

Background: Continuous adaptation of the bacterial membrane is required in response to changing environmental conditions.
Results: Pseudomonas aeruginosa ORF PA0919 codes for an alanyl-phosphatidylglycerol hydrolase that is anchored to the periplasmic surface of the inner membrane.
Conclusion: The elucidated enzymatic activity implies a new regulatory circuit for the fine tuning of cellular alanyl-phosphatidylglycerol concentrations.
Significance: Lipid homeostasis is crucial for understanding antimicrobial susceptibility.
Keywords: Antibiotic Resistance, Cationic Antimicrobial Peptide (CAMP), Lipid Metabolism, Membrane Proteins, Pseudomonas aeruginosa, MprF, Alanyl-Phosphatidylglycerol, Lipid Homeostasis, Lysyl-Phosphatidylglycerol
Abstract
Specific aminoacylation of the phospholipid phosphatidylglycerol (PG) with alanine (or with lysine) was shown to render various organisms less susceptible to antimicrobial agents and environmental stresses. In this study, we make use of the opportunistic pathogen Pseudomonas aeruginosa to decode ORF PA0919-dependent lipid homeostasis. Analysis of the polar lipid content of the deletion mutant ΔPA0919 indicated significantly enlarged levels of alanyl-PG. The resulting phenotype manifested an increased susceptibility to several antimicrobial compounds when compared with the wild type. A pH-dependent PA0919 promoter located within the upstream gene PA0920 was identified. Localization experiments demonstrated that the PA0919 protein is anchored to the periplasmic surface of the inner bacterial membrane. The recombinant overproduction of wild type and several site-directed mutant proteins in the periplasm of Escherichia coli facilitated a detailed in vitro analysis of the enzymatic PA0919 function. A series of artificial substrates (p-nitrophenyl esters of various amino acids/aliphatic acids) indicated enzymatic hydrolysis of the alanine, glycine, or lysine moiety of the respective ester substrates. Our final in vitro activity assay in the presence of radioactively labeled alanyl-PG then revealed hydrolysis of the aminoacyl linkage, resulting in the formation of alanine and PG. Consequently, PA0919 was termed alanyl-PG hydrolase. The elucidated enzymatic activity implies a new regulatory circuit for the appropriate tuning of cellular alanyl-PG concentrations.
Introduction
In microbes, the cytoplasmic membrane is probably the most important physical barrier to the surrounding habitat. Therefore, continuous adaptation of the lipid composition is required in response to changing environmental conditions (1). According to the growth temperature, the degree of fatty acid saturation is adopted to maintain the appropriate membrane fluidity (2). Besides this, the modification of the negatively charged headgroup of phosphatidylglycerol (PG)2 into the corresponding aminoacyl esters of PG (aa-PG) is a widely used strategy enabling bacteria to cope with molecules that are potentially harmful for the integrity of the cell membrane. Cationic antimicrobial peptides, but also various antibiotics, have the ability to interact directly with the negatively charged membrane as an antibacterial target (3). One important bacterial response to such compounds is the formation of aa-PG molecules which results in a reduction of the overall net negative charge of the membrane. The resulting lipids can either be the zwitterionic alanyl-PG (A-PG) or alternatively lysyl-PG (L-PG) possessing an overall positive net charge (4–9). The consequential charge characteristics of these molecules were shown to have a direct impact on the electrostatic interaction of antimicrobials with the bacterial membrane (10, 11). Besides this, it was also proposed that the synthesis of aa-PG molecules facilitates the adequate fine tuning of other biophysical properties like membrane fluidity and lipid headgroup interaction (12–14).
Biosynthesis of aa-PG at the bacterial plasma membrane is accomplished by dual function aa-PG synthases (aa-PGS). First, these transmembrane enzymes perform the tRNA-dependent synthesis of aa-PG molecules using alanyl-tRNAAla (Ala-tRNAAla) or lysyl-tRNALys as substrate, respectively. All key amino acid residues for catalysis have been found located at the C-terminal domain of aa-PGSs (15–17). Second, these proteins possess a flippase activity that is essential for the translocation of the newly synthesized phospholipid from the inner to the outer leaflet of the bilayer. Recent research on the Staphylococcus aureus L-PG synthase clearly indicated that this flippase activity located at the hydrophobic N-terminal transmembrane part of the protein is fundamental for the reduced antimicrobial susceptibility of the organism (17, 18).
The Gram-negative bacterium Pseudomonas aeruginosa is one of the dominant pathogens of chronic infections that are associated with cystic fibrosis, and it has long been known for its successful adaptation to several environmental niches (19). During all kinds of infectious and non-infectious states, P. aeruginosa is confronted with changing environmental conditions, which also include highly variable pH values. For example, under conditions of lung infections of cystic fibrosis patients, the liquid of the airway surface of the lung was found acidified due to the genetically caused defect in bicarbonate ion transport (20). It was proposed that this pH alteration contributes to cystic fibrosis pathogenesis. Moreover, a typical inflammatory response also includes an acidification of the corresponding site by the production of acids (21).
Only recently, our laboratory contributed a detailed biochemical and phenotypic characterization of A-PG synthesis in P. aeruginosa (9, 14, 15). The A-PG synthase (A-PGS) transmembrane protein was found located in the inner bacterial membrane (9). Analysis of the P. aeruginosa membrane composition revealed an A-PG content of up to 6% under acidic growth conditions localized in the inner and the outer membrane, whereas under neutral pH conditions, almost no A-PG was detectable. The specific synthesis of A-PG was also found to mediate resistance phenotypes in the presence of the antimicrobial compounds protamine sulfate, cefsulodin, and sodium lactate and also in the presence of CrCl3 (9). Therefore, aa-PGS enzymes might provide a new target to potentiate the efficacy of existing antimicrobial compounds.
In the P. aeruginosa genome, ORF PA0919 is closely located downstream the A-PGS gene (PA0920). Such a genomic organization is found in most Gram-negative organisms containing aa-PGS genes. Orthologous genes of PA0919 are, for example, atvA (acid tolerance and virulence) of Rhizobium tropici and acvB in Agrobacterium tumefaciens (22). Besides acvB, a paralogous gene is additionally found in A. tumefaciens in a completely differing genomic context; the virJ gene is localized on the Ti-plasmid encoding the Type IV secretion system components. The translocation of VirJ into the periplasm was demonstrated (23). For the P. aeruginosa PA0919 protein, solely the localization in the periplasm was experimentally demonstrated within a global protein localization study (24), whereas no additional biochemical data are available to date.
This study focuses on PA0919-dependent A-PG homeostasis of P. aeruginosa. We demonstrate that the accurate fine tuning of the cellular A-PG content is relevant for antimicrobial susceptibility. The transcriptional regulation and the biochemical function of a new lipid-modifying enzyme, PA0919, are demonstrated. Our theoretical analyses reveal a series of orthologous proteins involved in differing bacterial virulence mechanisms (22, 25, 26). This new aspect of lipid modification might be relevant for the understanding of such distantly related systems in the future.
EXPERIMENTAL PROCEDURES
Analysis of Orthologous Gene Clusters, Sequence Alignment
The genomic context of ORF PA0919 was analyzed using the Microbial Genome Database (MBGD) for the comparative analysis of orthologous groups of genes (27). Sequence identity values were calculated by using BLAST analyses (28), and sequence alignments were generated using ClustalW2 (29).
Media and Bacterial Strains
P. aeruginosa and Escherichia coli strains (supplemental Table S1) were grown either in LB medium (30) or in AB medium (31). The AB medium was adjusted to pH 7.3 or 5.3 using an appropriate phosphate buffer (9).
Construction of P. aeruginosa Deletion Strain ΔPA0919 and Complementation Variants
A markerless PA0919 gene deletion mutant was obtained using well established strategies based on sacB-based counterselection and FLP recombinase excision according to Hoang et al. (32). The construction of the suicide vector (pEX18Ap/ΔPA0919) for the replacement of the chromosomal PA0919 gene with a gentamicin resistance cassette was as follows. A 438-bp fragment of the PA0919 upstream region was amplified using primers 1 and 2 (supplemental Table S2). The amplification of the 673-bp downstream region was performed by using primers 3 and 4. For the construction of vector pEX18Ap/ΔPA0919, the BamHI-digested gentamicin resistance cassette of plasmid pPS858 was cloned between the two PCR fragments (upstream region and downstream region of PA0919) into pEX18Ap. This plasmid was then transferred into P. aeruginosa PAO1 by diparental mating using E. coli ST18 as a donor, and the double-crossover mutant was obtained by sacB-based counterselection according to Thoma and Schobert (33). Finally, the plasmid pFLP2 encoding the FLP recombinase was transferred into the modified P. aeruginosa strain, and the FRT-flanked gentamicin resistance cassette was excluded. Chromosomal deletion of the resulting strain ΔPA0919 was verified by DNA sequencing of an appropriate PCR product (using primers 1 and 4).
For the complementation of ΔPA0919, two differing strategies were employed. First, ORF PA0919 together with its 430-bp upstream region containing the putative PPA0919 promoter was integrated at the attB locus of strain ΔPA0919 (Δ19 P19-19). In the alternative complementation strain (Δ19 P20-19), PPA0919 is replaced by the promoter of the PA0920 gene (coding for the A-PGS) (9). Both complementation strains employ a rrnB terminator (14).
For the construction of Δ19 P19-19, a 1,714-bp PCR product covering 430 bp of the PA0919 upstream region and the PA0919 gene was amplified using primers 5 and 6 and inserted into mini-CTX2-PTC (14), resulting in mini-CTX2-PPA0919-PA0919-T (supplemental Table S1).
For the construction of Δ19 P20-19, a 1,284-bp fragment of ORF PA0919 was PCR-amplified (primers 7 and 6) and ligated downstream of the 187-bp PPA0920 promoter sequence (9) into the mini-CTX2-PTC (14). The resulting mini-CTX2-PPA0920-PA0919-T and also mini-CTX2-PPA0919-PA0919-T were transferred into P. aeruginosa ΔPA0919 by diparental mating as described above. The CTX integrase encoded on plasmids mini-CTX2-PPA0920-PA0919-T and mini-CTX2-PPA0919-PA0919-T promoted the vector integration into the attB site of ΔPA0919. The following removal of the FRT-flanked vector fragments by the pFLP2-encoded FLP recombinase provided the markerless complemented strains (32). Strains for specific negative control experiments were generated by insertion of the putative promoter PPA0919 (Δ19 P19) and the promoter PPA0920 (Δ19 P20) at the attB site of ΔPA0919 as described above. Respective primers for plasmid construction and all strains are summarized in supplemental Tables S1 and S2.
