

Background: E2F7 is a transcription factor that controls cell cycle by repressing the expression of G1/S genes in late S phase.
Results: E2F7 forms a heterodimer with E2F1, and it recruits the co-repressor CtBP to repress G1/S transcription.
Conclusion: E2F7 represses gene transcription by interacting with E2F1 and co-repressor CtBP.
Significance: These findings suggest a mechanism for the repression of transcription by E2F7.
Keywords: Cell Cycle, DNA-binding Protein, E2F Transcription Factor, Transcription Factors, Transcription Repressor
Abstract
Previous work has identified distinct functions for E2F proteins during a cellular proliferative response including a role for E2F1–3 in the activation of transcription at G1/S and a role for E2F4–8 in repressing the same group of E2F1–3 target genes as cells progress through S phase. We now find that E2F7 and E2F8, which are induced by E2F1–3 at G1/S, can form a heterodimer with E2F1 through interactions involving the DNA-binding domains of the two proteins. In vitro DNA interaction assays demonstrate the formation of an E2F1-E2F7 complex, as well as an E2F7-E2F7 complex on adjacent E2F-binding sites. We also show that E2F7 recruits the co-repressor C-terminal-binding protein (CtBP) and that CtBP2 is essential for E2F7 to repress E2F1 transcription. Taken together, these findings suggest a mechanism for the repression of transcription by E2F7.
Introduction
The E2F family of transcription factors (E2F1–8) is known to regulate many essential genes involved in cell proliferation, differentiation, DNA damage response, and apoptosis (1, 2). They can be simply categorized into two groups based on whether they function as transcription activators or repressors. E2F1, E2F2, and E2F3a can drive quiescent cells into S phase by activating target genes required for G1/S transition. The repressor subfamily consists of E2F3b, E2F4, E2F5, E2F6, E2F7, and E2F8. E2F4 and E2F5 are bound to retinoblastoma (Rb)2-related pocket proteins and associated co-repressors to repress target gene transcription during G0 phase. E2F6, as one component of a multimeric protein complex that contains Mga and Max, represses G1/S gene transcription in a manner independent of Rb family members (3, 4). All of these six classical E2Fs possess a DNA-binding domain (DBD) followed by a leucine zipper domain enabling interaction with the distantly related proteins from the transcription factor DP family. Heterodimerization with DPs enhances E2F DNA binding and transactivation activities (5–7).
E2F7 and E2F8 have the least similarity to other E2Fs because they lack leucine zipper and Rb-binding domains. Instead, they have two distinct DNA-binding domains (8). As targets of E2F1, they are highly expressed during mid to late S phase. Once expressed, E2F7 and E2F8 bind to the E2F1 promoter and repress its transcription (9–14). E2F7−/−, E2F8−/− mice fail to survive embryonic day 11.5, indicating that their overlapping functions are essential for cell survival and embryonic development (15). ChIP-sequencing analysis reveals that E2F7 binds to multiple G1/S-regulated genes and represses their transcription (16). E2F7 is induced by p53 and is involved in cell cycle arrest response to DNA damage and cellular senescence (17–19). Recent studies using mouse model systems also show that the atypical E2Fs are involved in angiogenesis, polyploidization, and placental development (20–23).
Despite the intense study of E2F function, the mechanism by which E2F7 or E2F8 represses gene transcription remains unclear. Here we conducted affinity purifications to identify cofactors for E2F1, E2F7, and E2F8. We now find that E2F7 and E2F8 can form a heterodimer with E2F1 through interactions involving the DBDs of the two proteins. Gel shift assays demonstrate the formation of an E2F1-E2F7 complex, as well as an E2F7-E2F7 complex, on adjacent E2F-binding sites. We also show that E2F7 recruits the co-repressor CtBP and that CtBP2 is essential for E2F7 to repress E2F1 transcription. Taken together, these findings suggest a mechanism by which E2F7 mediates transcriptional repression.
EXPERIMENTAL PROCEDURES
Adenovirus Cloning and Infection
FLAG-E2F1, FLAG-E2F7, and FLAG-E2F8, were cloned into Gateway® entry vector pENTRTM-1A (Invitrogen) by using BamHI and NotI sites. The inserts were then transferred into Gateway® adenovirus expression vector pAd/CMV/V5-DESTTM (Invitrogen). The expression clones were digested with PacI followed by transfection into 293A cells. The adenoviral stocks were produced and used for transduction according to the manufacturer's protocol.
Sample Preparation and LC-MS/MS Analysis
U2OS cells (2 × 108) were infected with adenovirus-FLAG-E2F1 (multiplicity of infection: 25), FLAG-E2F8 (multiplicity of infection: 100), and FLAG-E2F7 (multiplicity of infection: 30)for 48 h, respectively. Cells were collected and lysed in ∼30 ml of lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100). Cell lysates were incubated with anti-FLAG M2 affinity gel (Sigma) overnight, and proteins were eluted with FLAG peptides. An initial concentration was determined by mini-Bradford assay using bovine serum albumin as the calibrator. A 5-μg aliquot of each sample was pipetted out, solubilized in 0.1% RapiGest (Waters Corp.), and reduced, alkylated, and digested following a standard in-solution digestion protocol (Duke Proteomics Core Facility). Briefly, the samples were reduced in 10 mm dithiothreitol (VWR International, VW1506-02), alkylated in 20 mm iodoacetamide (Calbiochem 407710), and digested with 0.1 μg of sequencing grade modified trypsin (Promega V5111). After digestion overnight, samples were acidified to 1% trifluoroacetic acid, 2% acetonitrile and heated for 2 h at 60 °C to hydrolyze RapiGest. Samples were then desalted by solid phase extraction via C18 ZipTip (Millipore) using the manufacturer's protocol. Sample eluents were dried down and reconstituted in 1% trifluoroacetic acid, 2% acetonitrile and pipetted into recovery LC vials (Waters). Each sample was analyzed by injecting ∼1 μg of total digested protein onto a 75 μm × 250-mm BEH C18 column (Waters) and separated using a gradient of 5–40% acetonitrile with 0.1% formic acid, with a flow rate of 0.3 μl/min, in 90 min on a nanoACQUITY liquid chromatograph (Waters). Electrospray ionization was used to introduce the sample in real time to a Q-Tof Synapt G1 mass spectrometer (Waters), collecting data for each sample in singlicate in data-dependent acquisition mode. Data-dependent acquisition mode utilized a 0.6-s MS scan followed by MS/MS acquisition on the top 3 ion with charge greater than 1. MS/MS scans for each ion used an isolation window of ∼2.3 Da, 0.6-s scans with a maximum of 3 s per precursor, and dynamic exclusion for 120 s within 1.2 Da of the selected precursor m/z.
