

Background: Mapping heterogeneous nuclear ribonucleoprotein (hnRNPK) genome-wide binding could better define its role in transcription.
Results: ChIP-Seq revealed hnRNPK accumulation at 3′-ends of immediate early genes. HnRNPK knockdown altered degradation of RNA downstream of poly(A) at EGR1 by decreased binding of XRN2 exonuclease.
Conclusion: HnRNPK facilitates XRN2 recruitment influencing co-transcriptional 3′-end mRNA processing.
Significance: A novel hnRNPK-dependent process has been identified in the control of transcription termination.
Keywords: Chromatin Immunoprecipitation (ChiP), Gene Expression, Mass Spectrometry (MS), RNA Polymerase II, Transcription Termination, XRN2, hnRNPK
Abstract
The heterogeneous nuclear ribonucleoprotein K (hnRNPK) is a nucleic acid-binding protein that acts as a docking platform integrating signal transduction pathways to nucleic acid-related processes. Given that hnRNPK could be involved in other steps that compose gene expression the definition of its genome-wide occupancy is important to better understand its role in transcription and co-transcriptional processes. Here, we used chromatin immunoprecipitation followed by deep sequencing (ChIP-Seq) to analyze the genome-wide hnRNPK-DNA interaction in colon cancer cell line HCT116. 9.1/3.6 and 7.0/3.4 million tags were sequenced/mapped, then 1809 and 642 hnRNPK binding sites were detected in quiescent and 30-min serum-stimulated cells, respectively. The inspection of sequencing tracks revealed inducible hnRNPK recruitment along a number of immediate early gene loci, including EGR1 and ZFP36, with the highest densities present at the transcription termination sites. Strikingly, hnRNPK knockdown with siRNA resulted in increased pre-RNA levels transcribed downstream of the EGR1 polyadenylation (A) site suggesting altered 3′-end pre-RNA degradation. Further ChIP survey of hnRNPK knockdown uncovered decreased recruitment of the 5′-3′ exonuclease XRN2 along EGR1 and downstream of the poly(A) signal without altering RNA polymerase II density at these sites. Immunoprecipitation of hnRNPK and XRN2 from intact and RNase A-treated nuclear extracts followed by shotgun mass spectrometry revealed the presence of hnRNPK and XRN2 in the same complexes along with other spliceosome-related proteins. Our data suggest that hnRNPK may play a role in recruitment of XRN2 to gene loci thus regulating coupling 3′-end pre-mRNA processing to transcription termination.
Introduction
The heterogeneous nuclear ribonucleoprotein K (hnRNPK)2 is a nucleic acid-binding protein that acts as a docking platform integrating signal transduction pathways to nucleic acid-directed processes (1). When bound to DNA it assists other factors in activating or repressing transcription by recruiting gene-specific transcriptional factors, chromatin remodelers, and general components of the transcription machinery to promoters, such as SP1 (2), poly(ADP-ribose) polymerase 1 (3), and TATA-binding protein (4), respectively. HnRNPK has been shown to directly bind and regulate expression from multiple promoters including c-Myc (5), c-Src (6), thymidine kinase 1 (7), androgen receptor (8), eukaryotic translation initiation factor 4E (9), and p53 target genes (10). Using chromatin immunoprecipitation (ChIP) assays we have recently shown that hnRNPK recruitment along genes mirrors occupancy of RNA polymerase II (Pol2) suggesting it may also play a role beyond the initiation step of transcription (11). The advent of the next generation sequencing technique together with the ChIP method (ChIP-Seq) allows to study protein-DNA interactions genome-wide at single nucleotide resolutions (12). Further advancements in next generation sequencing and the ENCODE project are making it possible to define chromatin architecture and identify functional elements in the human genome sequence in greater detail (13, 14).
Here, we used ChIP-Seq to assess the repertoire of hnRNPK-bound genes in the colon cancer cell line HCT116. We detailed hnRNPK binding to chromatin in quiescent and serum-stimulated cells. For hnRNPK target genes identified in the ChIP-Seq survey we performed a RNA-IP (RIP) assay of cognate transcripts demonstrating both previously described and new hnRNPK mRNA partners. Reverse transcriptase (RT) and ChIP measurements after siRNA hnRNPK knockdown revealed alternated early growth response 1 (EGR1) 3′-end mRNA processing that was accompanied by decreased recruitment of 5′-3′ exonuclease XRN2, a factor involved in transcription termination (15), along this locus. Immunoprecipitation (IP) from nuclear extracts with anti-hnRNPK and anti-XRN2 antibodies followed by mass spectrometry (MS) and Western blot analysis revealed that both proteins co-IP in the same complex. In summary, our data suggest a novel mechanism whereby hnRNPK interacting with XRN2 regulates transcription termination.
EXPERIMENTAL PROCEDURES
Cells
HCT116 WT human colon carcinoma cells lines were grown in plastic 6-well plates in McCoy's media supplemented with 10% FBS (11). For sequencing, chromatin was isolated from 24-h serum-starved cells (0.5% FBS) and cells were stimulated for 30 min with 10% FBS. In addition, for more time point specific data chromatin, RNA, and total protein lysates at 0, 5, 15, 30, 60, and 180 min following FBS stimulation were prepared for ChIP-qPCR, quantitative (q)RT-PCR, and Western blot analyses, respectively.
HnRNPK/XRN2 Knockdown and Western Blotting
Cells were transfected with hnRNPK siRNA (sequences were described before (11)), XRN2 (Ambion, assay ID s22412), or non-complementary (NC) siRNA at a final concentration of 22 nm using Lipofectamine RNAiMAX. 24 h after transfection cells were switched to 0.5% FBS medium and 24 h after quiescence the cells were harvested after treatment with 10% FBS for 0, 5, 15, 30, 60, and 180 min. Experiments with at least 90% decrease in target mRNA levels were pursued further. For Western blots cells were lysed then resolved by SDS-PAGE and electrotransferred to a PVDF membrane. Blotted proteins were assessed by Western blot analysis using the following antibodies: hnRNPK #54, XRN2 (Abcam; ab72181), Histone H3 (Abcam; ab1791), β-Actin (Abcam; ab6276), and GST (Abcam; ab92).
Total RNA Extraction and qRT-PCR
Total RNA was extracted from cells using TRIzol® Plus RNA Purification Kit (Invitrogen) followed by on-column PureLinkTM DNase (Invitrogen) treatment. One μg of total RNA and random hexamers or alternatively 2 μm gene-specific primers were used in cDNA synthesis with SuperScript III according to the manufacturer's protocol (Invitrogen). Levels of specific mRNAs were assessed by qRT-PCR using primer pairs designed to the exon-exon junction (Table 1). qPCR for cDNA and ChIP samples was carried out on Applied Biosystems 7900HT Fast Real-time PCR System with Sensimix SYBR kit (Bioline). 60S acidic ribosomal protein P0 (RPLP0) mRNA expression was used as reference mRNA. Mean gene expression was calculated with Q-gene software (16). For cDNA generated with the gene-specific primer, the Δ-ΔCt (ddCt) method (17) was used to establish the relative expression ratio between control and hnRNPK-depleted cells.
TABLE 1.