Growth Curves and Lipid Analysis of P. aeruginosa Mutant Strains
P. aeruginosa strains were cultivated in 60 ml of AB medium (pH 7.3 and pH 5.3) at 37 °C and 200 rpm. Growth was monitored by turbidity measurement at 578 nm. Cells (15-ml culture with A578 = 0.6) were harvested in identical amounts (according to the culture turbidity) by centrifugation after 24 h (late stationary phase), and cell pellets were stored at −20 °C. Lipid extraction and analysis by two-dimensional TLC was performed as described elsewhere (9). Experiments were performed three times independently.
Quantification of the Polar Lipid Content
Overall amounts of polar lipids from two-dimensional TLC plates were analyzed by molybdatophosphoric acid staining and subsequent quantification using the Gelscan version 6.0 software (BioSciTec GmbH, Frankfurt, Germany). For mutant strains of P. aeruginosa, the amount of A-PG was related to the A-PG content of the wild type strain (wild type set as 100%).
Determination of Minimal Inhibitory Concentrations (MIC)
For MIC determination in the presence of the antimicrobials ampicillin, vancomycin (Roth, Karlsruhe, Germany), daptomycin (Novartis, Nürnberg, Germany), cefsulodin, poly-l-lysine, polymyxin B, polymyxin E, benzethonium chloride, and domiphen bromide (Sigma-Aldrich), a modified method according to Andrews (34) was used as described before (14). In brief, strains were cultivated in AB medium (pH 5.3) to late exponential phase (4 h), and 96-well plates containing the antimicrobial compounds were inoculated with the respective strains, covered with a gas-permeable membrane (Breathe-EasyTM sealing membrane, Diversified Biotech, Dedham, MA), and incubated at 37 °C and 350 rpm (PHMP-4, Grant Instruments, Cambridge, UK) for 18 h. Growth was monitored by measuring the A595 (model 680 microplate reader, Bio-Rad) in the absence of the sealing membrane.
This experimental setup allows for the accurate determination of the MIC by gradually narrowing the range of the employed antimicrobial concentrations. The lowest concentration resulting in an A595 of <0.05 was defined as the MIC of the respective compound. All values were reproduced on the basis of three independent sets of experiments. The relative S.D. values for MIC determinations were evaluated on the basis of the A595 concentration plots. Values of ∼5% (10% for daptomycin) were obtained. Significance of the presented data were calculated using a paired Student's t test.
Construction and Testing of Promoter-lacZ Reporter Gene Fusions
The employed chromosomal PA0919 promoter-lacZ reporter gene fusion, and alternatively, two truncated promoter variants were constructed using the mini-CTX-lacZ vector system (35). For this purpose, a 430-bp fragment (or alternatively a 206-bp or a 140-bp fragment) located upstream of ORF PA0919 was PCR-amplified and linked to KpnI and BamHI restriction sites using primers 8 and 2 (alternatively 9 and 2 or 10 and 2, respectively) (supplemental Table S2). The corresponding PCR products were each cloned into the mini-CTX-lacZ (see supplemental Table S1). Transfer of the respective plasmids into P. aeruginosa PAO1 was carried out by diparental mating (see above), and plasmid integration into the attB locus of the genome was promoted by the CTX integrase of the mini-CTX-lacZ vector system. In the resulting strains, the mini-CTX-lacZ vector sequences (containing the tetracycline resistance cassette) were deleted using an FLP recombinase as described above to yield strains PAO1 PPA0919-430-lacZ, PAO1 PPA0919-206-lacZ, and PAO1 PPA0919-140-lacZ (supplemental Table S1). The PAO1 KS11 strain, which is devoid of any promoter, was used in control experiments (36). All reporter gene fusion assays were performed in triplicate as outlined before (37, 38), and β-galactosidase activities were calculated according to Miller (39). Mean and S.D. values were calculated, and the significance of the presented data was assessed using a paired Student's t test.
Construction of P. aeruginosa Strain for PA0919 Localization
For subcellular localization experiments, the native PA0919 promoter sequence in combination with the PA0919 gene was fused to a C-terminal Strep-tag II sequence to generate a plasmid construct that ensures an identical promoter control in the genetic background of the ΔPA0919 strain. Therefore, the PA0919 gene together with its 430-bp upstream region was PCR-amplified using primers 11 and 12, which also encodes for the additional Strep-tag II sequence. The resulting fragment was cloned into the KpnI/BamHI site of pUCP20T (40) to generate plasmid pUCP20T/PPA0919-PA0919-Strep (see Tables S1 and S2), which was subsequently transferred into PAO1 ΔPA0919 by diparental mating using E. coli ST18 as a donor.
Construction of an E. coli Overproduction Vector for PA0919
The efficient periplasmatic production of PA0919 as a C-terminal Strep-tag II fusion was based on the commercially available pET22b(+) vector (Novagen, Merck). This plasmid facilitates protein secretion due to an N-terminal E. coli-specific PelB signal sequence. Vector pET22b(+) was modified by inserting a C-terminal Strep-tag II sequence using oligonucleotides 13 and 14 (supplemental Table S2) and termed pET22b(+)Strep (generously provided by T. Nicke). The predicted P. aeruginosa-specific signal sequence of PA0919 comprises the initial 34 amino acid residues. Therefore, the coding sequence of residues 35–427 was PCR-amplified (primers 15 and 16) and cloned into the NcoI/HindIII site of pET22b(+)Strep, resulting in pET22b(+)PA0919-Strep.
Mutagenesis of the P. aeruginosa PA0919 Protein
Three pET22b(+)PA0919-Strep variant plasmids were constructed by site-directed mutagenesis with the QuikChange kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions. The respective primers and plasmids are summarized in supplemental Tables S1 and S2.
Production and Purification of Recombinant PA0919 from the Periplasmic E. coli Fraction
E. coli BL21 (λDE3) (Stratagene) containing pET22b(+)PA0919-Strep (or the respective variants) was cultivated in 500 ml of LB (100 μg/ml ampicillin) to an A578 of 0.5. Protein production was induced by 25 μm isopropyl-β-d-thiogalactopyranoside (GERBU Biotechnik GmbH, Wieblingen, Germany), and the cultivation was continued at 17 °C for 24 h. Cells were harvested by centrifugation, suspended in 50 mm HEPES-NaOH, pH 8.0, 150 mm NaCl, 20% (w/v) d-(+)-sucrose, and pellets were stored at −20 °C. Cells were thawed and suspended in 50 mm HEPES-NaOH, pH 8.0, 150 mm NaCl, 20% (w/v) d-(+)-sucrose and 2 mg/ml polymyxin B at 4 °C. After a 1.5-h incubation, the periplasmic fraction was separated by centrifugation (1.5 h, 20,000 × g, 4 °C). Subsequently, 0.4 μm avidin (Calbiochem®, Merck) was added to mask biotinylated host proteins. This soluble protein fraction was loaded onto 1 ml of Strep-Tactin® Superflow® high capacity resin (IBA, Göttingen, Germany) equilibrated with washing buffer (50 mm HEPES-NaOH, pH 8.0, 100 mm NaCl). After two washing steps (2 × 5 column volumes of washing buffer), the protein was liberated using washing buffer containing 2.5 mm desthiobiotin (IBA). PA0919-containing fractions were dialyzed against 20 mm HEPES-NaOH, pH 6.8, concentrated to 3 mg/ml using an Amicon® Ultra-0.5 device (nominal molecular mass cut-off of 10 kDa; Merck). Protein concentrations were determined using Bradford reagent (Sigma-Aldrich) according to the manufacturer's instructions.
N-terminal Amino Acid Sequence Determination
Automated Edman degradation was used to confirm the identity of purified proteins.
Determination of Native Molecular Mass
Analytical gel permeation chromatography was performed as described elsewhere (41).
UV-visible Light Absorption Spectroscopy and Fluorescence Measurements
UV-visible light spectra of the purified PA0919 protein were recorded from 260 to 900 nm using a V-550 spectrometer (Jasco, Gross Umstadt, Germany). Fluorescence spectra via an LS50B luminescence spectrometer (PerkinElmer) were monitored to detect possible fluorescent cofactors. Therefore, a protein sample was excited from 250 to 450 nm. Fluorescence emission maxima were detected from 250 to 800 nm.
Analysis of PA0919 Substrate Recognition Using Artificial Chromogenic Substrates
The main determinants of PA0919 substrate recognition were investigated using a series of artificial chromogenic substrates. For this purpose, the following commercially available p-nitrophenol derivatives were employed: Ala-ONp, Gly-ONp, acetyl-glycine 4-nitrophenyl ester, t-butoxycarbonyl-Ala-ONp, and alanine p-nitroanilide (Bachem AG, Bubendorf, Switzerland) and 4-nitrophenyl butyrate, 4-nitrophenyl acetate, and 4-nitrophenyl phosphate (Sigma-Aldrich). The chromogenic substrates were dissolved in DMSO, and 4-nitrophenyl phosphate was dissolved in dialysis buffer (20 mm HEPES-NaOH, pH 6.8). PA0919 activity assays (containing 20 mm HEPES-NaOH, pH 6.8, protein concentration of 3–12 μm, in a total volume of 294 μl) were initiated by the addition of 6 μl of 2.5 mm of the respective substrate (2% (v/v) final DMSO concentration). p-Nitrophenolat formation at 22 °C was directly monitored at a wavelength of 400 nm using a V-550 absorption spectrometer (Jasco). In all cases, identical control experiments in the absence of the respective protein were performed to account for the spontaneous hydrolysis of the individual chromogenic substrate. All experiments were performed in triplicate.
Synthesis of Lys-ONp
In many organisms, aa-PGS-mediated lipid homeostasis is based on the biosynthesis of L-PG. Therefore, the analysis of an artificial Lys-ONp substrate is a promising task. To complete the analysis of the artificial substrate spectrum of PA0919, the de novo synthesis of Lys-ONp was performed (see Fig. 4D).
FIGURE 4.