LC-MS/MS Data Processing
Data-dependent acquisition was processed through Distiller and searched in Mascot 2.2 against the Uniprot/Swissprot database with human taxonomy filter enabled. Precursor ion mass tolerance was 20 ppm, product ion tolerance was 0.04 Da, and enzyme specificity was set to tryptic. A maximum of 2 missed cleavages was allowed. Carbamidomethyl cysteine was included as a fixed modification, and variable modifications included oxidized methionine and deamidated asparagine and glutamine. Curation of data was performed in Scaffold v3.5.1.
Cell Culture, Transfection, and Antibodies
Human embryonic kidney 293 cells and human osteosarcoma cell line U2OS cells were cultured in Dulbecco's modified Eagle's medium, supplemented with 10% fetal bovine serum (Sigma). Cells were transfected using Lipofectamine 2000 (Invitrogen). All the antibodies used in these studies are from Santa Cruz Biotechnology (Santa Cruz, CA). For Western blot, anti-c-Myc (A-14), anti-HA (Y-11), anti-CtBP1 (K-15), and anti-CtBP2 (E-16) were used. For immunoprecipitation and supershift experiments, anti-c-Myc (9E10), anti-HA (F-7), anti-E2F7 (R-17), anti-E2F7 (N-20), anti-E2F1 (KH95), and anti-E2F1 (c-20) were used.
Plasmids and Molecular Cloning
pcDNA3-HA-E2F1, pcDNA3-HA-E2F2, pcDNA3-HA-E2F3, and pGEX-GST-E2F1 plasmids were described previously (24). E2F7, E2F8, CtBP1, CtBP2 cDNA, and various E2F truncates were PCR-amplified and inserted into pcDNA3-3×Myc or pcDNA3-3×HA vectors. DNA mutagenesis was performed using the GeneTailer site-directed mutagenesis system (Invitrogen). Primer sequences for molecular cloning will be available upon request.
GST Pulldown Assay
Myc-E2F7 and Myc-E2F8 proteins were in vitro-transcribed and -translated (TnT in vitro translation kit; Promega). Recombinant GST-E2F1 was expressed and purified as described previously (25). Equal amounts of GST and GST-E2F1 were coupled to glutathione-Sepharose beads and incubated with in vitro-translated Myc-E2F7 or Myc-E2F8 for 2 h at 4 °C. Beads were washed four times with PBS buffer and boiled in SDS sample buffer, and the proteins were analyzed by 10% SDS-PAGE followed by Western blot.
Luciferase Reporter Assay
U2OS cells (2 × 104 cells/well, 24-well dishes) were co-transfected with pGL-E2F1-Luc (26), pGL-CMV-Renilla luciferase, and various amount of expression plasmids. Cells were harvested 24 h after transfection, and luciferase activities were determined using the Dual-Luciferase reporter assay system (Promega).
Immunoprecipitation
For detection of interactions with overexpressed proteins, cells (1 × 107) were lysed with 700 μl of lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100). Cell lysates (500 μg) were incubated with 1 μg of antibodies at 4 °C overnight. 30 μl of protein G-agarose (Calbiochem) were added and incubated for 2 h. The beads were washed with lysis buffer three times followed by immunoblotting. For detection of endogenous interactions, cells (7 × 107) were lysed with Nonidet P-40 buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40). Cell lysates were incubated with 3 μg of antibodies at 4 °C overnight. 50 μl of protein A/G-agarose (Calbiochem) were added and incubated for 2 h. The beads were washed followed by immunoblotting.
Gel Shift Assay
Gel shift procedures were described previously (27). The two-site probe (WT) contains E2F1 promoter sequence between positions −39 and +2 (5′-CGTGGCTCTTTCGCGGCAAAAAGGATTTGGCGCGTAAAAGT-3′). The single-site probe (mutant) contains only the proximal E2F-binding site and lacks the distal E2F site (5′-CGTGGCTCTTTCGATGCAAAAAGGATTTGGCGCGTAAAAGT-3′).
ChIP and Re-ChIP Assays
ChIP and Re-ChIP assays were conducted according to the protocol(28). Real time PCR was used to detect E2F1 promoter sequence by using the primers 5′-AGGAACCGCCGCCGTTGTTCCCG-3′ and 5′-CTGCCTGCAAAGTCCCGGCCACTT-3′.
Real Time PCR
RNA was prepared using RNeasy kit (Qiagen) and reverse-transcribed using SuperScript TM II reverse transcriptase (Invitrogen). Real time PCR was performed on StepOnePlusTM real time PCR machine (Applied Biosystems) using SYBR select master mix (Invitrogen). Expression levels were normalized across samples using GAPDH levels. E2F1 primers were: 5′-AGATGGTTATGGTGATCAAAGCC-3′, and 5′-ATCTGAAAGTTCTCCGAAGAGTCC-3′; GAPDH primers were: 5′-TGATGACATCAAGAAGGTGGTGAAG-3′, and 5′-TCCTTGGAGGCCATGTGGGCCAT-3′.