List of primer pairs used in cloning, ChIP, and qRT-PCR
| Gene name | Assay/location on the figures | Forward (5′ → 3′) | Reverse (5′ → 3′) |
|---|---|---|---|
| XRN2 | cloning | CACCATGGGAGTCCCGGCGTTCTTC | CAAAAGCTTAATTCCAATTGTATCTTCCTG |
| ATF3 | cDNA | ACTTCCGAGGCAGAGACCTG | GGCCAGACAAACAGCCC |
| ZFP36 | cDNA | GACTCAGTCCCTCCATGGTC | TCATGGCCAACCGTTACAC |
| FOS | cDNA | GTGGGAATGAAGTTGGCACT | CTACCACTCACCCGCAGACT |
| EGR1 | cDNA | CAGCACCTTCAACCCTCAG | AGCGGCCAGTATAGGTGATG |
| CYR61 | cDNA | CCCGTTTTGGTAGATTCTGG | GCTGGAATGCAACTTCGG |
| EREG | cDNA | GATGACCGCGGGGAGGAGGA | TGGAAACCCAGGCAGAGCAGC |
| RPLP0 | cDNA | GCAATGTTGCCAGTGTCTG | GCCTTGACCTTTTCAGCAA |
| HnRNPK | cDNA | AATGCCAGTGTTTCAGTCCC | AGGCCCTCTTCCAAGGTAGG |
| XRN2 | cDNA | AGGAATTAAGCGAAAAGCAGAAGA | TCCAGCCAGCTTCCCATAAC |
| HBB | ChIP/Promoter | TTATGCTGGTCCTGTCCTCC | CTGAAAGAGATGCGGTGG |
| ZFP36 | ChIP/ex1 | ATAAGTAGCCGGCTCTCGGT | CACCTCGTAGATGGCAGTCA |
| ZFP36 | ChIP/downstream poly(A) | TCTTCCTCTCCGAGCCCCGC | GGCTCCCCCGGTTCCCATCT |
| EGR1 | ChIP/ex1 | AGCTCTCCAGCCTGCTCGT | GGTAGTTGTCCATGGTGGGC |
| EGR1 | ChIP, cDNA/ex1-intr1 | CAGCACCTTCAACCCTCAG | GAAACCCGGCTCTCATTCTA |
| EGR1 | ChIP, cDNA/ex2 | GCTGAGCTGAGCTTCGGTTC | TCGCCGCCTACTCAGTAGGTA |
| EGR1 | ChIP, cDNA/A; 1kb | CAAGCCAAGAATCCTTCCAG | CCGGATGGGAACTTTAGACA |
| EGR1 | ChIP, cDNA/B | AGGAGGCCTGAGGTTCTAGG | CCCAACCAACAATCCACATT |
| EGR1 | ChIP, cDNA/C | TCAGAACCGCCCCTATCCCC | GGCTTCCCCTCTCCAACTGC |
| EGR1 | Gene-specific primer used in RT/# | NAa | TTTTGCCAGGAGGCTCACAG |
| U1_snDNA; RNU1–2 | cDNA-DNA | AAAGCGCGAACGCAGTCCCC | CGGATGTGCTGACCCCTGCG |
a NA, not applicable.
Chromatin Immunoprecipitation (ChIP) Assay
Chromatin cross-linking and cell harvesting was done as described before (18). Chromatin was sheared in a Bioruptor (Diagenode, Philadelphia, PA) (0.5-ml tubes) using a 30-s on-off cycles protocol for 15 min at high intensity. ChIP assays were done using Matrix-ChIP (18) or a modified Fast-ChIP protocol (19) for Chip-Seq libraries. For sequencing we performed ChIP on quiescent and 30-min FBS-stimulated cells with #54 antibody to hnRNPK and nonspecific rabbit IgG (I-1000, Vector Labs). The other antibodies used in ChIP studies were: Pol 2 (4H8) (Santa Cruz; sc-47701), Pol 2 (N-20) (Santa Cruz; sc-899), and XRN2 (Abcam; ab72181).
ChIP-seq
10 ng of the DNA from ChIP was amplified according to the ChIP Sequencing Sample Preparation Guide provided by Illumina, using adaptor oligo and primers from Illumina (IP-102-1001), enzymes from New England Biolabs, and a PCR Purification Kit and MinElute Kit from Qiagen. Deep sequencing was performed by Genomics Resource at the Fred Hutchinson Cancer Research Center, Seattle, WA, using Illumina Genome Analyzer with single-end 36-bp reads. Sequences were obtained using the Solexa Analysis Pipeline and then mapped to the human genome assembly (hg19) using Bowtie (20). The hnRNPK binding sites were detected with MACS (21) assuming at least 10-fold binding intensity change for highly confident peaks (quiescent/growing cells versus IgG).
Matrix-RIP Assay
HCT116 WT cells were made quiescent by lowering the FBS concentration to 0.5%. Cells were treated 24 h later in McCoy's media supplemented with 10% FBS for the given time points. Cells were harvested, lysates were prepared and used in RIP assay using rabbit IgG (I-1000, Vector Labs), hnRNPK #54, and XRN2 (ab72181) antibody as described (11).
In Vitro Pulldown Assay
A cDNA of XRN2 protein was amplified by PCR using Phusion High-Fidelity DNA Polymerase (Thermo Scientific) then cloned and expressed as His6-tagged recombinant protein using ChampionTM pET151 Directional TOPO® Expression Kit (Invitrogen) according to the manufacturer's protocol. Purification of XRN2-His protein was performed using nickel-nitrilotriacetic acid-agarose resin (Qiagen). For synthesis of recombinant hnRNPK protein we used a previously described GST-hnRNPK construct expressed in BL21(DE3) pLysS cells (25). Cells were lysed in extraction buffer (50 mm Tris-HCl, pH 8.5, 100 mm NaCl, 1 mm EDTA, 1 mm DTT) supplemented with 1 mg/ml of lysozyme and Halt Protease Inhibitor mixture (Thermo Scientific) for 1 h on ice. Sacrosyl was then added to a final 1% concentration and sonication on ice was performed until the solution was fluid and clear using a Cole-Parmer 500-watt Ultrasonic Homogenizer. GST and GST-hnRNPK fusion protein extracts were absorbed on 100 μl (50% slurry) glutathione-Sepharose 4B beads (GE Healthcare), washed in 10 bed volumes of extraction buffer with 0.5% Triton X-100 and used as bait in a pulldown assay. Prior to in vitro binding the XRN2-His solution was dialyzed against extraction buffer using a Slide-A-Lyzer 7000 MWCO unit (Thermo Scientific) for 2 h in a cold room. 200 μl of XRN2-His was loaded on either GST or GST-hnRNPK glutathione beads and incubated 1 h with rotation in a cold room. Beads were washed with 10 bed volumes of extraction buffer with 0.5% Triton X-100. Bound proteins were eluted with 1× Laemmli buffer resolved by SDS-PAGE and electrotransferred on PVDF membrane for immunostaining.
HnRNPK and XRN2 Immunoprecipitation from Nuclear Extracts (NE)
Protein IP was performed essentially as described (22) except here we used magnetic Dynabeads/Protein A to conjugate the antibodies. Briefly, 50 μl of Dynabeads/Protein A (Invitrogen) were conjugated to 10 μg of rabbit IgG (I-1000, Vector Labs), XRN2 (ab72181), and hnRNPK #54 antibody with bis(sulfosuccinimidyl)suberate (21585; Pierce) according to the manufacturer's protocol. 107 HCT116 cells were suspended in 2 ml of Buffer A (10 mm HEPES, pH 7.9, 2 mm MgCl2, 10 mm KCl) supplemented with phosphatase (Sigma; P2850 and P5726) and protease (Thermo; 78437) inhibitors and Dounce homogenized with 30 strokes then centrifuged at 1000 × g for 10 min at 4 °C. Nuclei pellet was fixed with freshly prepared 0.5% formaldehyde solution in PBS for 5 min and quenched with 125 mm glycine at room temperature for 5 min, washed once with plain PBS, then immediately resuspended in 300 μl of cold IP buffer spiked with phosphatase and protease inhibitors and alternatively incubated with 50 ng/ml of RNase A for 30 min on ice. Samples were disrupted in a Bioruptor for 10 min at high intensity using 30-s on/off duty. NE were centrifuged (12,000 × g, 4 °C, 10 min) and pre-cleared by incubation with 50 μl of IgG/Dynabeads A at 4 °C for 30 min with rotation followed by IP reaction on 50 μl of antibody 54/XRN2-Dynabeads A under the same conditions. Magnetic beads were washed separately three times with 300 μl of IP buffer and proteins were eluted from resin with 200 μl of 0.1% trifluoroacetic acid (TFA). Immediately after elution the pH sample was adjusted by adding 1/10 volume of 1 m NH4HCO3. Samples were either mixed with 4× Laemmli buffer and analyzed by immunoblotting with the indicated antibodies or subjected to mass spectrometry (MS) analysis.