Chromogenic compounds used to analyze PA0919 substrate recognition in vitro. A, chromogenic amino acid p-nitrophenol compounds being hydrolyzed by PA0919. B, p-nitrophenol derivatives not accepted as a PA0919 substrate. C, structural formula of A-PG. The results of the activity analysis using different chromogenic compounds are assigned to the putative PA0919 substrate. Determinants of PA0919 substrate recognition are highlighted in gray. D, chemical synthesis of Lys-ONp. For experimental details, see “Experimental Procedures.” E, in vitro activity assay using the artificial substrate Ala-ONp. Measurements were performed in the presence of 50 μm Ala-ONp at different protein concentrations at 22 °C, and the liberation of p-nitrophenolate was monitored at a wavelength of 400 nm. The spontaneous hydrolysis in the absence of protein was subtracted. 1, Ala-ONp; 2, Gly-ONp; 3, Lys-ONp; 4, 4-nitrophenyl acetate; 5, 4-nitrophenyl butyrate; 6, acetyl-glycine 4-nitrophenyl ester; 7, tert-butyloxycarbonyl alanine 4-nitrophenyl ester; 8, alanine p-nitroanilide; 9, 4-nitrophenyl phosphate; 10, A-PG.
Similar to the method of Ke et al. (42), l-lysine (4.4 g, 30 mmol) and KOH (2.3 g, 39 mmol) were dissolved in water (100 ml) and THF (12.5 ml). Di-tert-butyl dicarbonate (14.4 g, 66 mmol) was added in portions, and the mixture was stirred for 14 h at 50 °C. A 1 m solution of HCl was added dropwise to adjust the pH to 5.5, followed by extraction with EtOAc (3 × 100 ml). The combined extracts were dried with MgSO4 and concentrated in vacuo. Column chromatography on silica gel with hexane/EtOAc (2:1) yielded N,N′-di-(tert-butyloxycarbonyl)-l-lysine (4.6 g, 13.3 mmol, 45%) as a colorless solid.
1H NMR (400 MHz, CDCl3): δ = 1.43 (s, 18H, 6×CH3), 1.43–1.50 (m, 4H, 2×CH2), 1.70–1.80 (br s, 1H, CH2), 1.80–1.95 (br s, 1H, CH2), 3.05–3.15 (m, 2H, CH2), 4.11–4.28 (br m, 1H, NH), 4.75 (br m, 1H, NH), 5.30 (d, 1H, NH), 9.97 (br s, 1H, COOH) ppm. 13C NMR (100 MHz, CDCl3): δ = 22.4 (CH2), 28.3 (3×CH3), 28.4 (3×CH3), 29.5 (CH2), 32.0 (CH2), 40.0 (CH2), 53.2 (CH), 79.3 (C), 80.0 (C), 155.8 (C), 156.3 (C), 176.4 (C) ppm.
The obtained N,N′-di-(tert-butyloxycarbonyl)-l-lysine (4.6 g, 13.3 mmol) was dissolved in dry dichloromethane (130 ml). p-Nitrophenol (2.2 g, 15.9 mmol) and dicyclohexylcarbodiimide (3.3 g, 15.9 mmol) were added, and the mixture was stirred at room temperature overnight. The precipitated solid was filtered off and washed with dichloromethane. The solvents were removed under reduced pressure to obtain the pure p-nitrophenyl ester of N,N′-di-(tert-butyloxycarbonyl)-l-lysine (2.1 g, 4.6 mmol, 35%) as a colorless solid.
1H NMR (400 MHz, CDCl3): δ = 1.43 (s, 9H, 3×CH3), 1.45 (s, 9H, 3×CH3), 1.47–1.60 (m, 4H, 2×CH2), 1.80–1.92 (m, 1H, CH2), 1.95–2.05 (m, 1H, CH2), 3.10–3.20 (m, 2H, CH2), 4.43–4.50 (m, 1H, CH), 4.65 (br s, 1H, NH), 5.30 (br s, 1H, NH), 7.27–7.30 (m, 2H, 2×CH), 8.24–8.27 (m, 2H, 2×CH) ppm. 13C NMR (100 MHz, CDCl3): δ = 22.5 (CH2), 28.3 (3×CH3), 28.4 (3×CH3), 29.7 (CH2), 31.4 (CH2), 39.6 (CH2), 53.8 (CH), 79.3 (C), 80.3 (C), 122.3 (2×CH), 125.2 (2×CH), 145.5 (C), 155.2 (C), 155.6 (C), 156.3 (C), 170.8 (C) ppm.
As described by Schnabel (43), a solution of HBr in glacial acetic acid (1.6 m, 20 ml) was added dropwise to the p-nitrophenyl ester of N,N′-di-(tert-butyloxycarbonyl)-l-lysine (1.5 g, 3.3 mmol). The mixture was stirred for 2 h at room temperature. The precipitated product was filtered off, washed with EtOAc, and dried in vacuo to yield the pure l-lysine p-nitrophenylester dihydrobromide (Lys-ONp, 750 mg, 1.76 mmol, 55%) as a colorless solid.
1H NMR (400 MHz, D2O): δ = 1.63–1.80 (m, 2H, CH2), 1.83–1.91 (m, 2H, CH2), 2.14–2.25 (m, 1H, CH2), 2.26–2.37 (m, 1H, CH2), 3.12 (t, 3JH,H = 7.7 Hz, 2H, CH2), 4.59 (t, 1H, 3JH,H = 6.5 Hz, CH), 7.48–7.52 (m, 2H, 2×CH), 8.35–8.41 (m, 2H, 2×CH) ppm. 13C NMR (100 MHz, D2O): δ = 24.6 (CH2), 29.2 (CH2), 32.1 (CH2), 42.0 (CH2), 55.7 (CH), 125.3 (2×CH), 128.6 (2×CH), 148.8 (C), 157.0 (C), 171.2 (C) ppm.
Chemical Modification of PA0919
To characterize amino acid residues of key relevance for PA0919 catalysis, the purified protein was chemically modified with reagents showing a high degree of specificity for activated serine or histidine residues. Protein samples (30 μm) were incubated in the presence of 5 mm PMSF (Roth), 0.3 Nα-tosyl-l-lysine chloromethyl ketone (TLCK; Sigma-Aldrich), or 5 mm TLCK respectively, or alternatively in the presence of 5 mm EDTA as a metal ion-chelating agent (4 °C for 30 min). The modified protein samples were then analyzed with the in vitro activity assay (in the presence of Ala-ONp) at a final protein concentration of 6 μm. In all cases, control experiments in the absence of PA0919 (chemical modifications subsequently followed by activity measurements) were performed to account for the spontaneous hydrolysis of the chromogenic substrate under the specific conditions of the individual modification. Enzymatic activities were averaged from two independent experiments. All activity measurements were performed in triplicate.
PA0919 in Vitro Activity Assay Using [1-14C]A-PG
To demonstrate the enzymatic activity of PA0919, a coupled in vitro activity test was developed, which was mainly based on a recently published A-PGS assay (15). The main principle of the employed procedure is outlined in the scheme shown in Fig. 3C. Assay mixtures of 1 ml containing 10 μm purified A-PGS(543–881) (15), 20 μm l-[1-14C]alanine, 2 mm ATP, and an ATP-regenerating system (18 mm creatine phosphate, 35 units of creatine phosphokinase) in 50 mm HEPES-NaOH, pH 7.8, were supplemented with 700 μl of a crude cellular extract of E. coli Rosetta (DE3) pLysS pET28b(+)PA0903 (15). This extract functions as a source of PG and Ala-tRNA synthetase and ensures the biosynthesis of sufficient amounts of [1-14C]-Ala-tRNAAla. The coupled synthesis of [1-14C]A-PG as a potential PA0919 substrate was performed at 37 °C for 1 h under continuous agitation. The efficient formation of [1-14C]A-PG was monitored by scintillation counting as described elsewhere (15). The de novo synthesis of [1-14C]A-PG was stopped by the addition of 10 μl of 10 mg/ml RNase A (Invitrogen) for 15 min at 37 °C before the A-PG-containing mixtures were stored at −20 °C (as aliquots of 200 μl). For the PA0919 in vitro activity assay, polar lipids were extracted using the modified method of Bligh and Dyer (44) as described elsewhere (9), the organic phase was evaporated, and the obtained lipid fraction was dissolved in 190 μl of dialysis buffer containing 3.2 mg/ml TritonTM X-100 (Sigma-Aldrich) for 15 min at 37 °C under vigorous shaking. After a subsequent centrifugation step (10 min, 11,000 × g), the respective supernatant was employed as a substrate for the PA0919 in vitro activity assay. A typical assay mixture of 160 μl contained 80 μl of 80 μm PA0919 (or a mutant PA0919 protein) in HEPES-NaOH, pH 6.8, and 80 μl of solubilized lipid substrate. Assays were incubated for 30 min at 37 °C before the individual reactions were stopped by a heat inactivation step at 60 °C for 5 min. Subsequently, 12 μl of each sample was spotted onto a TLC plate (Macherey-Nagel, Düren, Germany). Analogously, 1 μl of a 10 mm l-alanine sample (Sigma-Aldrich) was analyzed as a reference. Amino acids and lipids were separated using n-butanol, acetic acid, and water (5:3:2, v/v/v) (45) as eluent. Plates were sprayed with ninhydrin solution (Merck) and incubated at 100 °C. Dried TLC plates were analyzed by autoradiography. In all cases, control experiments in the absence of PA0919 were performed. All activity assays were reproduced in at least three independent experiments.
FIGURE 3.