RNAi
pGIPZ-based lentiviral shRNA constructs against CtBP1 and CtBP2 were obtained from Open Biosystems. Lentivirus was packaged according to the manufacturer's protocol.
RESULTS
E2F7 and E2F8 Interact with E2F1 and CtBP
To explore a mechanism for E2F7- and E2F8-mediated transcription repression, we sought to identify E2F7- or E2F8-interacting proteins. FLAG-tagged E2F7 or FLAG-tagged E2F8 was expressed in U2OS cells, and cell extracts were subjected to purification with anti-FLAG antibodies. The identities of polypeptides associated with E2F7 or E2F8 were determined by mass spectrometry (Table 1). Rb was not detected as an E2F7- or E2F8-interacting protein, consistent with previous observations (9, 10). We did, however, detect an interaction of E2F7 with CtBP1 and CtBP2, proteins previously demonstrated to be involved in transcription repression through recruitment of histone-modifying activities (29). These proteins have also been identified as interactions with Rb family proteins, again in the context of transcriptional repression mediated by E2F-Rb (30).
TABLE 1.
List of selected proteins identified by mass spectrometry
Numbers are distinct peptides identified for each protein; IP, immunoprecipitation.
| Protein | Peptides identified |
||
|---|---|---|---|
| E2F1 IP | E2F8 IP | E2F7 IP | |
| E2F1 | 22 | 8 | 0 |
| RB | 21 | 0 | 0 |
| DP-1 | 6 | 0 | 0 |
| DP-2 | 3 | 0 | 0 |
| p107 | 6 | 0 | 0 |
| p130 | 2 | 0 | 0 |
| CDK2 | 8 | 0 | 0 |
| CDC2 | 6 | 0 | 0 |
| CCNA2 | 4 | 0 | 0 |
| E2F8 | 0 | 32 | 0 |
| E2F7 | 2 | 0 | 42 |
| CTBP1 | 0 | 0 | 1 |
| CTBP2 | 0 | 0 | 4 |
In addition to the interactions noted above, we also identified E2F1 as an interacting protein with E2F8. To verify and extend this finding, we reversed the strategy to purify proteins interacting with E2F1. This analysis revealed a collection of well known E2F1 partners including Rb, DP-1, DP-2, CCNA2, CDK2, and CDC2 (Table 1). Notably, this analysis also identified E2F7 as an interacting protein, thus confirming that E2F1 interacts with atypical E2F transcription factors.
Co-immunoprecipitation experiments were used to further confirm that ectopically expressed HA-tagged E2F1 interacts with Myc-tagged E2F7 or E2F8 (Fig. 1A). The same was true when the assay was reversed using Myc-tagged E2F7 or E2F8 for the immunoprecipitation (Fig. 1A). Importantly, we also detected the endogenous interaction between E2F1 and E2F7 (Fig. 1B). Further, HA-tagged E2F2 or E2F3 co-immunoprecipitated with E2F7 or E2F8 (Fig. 1C), suggesting that E2F2 and E2F3 also interact with E2F7 and E2F8. Finally, in vitro GST pulldown assays demonstrated that E2F1 directly bound to E2F7 or E2F8 (Fig. 1D).
FIGURE 1.

E2F1–3 interact with E2F7 and E2F8. A, E2F1 interacts with E2F7 or E2F8. Lysate from 293 cells transfected with both HA-E2F1 and Myc-E2F7 or Myc-E2F8 was immunoprecipitated (IP) followed by immunoblotting with anti-HA and anti-Myc antibodies as indicated. Immunoprecipitation with normal mouse IgG was used as negative control. B, endogenous E2F1 interacts with E2F7. Lysate from HeLa cells was immunoprecipitated (IP) with anti-E2F1 antibody (KH95, Santa Cruz Biotechnology) followed by immunoblotting with anti-E2F7 antibody (N-20, Santa Cruz Biotechnology). C, E2F2 and E2F3 interact with E2F7 and E2F8. HA-E2F2 or HA-E2F3 was co-transfected with Myc-E2F7 or Myc-E2F8 into 293 cells. Cell lysate was immunoprecipitated (IP) and immunoblotted with anti-HA and anti-Myc antibodies as indicated. D, GST pulldown assay. Recombinant GST or GST-E2F1 (shown by Ponceau staining) was bound to the glutathione-Sepharose beads, incubated with in vitro-translated Myc-E2F7 or Myc-E2F8, washed with PBS buffer, and boiled in SDS sample buffer followed by immunoblotting (IB) using antibodies as indicated.
To better characterize the nature of the interactions, we mapped the domain of E2F1 that mediates the interaction with E2F7 or E2F8. Several truncated forms of E2F1 (Fig. 2A) were expressed together with full-length E2F7 or E2F8 in 293 cells. Co-immunoprecipitation experiments demonstrated that the shortest form of E2F1 that is sufficient for binding to E2F7 or E2F8 contains amino acids 126–251, which consists of the E2F1 DBD and leucine zipper (Fig. 2A).
FIGURE 2.