MS Proteomic Analysis
MS analysis proteins were reduced and alkylated by incubating the sample in 10 mm DTT for 30 min at 56 °C and 50 mm iodoacetamide for 45 min at ambient temperature, respectively. Proteins were digested overnight with trypsin (sequencing grade; Promega) at 37 °C. MS analysis was performed using the LTQ Orbitrap (Thermo) mass spectrometer coupled to a nanoAcquity (Waters) LC system (23). MS/MS raw data files were processed to peak lists with the Mascot Distiller software (version 2.2.1, Matrix Science). The resulting ion lists were searched using the Mascot search engine (version 2.2.03, Matrix Science) against a composite database containing human protein entries from the Swissprot database and reversed versions of these entries. The search parameters were set as follows: enzyme specificity, trypsin; fixed modification, carbamidomethylation (C); variable modifications, oxidation (M), phosphorylation (P); protein mass, unrestricted. The statistical significance of peptide identifications was assessed as described (23, 24) and peptides with q-values less than or equal to 0.01 were regarded as confidently identified. Mascot search result processing were carried out using a proprietary software tool implemented in Java programming language (MScan, available at http://proteom.ibb.waw.pl/). The list of peptides identified in the IgG sample (background) was subtracted from hnRNPK and XRN2 specific results.
RESULTS
HnRNPK is a nucleic acid-binding protein that integrates cellular signaling at sites of nucleic acid-related processes. We have recently found that hnRNPK is recruited along several inducible genes with a pattern mirroring the Pol2 complex, an event that regulates the activity of the MAPK pathway components at these sites (11). To gain more insight about the role of its inducible chromatin binding we compared ChIP-Seq hnRNPK analyses of the human colon carcinoma HCT-116 line in quiescent and proliferative states.
HnRNPK Binds to Multiple IEGs Loci and Their mRNAs
In ChIP-seq 9.1, 7.0, and 5.1 million tags were sequenced for quiescent, 30-min serum-treated cells, and mock IgG libraries, respectively. After filtering out tags overlapping with the IgG library 3.6 and 3.4 million tags were successfully mapped. 1809 and 642 hnRNPK binding sites were detected (supplemental Tables S1 and S2) and of these, 1064 and 285 were located in proximity to the genes for quiescent and serum-stimulated cells, respectively (supplemental Tables S3 and S4). HnRNPK binds CT-rich stretches, therefore we searched for CT-like motifs within the hnRNPK ChIP-Seq binding sites. To do this, we used a consensus nucleotide motif, C(T/C)C(C/T)(T/C)(C/G)CC(C/A), derived from c-Myc (5), thymidine kinase 1 (7), androgen receptor (8), eukaryotic translation initiation factor 4E (9), osteocalcin (26), and CD43 (27) promoters (MEME software (28)). We found that this consensus sequence was present within 49 and 34% ChIP-seq sites in quiescent and serum-stimulated cells, respectively (supplemental Tables S1 and S2). As shown in Fig. 1A, the hnRNPK ChIP signal was enriched around the transcription start site (TSS) regions. To establish functional similarities among hnRNPK target genes we used Gene Ontology (GO) annotation in the biological processes category implemented in the DAVID program (29). The two top most significant GO categories associated were the GO:0006414 translational elongation and GO:0006412 translation (5% false discovery rate threshold, supplemental Table S5). The inspection of sequencing tracks on University of California, Southern California (UCSC) browser revealed serum-inducible hnRNPK recruitment along a number of IEG loci including EGR1 and ZFP36 with the highest densities observed downstream of the polyadenylation (A) sites (Fig. 1B) suggesting a role of hnRNPK in transcription termination (30). ChIP-qPCR measurements confirmed inducible hnRNPK recruitment near the TSS and to the sites of highest ChIP-Seq signals downstream from the poly(A) sites (Fig. 1C) with a pattern resembling Pol2. As a control we also analyzed β-globin (HBB) promoter where ChIP signals for both hnRNPK and Pol2 were, expectedly, low. Interestingly, we also found high constitutive binding to the genes encoding different classes of short non-coding RNAs, i.e. U1 and U2 (data not shown), indicating a potential role of the hnRNPK protein in pre-snRNA processing. HnRNPK is an RNA-binding protein. Next, we used the Matrix-RIP assay to test if K protein binds to RNAs encoded by genes identified in the ChIP-seq survey. RIP assay revealed both transient and constitutive hnRNPK-RNA association upon serum stimulation (Fig. 2, RIP). For example, transient IEG mRNAs binding of EGR1, ZFP36, ATF3, CYR61, and EREG paralleled their induction pattern after serum stimulation (Fig. 2, NC siRNA), whereas levels of U1 snRNA remained largely unchanged during the serum time course. Interestingly, EGR1 and ZFP36 transcripts were decreased. ATF3 and CYR61 transcripts levels were unaffected, whereas EREG and U1 snRNA transcripts were increased by K protein knockdown (Fig. 2, siRNA). Consistent with its diverse function, hnRNPK knockdown revealed differential effects of lowered hnRNPK protein levels on previously known (EGR1) and now newly identified (ZFP36, ATF3, CYR61, EREG, and U1 snRNA) transcripts.
FIGURE 1.

Human colon HCT116 carcinoma cell global and local hnRNPK binding distribution in relationship to TSSs and at two immediate early genes, respectively. A, genome-wide hnRNPK occupancy (blue) plotted relative to the nearest TSSs in a region spanning ±1 kb. IgG (red) denotes the unspecific ChIP-Seq signal in the same regions. The y axis indicates a sum of tags aligned at each position, for all TSSs in the ENSEMBL database. The signal was normalized to the equalize component from each TSS, regardless of the total number of tags in its vicinity. Binding profiles were generated with jChIP software (available at www.ire.pw.edu.pl/∼kchojnowski/jChIP/). B, hnRNPK ChIP-Seq signal at EGR1 and ZFP36 genes. After filtering out tags overlapping with the IgG library sequence, reads were mapped on human genome (version; hg19) and linked with the UCSC genome browser (genome.ucsc.edu). The y axis indicates the number of tags aligned at each position in the genome. C, ChIP analysis (11) of serum HCT116 time course binding profile of Pol2 and hnRNPK to EGR1, ZFP36 loci, and β-globin (HBB) promoter. ChIP analysis of sheared chromatin from a time course of serum-treated primers (10% FBS for 0, 5, 15, 30, 60, and 180 min). qPCR was done using primers (Table 1) spanning an area as shown in B. ChIP data are expressed as DNA recovery in percentage (%) of input (mean ± S.D., n = 3).
FIGURE 2.