In vitro analysis of PA0919. A, partial sequence alignment of homologous PA0919 proteins. Gene names or locus tags are given as follows. PA0919, P. aeruginosa PAO1; PP_1201, Pseudomonas putida KT2440; Atu_AcvB, A. tumefaciens C58; Atu_VirJ, A. tumefaciens octopine-type Ti-plasmid pTiA6; Rtr_AtvA, R. tropici CIAT 899; Bphy_4018, B. phymatum STM815; Rpic_3994, R. pickettii 12J; XAC1368 and XAC2411, Xanthomonas axonopodis 306; Phep_2436, Phep_2437, and Phep_2737, P. heparinus DSM2366; XF0754 and XF2679, X. fastidiosa 9a5c; CJA_1309, Cellvibrio japonicus Ueda107. Conserved and partly conserved residues are indicated by an asterisk, colon, or period, respectively. The related conservation pattern of different lipase active site motifs (22) is indicated as a sequence logo. Amino acid positions subjected to site-directed mutagenesis are highlighted boldface type. B, recombinant production and purification of Strep-tagged PA0919 protein(s). The periplasmic E. coli fraction was isolated (lane 1), PA0919 was chromatographically purified (lane 2, column flow-through; lane 3, washing step 1; lane 4, washing step 2; lane 5, elution) and subsequently dialyzed (lane 6). The purified mutant proteins S240A, S307A, and H401A (after buffer exchange) are indicated in lanes 7–9, respectively. Samples were analyzed by 12% SDS-PAGE and Coomassie Brilliant Blue staining. Lane M, molecular mass marker; relative molecular masses (× 1,000) are indicated. C, schematic representation of the in vitro PA0919 activity assay. Top, summary of the synthesis of the 14C-labeled A-PG substrate. An E. coli extract after recombinant overproduction of Ala-tRNA synthetase additionally provides tRNAAla and PG as substrate for the synthesis of [1-14C]Ala-tRNAAla (the [1-14C]-Ala is added after cell disruption). The addition of purified A-PGS (A-PGS(543–881)) then ensures the synthesis of [1-14C]A-PG. A-PG synthesis is stopped by RNase A treatment, and the polar lipids are extracted and subsequently solubilized. Bottom, PA0919-dependent catalysis. [1-14C]A-PG is hydrolyzed by PA0919, and the reaction products [1-14C]Ala and PG are analyzed by TLC in combination with audioradiography. D, activity of PA0919 variant proteins using Ala-ONp and [1-14C]A-PG as substrates. The PA0919-dependent p-nitrophenolate formation was monitored spectrophotometrically at 400 nm. All enzymatic activities (after subtraction of the spontaneous substrate hydrolysis) were related to the wild type enzyme (S.D. ± 15%). Relative PA0919 activities in the presence of [1-14C]A-PG were determined as follows. E, autoradiography of the PA0919 in vitro activity assay in the presence of [1-14C]A-PG. The radioactively labeled substrate was synthesized as outlined (see C). PA0919 dependent hydrolysis of [1-14C]A-PG (after a 30-min incubation at 37 °C) was analyzed by TLC followed by ninhydrin staining and autoradiography (for experimental details, see “Experimental Procedures”). Lane 1 contains an l-alanine reference sample stained with ninhydrin, and lane 2 shows the negative control incubated in the absence of PA0919 protein. Activity assays in the presence of 40 μm wild type PA0919 protein and of mutant proteins S240A, S307A, and H401A are analyzed in lanes 3 and 4–6, respectively. F, enzymatic activity of PA0919 after chemical modification. The PA0919 protein was incubated in the presence of PMSF, TLCK, and EDTA, respectively. Subsequently, in vitro activity assays using Ala-ONp were performed to determine residual PA0919 activity (after subtraction of spontaneous substrate hydrolysis; see “Experimental Procedures”). The activity of the untreated wild type protein was set as 100%, and all other values were related to that. G, subcellular localization of P. aeruginosa A-PGH. Plasmid-encoded A-PGH protein fused to a C-terminal Strep-tag II sequence was analyzed in the P. aeruginosa ΔPA0919 strain by Western blotting. Lane M, molecular size marker, relative molecular masses as indicated × 1,000; lane 1, whole cells; lane 2, soluble periplasmic fraction after polymyxin B treatment; lane 3, soluble cytoplasmic fraction; lane 4, preparation of a total membrane fraction, whole cells; lane 5, supernatant after French press and low speed centrifugation; lane 6, unbroken cells; lane 7, supernatant after high speed centrifugation; lane 8, pelleted (inner and outer membrane) fraction after TritonTM X-100 solubilization. Separation of the inner and the outer membrane fraction via sucrose gradient centrifugation is shown: inner membrane proteins (lane 9) and outer membrane proteins (lane 10). Lane 11, Sarkosyl-solubilized inner membrane proteins; lane 12, outer membrane proteins after the Sarkosyl solubilization method. n.d., not detectable.
Inductively Coupled Plasma-MS
To determine protein-bound metal ions (Cu2+, Fe2+, Mg2+, Mn2+, Ni2+, and Zn2+), inductively coupled plasma-MS was performed (CURRENTA Bayer-Analytics, Leverkusen, Germany) using a PA0919 protein sample of 8 mg/ml (180 μm).
PA0919 Localization Studies
All cellular localization experiments were performed in the presence of the ΔPA0919 mutant strain carrying plasmid pUCP20T/PPA0919-PA0919-Strep, which encodes for the native PA0919 protein fused to a C-terminal Strep-tag II sequence. Subcellular fractions were analyzed by Western blotting using a Strep-Tactin® alkaline phosphatase conjugate according to the manufacturer's instructions (IBA). In all cases, SDS-PAGE samples of membrane protein fractions were incubated with standard SDS-PAGE loading buffer to prevent protein aggregation at 40 °C for 30 min. 1 liter of bacterial cells were cultivated in AB medium at pH 5.3 (to ensure optimal promoter induction) supplemented with 250 μg/ml carbenicillin to an A578 of 0.5 and harvested, and the soluble periplasmic protein fraction was released from intact cells by polymyxin B treatment for 3 h according to the method described by Jensch and Fricke (46). An identical culture was employed to prepare a total membrane fraction. P. aeruginosa cells were harvested, resuspended in 50 mm Tris-HCl, pH 8.0, and subsequently disrupted by passage through a French press cell at 19,200 p.s.i. This cell-free extract was centrifuged for 20 min at 2,000 × g and 4 °C to remove cell wall fragments (low speed centrifugation). The obtained supernatant was then subjected to high speed centrifugation for 1 h at 100,000 × g to obtain a membrane pellet, which was washed three times using resuspension buffer containing 20 mm MgCl2 (washing buffer). Subsequently, membrane proteins were solubilized in washing buffer containing 2% (v/v) TritonTM X-100. The solubilized proteins were then separated from the insoluble fraction by centrifugation at 100,000 × g for 1 h. To separate the inner and outer membrane fraction, 3 liters of a P. aeruginosa culture was cultivated in AB medium at pH 5.3 to an A578 of 0.5. Harvested cells were suspended in membrane separation buffer (100 mm potassium acetate, 5 mm magnesium acetate, 50 mm HEPES-NaOH, pH 8.0, 0.05% (v/v) β-mercaptoethanol) containing a protease-inhibiting mixture (Complete protease inhibitor mixture EDTA-free, Roche Applied Sciences) and disrupted by two passages through a French press at 19,200 p.s.i. Unbroken cells were sedimented by centrifugation at 2,000 × g at 4 °C for 20 min. The obtained cell-free extract was loaded on top of a three-step sucrose gradient (2 m sucrose, 1.5 m sucrose, and 0.5 m sucrose) and separated by isopycnic ultracentrifugation at 100,000 × g at 4 °C for 1 h. The enriched membrane fractions were collected and analyzed by SDS-PAGE and Western blotting. An alternative experimental strategy was employed to analyze PA0919 localization (2-liter culture volume, AB medium, pH 5.3) according to Eitel and Dersch (47), using Sarkosyl as a detergent allowing for the selective solubilization of proteins localized in the inner membrane.
RESULTS
The aa-PGS Gene of P. aeruginosa and ORF PA0919 Are Located in an Orthologous Gene Cluster
The MBGD database (27) was employed as a powerful tool to investigate aa-PG-dependent lipid homeostasis of the bacterial kingdom. Our comparative genomics investigation revealed an orthologous grouping of the aa-PGS gene (PA0920) and ORF PA0919 of P. aeruginosa. Such downstream localization of PA0919 homologues was found inter alia in A. tumefaciens (AcvB; 61% sequence identity), in R. tropici (AcvB; 38% sequence identity), in Burkholderia phymatum (AcvB; 36% sequence identity), and also in Ralstonia pickettii (AcvB; 60% sequence identity). Some organisms also carry additional paralogous ORFs distantly located in the genome (e.g. in Sinorhizobium meliloti, Xylella fastidiosa, and Pedobacter heparinus). In contrast, the paralogous gene virJ of A. tumefaciens is localized on the virulence region of the octopine-type Ti-plasmid (33% sequence identity).
Our investigation did not reveal any PA0919-related genes in Gram-positive organisms. This theoretical result would match with the potential secretion of the PA0919 protein into the periplasm as it was proposed on the basis of a global protein localization study for P. aeruginosa (24). Besides this initial result, no biochemical data concerning the function of PA0919 (or of a related homolog) is available from the literature to date. The prevalent operon structure of orthologous PA0920 and PA0919 genes might indicate a potential role of ORF PA0919 in P. aeruginosa lipid homeostasis.
Deletion of ORF PA0919 Results in Significantly Increased Amounts of A-PG
Well established techniques (32) were employed to construct the markerless P. aeruginosa gene deletion mutant ΔPA0919. A-PG synthesis of P. aeruginosa has been found up-regulated at pH 5.3, as indicated by a cellular A-PG content of up to 6% in comparison with the overall lipid content (9). The relevance of ORF PA0919 under acidic growth conditions was indicated by a significant stationary phase growth phenotype of the deletion mutant ΔPA0919 in AB medium (pH 5.3) when compared with the wild type (Fig. 1A), whereas identical growth curves were obtained under neutral pH conditions (not shown). The A-PG level of the wild type strain (measured in the late stationary phase, pH 5.3) was defined as 100%, and the A-PG content of all mutant strains was related to that value (see Fig. 1B). For the deletion strain ΔPA0919, a significantly increased A-PG content of 200% was determined, whereas the cellular content of all other polar lipids was unaltered. These results indicate that ORF PA0919 is relevant for the tuning of cellular A-PG concentrations.
FIGURE 1.

Growth curves (A) and lipid analysis (B) of complemented P. aeruginosa mutant strains. A, growth curves of P. aeruginosa PAO1 wild type and PAO1 ΔPA0919 deletion mutant strains, cultivated in AB medium (pH 5.3), were plotted against time. S.D. values (error bars) are indicated. B, P. aeruginosa variants were cultivated in AB medium (pH 5.3) for 24 h. After harvesting, polar lipids were extracted, analyzed by two-dimensional TLC, and quantified using Gelscan version 6.0 after molybdatophosphoric acid staining. All A-PG levels were related to the wild type strain. The specific position of A-PG is indicated by an arrow. PE, phosphatidylethanolamine.