Map regions that are required for the interaction between E2F7/8 and E2F1. A, map regions of E2F1 to interact with E2F7/8. Left panel, illustration of E2F1 fragments used for binding assays. Right panel, co-immunoprecipitation experiments. 293 cells were transfected with expression vectors encoding HA-E2F7 (upper panel) or HA-E2F8 (lower panel) and Myc-tagged E2F1 fragments as indicated, and immunoprecipitation (IP) with anti-Myc antibody, followed by immunoblotting using antibodies as indicated, was performed. B, dimerization and DNA-binding residues are required for E2F7/8 to bind to E2F1. Left panel, illustration of residues in DBDs used for mutagenesis. R185A,R334R and R156A,R314A represent E2F7 and E2F8 DNA-binding mutants, respectively; NVL191–193AAA/NVL340–342AAA and NVL162–164AAA/NVL320–322AAA represent E2F7 and E2F8 dimerization mutants, respectively (19). Right panel, 293 cells were transfected with overexpression vectors encoding HA-E2F1 and Myc-E2F7 or Myc-E2F8 mutants as indicated, and immunoprecipitation (IP) with anti-HA antibody, followed by immunoblotting with antibodies as indicated, was performed. C, repression activities of the E2F7 or E2F8 mutants. U2OS cells were transfected with 100 ng of the reporter plasmid pGL2-E2F1-Luc, 5 ng of pGL2-Renilla, 100 ng of pCDNA3-HA-E2F1, and increasing amounts (25 and 100 ng) of E2F7 or E2F8 expression plasmid. After a 24-h transfection, cells were collected for luciferase assay. All luciferase values were corrected for transfection efficiency by normalizing with Renilla luciferase activity. Error bars represent ± S.D. from three independent experiments.
We next mapped the domain(s) of E2F7 required for the interaction with E2F1. The crystal structure of an E2F4-DP2-DNA complex shows that the DBDs of the E2F and DP proteins form an extensive protein-protein interface (31). Because residues at the heterodimerization interface are well conserved within E2F/DP families, it has been suggested that the duplicated DBDs of E2F7 (or E2F8) structurally mimic the DNA-binding interface of E2F-DP heterodimers, with an intramolecular interaction facilitating binding to the E2F site (11). E2F7 and E2F8 can also form homodimers or heterodimers probably through intermolecular interactions between DBDs (9, 11, 15, 19). To focus on the role of the DNA-binding domain, we analyzed the effect of mutation of potential dimerization residues (NVL191–193AAA/NVL340–342AAA and NVL162–164AAA/NVL320–322AAA)3 or adjacent DNA-binding residues (R185A,R334R and R156A,R314A). As shown in Fig. 2B, these mutations largely abolished the interaction with E2F1.
To further explore the functional consequence of an interaction between E2F7, or E2F8 and E2F1, we made use of reporter assays to measure the transcriptional repressing function of atypical E2Fs. As shown in Fig. 2C, expression of E2F1 resulted in a robust activation of a reporter under the control of the E2F1 promoter. Co-expression of increasing amounts of a wild type E2F7 or E2F8 vector resulted in a near complete repression of the E2F1-induced expression. Assay of mutants altered in the E2F7 or E2F8 DNA-binding domain abolished the capacity of E2F7 or E2F8 to repress transcription. The loss of inhibitory effect of these mutants might be due to their defect of E2F1 interaction, although it might also due to their defect of DNA binding because the two dimerization motifs within E2F7 or E2F8 can interact with each other, forming an intramolecular dimer to bind to DNA (9, 11, 15, 19). Taken together, these findings demonstrate that the E2F7 and E2F8 transcriptional repressors interact with the E2F1–3 activator proteins, dependent on DBDs that are also essential for transcriptional repression by E2F7 and E2F8.
Binding of E2F7 and E2F1 to the E2F1 Promoter
In considering the specificity of E2F7-mediated repression that appears to be selective for genes induced by E2F1 at G1/S, and considering the finding that E2F7 and E2F8 directly interact with E2F1, we have noted previous observations that established a role for cooperative binding of E2F to the adenovirus E2 promoter (32). This work demonstrated that E2F proteins bind with low affinity to the viral promoter, occupying only one of the two possible recognition sites in the promoter. In contrast, in the presence of the adenovirus E4 6/7 gene product, a protein shown to directly bind to E2F proteins, a very stable E2F-promoter complex is formed, involving a dimeric E2F-E4 complex bound to both E2F recognition sites in the promoter. We have used a similar approach to assess the functional significance of the interaction of E2F7 with E2F1.
Gel shift assays were performed using a probe representing the E2F1 promoter and cell extract derived from 293 cells overexpressing E2F1, E2F7, or both proteins. We used two probes for the assays, one containing the wild type promoter sequence with the normal arrangement of two adjacent E2F sites derived from E2F1 promoter and the other bearing mutations in the distal E2F site, thus leaving only one functional binding site (Fig. 3A). As shown in Fig. 3B, expression of E2F1 alone results in formation of a DNA-protein complex (E2F1-DNA) in which E2F1 is bound to only one of the two E2F sites, as indicated by the formation of the same complex on either the probe containing two sites (lane 1) or the probe containing one site (lane 10). The involvement of E2F1 in the complex was confirmed by an antibody supershift assay (lane 2 and lane 11).
FIGURE 3.
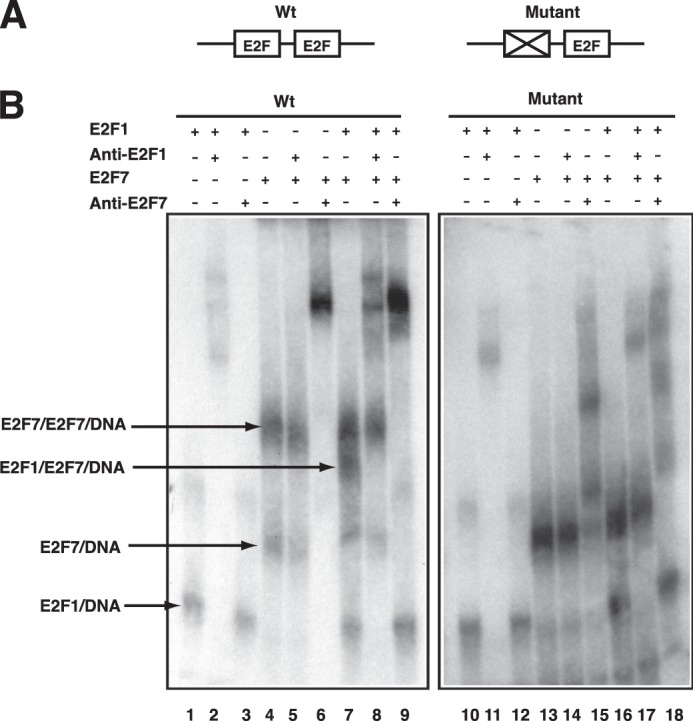
Gel shift assay for E2F DNA-binding properties. A, probes used for gel shift assay. Probe Wt has two E2F sites derived from E2F1 promoter; Probe Mutant has the distal E2F site mutated. B, binding reactions contained the indicated probe along with cell lysate from 293 cells transfected with overexpression vectors encoding E2F1-(HA-DP1), Myc-E2F7, and E2F1-(HA-DP1)-(Myc-E2F7), respectively. Various antibodies were added to the reactions for distinguishing different protein-DNA complexes as indicated.