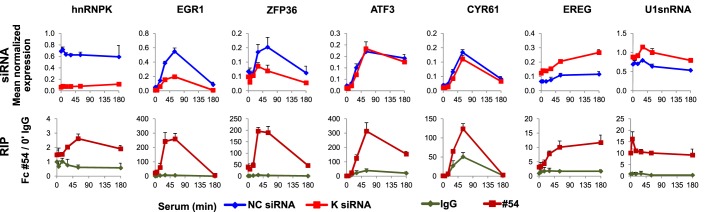
Diverse effects of hnRNPK protein knockdown on its RNA partners. siRNA, the HCT116 WT human colon carcinoma cell line was transfected with α-hnRNPK siRNA and control (NC) siRNA, and subjected to serum time course followed by RNA extraction, DNase I treatment, and qRT-PCR. The results were normalized for RPLP0 mRNA (mean ± S.D., n = 3). RIP, whole cell lysates of HCT116 WT treated with serum were used in a microplate-based RIP assay with the antibody to rabbit IgG and hnRNPK (#54). The results are expressed as a fold-change of RNA binding to #54 antibody over IgG at 0′ time point (mean ± S.D., n = 3).
HnRNPK Knockdown Increases Abundance of Pre-mRNA Levels Downstream of EGR1 Gene
We have previously shown that siRNA knockdown of hnRNPK is associated with decreased levels of elongating pre-mRNA and less efficient splicing of EGR1 and Nr4A3 transcripts (11). This and the current ChIP-Seq data showing hnRNPK accumulation downstream of the poly(A) signal (Fig. 1B) prompted us to test if hnRNPK may also play a role in transcription termination. To map the EGR1 transcription termination site we used the ChIP-Seq and RNA-Seq datasets for Pol2 and EGR1 mRNA, respectively, generated for the HCT116 cell line (ENCODE) (13). First, direct ChIP-Seq track comparison in the UCSC browser revealed an overlap between Pol2 and hnRNPK occupancy and the region downstream of the poly(A) site. Second, the RNA-Seq signal downstream of the poly(A) site becomes discontinued, marking a cleft, a region where primer A at 1-kb site is located (Fig. 3A, RNA-Seq zoomed), suggesting a site where Pol2 elongation of pre-mRNA is terminated. An interesting observation is that the stretch covered by the A 1-kb and B primer pairs overlaps with a high level of DNA sequence homology found in mammals, which could represent a cis-acting DNA/RNA element (Fig. 3A, Mammal Cons, boxed). Such a regulatory element could serve to recruit factor(s) that trigger Pol2 to pause and terminate.
FIGURE 3.

HnRNPK knockdown increases abundance of pre-mRNA downstream EGR1. A, occupancy profiles of hnRNPK and Pol2 at the EGR1 locus in HCT116 cell line. UCSC browser (hg19) profile of hnRNPK (red), Pol2 (green), ChIP-seq and RNA-Seq (blue) track (y axis; a number of tags aligned at each position in the genome, hnRNPK; reads per million, Pol2) together with locations of amplicons from qRT-PCR and ChIP studies. Coordinates shown represent the chr5:137,798,894–137,808,801 chromosomal region. The tracks for Pol2 and RNA-Seq were taken from the ENCODE dataset deposited at the UCSC browser under accession codes wgEncodeEH001627 and wgEncodeEH001425, respectively. B, effect of hnRNPK knockdown on nascent pre-mRNA levels along EGR1 and the downstream poly(A) site (see Fig. 2 siRNA for experimental details), primer sequences are listed in Table 1. Insert, RT with gene-specific primer (#, denotes its location) in quiescent cells. Pre-mRNA levels were calculated using the relative ΔΔ (dd) Ct method. Statistical analysis of differences between mean cDNA levels for control and K-depleted cells was performed using t tests. A p value of < 0.05 (*) was considered significant. Red square points to the 1-kb region that overlaps with a high level of DNA sequence homology found in mammals.
Based on these considerations we measured pre-cDNA generated with random hexamers along EGR1 and the downstream poly(A) site using qRT-PCR after K protein knockdown. In cells transfected with hnRNPK siRNA, the inducible EGR1 pre-mRNA levels probed at the A 1-kb site were lower compared with cells transfected with NC siRNA (Fig. 3B). However, the pre-mRNA measurement just beyond the “1-kb” site in quiescent cells revealed significantly increased (p value < 0.05, t test) constitutive local transcript levels in hnRNPK-knockdown cells that were not responsive to mitogen stimulation. This observation was confirmed at that site using cDNA templates generated with gene-specific primers from quiescent cells (Fig. 3B, insets). This result suggested that with hnRNPK knockdown there is a Pol2 complex “escape” from termination manifested by an increased leak further downstream of the 1-kb site and/or an alternation in RNA degradation. These possibilities were tested next.
XRN2 Knockdown Augments Inducible Pre-mRNA Transcription Downstream of EGR1 Gene
The relative increase in RNA levels downstream of the poly(A) site with K knockdown (Fig. 3B) could result from altered RNA degradation. In human cells, loss of 5′-3′ exonuclease XRN2 stabilizes transcripts downstream of the poly(A) site (31). Moreover, XRN2 is thought to be major player in coupling the 3′-end RNA processing to transcription termination. In our previous hnRNPK protein interactome studies in rat hepatocytes in the HTC-IR line, we found XRN2 among proteins with the strongest binding affinity for hnRNPK (22). We next tested the effects of XRN2 depletion on EGR1 pre-mRNA abundance using qRT-PCR. XRN2 knockdown (Fig. 4, A and B) elevated (p value < 0.05, t test) the pre-mRNA levels downstream of the EGR1 poly(A) site (Fig. 4C). XRN2 knockdown had no effect on hnRNPK mRNA levels. These results confirm the role of XRN2 in mRNA degradation. Given that hnRNPK and XRN2 may interact (see Ref. 22 and results below), these results also suggest that hnRNPK is required for efficient decay of the EGR1 pre-mRNA downstream 1-kb site by recruiting XRN2. This possibility was investigated next.
FIGURE 4.
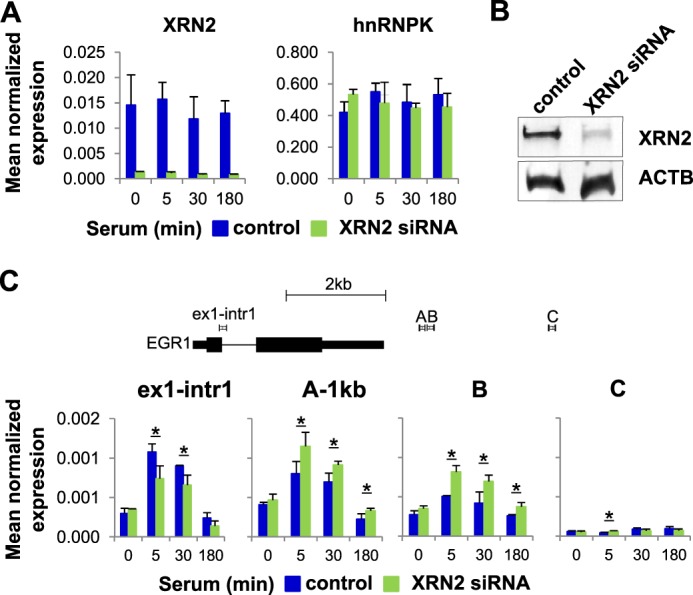
XRN2 depletion augments inducible pre-mRNA levels downstream of the EGR1 gene. A and C, HCT116 WT human colon carcinoma cells line were transfected with either XRN2 siRNA or NC siRNA in the presence of Lipofectamine RNAiMAX. 24 h after transfection, cells were switched to 0.5% FBS medium, and 24 h after quiescence, cells were treated with 10% FBS for 0, 5, 30, and 180 min followed by RNA extraction, DNase I treatment, and qRT-PCR. The results were normalized for RPLP0 mRNA (mean ± S.D., n = 4). A p value of <0.05 (*) was considered significant. Graphical depiction of amplicons localization at the EGR1 gene corresponds to coordinates presented at Fig. 3A. B, quiescent XRN2-depleted HCT116 cells prepared the same as in A; C were harvested and lysates were resolved by SDS-PAGE and electrotransferred to PVDF membrane. Blotted proteins were assessed by Western blot analysis using the antibodies to XRN2 (ab72181) and β-Actin (ab6276).