The deletion mutant strain was specifically complemented by introducing ORF PA0919 together with its 430-bp upstream region carrying a hypothetical PA0919-specific promoter (strain Δ19 P19-19). This hypothetical promoter region is located within the preceding PA0920 gene. An alternative complementation strategy was employed using the well characterized PA0920 promoter that is located in front of the PA0920/PA0919 operon (9). This complemented mutant containing promoter PA0920 in conjunction with ORF PA0919 was termed Δ19 P20-19. Adequate control strains were complemented solely with the PA0920 promoter or alternatively with the putative PA0919 promoter (Δ19 P20 or Δ19 P19). Both complemented strains (Δ19 P20-19 and Δ19 P19-19) almost restored the wild type lipid concentrations (A-PG levels of ∼110%, respectively) and revealed identical growth curves in comparison with the wild type strain (data not shown). The corresponding control strains (Δ19 P20 or Δ19 P19) showed the same growth phenotype (not shown) and a lipid composition identical to that of the ΔPA0919 strain (see Fig. 1B).
Artificially Elevated A-PG Levels Result in a Reduced Resistance against Antimicrobial Compounds
It was demonstrated that the formation of aa-PG molecules renders various organisms less susceptible to antimicrobial agents (9, 48). Only recently, we employed the model organism P. aeruginosa to investigate sophisticated phenotypes in response to several PA0920 complementation strategies (14). In the present investigation, the deletion of ORF PA0919 (strain ΔPA0919) resulted in a characteristic phenotype (MIC reduction) when compared with the wild type strain: ampicillin (50% MIC reduction), cefsulodin (10%), daptomycin (40%), poly-l-lysine (25%), polymyxin E (50%), polymyxin B (29%), benzethonium chloride (50%), domiphene bromide (26%), and vancomycin (13%). The corresponding MIC values and the results of the related control experiments are summarized in Table 1. Obviously, the elevated A-PG content (200%) of the deletion mutant results in a significantly increased susceptibility in the presence of various antimicrobial agents. In most cases, both complementation strategies (Δ19 P19-19 and Δ19 P20-19) enabled the efficient reconstitution of the MIC values observed for the wild type strain (ampicillin, daptomycin, poly-l-lysine, polymyxin E, polymyxin B, domiphene bromide, and vancomycin). However, in the presence of cefsulodin and benzethonium chloride, both complemented strains only showed partially restored MIC values, which might be attributed to the slightly increased A-PG content of ∼110% (see Fig. 1B). From these results, we conclude that PA0919-dependent lipid homeostasis is responsible for the appropriate “down-modulation” of cellular A-PG levels. Obviously, such “fine tuning” of the cellular lipid composition is relevant for the susceptibility to antimicrobial agents. The presented complementation experiments further substantiate the involvement of the newly identified PA0919 promoter.
TABLE 1.
MIC determination for complemented ΔPA0919 strains
MICs were determined by treating the different P. aeruginosa strains with gradually varying inhibitor concentrations in AB medium (pH 5.3). The relative S.D. is approximately 5% (daptomycin 10%). Significance analysis revealed p values in the footnotes below. For details, see “Experimental Procedures.”
| Antimicrobials | Wild type | ΔPA0919 | Δ19 P20 | Δ19 P20-19 | Δ19 P19 | Δ19 P19-19 |
|---|---|---|---|---|---|---|
| μg/ml | μg/ml | μg/ml | μg/ml | μg/ml | μg/ml | |
| Ampicillin | 360 ± 18 | 180a ± 9 | 150a ± 8 | 360 ± 18 | 150b ± 8 | 360 ± 18 |
| Cefsulodin | 61 ± 3 | 55 ± 3 | 33a ± 2 | 44c ± 2 | 39b ± 2 | 44c ± 2 |
| Daptomycin | >15,000 | 9,000c ± 900 | 9,000c ± 900 | >15,000 | 9,000a ± 900 | >15,000 |
| Poly-l-lysine | 8 ± 0.4 | 6c ± 0.3 | 6c ± 0.3 | 8 ± 0.4 | 6c ± 0.3 | 8 ± 0.4 |
| Polymyxin E | 4 ± 0.2 | 2c ± 0.1 | 2c ± 0.1 | 4 ± 0.2 | 2c ± 0.1 | 4 ± 0.2 |
| Polymyxin B | 2.4 ± 0.1 | 1.7c ± 0.1 | 1.7c ± 0.1 | 2.4 ± 0.1 | 1.7c ± 0.1 | 2.4 ± 0.1 |
| Benzethonium chloride | 22 ± 1.1 | 11b ± 0.6 | 11b ± 0.6 | 18a ± 0.9 | 11b ± 0.6 | 18a ± 0.9 |
| Domiphen bromide | 6.8 ± 0.3 | 5.0b ± 0.3 | 5.0c ± 0.3 | 6.2 ± 0.3 | 5.0a ± 0.3 | 6.2 ± 0.3 |
| Vancomycin | 23,800 ± 1,200 | 20,800b ± 1,000 | 20,800b ± 1,000 | 23,800 ± 1,200 | 20,800b ± 1,000 | 23,800 ± 1,200 |
a p ≤ 0.05.
b p ≤ 0.01.
c p ≤ 0.1.
The Promoter of Gene PA0919 Is Induced under Acidic Conditions
To further corroborate the newly identified PA0919 promoter, the reporter gene fusion PPA0919-430-lacZ was inserted into the genome of the PAO1 wild type strain and studied under various growth conditions. The promoter activity in cells grown in AB medium (pH 7.3) remained constant at ∼80 Miller units (Fig. 2, dark gray bars). However, lowering the pH value to 5.3 resulted in an increase of β-galactosidase activity up to 170 ± 20 Miller units (Fig. 2, light gray bars) in the stationary phase. Truncated promoter variants confirmed the presence of a pH-controlled promoter located in the region 430 bp upstream of ORF PA0919 (see Fig. 2). Recent P. aeruginosa A-PGS investigations have indicated that the pH value most dominantly influences cellular A-PG levels (9, 14). However, one might expect that several other stimuli mediate the promoter activities for genes PA0919 and PA0920 under different environmental conditions to sustain lipid homeostasis in P. aeruginosa.
FIGURE 2.

Analysis of the PA0919 promoter. The employed P. aeruginosa strains containing different PPA0919 promoter versions were cultivated in AB medium, pH 5.3 and 7.3, respectively. β-Galactosidase activities were determined at different time points, and values of the promoterless control strain KS11 were subtracted. Mean values of three independent experiments are presented together with the respective S.D. values (error bars). *, significance with p < 0.001. A, the activity of a genome-integrated PPA0919 promoter-lacZ fusion (PAO1 PPA0919-430-lacZ) at pH 5.3 (light gray bars) and pH 7.3 (dark gray bars). B, the activities of truncated PPA0919 promoter-lacZ fusion variants PAO1 PPA0919-430-lacZ (light gray bars), PAO1 PPA0919-206-lacZ (medium gray bars), and PAO1 PPA0919-140-lacZ (dark gray bars) analyzed at pH 5.3 and pH 7.3. C, chromosomal organization of ORFs PA0920 and PA0919. Predicted promoter regions −35 and −10 and the ribosome binding site (RBS) are indicated. Different genome-integrated promoter fragment lacZ fusions (PAO1 PPA0919-430-lacZ, PAO1 PPA0919-206-lacZ, and PAO1 PPA0919-140-lacZ) are depicted schematically.
Toward a Catalytic Function of the PA0919 Protein
Based on the obtained results, a catalytic function of PA0919 was assumed. This is verified by a cellular localization study, by using a series of artificial substrates, and by establishing an in vitro PA0919 activity assay.
Manual sequence analyses indicated a characteristic amino acid sequence pattern also found in lipases (active site serine motif) as described before by Vinuesa et al. (22). The postulated lipase signature and the related sequence logo of lipases (accession number PS00120, PROSITE (49)) is depicted in Fig. 3A together with two sequence regions carrying a highly conserved serine and histidine residue, respectively. The presented experimental data and also these theoretical results might indicate that the PA0919 protein is responsible for the turnover of the phospholipid A-PG.
Overproduction and Purification of Wild Type and Mutant PA0919 Proteins
For subsequent PA0919 in vitro analyses, a recombinant production system for the periplasmic overproduction in E. coli was established. The SDS-PAGE analysis of Fig. 3B shows the affinity purification (Strep-Tactin® chromatography) of wild type PA0919 (employing the N-terminal PelB leader sequence and a C-terminal Strep-tag II, respectively). A soluble periplasmic fraction (lane 1) was applied to the affinity matrix (flow-through fraction, lane 2), and after two washing steps (lanes 3 and 4), the protein was eluted with desthiobiotin (lane 5). Subsequently, the purified protein was dialyzed (lane 6), and the integrity of the mature N terminus after signal sequence cleavage was confirmed by Edman degradation (MAKLEALDLDDGGHA). Analytical gel permeation chromatography revealed a monomeric protein with a relative molecular mass of ∼57 kDa (calculated molecular mass 44,626 Da; data not shown). Absorption and fluorescence spectroscopic analyses (not shown) of the apparently homogenous protein fraction did not reveal the presence of any chromogenic cofactor. In the SDS-PAGE shown in Fig. 3B (lanes 6–9), the purified wild type protein and the site-directed mutant proteins S307A (carrying a mutation in the hypothetical active site lipase motif; see Fig. 3A) and S240A and H401A (mutation of a highly conserved serine or histidine residue; see Fig. 3A) are depicted, respectively. A typical protein production revealed 3 mg of purified protein from a 1-liter culture of E. coli.
Substrate Specificity Using Artificial Nitrophenyl Ester Substrates
To initially characterize the substrate specificity of the PA0919-dependent catalysis, a series of commercially available p-nitrophenyl esters of various amino acids/aliphatic acids were employed as potential substrates (compounds 1, 2, and 4–9; Fig. 4). The p-nitrophenyl ester of lysine (3; Lys-ONp) was obtained by chemical synthesis from l-lysine (Fig. 4D). For this purpose, both amino groups of lysine were protected with tert-butyloxycarbonyl protecting groups, followed by esterification with p-nitrophenol under carboxylic acid activation using dicyclohexylcarbodiimide and deprotection of the obtained ester with hydrobromic acid, yielding compound 3 as bis-hydrobromide.
