

Background: Mtbhsp60 induces TLR2-mediated anti-inflammatory response in macrophages, but the mechanisms are not well understood.
Results: Clathrin-dependent TLR2-mediated endocytosis of Mtbhsp60 is required to induce anti-inflammatory response via p38 MAPK activation.
Conclusion: Mtbhsp60 induces anti-inflammatory response upon endocytosis. Blockage of endocytosis predominantly leads to pro-inflammatory cytokine production.
Significance: This information is important to tailor the Mtbhsp60-triggered IL-10 signaling to specifically block the excess nonprotective Th2-type response.
Keywords: Cytokine, Endocytosis, MAP Kinases (MAPKs), Mycobacterium tuberculosis, Toll-like Receptors (TLR)
Abstract
Understanding the signaling pathways involved in the regulation of anti-inflammatory and pro-inflammatory responses in tuberculosis is extremely important in tailoring a macrophage innate response to promote anti-tuberculosis immunity in the host. Although the role of toll-like receptors (TLRs) in the regulation of anti-inflammatory and pro-inflammatory responses is known, the detailed molecular mechanisms by which the Mycobacterium tuberculosis bacteria modulate these innate responses are not clearly understood. In this study, we demonstrate that M. tuberculosis heat shock protein 60 (Mtbhsp60, Cpn60.1, and Rv3417c) interacts with both TLR2 and TLR4 receptors, but its interaction with TLR2 leads to clathrin-dependent endocytosis resulting in an increased production of interleukin (IL)-10 and activated p38 MAPK. Blockage of TLR2-mediated endocytosis inhibited IL-10 production but induced production of tumor necrosis factor (TNF)-α and activated ERK1/2. In contrast, upon interaction with TLR4, Mtbhsp60 remained predominantly localized on the cell surface due to poorer endocytosis of the protein that led to decreased IL-10 production and p38 MAPK activation. The Escherichia coli homologue of hsp60 was found to be retained mainly on the macrophage surface upon interaction with either TLR2 or TLR4 that triggered predominantly a pro-inflammatory-type immune response. Our data suggest that cellular localization of Mtbhsp60 upon interaction with TLRs dictates the type of polarization in the innate immune responses in macrophages. This information is likely to help us in tailoring the host protective immune responses against M. tuberculosis.
Introduction
The host innate immune response to tuberculosis is highly complex and is associated with induction of both anti-inflammatory and pro-inflammatory type of cytokines (1, 2). The pro-inflammatory cytokines like IL-12 and TNF-α, secreted by the macrophages, are considered to be critical for conferring protection against infection with Mycobacterium tuberculosis (3–5). However, anti-inflammatory cytokines like IL-10 produced by the activated macrophages favor a T-helper 2 (Th2)4-type of immune response by inhibiting production of protective cytokines, promoting bacterial survival, and associating with chronic progression of tuberculosis (6–10).
The Toll-like receptors (TLR) are key sensors of M. tuberculosis infection and play an important role in shaping up the innate immune responses of the host (11–14). A number of mycobacterial proteins and lipids are found to be involved in mediating signals through the TLRs (11). TLRs are a group of pattern recognition receptors that are capable of recognizing several pathogen-specific ligands and produce diverse arrays of cytokines that regulate the effector functions of the macrophages (12–14). Among the TLRs, the TLR2 and the TLR4 receptors have been implicated to play a major role in the activation of macrophages by mycobacteria (15). These receptors can regulate the effector functions differently in response to various mycobacterial ligands by producing different levels of counteractive anti-inflammatory and pro-inflammatory cytokines (11, 16–21). However, the exact molecular mechanisms by which the TLR-mediated anti-inflammatory and pro-inflammatory responses are regulated during mycobacterial infection are not clearly understood.
Several microbial heat shock proteins (HSPs) have been demonstrated to modulate the macrophage effector function by inducing various cytokines in both TLR-dependent and -independent ways in addition to their classical chaperonin functions (22–27). Among the M. tuberculosis heat shock proteins, HSP65 and HSP70 were found to induce pro-inflammatory cytokines in human dermal endothelial cells in TLR2- and TLR4-dependent ways (28). Another M. tuberculosis heat shock protein Mtbhsp60 (Cpn60.1, Rv3417c) is also known to be a very potent stimulator of cytokine production in macrophages and is believed to play an important role in M. tuberculosis virulence (26).