HnRNPK Depletion Decreases XRN2 Recruitment to EGR1
Elevated RNA abundance downstream of the poly(A) site (Fig. 3B) could reflect a deficiency of XRN2 bound to this locus, using a ChIP assay we tested XRN2 occupancy along EGR1 after hnRNPK knockdown (Fig. 5A). These measurements revealed serum inducible recruitment of XRN2 along as well as downstream of the EGR1 gene that was significantly decreased (p value < 0.05) with hnRNPK knockdown (Fig. 5A, XRN2). There was no indication of change in the Pol2 complex density in K protein-depleted chromatin (Fig. 5A, Pol2). We next ascertained that K protein-depleted cells have unchanged levels of XRN2 protein and mRNA levels using Western blot (Fig. 5B) and qRT-PCR analysis (Fig. 5C), respectively. Taken together these results suggest that hnRNPK tethers XRN2 to EGR1 sites. If so, hnRNPK and XRN2 might be present in the same complex in HCT116 cells. This possibility was examined next.
FIGURE 5.

HnRNPK knockdown decreases the 5′-3′ exonuclease XRN2 recruitment do EGR1 locus. A, HCT116 WT human colon carcinoma cell lines were transfected with either hnRNPK siRNA or NC siRNA in the presence of Lipofectamine RNAiMAX. 24 h after transfection, cells were switched to 0.5% FBS medium, and 24 h after quiescence, cells were treated with 10% FBS for 0, 5, 15, and 180 min then fixed, chromatin isolated, and sheared. Matrix ChIP assay was done using antibodies to Pol2, hnRNPK, and XRN2. ChIP results are shown for the PCR product depicted on the gene schematic. ChIP data are expressed as DNA recovery in percentage (%) of input (mean ± S.D., n = 3). Statistical analysis of differences between mean DNA recovery for control and K-depleted chromatin was performed using t tests. A p value of <0.05 (*) was considered significant. B, hnRNPK-depleted HCT116 cells prepared as in A were treated with 10% FBS for 0, 5, 15, 30, 60, and 180 min, then harvested, and lysates were resolved by SDS-PAGE and electrotransferred to PVDF membrane. Blotted proteins were assessed by Western blot analysis using the antibodies to hnRNPK #54, XRN2 (ab72181), and H3 (ab1791). C, cells prepared as in A were subjected to a serum time course followed by RNA extraction, DNase I treatment, and qRT-PCR. Results were normalized for RPLP0 mRNA (mean ± S.D., n = 3).
HCT116 Cell HnRNPK and XRN2 Are Present in the Same RNP Complex and Directly Interact
The RIP assay showed that hnRNPK and XRN2 co-IP IEG mRNAs with similar patterns upon serum stimulation of HCT116 cells (Fig. 6A) suggesting that the two proteins are molecular partners. To confirm the interaction between hnRNPK and XRN2 we performed in vitro binding of recombinant proteins. As shown in Fig. 6B, the GST-hnRNPK protein used as a bait captured His-tagged XRN2 (lane 2). There was no XRN2 binding to GST alone (lane 1). Repricocal co-IP of hnRNPK and XRN2 from HCT116 nuclear extracts followed by immunoblotting confirmed their co-existence in the same protein complex (Fig. 6C). To better understand the molecular context of this interaction we next searched for other proteins that exist in complexes with hnRNPK, XRN2 or both.
FIGURE 6.

HnRNPK and XRN2 are present in the same RNP complex. A, RIP assay, HCT116 whole cell lysates from time course experiments were used in microplate-based RIP assay with the antibody to rabbit IgG, hnRNPK (#54), and XRN2. Results are expressed as a fold-change of RNA binding over IgG reference at 0 min time point (mean ± S.D., n = 3). B, bacterially expressed recombinant hnRNPK and XRN2 protein interactions. Purified GST and GST-hnRNPK proteins bound to glutathione beads were incubated with His-XRN2 protein. Proteins eluted from the beads were analyzed by SDS-PAGE, transferred to PVDF membrane, and visualized by immunostaining using antibodies to hnRNPK #54, XRN2 (ab72181), and GST (ab92). C, reciprocal co-IPs with mock IgG, anti-XRN2, and anti-hnRNPK antibody. IPs were resolved by SDS-PAGE, then electrotransferred to PVDF membrane and visualized by immunoblot using anti-XRN2 and anti-hnRNPK antibody. 10 μg of NE was loaded to denote the position of proteins.
Shotgun Mass Spectrometry (MS) Revealed That HnRNPK and XRN2 Co-exist in the Same Spliceosome-associated Complexes
MS analysis NE immunoprecipitates with or without RNase treatment identified 66 proteins with at least two peptides (supplemental Table S6); 20 and 25 were specific for anti-hnRNPK and XRN2 antibodies (Fig. 7A), respectively, whereas 17 proteins, including hnRNPK and XRN2 were present in all four IP reactions (Table 2). Analysis of these 17 proteins with STRING, a protein-protein interaction database (32), showed that most of them are highly interconnected with K protein (Fig. 7B), whereas XRN2 was not networked in the most recent human version of this database. However, our previous work in rat hepatocytes (22) and studies by Kaneko et al. (33) in HeLa showed a connection of XRN2 to this network through hnRNPK and the SFPQ protein, respectively. GO annotation with DAVID for all identified proteins revealed significant overrepresentation of biological processes entirety related to RNA metabolism (5% false discovery rate threshold, supplemental Table S7). Of 66 proteins identified 39 and 30 were involved in RNA splicing (GO:0008380) and RNA splicing via a spliceosome (GO:0000398), respectively. Together, the RIP assay and MS analyses suggest that hnRNPK and XRN2 coexist in the same spliceosome-associated protein complex in the human HCT-116 cell line, interactions that are not mediated by RNA.
FIGURE 7.
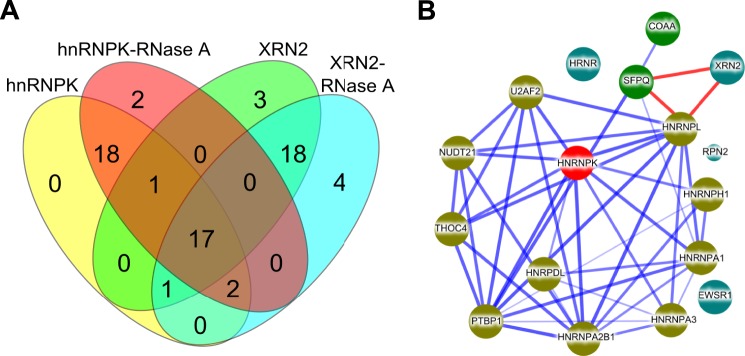
Shotgun mass spectrometry reveals the presence of hnRNPK and XRN2 in the same spliceosome complexes. A, nuclei pellets were fixed with formaldehyde and quenched with glycine, then resuspended in IP buffer with or without RNase A treatment followed by sonication. Nuclear extracts precleared with mock IgG and IP reactions using either anti-XRN2 or anti-hnRNPK antibody were performed. Mock IgG, hnRNPK, and XRN2 immunoprecipitates were reduced, alkylated, digested with trypsin, and subjected to MS analyses The Venn diagram depicts a number of proteins identified in IP reactions using anti-hnRNPK and anti-XRN2 antibody. B, a protein interaction network constructed with STRING (32) for a set of 17 human proteins common for hnRNPK and XRN2 IP reactions. Stronger associations are represented by thicker lines. Lines shown in light blue represent protein-protein interactions deposited in STRING database, lines shown in red represent XRN2 interactions shown by Kaneko et al. (33). Node color depicts the type of interaction with hnRNPK in the STRING database: dark yellow, green, and dark blue represent direct, indirect, and no interaction, respectively.