All activity assays using these chromogenic substrates were completed with the corresponding control experiments to determine the spontaneous substrate hydrolysis of the individual compound under conditions of the respective assay. Among these ester substrates, the aminoacyl compounds 1–3 (Ala-ONp, Gly-ONp, and Lys-ONp) were accepted as substrate (Fig. 4A). Kinetic Ala-ONp measurements for different protein concentrations are summarized in Fig. 4E. Specific activities of 11 ± 3 nmol/(min mg), 13 ± 4 nmol/(min mg), and 16 ± 4 nmol/(min mg) were determined, respectively (spontaneous substrate hydrolysis subtracted). In contrast, compound 8, carrying a more stable amide bond; compounds 4, 5, and 9, devoid of an amino group; and also the more bulky substrates (6 and 7) were not accepted (Fig. 4B). These results might indicate that the aminoacyl linkage (rather than the phosphodiester or the fatty acid acyl ester linkage) of A-PG is a determinant for PA0919 catalysis (see gray highlighting in Fig. 4C).
PA0919 in Vitro Activity Assay Using 14C-Labeled A-PG
To date, the organic synthesis of A-PG has not been described in the literature. Therefore, it was necessary to establish a coupled PA0919 in vitro activity assay that is mainly based on our recently published A-PGS in vitro assay (see Ref. 15). Main principles of the employed methodology are summarized in the scheme shown in Fig. 3C; an E. coli extract containing recombinantly overproduced Ala-tRNA synthetase in the presence of l-[1-14C]alanine ([1-14C]Ala) supplies sufficient amounts of radioactively labeled [1-14C]Ala-tRNAAla. The addition of purified A-PGS facilitates the synthesis of [1-14C]A-PG. Then RNase A treatment ensures that no de novo A-PG synthesis takes place, and polar lipids are extracted and solubilized in the presence of TritonTM X-100 before the purified PA0919 protein is added. Specific conversion of A-PG or de novo synthesis of polar lipids can be easily monitored using well established TLC analyses in combination with autoradiography (15, 45, 48).
Control experiments in the absence of PA0919 revealed the formation of [1-14C]A-PG, as indicated by a dominant spot with an Rf of 0.70 (see Fig. 3, C and E, lane 2). The observed amount of [1-14C]A-PG in the presence of a negligible background is a good prerequisite for the in vitro investigation of PA0919 catalysis. When the established assay was incubated in the presence of 6.4 nmol of a homogeneous PA0919 protein fraction, an additional radioactively labeled spot with an Rf of 0.46 was determined (see one-dimensional TLC; Fig. 3E, lane 3), showing identical migration characteristics as an l-alanine reference sample (lane 1), whereas two-dimensional TLC analysis did not reveal any additional radioactively labeled polar lipid. These results indicate that the 14C-labeled alanine moiety of the A-PG substrate is hydrolyzed in a PA0919-dependent catalysis. Accordingly, the corresponding enzyme was termed A-PG hydrolase (A-PGH).
Toward an Enzymatic Mechanism of A-PGH
Chemical modification experiments in combination with Ala-ONp activity assays were performed to initially characterize the main principles of A-PGH catalysis. Protein samples were incubated with the respective agent, and residual A-PGH activity was determined. The serine-specific inhibitor PMSF was employed to study the critical involvement of a serine residue potentially originating from the hypothetical active site lipase motif (see Fig. 3A). No residual A-PGH activity was detectable when a PMSF concentration of 1 mm was applied (Fig. 3F). Besides this, the histidine-specific inhibitor TLCK was also tested because our theoretical sequence analysis points toward the involvement of catalytically relevant histidine residues. Inactivating concentrations of 60 μm and 1 mm only revealed residual A-PGH activities of 90 and 10% when compared with the respective control experiment. According to this, highly conserved histidine residues must be considered as key amino acids of A-PGH. Identical experiments in the presence of high concentrations (1 mm) of the chelating agent EDTA did not indicate a reduced enzymatic activity. Furthermore, inductively coupled plasma-MS experiments did not reveal any ferrous, nickel, zinc, copper, manganese, or magnesium ions above the background level of the employed buffer system (data not shown). Therefore, metal dependence of the A-PGH enzyme was ruled out.
To substantiate the idea of a serine- and/or histidine-dependent enzymatic mechanism, a site-directed mutagenesis approach was employed. In terms of histidine and serine, an overall total of three amino acid positions have been found to be highly conserved in a sequence comparison of the orthologous A-PGH sequences of the database (not shown). The respective purified mutant proteins (depicted in the SDS-PAGE analysis of Fig. 3B, lanes 6–9) were subjected to the newly developed A-PGH in vitro activity assays (Ala-ONp assay and A-PGH assay in the presence of [1-14C]A-PG). The mutant S240A only revealed a residual activity of 45% when compared with the wild type enzyme (standard deviation ±15%), whereas the “highly conservative” point mutation of the S307A totally abolished any detectable A-PGH activity. Analogously, no residual activity was observed in experiments using mutant protein H401A. These results of the Ala-ONp assay were confirmed with the A-PGH assay in the presence of [1-14C]A-PG (compare the table in Fig. 3D and autoradiography in Fig. 3E). From these experiments, it was concluded that residues serine 307 and histidine 401 are key residues for the enzymatic mechanism of A-PGH.
P. aeruginosa A-PGH Is Anchored to the Periplasmic Surface of the Cytoplasmic Membrane
Recent investigation of the N-terminal A-PGH signal peptide using the alkaline phosphatase (PhoA) fusion method indicated periplasmic secretion via a Type I signal peptide (24). To further specify this cellular localization, the plasmid-encoded A-PGH protein fused to a C-terminal Strep-tag II sequence was analyzed in the P. aeruginosa ΔPA0919 mutant background. The localization of the employed Strep-tag II was analyzed using the Strep-Tactin® AP conjugate (Strep-Tactin® labeled with alkaline phosphatase; IBA) in combination with four independent subcellular fractionation techniques (Fig. 3G) as follows. 1) Soluble periplasmic proteins were released from intact P. aeruginosa cells (lane 1) by polymyxin B treatment according to well established procedures. However, neither the resulting periplasmic fraction (lane 2) nor the soluble cytoplasmic fraction (lane 3) revealed significant amounts of the A-PGH fusion protein. Accordingly, an alternative methodology to specify a potential membrane association was employed. 2) The total membrane fraction was isolated. Whole cells (see lane 4) were disrupted by using a French press, and residual unbroken cells were removed by low speed centrifugation (lane 6). The obtained supernatant (lane 5) was then subjected to high speed centrifugation (supernatant; see lane 7), and the sedimented (membrane) fraction was extracted using TritonTM X-100. This solubilized (inner and outer) membrane fraction indicated a dominant A-PGH protein band (lane 8). 3) The separation of the inner and the outer membrane fraction was performed via sucrose gradient centrifugation. A-PGH was found located in the inner membrane fraction (compare lanes 9 and 10). 4) An alternative method is based on the Sarkosyl treatment of isolated total membranes, which results in the specific release of inner membrane proteins. This cytoplasmic membrane fraction (lane 11) dominantly indicates the presence of A-PGH (compare lanes 11 and 12). However, the recombinant overproduction of A-PGH in E. coli (using a cleavable E. coli-specific PelB leader sequence) resulted in an enzymatically active protein that is soluble in the absence of any detergent molecules. Taking into account the results of the present localization study, we conclude that the native A-PGH protein is anchored to the periplasmic surface of the cytoplasmic membrane via its uncleaved N-terminal signal sequence. This is a good prerequisite for the specific interaction with A-PG substrate molecules of the inner bacterial membrane.
DISCUSSION
The understanding of bacterial resistance mechanisms is of central importance for the development of new antimicrobial strategies. Deletion of orthologous aa-PGS genes has been correlated with an attenuated virulence of pathogens, like S. aureus, Mycobacterium tuberculosis, and Listeria monocytogenes (48, 50, 51). For these Gram-positive organisms, experimental work indicated a “charge repulsion mechanism” as a plausible model to understand, for example, the L-PG-mediated resistance against positively charged cationic antimicrobial peptides or defensin-like peptides (48). However, a recent P. aeruginosa investigation demonstrated that the artificial increase of the overall net charge of the bacterial membrane (due to substitution of A-PG with L-PG) does not result in an increased resistance of such a mutant organism (14). These initial experimental data for a Gram-negative model system might accentuate the central relevance of lipid homeostasis. The present investigation provides clear evidence for the importance of an accurate fine tuning of cellular A-PG levels. The significantly elevated A-PG concentrations of the employed ΔPA0919 mutant strain resulted in a reduced resistance against several antimicrobial compounds (ampicillin, daptomycin, poly-l-lysine, polymyxin E, polymyxin B, domiphene bromide, vancomycin, cefsulodin, and benzethonium chloride). Obviously, in the context of the employed P. aeruginosa model system, further aspects of phospholipid function must be taken into account. Besides the alteration of the charge characteristics, also the variation of membrane fluidity and/or permeability must be considered (52). Furthermore, individual phospholipids might be arranged with higher local concentrations in so-called microdomains (53), which in turn influence the functional properties of membrane-associated processes (54).
The elucidated A-PGH activity in P. aeruginosa implies a new regulatory circuit for the appropriate tuning of cellular A-PG concentrations. The determined A-PGH function ensures the adequate “down-modulation” of A-PG levels, a process that might be of special importance under conditions of antimicrobial treatment. These results are in agreement with the outcome of a recent investigation that focused on P. aeruginosa mutant strains showing an artificially altered aa-PG content (14). According to these new findings, one might propose that the unbalancing of the bacterial lipid composition (e.g. also by up-regulation of a specific phospholipid) must be considered as an attractive antimicrobial strategy in the future.
The A-PGS- and A-PGH-dependent physiology of the P. aeruginosa cell envelope has been summarized in the schematic representation of Fig. 5. A-PGS, a transmembrane protein of the inner bacterial membrane, catalyzes the transfer of the alanine moiety from the unusual Ala-tRNAAla substrate (Fig. 5A). The resulting A-PG synthesis mainly depends on the C-terminal A-PGS domain (dark gray) that is located on the cytoplasmatic side of the membrane (15). Besides this, the proposed flippase activity of A-PGS (localized on the transmembrane domain; light gray) then facilitates the transfer of the reaction product into the outer leaflet of the inner bacterial membrane, a process that might also be fundamental for the subsequent translocation of A-PG into the outer bacterial membrane (9).