In our previous studies, we found that Mtbhsp60 primarily targeted the TLR2 signaling cascades to inhibit nuclear translocation of c-REL and consequently decreased the production of IL-12 p40 in purified protein derivative (PPD)-activated macrophages (29). It appears that interaction of Mtbhsp60 with TLR2 plays an important role in inducing a dominant Th2-type response during M. tuberculosis infection that favors the intracellular survival of the bacilli (8, 9). One of the possible mechanisms by which Mtbhsp60 blocks IL-12 p40 induction could be through induction of anti-inflammatory mediators like IL-10. The IL-10 cytokine has been shown to inhibit IL-12 p40 induction in macrophages (6, 30, 31) primarily by targeting the c-Rel transcription factor (32). Therefore, it may be possible that Mtbhsp60 predominantly activates IL-10 production through its interaction with TLR2, which subsequently inhibits c-REL vis à vis IL-12 p40 in activated macrophages (29).We also found that interaction of Mtbhsp60 with TLR4 but not TLR2 resulted in increased production of IL-12 (29). Therefore, it appears that Mtbhsp60 is unique in orchestrating anti-inflammatory and pro-inflammatory responses depending on its specific interaction with TLR2 or TLR4.
In this study, we present evidence that may explain how Mtbhsp60 induces opposing inflammatory cytokines upon its interaction with TLR2 or TLR4. We found that upon engagement with TLR2, clathrin-dependent endocytosis is required for Mtbhsp60 to activate p38 MAPK and production of the anti-inflammatory IL-10 cytokine. However, induction of pro-inflammatory cytokines such as TNF-α by Mtbhsp60 required its sequestration to the membrane either through TLR2 or TLR4 resulting in activation of ERK1/2 signaling cascades. To the best of our knowledge, herein we demonstrate for the first time that differential localization of Mtbhsp60 in the endosome or the cell surface upon interaction with TLRs can dictate two counteracting effector functions in macrophages. These findings may be useful in devising strategies to regulate macrophage innate responses to engineer a host protective immunity against the M. tuberculosis infection.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of Mtbhsp60 and Ecolihsp60
The M. tuberculosis hsp60 (Mtbhsp60) and Escherichia coli hsp60 (Ecolihsp60) clones were the kind gifts from Dr. Shekhar C. Mande, Centre for DNA Fingerprinting and Diagnostics, Hyderabad, India. The recombinant Mtbhsp60 protein was purified by nickel-nitrilotriacetic acid affinity chromatography as described earlier (29, 33). The protein was dialyzed extensively against 50 mm Tris-Cl, pH 8.0, and concentrated using Amicon ultrafiltration assembly. The Ecolihsp60 protein was purified as described elsewhere (34). Briefly, the crude extract was salted in at 30% and salted out at 65% and further purified by ion exchange chromatography, using HiLoad 16/60 Q-Sepharose HP column (GE Healthcare). The protein sample was equilibrated in a loading buffer (50 mm Tris-Cl, pH 8.0, and 1 mm EDTA) by dialysis and was loaded onto the column. A step gradient of 50 mm NaCl increments was applied to the column, and the Ecolihsp60 protein was eluted at 400 mm NaCl. The protein was dialyzed extensively against 50 mm Tris-Cl, pH 8.0. Endotoxin contamination was removed by incubating the protein with 10% v/v polymyxin B-agarose (Sigma) as described by us earlier (35). The endotoxin content of the recombinant protein was measured by Limulus amebocyte lysate assay (E-toxate kit from Sigma).
Isolation of Peritoneal Macrophages
Peritoneal macrophages were harvested from C57Bl/6 mice obtained from National Institute of Nutrition, Hyderabad, India. The mice were injected intraperitoneally with 1 ml of 4% sterile thioglycolate broth, and after 3 days, peritoneal macrophages were isolated as described earlier (35, 36). The animal experiments were conducted at the National Institute of Nutrition, Hyderabad, India, upon approval by the Institutional Animal Ethics Committee of the National Institute of Nutrition, Hyderabad, India. Experiments using TLR2 and TLR4 knock-out (KO) mice were conducted at the animal house facility of International Centre for Genetic Engineering and Biotechnology, New Delhi, India, according to the guidelines of the Institutional Animal Ethics Committee.