Expression of E2F7 also resulted in the formation of a complex (E2F7-DNA) that again appeared to involve binding to only one site, given the formation of the same complex on either the two-site probe (lane 4) or the one-site probe (lane 13). Again, the involvement of E2F7 was confirmed by antibody supershift (lane 6, lane 15). However, in addition to this complex, the expression of E2F7 also led to the formation of a larger, more slowly migrating complex (E2F7-E2F7-DNA) using the probe containing both sites (lane 4) that was not apparent using the probe with only one intact E2F-binding site (lane 13), indicating that both sites are occupied using the WT probe.
Co-expression of E2F1 and E2F7 also results in the formation of the same E2F7-E2F7-DNA complex but also a smaller sized complex that contains both E2F1 and E2F7 as demonstrated by antibody supershifts (lanes 7–9). Once again, the formation of this complex required the presence of both E2F-binding sites in the probe because it was not observed with the single-site probe (lane 16). Taken together, these results demonstrate a capacity of E2F7 to bind in a cooperative manner, either together with E2F1 or involving only E2F7, to the two adjacent binding sites in the E2F1 promoter.
E2F7 Mediates Transcriptional Repression through Association with the Co-repressor CtBP
In considering the mechanism for repression by E2F7, we returned to the observation that mass spectrometry analysis identified CtBP1 and CtBP2 as E2F7-interacting proteins (Table 1). Previous studies show that CtBPs are recruited to DNA by transcription factors that contain a PXDLS motif (33). Therefore, we examined E2F7 sequence and found that the N terminus has a potential CtBP-binding motif PIDLS (Fig. 4A). The interaction between E2F7 and CtBP1 or CtBP2 was demonstrated by co-immunoprecipitation experiments, and it was abolished if PIDLS motif was replaced by ASASA or PIAAS (Fig. 4B). These interactions are further confirmed by endogenous immunoprecipitation (Fig. 4C). We then investigated whether CtBPs and E2F7 were bound together on E2F1 promoter in a ChIP-reChIP assay, where two consecutive immunoprecipitations using anti-HA-E2F7 and anti-Myc-CtBP2 antibodies were performed. As shown in Fig. 4D, both E2F7 and CtBP2 were detected in ChIP assays. Further, a re-ChIP of the initial E2F7 ChIP identified CtBP2, indicating that both proteins co-occupy the same promoter.
FIGURE 4.

E2F7 mediates transcription repression through CtBPs. A, illustration of potential CtBP-binding motif (PIDLS) located at the N terminus of E2F7. B, E2F7 interacts with CtBP1 and CtBP2. 293 cells were transfected with overexpression plasmids encoding Myc-tagged CtBPs and HA-E2F7 as indicated. Cell lysate was immunoprecipitated (IP) with anti-Myc antibody followed by immunoblotting with anti-HA or anti-Myc antibodies as indicated. Immunoprecipitation with normal mouse IgG was used as negative control. C, endogenous E2F7 interacts with CtBPs. Lysate from HeLa cells was immunoprecipitated (IP) with anti-E2F7 antibody followed by immunoblotting with anti-CtBP1 or anti-CtBP2 antibody as indicated. Note that the lower band of CtBP1 blot is nonspecific (NS). D, ChIP-reChIP assays. 293 cells were co-transfected with HA-E2F7 and Myc-CtBP2. Cells were cross-linked after 24 h, lysed, sonicated, precleared, and immunoprecipitated with the indicated antibodies overnight followed by incubation with protein A/G beads for 2 h. After stringent washes, chromatin-bound proteins were eluted with 1 mm dithiothreitol followed by a second immunoprecipitation with the indicated antibodies. Chromatin was eluted from beads, uncross-linked, purified, and subjected to real time PCR for E2F1 promoter. Two-tailed t test was performed for statistical analysis. The asterisk indicates that the difference is significant (p < 0.05). E, CtBP2 is required for E2F7-mediated repression. 293T cells (5 × 105 cells) were infected with lentivirus harboring short hairpin RNAs (shRNA) targeting CtBP2 (two different hairpin sequences per gene were chosen). Cells were selected with 2 μg/ml puromycin for 48 h, transfected with 500 ng of HA-E2F7 for 14 h, and harvested for assays. Western blot was conducted to confirm the depletion of CtBP2 proteins (upper panel). RNA was isolated and reverse-transcribed, and the E2F1 level was measured by real time PCR. Cell line integrated with a nonsilencing shRNA was used as a control (Ctrl). All values across samples were normalized with glyceraldehyde phosphate dehydrogenase level.
To assess the functional importance of interactions between CtBPs and E2F7, we knocked down CtBP1 or CtBP2 expression by RNAi and examined E2F7-mediated E2F1 transcription. CtBP2 (Fig. 4E) but not CtBP1 (data not shown) knockdown significantly decreased the ability of E2F7 to repress the E2F1 transcription, suggesting that CtBP2 is required for E2F7 repression.