FIGURE 5.
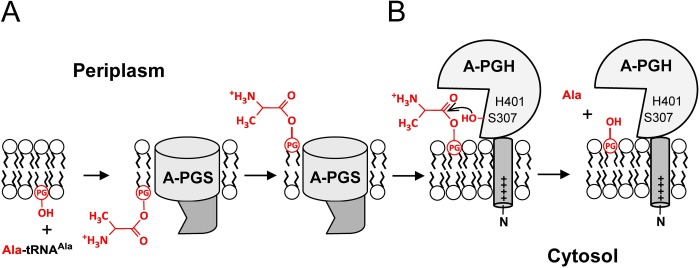
A-PG-dependent lipid homeostasis in P. aeruginosa. Schematic representation of the A-PG metabolism at the cytoplasmic membrane of P. aeruginosa. A, the C-terminal cytoplasmic domain of A-PGS (dark gray) is responsible for the Ala-tRNAAla-dependent synthesis of A-PG. The N-terminal transmembrane domain anchoring the overall enzyme in the inner bacterial membrane also provides flippase activity (57). B, lipid homeostasis in response to different environmental conditions requires accurate fine tuning of cellular A-PG levels. Therefore, the periplasmic protein PA0919 (A-PGH) is anchored to the surface of the inner bacterial membrane via its non-cleaved N-terminal signal sequence to facilitate the hydrolysis of A-PG. The proposed catalytic reaction mechanism involves serine 307 nucleophilically attacking the α-carbonyl group of the alanine moiety of A-PG. Residue histidine 401, possibly involved in catalytic diad/triad formation, is highlighted.
The newly identified A-PGH protein is efficiently anchored to the periplasmic surface of the inner bacterial membrane via the non-cleaved N-terminal leader sequence (Fig. 5B). The proposed transmembrane anchor sequence shows pronounced accumulation of positively charged amino acid residues at the cytoplasmic part (1MSVVKRHWRR10) when compared with the non-cytoplasmic part, in agreement with the “positive inside rule” (55). This cellular topology of A-PGH might facilitate solely the conversion of A-PG molecules located in the outer leaflet of the cytoplasmic membrane.
The initial characterization of the substrate specificity of A-PGH has been mainly evidenced on artificial nitrophenyl ester substrates. These experiments clearly indicate that the aminoacyl linkage of the respective substrates is the fundamental determinant for A-PGH substrate recognition. However, with respect to the natural substrate A-PG, not only were Ala-ONp and the more minimal substrate Gly-ONp accepted as a substrate, but also Lys-ONp, a molecule carrying a significantly more bulky aminobutyl moiety, was efficiently converted in the employed in vitro assay (see Fig. 4A). This result might be interesting in a more general context of aa-PG-dependent lipid homeostasis because several orthologous A-PGH proteins can be found in organisms that rely on the synthesis of L-PG (instead of A-PG) (22). The observed activity in the presence of Lys-ONp might be an evolutionary relic from an ancestral enzyme with a dual specificity for A-PG and L-PG. It will be interesting to see if future experiments in the presence of L-PG will confirm this hypothesis.
Besides the currently analyzed A-PGS/A-PGH gene cluster of P. aeruginosa, several other orthologous A-PGH sequences can be found in a completely different genomic context. The most prominent examples are VirJ and AcvB from A. tumefaciens. The virJ gene is localized on the Ti-plasmid directly surrounded by the vir genes coding for components of the Type IV secretion system. The specific inactivation of virJ/acvB genes resulted in avirulent mutant strains. The ability of virJ to complement the avirulent phenotype of an acvB mutant pointed toward an identical role for AcvB and VirJ during A. tumefaciens tumorigenesis (56). Nevertheless, the precise biological function of VirJ in A. tumefaciens DNA transfer is still unclear, so future analysis of a potential lipid-modifying activity in A. tumefaciens will be a promising task.
The biochemical characterization of enzymatic mechanisms is still an important methodology for the subsequent development of efficient inhibitor molecules. The present investigation revealed important insights into A-PGH catalysis. Chemical modification and site-directed mutagenesis experiments in combination with two different activity assays revealed two key residues of A-PGH. Ser-307 is located in a sequence region showing the highest degree of sequence conservation (see Fig. 3A). This part of A-PGH aligns well with the so-called active site lipase motif (PROSITE accession number PS00120) carrying the active site nucleophile serine as part of the active site triad (Ser, Asp/Glu, and His) of lipases (triacylglycerol acylhydrolases). Our inhibition experiments using the common active site serine-specific inhibitor PMSF at a concentration of 1 mm did not retain detectable A-PGH activity, which supports the idea of a Ser-307 nucleophile. Such a fundamental role was ruled out for the highly conserved residue Ser-240 because the mutant protein S240A retained a substantial activity of 45%. Besides this, the residual activity of only 10% in the presence of a 1 mm concentration of the histidine-specific active site inhibitor TLCK might indicate a potential role of His-401 in activating the hydroxyl group of Ser-307. The theoretical involvement of residues Ser-307 and His-401 in such a catalytic diad/triad would account for the complete loss of enzymatic activity that was observed for the respective mutant proteins S307A and H401A. However, further experimental studies, preferably the x-ray crystallographic investigation of covalently inhibited A-PGH, are required to prove the assumed enzymatic mechanism.
Acknowledgments
We thank Tristan Nicke, Tobias Schnitzer, and Henning Kuhz for experimental assistance. We especially thank our mentor Dieter Jahn for continuous support.
This work was supported by Deutsche Forschungsgemeinschaft Grant MO 1749/1-1.

This article contains supplemental Tables S1 and S2.
- PG
- phosphatidylglycerol
- aa-PG
- aminoacyl-phosphatidylglycerol
- aa-PGS
- aminoacyl-phosphatidylglycerol synthase
- A-PG
- alanyl-phosphatidylglycerol
- A-PGH
- alanyl-phosphatidylglycerol hydrolase
- A-PGS
- alanyl-phosphatidylglycerol synthase
- L-PG
- lysyl-phosphatidylglycerol
- MIC
- minimal inhibitory concentration(s)
- TLCK
- Nα-tosyl-l-lysine chloromethyl ketone
- Ala-ONp
- alanine 4-nitrophenyl ester
- Gly-ONp
- glycine 4-nitrophenyl ester
- Lys-ONp
- lysine 4-nitrophenyl ester.
REFERENCES
- 1. Zhang Y. M., Rock C. O. (2008) Membrane lipid homeostasis in bacteria. Nat. Rev. Microbiol. 6, 222–233 [DOI] [PubMed] [Google Scholar]
- 2. Weber M. H., Klein W., Müller L., Niess U. M., Marahiel M. A. (2001) Role of the Bacillus subtilis fatty acid desaturase in membrane adaptation during cold shock. Mol. Microbiol. 39, 1321–1329 [DOI] [PubMed] [Google Scholar]
- 3. Pogliano J., Pogliano N., Silverman J. A. (2012) Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J. Bacteriol. 194, 4494–4504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roy H., Ibba M. (2008) RNA-dependent lipid remodeling by bacterial multiple peptide resistance factors. Proc. Natl. Acad. Sci. U.S.A. 105, 4667–4672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. MacFarlane M. G. (1962) Characterization of lipoamino-acids as O-amino-acid esters of phosphatidyl-glycerol. Nature 196, 136–138 [Google Scholar]
- 6. Houtsmuller U. M., van Deenen L. (1963) Identification of a bacterial phospholipid as an O-ornithine ester of phosphatidyl glycerol. Biochim. Biophys. Acta 70, 211–213 [DOI] [PubMed] [Google Scholar]
- 7. Fischer W., Leopold K. (1999) Polar lipids of four Listeria species containing l-lysylcardiolipin, a novel lipid structure, and other unique phospholipids. Int. J. Syst. Bacteriol. 49, 653–662 [DOI] [PubMed] [Google Scholar]
- 8. Sohlenkamp C., Galindo-Lagunas K. A., Guan Z., Vinuesa P., Robinson S., Thomas-Oates J., Raetz C. R., Geiger O. (2007) The lipid lysyl-phosphatidylglycerol is present in membranes of Rhizobium tropici CIAT899 and confers increased resistance to polymyxin B under acidic growth conditions. Mol. Plant Microbe Interact. 20, 1421–1430 [DOI] [PubMed] [Google Scholar]
- 9. Klein S., Lorenzo C., Hoffmann S., Walther J. M., Storbeck S., Piekarski T., Tindall B. J., Wray V., Nimtz M., Moser J. (2009) Adaptation of Pseudomonas aeruginosa to various conditions includes tRNA-dependent formation of alanyl-phosphatidylglycerol. Mol. Microbiol. 71, 551–565 [DOI] [PubMed] [Google Scholar]
- 10. Peschel A., Otto M., Jack R. W., Kalbacher H., Jung G., Götz F. (1999) Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J. Biol. Chem. 274, 8405–8410 [DOI] [PubMed] [Google Scholar]
- 11. Muraih J. K., Pearson A., Silverman J., Palmer M. (2011) Oligomerization of daptomycin on membranes. Biochim. Biophys. Acta 1808, 1154–1160 [DOI] [PubMed] [Google Scholar]
- 12. Roy H. (2009) Tuning the properties of the bacterial membrane with aminoacylated phosphatidylglycerol. IUBMB Life 61, 940–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sacré M. M., El Mashak E. M., Tocanne J. F. (1977) A monolayer (π,ΔV) study of the ionic properties of alanylphosphatidylglycerol. Effects of pH and ions. Chem. Phys. Lipids 20, 305–318 [DOI] [PubMed] [Google Scholar]