TABLE 2.
List of common proteins identified in immunoprecipitation assay followed by MS analysis using antibodies against either hnRNPK or XRN2 proteins
Nuclei from proliferating HCT116 cells were fixed with formaldehyde, quenched with glycine, and the nuclear extract was prepared in IP buffer treated with or without RNase A. Samples were first precleared with IgG rabbit antibody and then followed by IP reaction using either hnRNPK or XRN2 specific antibody. The list of proteins is based on peptides were found with all IP reactions.
| ID | Name | Gene | Number of peptides identified in IP reaction |
|||
|---|---|---|---|---|---|---|
| HnRNPK | HnRNPK-RNase A | XRN2 | XRN2-RNase A | |||
| Q9H0D6 | 5′-3′ Exoribonuclease 2 | XRN2 | 4 | 5 | 21 | 18 |
| O43809 | Cleavage and polyadenylation specificity factor subunit 5 | NUDT21 | 1 | 2 | 2 | 3 |
| P04844 | Dolichyl-diphosphooligosaccharide--protein glycosyltransferase subunit 2 | RPN2 | 2 | 1 | 1 | 2 |
| P09651 | Heterogeneous nuclear ribonucleoprotein A1 | HNRNPA1 | 5 | 2 | 2 | 1 |
| P51991 | Heterogeneous nuclear ribonucleoprotein A3 | HNRNPA3 | 2 | 3 | 1 | 2 |
| O14979 | Heterogeneous nuclear ribonucleoprotein d-like | HNRPDL | 2 | 3 | 2 | 2 |
| P31943 | Heterogeneous nuclear ribonucleoprotein H | HNRNPH1 | 3 | 3 | 2 | 2 |
| P61978 | Heterogeneous nuclear ribonucleoprotein K | HNRNPK | 9 | 13 | 2 | 2 |
| P14866 | Heterogeneous nuclear ribonucleoprotein L | HNRNPL | 4 | 5 | 3 | 3 |
| P22626 | Heterogeneous nuclear ribonucleoproteins A2/B1 | HNRNPA2B1 | 5 | 1 | 3 | 2 |
| Q86YZ3 | Hornerin | HRNR | 1 | 8 | 1 | 3 |
| P26599 | Polypyrimidine tract-binding protein 1 | PTBP1 | 1 | 2 | 1 | 2 |
| Q96PK6 | RNA-binding protein 14 | RBM14 | 3 | 1 | 1 | 2 |
| Q01844 | RNA-binding protein EWS | EWSR1 | 2 | 3 | 1 | 2 |
| P26368 | Splicing factor U2AF 65 kDa subunit | U2AF2 | 1 | 2 | 2 | 2 |
| P23246 | Splicing factor, proline- and glutamine-rich | SFPQ | 3 | 5 | 2 | 4 |
| Q86V81 | THO complex subunit 4 | ALYREF | 1 | 2 | 1 | 2 |
DISCUSSION
Our results show that hnRNPK may facilitate XRN2 recruitment to sites of active transcription and thereby influence both co-transcriptional degradation of poly(A)-downstream RNA and transcription termination.
The cycle of Pol2 transcription, from initiation through elongation to termination is a highly regulated process (34). Although less well studied compared with Pol2 initiation and elongation, transcriptional termination has recently also emerged as an important nexus of molecular events that couple pre-mRNA 3′ end processing, nascent RNA release, Pol2 pausing and recycling (35). Three overlapping mechanisms acting in the vicinity of the transcription termination area are thought to define Pol2 termination on protein coding genes: (i) allosteric, where 3′ end processing factors like polyadenylation specificity factor (CPSF) and cleavage stimulatory factor (CstF) induce conformational changes in the Pol2 complex; (ii) torpedo, where XRN2 collides with Pol2 driving its dissociation from the template; and (iii) the RNA-DNA hybrid unwinding by helicases, such as senataxin (15). Although the key termination steps have been described, molecular details remain largely unknown (35).
HnRNPK is involved in an array of molecular processes that comprise cell signaling and gene expression (1). In the nucleus pre-mRNA capping, splicing, and 3′-end processing occurs co-transcriptionally (36) therefore the presence of RNA-binding proteins, including hnRNPs, in the vicinity of chromatin is expected. Here we provide evidence for a new transcription termination step where hnRNPK recruits the XRN2 exonuclease to the EGR1 locus. The hint that prompted us to investigate mechanistically came from the ChIP-Seq survey where we noticed 3′-end gene accumulation of hnRNPK upon serum stimulation (Fig. 1B). Higher densities in this area overlapped with dense Pol2 signals deposited in the ENCODE database (Fig. 3A). To ascertain the role of the hnRNPK protein within the transcription termination genomic region we silenced hnRNPK and then induced mitogen response with serum. Comparing levels of transcripts downstream of the EGR1 poly(A) site we found increased constitutive RNA levels associated with hnRNPK knockdown (Fig. 3B). Given this finding and the fact that there was no effect of hnRNPK knockdown on Pol2 density at these sites (Fig. 5A, Pol2) we reasoned that the higher RNA levels could result from the absence of local exonuclease activity. Our previous studies suggested that hnRNPK and the endonuclease XRN2 might exit in the same complex (22). Therefore we focused our studies on XRN2. Our pulldown experiments using nuclear extracts (Fig. 6C) as well as recombinant proteins (Fig. 6B) provide evidence that hnRNPK and XRN2 interact directly. The MS survey of immunoprecipitates from NE confirmed RNA-independent hnRNPK-XRN2 co-localization (Fig. 7, A and B, and Table 2). The result of the hnRNPK-XRN2 interaction is in concordance with our previous study (22) performed on rat hepatocytes, the HTC-IR cell line, and the observation from Caenorhabditis elegans where proteins F26B1.2 and Y48B6A.3, hnRNPK and XRN2 orthologs, respectively, have been shown to interact directly in a yeast two-hybrid screen (37). The RIP assay revealed that both XRN2 and hnRNPK bind to mRNA of IEGs with similar dynamics, suggesting their coexistence in the same RNP complex (Fig. 6A). Their association may be initiated co-transcriptionally as hnRNPK knockdown decreased the XRN2 level along the EGR1 gene as well as downstream of this locus. Our results suggest that XRN2 is loaded on the promoter and moves along the chromatin with the Pol2 complex to elicit other functions in addition to 3′-end transcription termination. Indeed, it has been proposed that XRN2 also co-transcriptionally degrades nascent pre-mRNA in HeLa (38) as well as being involved in decay of transcripts that fail to get a cap thereby exerting premature termination of transcription within the body of the gene (39). These observations on XRN2 location and function within the genes bodies were recently confirmed by Brannan et al. (40), where XRN2 occupancy along transcribed genes mirrored bimodal distribution of the Pol2 complex with higher levels at TSS and downstream of poly(A) sites where 3′-end termination occurs. That study also revealed that XRN2 positioned at TSS associates with the transcription termination factors as well as decapping proteins, hence playing a role in decision making on premature transcription termination. It would be interesting to test if hnRNPK also regulates the recruitment and activity of XRN2 at sites other than EGR1 loci, a question that will be addressed in future studies. In the EGR1 downstream 1-kb region, XRN2 knockdown increased constitutive and inducible RNA levels (Fig. 4C), an observation consistent with its role as a transcription termination exonuclease. In this region hnRNPK knockdown increased RNA constitutively (Fig. 3B) providing evidence for its involvement in termination. In addition to its involvement in promoting co-transcriptional RNA processing, hnRNPK also plays a role in signaling these processes. Thus, on the one hand hnRNPK knockdown may diminish the amount of transcript synthesized along EGR1 in response to serum treatment and delivered for terminal processing, whereas on the other hnRNPK knockdown may slow termination. Given hnRNPK involvement in multiple steps could explain the decreased inducible EGR1 mRNA with hnRNPK knockdown at the termination site.