DISCUSSION
A role for E2F1, E2F2, and E2F3 in the activation of transcription at G1/S, inducing the expression of the program of genes encoding DNA replication and cell cycle regulatory activities, has now been well established. In addition to the activation of genes that orchestrate the DNA replication process and enable progression through the cell cycle, it is also clear that there is tight control of these events that includes negative feedback limiting the extent of activator E2F function. One component is an induced destruction of E2F proteins through the action of SCF/Skp2 (34, 35) as well as the negative control of E2F activity by cyclin A-cdk2 (36, 37).
In addition, recent work has shown that the activator E2Fs also induce the expression of genes encoding atypical E2Fs including E2F7 and E2F8, which function as repressors of transcription (9–14). Further, these E2Fs have now been shown to function as repressors of the very genes that are induced by the activator E2Fs (16, 17). Such a regulatory relationship has been described as an incoherent feed-forward mechanism in which an activator induces a target but at the same time also induces a repressor of the target, thus limiting the extent of the induction (39). The net result of this regulatory action is to limit the accumulation of E2F activity during S phase. Recent work has highlighted the importance of this control and the adverse consequences of over-accumulation of E2F activity in S phase (40). Further, this form of negative feedback is also a key component of oscillatory systems, consistent with the role of E2F function during the cell cycle (41).
Although the relationship of the atypical E2Fs with respect to control of genes induced at G1/S has been established in past work, the basis by which specificity of this control is established has not been addressed. The work we present here identifies a protein interaction involving E2F1 with E2F7 and E2F8. The characteristics of DNA binding of complexes involving either E2F7 or E2F7 together with E2F1 suggest a novel mechanism by which the specificity of negative control is achieved. We speculate that E2F7 may be directed to the promoters activated by E2F1 (as well as E2F2 and E2F3) as a result of the direct interaction between E2F1 and E2F7 (Fig. 5). We have attempted to do a ChIP-reChIP experiment to determine whether E2F1 and E2F7 co-occupy the same E2F1 promoter, and although there is an enrichment of E2F1 promoter sequence after a sequential E2F1 and E2F7 ChIP, it is only a low level that might indicate that E2F1 and E2F7 might transiently form a complex on E2F1 promoter in vivo (data not shown).
FIGURE 5.
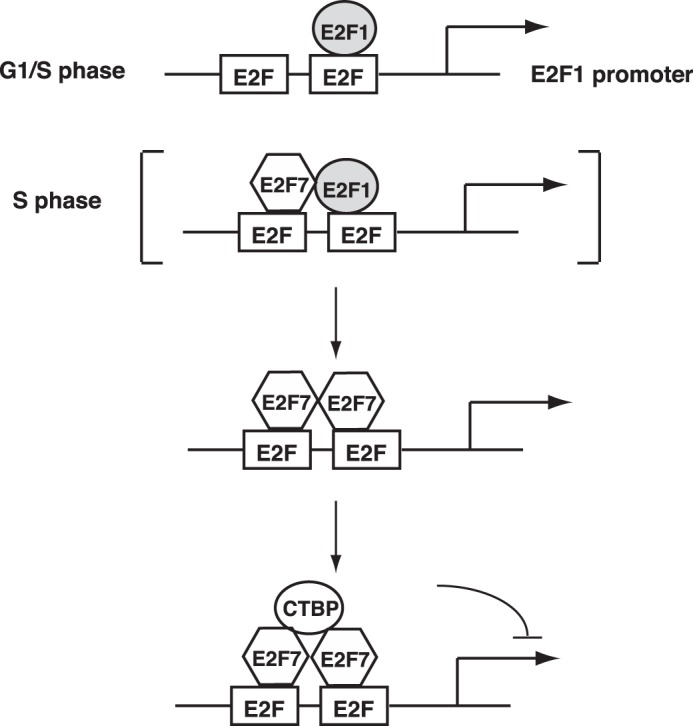
Possible model of E2F7-mediated transcription repression. E2F1 promoter contains two overlapping E2F-binding sites between which the spacing is 13 bp. It is activated in G1/S transition in an E2F-dependent manner when E2Fs are released from Rb. Note that E2F1 only binds to one of the two E2F sites on the promoter (38). E2F1 subsequently activates the transcription of E2F7, which in turn binds to the E2F1 promoter, possibly through occupying the other E2F-binding site. E2F7 might interact with E2F1 on the promoter but eventually E2F1 dissociates from the E2F-binding site. Two E2F7 molecules interact with each other and bind to the two E2F sites, forming a very stable protein-DNA complex. E2F7 subsequently recruits transcription co-repressor CtBPs for repressing the transcription of E2F1.
An initial E2F1-E2F7 promoter complex may then give way to the more stable E2F7-E2F7 complex observed in the promoter interaction assays, together with the recruitment of CtBP proteins that mediate the repression of transcription. In this scenario, the interaction of E2F1 with E2F7 provides the necessary specificity to direct the repression of transcription to those promoters that were initially activated.
Previous studies have shown that CtBPs are often recruited to transcription factors along with histone deacetylases (HDACs) (29). Indeed, in addition to the interaction of E2F7 with CtBP, we also find that E2F7 can co-precipitate with HDAC1(data not shown). The pan-HDAC inhibitor, trichostatin A, partially reversed the ability of E2F7 to repress the E2F1 and Cdc6 promoters (data not shown). However, E2F7 repression of E2F1 transcription is not alleviated in HDAC1 RNAi cell lines, indicating that complete E2F7 repression might depend on other HDACs. Further studies are needed to test these possibilities.
Acknowledgments
We thank Laszlo Jakoi for technical assistance and all the members of the Nevins laboratory for critical feedback and suggestions. We are grateful to Dr. J. Will Thompson and Laura G. Dubois at the Duke Proteomics Core Facility, who provided mass spectrometry service. We thank Kaye Culler for assistance in submitting the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants CA104663 and CA106520 (to J. R. N.).