- 14. Arendt W., Hebecker S., Jäger S., Nimtz M., Moser J. (2012) Resistance phenotypes mediated by aminoacyl-phosphatidylglycerol synthases. J. Bacteriol. 194, 1401–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hebecker S., Arendt W., Heinemann I. U., Tiefenau J. H., Nimtz M., Rohde M., Söll D., Moser J. (2011) Alanyl-phosphatidylglycerol synthase: mechanism of substrate recognition during tRNA-dependent lipid modification in Pseudomonas aeruginosa. Mol. Microbiol. 80, 935–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roy H., Ibba M. (2009) Broad range amino acid specificity of RNA-dependent lipid remodeling by multiple peptide resistance factors. J. Biol. Chem. 284, 29677–29683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ernst C. M., Staubitz P., Mishra N. N., Yang S. J., Hornig G., Kalbacher H., Bayer A. S., Kraus D., Peschel A. (2009) The bacterial defensin resistance protein MprF consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion. PLoS Pathog. 5, e1000660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Slavetinsky C. J., Peschel A., Ernst C. M. (2012) Alanyl-phosphatidylglycerol and lysyl-phosphatidylglycerol are translocated by the same MprF flippases and have similar capacities to protect against the antibiotic daptomycin in Staphylococcus aureus. Antimicrob. Agents Chemother. 56, 3492–3497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stover C. K., Pham X. Q., Erwin A. L., Mizoguchi S. D., Warrener P., Hickey M. J., Brinkman F. S., Hufnagle W. O., Kowalik D. J., Lagrou M., Garber R. L., Goltry L., Tolentino E., Westbrock-Wadman S., Yuan Y., Brody L. L., Coulter S. N., Folger K. R., Kas A., Larbig K., Lim R., Smith K., Spencer D., Wong G. K., Wu Z., Paulsen I. T., Reizer J., Saier M. H., Hancock R. E., Lory S., Olson M. V. (2000) Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959–964 [DOI] [PubMed] [Google Scholar]
- 20. Coakley R. D., Grubb B. R., Paradiso A. M., Gatzy J. T., Johnson L. G., Kreda S. M., O'Neal W. K., Boucher R. C. (2003) Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc. Natl. Acad. Sci. U.S.A. 100, 16083–16088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simmen H. P., Battaglia H., Giovanoli P., Blaser J. (1994) Analysis of pH, pO2 and pCO2 in drainage fluid allows for rapid detection of infectious complications during the follow-up period after abdominal surgery. Infection 22, 386–389 [DOI] [PubMed] [Google Scholar]
- 22. Vinuesa P., Neumann-Silkow F., Pacios-Bras C., Spaink H. P., Martínez-Romero E., Werner D. (2003) Genetic analysis of a pH-regulated operon from Rhizobium tropici CIAT899 involved in acid tolerance and nodulation competitiveness. Mol. Plant Microbe Interact. 16, 159–168 [DOI] [PubMed] [Google Scholar]
- 23. Pan S. Q., Jin S., Boulton M. I., Hawes M., Gordon M. P., Nester E. W. (1995) An Agrobacterium virulence factor encoded by a Ti plasmid gene or a chromosomal gene is required for T-DNA transfer into plants. Mol. Microbiol. 17, 259–269 [DOI] [PubMed] [Google Scholar]
- 24. Lewenza S., Gardy J. L., Brinkman F. S., Hancock R. E. (2005) Genome-wide identification of Pseudomonas aeruginosa exported proteins using a consensus computational strategy combined with a laboratory-based PhoA fusion screen. Genome Res. 15, 321–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujiwara A., Takamura H., Majumder P., Yoshida H., Kojima M. (1998) Functional analysis of the protein encoded by the chromosomal virulence gene (acvB) of Agrobacterium tumefaciens. Ann. Phytopathol. Soc. Jpn. 64, 191–193 [Google Scholar]
- 26. Majumder P., Takagi K., Shioiri H., Nozue M., Kojima M. (1999) Functional analysis of two chromosomal virulence genes chvA and acvB of Agrobacterium tumefaciens using avirulent mutants with transposon 5 insertion in the respective gene. Ann. Phytopathol. Soc. Jpn. 65, 254–263 [Google Scholar]
- 27. Uchiyama I., Higuchi T., Kawai M. (2010) MBGD update 2010. Toward a comprehensive resource for exploring microbial genome diversity. Nucleic Acids Res. 38, D361–D365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 29. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) ClustalW and ClustalX version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 30. Sambrook J., Russell D. W. (2001) Molecular Cloning: A Laboratory Manual, pp. 1.51–1.54, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 31. Heydorn A., Nielsen A. T., Hentzer M., Sternberg C., Givskov M., Ersbøll B. K., Molin S. (2000) Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology 146, 2395–2407 [DOI] [PubMed] [Google Scholar]
- 32. Hoang T. T., Karkhoff-Schweizer R. R., Kutchma A. J., Schweizer H. P. (1998) A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences. Application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212, 77–86 [DOI] [PubMed] [Google Scholar]
- 33. Thoma S., Schobert M. (2009) An improved Escherichia coli donor strain for diparental mating. FEMS Microbiol. Lett. 294, 127–132 [DOI] [PubMed] [Google Scholar]
- 34. Andrews J. M. (2001) Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 48, 5–16 [DOI] [PubMed] [Google Scholar]
- 35. Becher A., Schweizer H. P. (2000) Integration-proficient Pseudomonas aeruginosa vectors for isolation of single-copy chromosomal lacZ and lux gene fusions. BioTechniques 29, 948–950, 952 [DOI] [PubMed] [Google Scholar]
- 36. Schreiber K., Boes N., Eschbach M., Jaensch L., Wehland J., Bjarnsholt T., Givskov M., Hentzer M., Schobert M. (2006) Anaerobic survival of Pseudomonas aeruginosa by pyruvate fermentation requires an Usp-type stress protein. J. Bacteriol. 188, 659–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rompf A., Hungerer C., Hoffmann T., Lindenmeyer M., Römling U., Gross U., Doss M. O., Arai H., Igarashi Y., Jahn D. (1998) Regulation of Pseudomonas aeruginosa hemF and hemN by the dual action of the redox response regulators Anr and Dnr. Mol. Microbiol. 29, 985–997 [DOI] [PubMed] [Google Scholar]
- 38. Eschbach M., Schreiber K., Trunk K., Buer J., Jahn D., Schobert M. (2004) Long-term anaerobic survival of the opportunistic pathogen Pseudomonas aeruginosa via pyruvate fermentation. J. Bacteriol. 186, 4596–4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miller J. M. (1992) A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria, pp. 71–74, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 40. West S. E., Schweizer H. P., Dall C., Sample A. K., Runyen-Janecky L. J. (1994) Construction of improved Escherichia-Pseudomonas shuttle vectors derived from pUC18/19 and sequence of the region required for their replication in Pseudomonas aeruginosa. Gene 148, 81–86 [DOI] [PubMed] [Google Scholar]
- 41. Moser J., Lorenz S., Hubschwerlen C., Rompf A., Jahn D. (1999) Methanopyrus kandleri glutamyl-tRNA reductase. J. Biol. Chem. 274, 30679–30685 [DOI] [PubMed] [Google Scholar]
- 42. Ke D., Zhan C., Li X., Li A. D. Q., Yao J. (2009) The urea-dipeptides show stronger H-bonding propensity to nucleate β-sheetlike assembly than natural sequence. Tetrahedron 65, 8269–8276 [Google Scholar]
- 43. Schnabel E. (1964) Eine weitere Synthese der Insulinsequenz B 21–30. Liebigs Ann. Chem. 674, 218–225 [Google Scholar]
- 44. Bligh E. G., Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 45. Qiu T., Li H., Cao Y. (2010) Pre-staining thin layer chromatography method for amino acid detection. Afr. J. Biotechnol. 9, 8679–8681 [Google Scholar]
- 46. Jensch T., Fricke B. (1997) Localization of alanyl aminopeptidase and leucyl aminopeptidase in cells of Pseudomonas aeruginosa by application of different methods for periplasm release. J. Basic Microbiol. 37, 115–128 [DOI] [PubMed] [Google Scholar]
- 47. Eitel J., Dersch P. (2002) The YadA protein of Yersinia pseudotuberculosis mediates high-efficiency uptake into human cells under environmental conditions in which invasin is repressed. Infect. Immun. 70, 4880–4891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Peschel A., Jack R. W., Otto M., Collins L. V., Staubitz P., Nicholson G., Kalbacher H., Nieuwenhuizen W. F., Jung G., Tarkowski A., van Kessel K. P., van Strijp J. A. (2001) Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J. Exp. Med. 193, 1067–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sigrist C. J., de Castro E., Cerutti L., Cuche B. A., Hulo N., Bridge A., Bougueleret L., Xenarios I. (2013) New and continuing developments at PROSITE. Nucleic Acids Res. 41, D344–D347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Maloney E., Stankowska D., Zhang J., Fol M., Cheng Q. J., Lun S., Bishai W. R., Rajagopalan M., Chatterjee D., Madiraju M. V. (2009) The two-domain LysX protein of Mycobacterium tuberculosis is required for production of lysinylated phosphatidylglycerol and resistance to cationic antimicrobial peptides. PLoS Pathog. 5, e1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thedieck K., Hain T., Mohamed W., Tindall B. J., Nimtz M., Chakraborty T., Wehland J., Jänsch L. (2006) The MprF protein is required for lysinylation of phospholipids in listerial membranes and confers resistance to cationic antimicrobial peptides (CAMPs) on Listeria monocytogenes. Mol. Microbiol. 62, 1325–1339 [DOI] [PubMed] [Google Scholar]
- 52. Roy H., Dare K., Ibba M. (2009) Adaptation of the bacterial membrane to changing environments using aminoacylated phospholipids. Mol. Microbiol. 71, 547–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. López D., Kolter R. (2010) Functional microdomains in bacterial membranes. Genes Dev. 24, 1893–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dowhan W., Bogdanov M. (2002) Functional roles of lipids in membranes. in New Comprehensive Biochemistry (Dennis E., Vance J. E. V., ed) pp. 1–35, Elsevier, Amsterdam [Google Scholar]
- 55. von Heijne G. (2006) Membrane-protein topology. Nat. Rev. Mol. Cell Biol. 7, 909–918 [DOI] [PubMed] [Google Scholar]
- 56. Wirawan I. G., Kang H. W., Kojima M. (1993) Isolation and characterization of a new chromosomal virulence gene of Agrobacterium tumefaciens. J. Bacteriol. 175, 3208–3212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ernst C. M., Peschel A. (2011) Broad-spectrum antimicrobial peptide resistance by MprF-mediated aminoacylation and flipping of phospholipids. Mol. Microbiol. 80, 290–299 [DOI] [PubMed] [Google Scholar]