Macrophage Stimulation Assay
The human monocyte/macrophage cell line THP-1 was obtained from the National Centre for Cell Science, Pune, India, and maintained in RPMI 1640 medium (Invitrogen) supplemented with 10% (v/v) heat-inactivated FBS, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin (all from Invitrogen) at 37 °C in a humidified incubator maintaining 5% CO2. THP-1 cells were treated with 20 ng/ml PMA (Sigma) for 24 h followed by overnight rest. The PMA-differentiated THP-1 macrophages or peritoneal macrophages from C57Bl/6 mice (3 × 106 cells per ml) were treated with either various concentrations or a fixed concentration of 3 μg/ml of Mtbhsp60 or Ecolihsp60. In some experiments, the PMA-differentiated THP-1 macrophages were pretreated for 1 h with 10 μg/ml of either anti-human TLR2 mAb (mouse immunoglobulin G2a (IgG2a)), anti-human TLR4 mAb (mouse IgG2a), anti-mouse TLR2 Ab, or anti-mouse TLR4 Ab (all from Imgenex) or with both anti-TLR2 Ab and anti-TLR4 Ab for 60 min at 37 °C and subsequently exposed to 3 μg/ml of purified Mtbhsp60 or Ecolihsp60 protein. In control groups, isotype-matched antibody of IgG2a isotype (BD Biosciences) was added at the same concentrations. Cells were either harvested after 1, 2, and 5 h for checking IL-10 mRNA expression by semi-quantitative RT-PCR or cultured for 48 h for measuring IL-10/TNF-α levels secreted in the culture supernatants by enzyme immunoassay (EIA). In some experiments, macrophages were pretreated with 50 or 100 μm monodansylcadaverine (MDC) (Sigma) prior to exposure with Mtbhsp60.
Cytokine EIA
The levels of IL-10 and TNF-α cytokines were quantified by two-site sandwich EIA (BD Biosciences) as described earlier (35, 37). Standard curve for these cytokines were obtained using the recombinant standard proteins provided by the manufacturer.
Semi-quantitative Reverse Transcription PCR (RT-PCR) Analysis of IL-10 and TNF-α
For semi-quantitative RT-PCR, PMA-differentiated THP-1 macrophages or peritoneal macrophages from either C57Bl/6 wild-type (WT), TLR2 KO, or TLR4 KO mice were incubated with Mtbhsp60 (3 μg/ml) for various time points or for a fixed time point of 1 or 2 h. In some experiments, macrophages were pretreated with neutralizing antibody to TLR2 or TLR4 or isotype control antibody for 1 h at 37 °C followed by incubation with Mtbhsp60. Total RNA was extracted from these groups using TRIzol, and about 2 μg of total RNA was reverse-transcribed using Moloney murine leukemia virus reverse transcriptase as per the manufacturer's instruction (Invitrogen). Semi-quantitative PCR was performed to measure the mRNA levels of various experimental groups. For human IL-10, the forward primer 5′-GCAACCTGCCTAACATGCTTCG-3′ and reverse primer 5′-GAAGATGTCAAACTCACTCATGGC-3′ were used where the annealing temperature was 65 °C. β-Actin was used as an internal control with the forward primer 5′-GTGATGGTGGGCATGGGTCA-3′ and reverse primer 5′-TTAATGTCACGCACGATTTCCC-3′, and the annealing temperature used was 58 °C. The amplification conditions were as follows: denaturation at 94 °C for 30 s, annealing for 1 min at the appropriate temperature, and extension at 72 °C for 2 min. For mouse IL-10, TNF-α, and GAPDH the forward primers 5′-TGCTATGCTGCCTGCTCTTA-3′, 5′-ACGTCGTAGCAAACCACCAAG-3′, and 5′-ACTTTGGCATTGTGGAAGG-3′ and the reverse primers 5′-TCATTTCCGATAAGGCTTGG-3′, 5′-CTCTTGACGGCAGAGAGGAGG-3′, and 5′-ACACATTGGGGGTAGGAACA-3′, were used, respectively. The amplification conditions were as follows: denaturation at 94 °C for 30 s, annealing for 45 s at 55 °C, and extension at 72 °C for 1 min. After 30 cycles, the amplicons for human IL-10 (388 bp) and β-actin (510 bp) as well as mouse IL-10 (242 bp), TNF-α (249 bp), and GAPDH (221 bp) were resolved by electrophoresis on 1.2% agarose gels and visualized by ethidium bromide staining. Quantification of the mRNA was performed by densitometric analysis using AlphaEaseFC software and the Spot Denso tool (version 7.0.1; Alpha Innotech, San Leandro, CA).