The following multiple mutant designations are used: NVL191–193AAA/NVL340–342AAA, N191A,V192A,L193A-N340A,V341A,L342A; NVL162–164AAA/NVL320–322AAA, N162A,V163A,L164A-N320A,V321A,L322A.
- Rb
- retinoblastoma
- DBD
- DNA-binding domain
- CtBP
- C-terminal-binding protein
- HDAC
- histone deacetylase.
REFERENCES
- 1. Dimova D. K., Dyson N. J. (2005) The E2F transcriptional network: old acquaintances with new faces. Oncogene 24, 2810–2826 [DOI] [PubMed] [Google Scholar]
- 2. DeGregori J., Johnson D. G. (2006) Distinct and overlapping roles for E2F family members in transcription, proliferation, and apoptosis. Curr. Mol. Med. 6, 739–748 [DOI] [PubMed] [Google Scholar]
- 3. Giangrande P. H., Zhu W., Schlisio S., Sun X., Mori S., Gaubatz S., Nevins J. R. (2004) A role for E2F6 in distinguishing G1/S- and G2/M-specific transcription. Genes Dev. 18, 2941–2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ogawa H., Ishiguro K., Gaubatz S., Livingston D. M., Nakatani Y. (2002) A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science 296, 1132–1136 [DOI] [PubMed] [Google Scholar]
- 5. Helin K., Wu C. L., Fattaey A. R., Lees J. A., Dynlacht B. D., Ngwu C., Harlow E. (1993) Heterodimerization of the transcription factors E2F-1 and DP-1 leads to cooperative trans-activation. Genes Dev. 7, 1850–1861 [DOI] [PubMed] [Google Scholar]
- 6. Krek W., Livingston D. M., Shirodkar S. (1993) Binding to DNA and the retinoblastoma gene product promoted by complex formation of different E2F family members. Science 262, 1557–1560 [DOI] [PubMed] [Google Scholar]
- 7. Bandara L. R., Buck V. M., Zamanian M., Johnston L. H., La Thangue N. B. (1993) Functional synergy between DP-1 and E2F-1 in the cell cycle-regulating transcription factor DRTF1/E2F. EMBO J. 12, 4317–4324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lammens T., Li J., Leone G., De Veylder L. (2009) Atypical E2Fs: new players in the E2F transcription factor family. Trends Cell Biol. 19, 111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Di Stefano L., Jensen M. R., Helin K. (2003) E2F7, a novel E2F featuring DP-independent repression of a subset of E2F-regulated genes. EMBO J. 22, 6289–6298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de Bruin A., Maiti B., Jakoi L., Timmers C., Buerki R., Leone G. (2003) Identification and characterization of E2F7, a novel mammalian E2F family member capable of blocking cellular proliferation. J. Biol. Chem. 278, 42041–42049 [DOI] [PubMed] [Google Scholar]
- 11. Logan N., Delavaine L., Graham A., Reilly C., Wilson J., Brummelkamp T. R., Hijmans E. M., Bernards R., La Thangue N. B. (2004) E2F-7: a distinctive E2F family member with an unusual organization of DNA-binding domains. Oncogene 23, 5138–5150 [DOI] [PubMed] [Google Scholar]
- 12. Logan N., Graham A., Zhao X., Fisher R., Maiti B., Leone G., La Thangue N. B. (2005) E2F-8: an E2F family member with a similar organization of DNA-binding domains to E2F-7. Oncogene 24, 5000–5004 [DOI] [PubMed] [Google Scholar]
- 13. Maiti B., Li J., de Bruin A., Gordon F., Timmers C., Opavsky R., Patil K., Tuttle J., Cleghorn W., Leone G. (2005) Cloning and characterization of mouse E2F8, a novel mammalian E2F family member capable of blocking cellular proliferation. J. Biol. Chem. 280, 18211–18220 [DOI] [PubMed] [Google Scholar]
- 14. Christensen J., Cloos P., Toftegaard U., Klinkenberg D., Bracken A. P., Trinh E., Heeran M., Di Stefano L., Helin K. (2005) Characterization of E2F8, a novel E2F-like cell-cycle regulated repressor of E2F-activated transcription. Nucleic Acids Res. 33, 5458–5470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li J., Ran C., Li E., Gordon F., Comstock G., Siddiqui H., Cleghorn W., Chen H. Z., Kornacker K., Liu C. G., Pandit S. K., Khanizadeh M., Weinstein M., Leone G., de Bruin A. (2008) Synergistic function of E2F7 and E2F8 is essential for cell survival and embryonic development. Dev. Cell 14, 62–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Westendorp B., Mokry M., Groot Koerkamp M. J., Holstege F. C., Cuppen E., de Bruin A. (2012) E2F7 represses a network of oscillating cell cycle genes to control S-phase progression. Nucleic Acids Res. 40, 3511–3523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aksoy O., Chicas A., Zeng T., Zhao Z., McCurrach M., Wang X., Lowe S. W. (2012) The atypical E2F family member E2F7 couples the p53 and RB pathways during cellular senescence. Genes Dev. 26, 1546–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carvajal L. A., Hamard P. J., Tonnessen C., Manfredi J. J. E2F7, a novel target, is up-regulated by p53 and mediates DNA damage-dependent transcriptional repression. Genes Dev. 26, 1533–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zalmas L. P., Zhao X., Graham A. L., Fisher R., Reilly C., Coutts A. S., La Thangue N. B. (2008) DNA-damage response control of E2F7 and E2F8. EMBO Rep. 9, 252–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ouseph M. M., Li J., Chen H. Z., Pécot T., Wenzel P., Thompson J. C., Comstock G., Chokshi V., Byrne M., Forde B., Chong J. L., Huang K., Machiraju R., de Bruin A., Leone G. (2012) Atypical E2F repressors and activators coordinate placental development. Dev. Cell 22, 849–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pandit S. K., Westendorp B., Nantasanti S., van Liere E., Tooten P. C., Cornelissen P. W., Toussaint M. J., Lamers W. H., de Bruin A. (2012) E2F8 is essential for polyploidization in mammalian cells. Nat. Cell Biol. 14, 1181–1191 [DOI] [PubMed] [Google Scholar]