Transcription termination requires both cleavage and polyadenylation of the RNA transcripts as Pol2 elongates beyond the poly(A) site. The 3′-end processing complex consists of more than 20 proteins, several of which interact with the carboxyl-terminal domain of Pol2 (41). Among the proteins shared between hnRNPK and XRN2, NUDT21 (Fig. 7B, Table 2) is a component of the cleavage factor Im (CFIm) complex involved in initiating the 3′ pre-mRNA complex assembly and cleavage reaction. NUDT21/CFIm25 has been implicated in transcription termination where its action is mediated by binding to UGUA elements providing a platform for poly(A) polymerase recruitment (42). NUDT21-depleted HeLa cells exhibited changes in the selection of the mRNAs poly(A) site (43) and this observation was recently confirmed in structural studies showing the role of the CFIm complex in poly(A) site selection through looping of RNA sequences (44). Noteworthy, a study by Naganuma et al. (45) published when this work was being completed has shown that hnRNPK arrested binding of NUDT21 in the proximity of the alternative poly(A) site of NEAT1 non-coding RNA. This resulted in a production of a long NEAT1 isoform involved in a formation of nuclear paraspeckles. In analogy to the escape from the termination phenomenon we observed (Fig. 3) production of the long NEAT1 isoform requires inhibiting the 3′-end termination events enabling further Pol2 elongation. Therefore, the findings of Naganuma et al. (45) support a role of hnRNPK in transcription termination through regulating cleavage and polyadenylation.
Previous (22) and our proteomic MS analysis (Fig. 7) underscore the highly diverse and interactive nature of hnRNPK. HnRNPK has the unique property where on one hand it binds RNA/DNA, whereas on the other it interacts with kinases and other signal transducers (1). Given its functions as a docking platform at the site of RNA/DNA-directed processes hnRNPK could serve as an information conduit that coordinates transcription termination with other processes that compose gene expression as well as the signal transduction pathway that control these events. Current knowledge about signaling pathways and specific kinases involved in transcription termination is limited (15, 41). Thus, identification of hnRNPK as a player in transcription termination opens new avenues to study regulation of this less well studied gene expression step in response to signaling pathways.
Acknowledgments
We thank Steve Flanagin for developing GraphGrid software tools. We thank Agnieszka Paziewska and Jakub Karczmarski for technical assistance with immunoprecipitation and mass spectrometry analyses. We thank Joanna Ledwon for technical assistance with in vitro pulldown assay.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DK083310 and R37 DK45978 (to K. B.) and Ministry of Science and Higher Education Grant IP2010 026770.

This article contains supplemental Tables S1–S7.
- hnRNPK
- heterogeneous nuclear ribonucleoprotein K
- NE
- nuclear extract
- TSS
- transcription start site
- qRT-PCR
- quantitative reverse transcription PCR
- CFIm
- cleavage factor Im
- RIP
- RNA-immunoprecipitation
- NC
- non-complementary
- IEG
- immediate early gene.
REFERENCES
- 1. Bomsztyk K., Denisenko O., Ostrowski J. (2004) HnRNP K. One protein multiple processes. Bioessays 26, 629–638 [DOI] [PubMed] [Google Scholar]
- 2. Du Q., Melnikova I. N., Gardner P. D. (1998) Differential effects of heterogeneous nuclear ribonucleoprotein K on Sp1- and Sp3-mediated transcriptional activation of a neuronal nicotinic acetylcholine receptor promoter. J. Biol. Chem. 273, 19877–19883 [DOI] [PubMed] [Google Scholar]
- 3. Shi L., Ko S., Kim S., Echchgadda I., Oh T. S., Song C. S., Chatterjee B. (2008) Loss of androgen receptor in aging and oxidative stress through Myb proto-oncoprotein-regulated reciprocal chromatin dynamics of p53 and poly(ADP-ribose) polymerase PARP-1. J. Biol. Chem. 283, 36474–36485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Michelotti E. F., Michelotti G. A., Aronsohn A. I., Levens D. (1996) Heterogeneous nuclear ribonucleoprotein K is a transcription factor. Mol. Cell. Biol. 16, 2350–2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Takimoto M., Tomonaga T., Matunis M., Avigan M., Krutzsch H., Dreyfuss G., Levens D. (1993) Specific binding of heterogeneous ribonucleoprotein particle protein K to the human c-myc promoter, in vitro. J. Biol. Chem. 268, 18249–18258 [PubMed] [Google Scholar]
- 6. Ritchie S. A., Pasha M. K., Batten D. J., Sharma R. K., Olson D. J., Ross A. R., Bonham K. (2003) Identification of the SRC pyrimidine-binding protein (SPy) as hnRNP K. Implications in the regulation of SRC1A transcription. Nucleic Acids Res. 31, 1502–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lau J. S., Baumeister P., Kim E., Roy B., Hsieh T. Y., Lai M., Lee A. S. (2000) Heterogeneous nuclear ribonucleoproteins as regulators of gene expression through interactions with the human thymidine kinase promoter. J. Cell Biochem. 79, 395–406 [DOI] [PubMed] [Google Scholar]
- 8. Wang L. G., Johnson E. M., Kinoshita Y., Babb J. S., Buckley M. T., Liebes L. F., Melamed J., Liu X.-M., Kurek R., Ossowski L., Ferrari A. C. (2008) Androgen receptor overexpression in prostate cancer linked to Pur α loss from a novel repressor complex. Cancer Res. 68, 2678–2688 [DOI] [PubMed] [Google Scholar]
- 9. Lynch M., Chen L., Ravitz M. J., Mehtani S., Korenblat K., Pazin M. J., Schmidt E. V. (2005) HnRNP K binds a core polypyrimidine element in the eukaryotic translation initiation factor 4E (eIF4E) promoter, and its regulation of eIF4E contributes to neoplastic transformation. Mol. Cell. Biol. 25, 6436–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moumen A., Masterson P., O'Connor M. J., Jackson S. P. (2005) HnRNP K. An HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell 123, 1065–1078 [DOI] [PubMed] [Google Scholar]
- 11. Mikula M., Bomsztyk K. (2011) Direct recruitment of ERK cascade components to inducible genes is regulated by heterogeneous nuclear ribonucleoprotein (hnRNP) K. J. Biol. Chem. 286, 9763–9775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnson D. S., Mortazavi A., Myers R. M., Wold B. (2007) Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497–1502 [DOI] [PubMed] [Google Scholar]
- 13. Rosenbloom K. R., Dreszer T. R., Long J. C., Malladi V. S., Sloan C. A., Raney B. J., Cline M. S., Karolchik D., Barber G. P., Clawson H., Diekhans M., Fujita P. A., Goldman M., Gravell R. C., Harte R. A., Hinrichs A. S., Kirkup V. M., Kuhn R. M., Learned K., Maddren M., Meyer L. R., Pohl A., Rhead B., Wong M. C., Zweig A. S., Haussler D., Kent W. J. (2012) ENCODE whole-genome data in the UCSC Genome Browser. Update 2012. Nucleic Acids Res. 40, D912-D917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. ENCODE Project Consortium (2011) A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 9, e1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuehner J. N., Pearson E. L., Moore C. (2011) Unravelling the means to an end. RNA polymerase II transcription termination. Nat. Rev. Mol. Cell Biol. 12, 283–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Simon P. (2003) Q-Gene. Processing quantitative real-time RT-PCR data. Bioinformatics 19, 1439–1440 [DOI] [PubMed] [Google Scholar]
