitments, what to leave in and what to leave out. Genes Dev. 8, 1–8 [DOI] [PubMed] [Google Scholar]
- 32. Wright W. E. (1992) Muscle basic helix-loop-helix proteins and the regulation of myogenesis. Curr. Opin. Genet. Dev. 2, 243–248 [DOI] [PubMed] [Google Scholar]
- 33. Atherton G. T., Travers H., Deed R., Norton J. D. (1996) Regulation of cell differentiation in C2C12 myoblasts by the Id3 helix-loop-helix protein. Cell Growth Differ. 7, 1059–1066 [PubMed] [Google Scholar]
- 34. Nakada C., Tsukamoto Y., Oka A., Nonaka I., Sato K., Mori S., Ito H., Moriyama M. (2004) Altered expression of ARPP protein in skeletal muscles of patients with muscular dystrophy, congenital myopathy and spinal muscular atrophy. Pathobiology 71, 43–51 [DOI] [PubMed] [Google Scholar]
- 35. Tsukamoto Y., Hijiya N., Yano S., Yokoyama S., Nakada C., Uchida T., Matsuura K., Moriyama M. (2008) Arpp/Ankrd2, a member of the muscle ankyrin repeat proteins (MARPs), translocates from the I-band to the nucleus after muscle injury. Histochem. Cell Biol. 129, 55–64 [DOI] [PubMed] [Google Scholar]
- 36. Kumar D., Shadrach J. L., Wagers A. J., Lassar A. B. (2009) Id3 is a direct transcriptional target of Pax7 in quiescent satellite cells. Mol. Biol. Cell 20, 3170–3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clever J. L., Sakai Y., Wang R. A., Schneider D. B. (2010) Inefficient skeletal muscle repair in inhibitor of differentiation knockout mice suggests a crucial role for BMP signaling during adult muscle regeneration. Am. J. Physiol. Cell Physiol. 298, C1087–C1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Blau H. M., Pavlath G. K., Hardeman E. C., Chiu C. P., Silberstein L., Webster S. G., Miller S. C., Webster C. (1985) Plasticity of the differentiated state. Science 230, 758–766 [DOI] [PubMed] [Google Scholar]
- 39. Benezra R., Davis R. L., Lockshon D., Turner D. L., Weintraub H. (1990) The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell 61, 49–59 [DOI] [PubMed] [Google Scholar]
- 40. Ruzinova M. B., Benezra R. (2003) Id proteins in development, cell cycle and cancer. Trends Cell Biol. 13, 410–418 [DOI] [PubMed] [Google Scholar]
- 41. Miller M. K., Bang M. L., Witt C. C., Labeit D., Trombitas C., Watanabe K., Granzier H., McElhinny A. S., Gregorio C. C., Labeit S. (2003) The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. J. Mol. Biol. 333, 951–964 [DOI] [PubMed] [Google Scholar]
- 42. Kojic S., Medeot E., Guccione E., Krmac H., Zara I., Martinelli V., Valle G., Faulkner G. (2004) The Ankrd2 protein, a link between the sarcomere and the nucleus in skeletal muscle. J. Mol. Biol. 339, 313–325 [DOI] [PubMed] [Google Scholar]
- 43. Rotwein P., Wilson E. M. (2009) Distinct actions of Akt1 and Akt2 in skeletal muscle differentiation. J. Cell Physiol. 219, 503–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wilson E. M., Rotwein P. (2007) Selective control of skeletal muscle differentiation by Akt1. J. Biol. Chem. 282, 5106–5110 [DOI] [PubMed] [Google Scholar]