- 22. Weijts B. G., Bakker W. J., Cornelissen P. W., Liang K. H., Schaftenaar F. H., Westendorp B., de Wolf C. A., Paciejewska M., Scheele C. L., Kent L., Leone G., Schulte-Merker S., de Bruin A. (2012) E2F7 and E2F8 promote angiogenesis through transcriptional activation of VEGFA in cooperation with HIF1. EMBO J. 31, 3871–3884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen H. Z., Ouseph M. M., Li J., Pécot T., Chokshi V., Kent L., Bae S., Byrne M., Duran C., Comstock G., Trikha P., Mair M., Senapati S., Martin C. K., Gandhi S., Wilson N., Liu B., Huang Y. W., Thompson J. C., Raman S., Singh S., Leone M., Machiraju R., Huang K., Mo X., Fernandez S., Kalaszczynska I., Wolgemuth D. J., Sicinski P., Huang T., Jin V., Leone G. (2012) Canonical and atypical E2Fs regulate the mammalian endocycle. Nat. Cell Biol. 14, 1192–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schlisio S., Halperin T., Vidal M., Nevins J. R. (2002) Interaction of YY1 with E2Fs, mediated by RYBP, provides a mechanism for specificity of E2F function. EMBO J. 21, 5775–5786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burke T. W., Cook J. G., Asano M., Nevins J. R. (2001) Replication factors MCM2 and ORC1 interact with the histone acetyltransferase HBO1. J. Biol. Chem. 276, 15397–15408 [DOI] [PubMed] [Google Scholar]
- 26. Sears R., Ohtani K., Nevins J. R. (1997) Identification of positively and negatively acting elements regulating expression of the E2F2 gene in response to cell growth signals. Mol. Cell. Biol. 17, 5227–5235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nevins J. R., DeGregori J., Jakoi L., Leone G. (1997) Functional analysis of E2F transcription factor. Methods Enzymol. 283, 205–219 [DOI] [PubMed] [Google Scholar]
- 28. Furlan-Magaril M., Rincón-Arano H., Recillas-Targa F. (2009) Sequential chromatin immunoprecipitation protocol: ChIP-reChIP. Methods Mol. Biol. 543, 253–266 [DOI] [PubMed] [Google Scholar]
- 29. Shi Y., Sawada J., Sui G., Affar el B., Whetstine J. R., Lan F., Ogawa H., Luke M. P., Nakatani Y., Shi Y. (2003) Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422, 735–738 [DOI] [PubMed] [Google Scholar]
- 30. Meloni A. R., Smith E. J., Nevins J. R. (1999) A mechanism for Rb/p130-mediated transcription repression involving recruitment of the CtBP corepressor. Proc. Natl. Acad. Sci. U.S.A. 96, 9574–9579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zheng N., Fraenkel E., Pabo C. O., Pavletich N. P. (1999) Structural basis of DNA recognition by the heterodimeric cell cycle transcription factor E2F-DP. Genes Dev. 13, 666–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Raychaudhuri P., Bagchi S., Neill S. D., Nevins J. R. (1990) Activation of the E2F transcription factor in adenovirus-infected cells involves E1A-dependent stimulation of DNA-binding activity and induction of cooperative binding mediated by an E4 gene product. J. Virol. 64, 2702–2710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chinnadurai G. (2002) CtBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol. Cell 9, 213–224 [DOI] [PubMed] [Google Scholar]
- 34. Zhang L., Wang C. (2006) F-box protein Skp2: a novel transcriptional target of E2F. Oncogene 25, 2615–2627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marti A., Wirbelauer C., Scheffner M., Krek W. (1999) Interaction between ubiquitin-protein ligase SCFSKP2 and E2F-1 underlies the regulation of E2F-1 degradation. Nat. Cell Biol. 1, 14–19 [DOI] [PubMed] [Google Scholar]
- 36. Krek W., Ewen M. E., Shirodkar S., Arany Z., Kaelin W. G., Jr., Livingston D. M. (1994) Negative regulation of the growth-promoting transcription factor E2F-1 by a stably bound cyclin A-dependent protein kinase. Cell 78, 161–172 [DOI] [PubMed] [Google Scholar]
- 37. Xu M., Sheppard K. A., Peng C. Y., Yee A. S., Piwnica-Worms H. (1994) Cyclin A/CDK2 binds directly to E2F-1 and inhibits the DNA-binding activity of E2F-1/DP-1 by phosphorylation. Mol. Cell. Biol. 14, 8420–8431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Araki K., Nakajima Y., Eto K., Ikeda M. A. (2003) Distinct recruitment of E2F family members to specific E2F-binding sites mediates activation and repression of the E2F1 promoter. Oncogene 22, 7632–7641 [DOI] [PubMed] [Google Scholar]
- 39. Alon U. (2007) Network motifs: theory and experimental approaches. Nat. Rev. Genet. 8, 450–461 [DOI] [PubMed] [Google Scholar]
- 40. Wong J. V., Dong P., Nevins J. R., Mathey-Prevot B., You L. (2011) Network calisthenics: control of E2F dynamics in cell cycle entry. Cell Cycle 10, 3086–3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ferrell J. E., Jr., Tsai T. Y., Yang Q. (2011) Modeling the cell cycle: why do certain circuits oscillate? Cell 144, 874–885 [DOI] [PubMed] [Google Scholar]