- 17. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 18. Flanagin S., Nelson J. D., Castner D. G., Denisenko O., Bomsztyk K. (2008) Microplate-based chromatin immunoprecipitation method, Matrix ChIP. A platform to study signaling of complex genomic events. Nucleic Acids Res. 36, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nelson J. D., Denisenko O., Bomsztyk K. (2006) Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc. 1, 179–185 [DOI] [PubMed] [Google Scholar]
- 20. Langmead B., Trapnell C., Pop M., Salzberg S. L. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mikula M., Dzwonek A., Karczmarski J., Rubel T., Dadlez M., Wyrwicz L. S., Bomsztyk K., Ostrowski J. (2006) Landscape of the hnRNP K protein-protein interactome. Proteomics 6, 2395–2406 [DOI] [PubMed] [Google Scholar]
- 23. Mikula M., Gaj P., Dzwonek K., Rubel T., Karczmarski J., Paziewska A., Dzwonek A., Bragoszewski P., Dadlez M., Ostrowski J. (2010) Comprehensive analysis of the palindromic motif TCTCGCGAGA, A regulatory element of the HNRNPK promoter. DNA Res. 17, 245–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Käll L., Storey J. D., MacCoss M. J., Noble W. S. (2008) Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res. 7, 29–34 [DOI] [PubMed] [Google Scholar]
- 25. Van Seuningen I., Ostrowski J., Bustelo X. R., Sleath P. R., Bomsztyk K. (1995) The K protein domain that recruits the interleukin 1-responsive K protein kinase lies adjacent to a cluster of c-Src and Vav SH3-binding sites. Implications that K protein acts as a docking platform. J. Biol. Chem. 270, 26976–26985 [DOI] [PubMed] [Google Scholar]
- 26. Stains J. P., Lecanda F., Towler D. A., Civitelli R. (2005) Heterogeneous nuclear ribonucleoprotein K represses transcription from a cytosine/thymidine-rich element in the osteocalcin promoter. Biochem. J. 385, 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Da Silva N., Bharti A., Shelley C. S. (2002) hnRNP-K and Pur(α) act together to repress the transcriptional activity of the CD43 gene promoter. Blood 100, 3536–3544 [DOI] [PubMed] [Google Scholar]
- 28. Bailey T. L., Boden M., Buske F. A., Frith M., Grant C. E., Clementi L., Ren J., Li W. W., Noble W. S. (2009) MEME SUITE. Tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 30. Glover-Cutter K., Kim S., Espinosa J., Bentley D. L. (2008) RNA polymerase II pauses and associates with pre-mRNA processing factors at both ends of genes. Nat. Struct. Mol. Biol. 15, 71–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. West S., Gromak N., Proudfoot N. J. (2004) Human 5′ → 3′ exonuclease Xrn2 promotes transcription termination at co-transcriptional cleavage sites. Nature 432, 522–525 [DOI] [PubMed] [Google Scholar]
- 32. Szklarczyk D., Franceschini A., Kuhn M., Simonovic M., Roth A., Minguez P., Doerks T., Stark M., Muller J., Bork P., Jensen L. J., von Mering C. (2011) The STRING database in 2011. Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, D561–D568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kaneko S., Rozenblatt-Rosen O., Meyerson M., Manley J. L. (2007) The multifunctional protein p54nrb/PSF recruits the exonuclease XRN2 to facilitate pre-mRNA 3′ processing and transcription termination. Genes Dev. 21, 1779–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gilmour D. S., Fan R. (2008) Derailing the locomotive. Transcription termination. J. Biol. Chem. 283, 661–664 [DOI] [PubMed] [Google Scholar]
- 35. Richard P., Manley J. L. (2009) Transcription termination by nuclear RNA polymerases. Genes Dev. 23, 1247–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Perales R., Bentley D. (2009) “Cotranscriptionality.” The transcription elongation complex as a nexus for nuclear transactions. Mol. Cell 36, 178–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li S., Armstrong C. M., Bertin N., Ge H., Milstein S., Boxem M., Vidalain P. O., Han J.-D., Chesneau A., Hao T., Goldberg D. S., Li N., Martinez M., Rual J. F., Lamesch P., Xu L., Tewari M., Wong S. L., Zhang L. V., Berriz G. F., Jacotot L., Vaglio P., Reboul J., Hirozane-Kishikawa T., Li Q., Gabel H. W., Elewa A., Baumgartner B., Rose D. J., Yu H., Bosak S., Sequerra R., Fraser A., Mango S. E., Saxton W. M., Strome S., Van Den Heuvel S., Piano F., Vandenhaute J., Sardet C., Gerstein M., Doucette-Stamm L., Gunsalus K. C., Harper J. W., Cusick M. E., Roth F. P., Hill D. E., Vidal M. (2004) A map of the interactome network of the metazoan C. elegans. Science 303, 540–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Davidson L., Kerr A., West S. (2012) Co-transcriptional degradation of aberrant pre-mRNA by Xrn2. EMBO J. 31, 2566–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jimeno-González S., Haaning L. L., Malagon F., Jensen T. H. (2010) The yeast 5′-3′ exonuclease Rat1p functions during transcription elongation by RNA polymerase II. Mol. Cell 37, 580–587 [DOI] [PubMed] [Google Scholar]
- 40. Brannan K., Kim H., Erickson B., Glover-Cutter K., Kim S., Fong N., Kiemele L., Hansen K., Davis R., Lykke-Andersen J., Bentley D. L. (2012) mRNA decapping factors and the exonuclease Xrn2 function in widespread premature termination of RNA polymerase II transcription. Mol. Cell 46, 311–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chan S., Choi E.-A., Shi Y. (2011) Pre-mRNA 3′-end processing complex assembly and function. Wiley Interdiscip. Rev. RNA 2, 321–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang Q., Doublié S. (2011) Structural biology of poly(A) site definition. Wiley Interdiscip. Rev. RNA 2, 732–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kubo T., Wada T., Yamaguchi Y., Shimizu A., Handa H. (2006) Knock-down of 25-kDa subunit of cleavage factor Im in HeLa cells alters alternative polyadenylation within 3′-UTRs. Nucleic Acids Res. 34, 6264–6271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang Q., Coseno M., Gilmartin G. M., Doublié S. (2011) Crystal structure of a human cleavage factor CFI(m)25/CFI(m)68/RNA complex provides an insight into poly(A) site recognition and RNA looping. Structure 19, 368–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Naganuma T., Nakagawa S., Tanigawa A., Sasaki Y. F., Goshima N., Hirose T. (2012) Alternative 3′-end processing of long noncoding RNA initiates construction of nuclear paraspeckles. EMBO J. 31, 4020–4034 [DOI] [PMC free article] [PubMed] [Google Scholar]