TLR2 and TLR4 siRNA
The negative control scrambled siRNA, TLR2 targeting siRNA (sense, 5′-GCCUUGACCUGUCCAACAtt-3′, where the lowercase letters represent two deoxy bases that serve as overhangs for cleavage by dicer), and TLR4 targeting siRNA (sense 5′-CCAAUCUAGAGCACUUGGAtt-3′) were purchased from Ambion Inc. and were used according to the manufacturer's instructions as described earlier (29). Transfection of PMA-differentiated THP-1 macrophages was carried out using Lipofectamine 2000 (Invitrogen). Macrophages were seeded at a density of 2 × 106 cells per well in a 12-well plate and were transfected with 100 nm siRNAs. After 6 h, the culture medium was replaced, and the cells were kept in culture for an additional 24 h. The transfected cells were activated with 3 μg/ml of Mtbhsp60 for 48 h, and IL-10 levels were quantified in the culture supernatants by EIA.
Biotinylation of Mtbhsp60
Mtbhsp60 protein was biotinylated using a commercially available biotinylation kit from Pierce as described earlier (38). Briefly, Mtbhsp60 was incubated with 5-fold molar excess of sulfo NHS-biotin reagent (sulfosuccinimidyl-2-[biotinamido] ethyl-1,3-dithiopropionate) for about 1 h at room temperature. The unconjugated biotin was removed from the biotinylated protein sample by desalting using Amicon ultracentrifugal filter units. Biotinylation of the protein was confirmed by EIA using streptavidin conjugated to HRP.
Labeling of Mtbhsp60 and Ecolihsp60 with FITC
FITC-labeled Mtbhsp60 (Mtbhsp60-FITC) or Ecolihsp60 (Ecolihsp60-FITC) was prepared by incubating the purified recombinant Mtbhsp60 or Ecolihsp60 protein with FITC using a commercially available FITC antibody labeling kit from Pierce following the manufacturer's protocol.
Flow Cytometry Analyses
To examine surface expression of TLR2/TLR4, the PMA-differentiated THP-1 macrophages were incubated with 10 μg/ml mouse anti-TLR2 mAb or mouse anti-TLR4 mAb or isotype-matched IgG2a antibody for 60 min at 4 °C followed by incubation with FITC-conjugated goat anti-mouse IgG (Sigma) for another 45 min at 4 °C. Cells were washed and resuspended in sheath fluid. Cell-bound fluorescence was measured with a BD FACSVantage SE (BD Biosciences) using CellQuest data acquisition and analysis software (BD Biosciences). In some experiments, PMA-differentiated THP-1 macrophages (0.5–1 × 106 cells) were pretreated for 60 min with 10 μg/ml of either anti-TLR2 mAb or anti-TLR4 mAb or IgG2a isotype control antibody followed by incubation with biotin-labeled Mtbhsp60 protein for 5 and 30 min on ice and then incubated with streptavidin-FITC for 45 min at 4 °C. The fluorescence was measured by flow cytometry.
Confocal Microscopy
PMA-differentiated THP-1 macrophages were seeded in a chamber slide (BD Biosciences) and incubated with 10 μg/ml Mtbhsp60-FITC at 37 °C for 15 min. In experiments with inhibitor, cells were preincubated for 30 min with 100 μm concentration of MDC. In some experiments, macrophages were pretreated with 10 μg/ml neutralizing anti-TLR2, anti-TLR4 mAb, or isotype control Ab and then incubated with Mtbhsp60-FITC/Ecolihsp60-FITC for 15 min. The cells were washed and fixed with 3% paraformaldehyde (Sigma). Confocal microscopy was performed on a Zeiss LSM 510META laser scanning microscope (Carl Zeiss).
Measurement of Phospho-p38 and Phospho-ERK1/2 by Western Blotting and Flow Cytometry
The PMA-differentiated THP-1 macrophages (1–2 × 106) were preincubated with anti-TLR2 mAb or anti-TLR4 mAb or isotype-matched control antibody (10 μg/ml) and then treated with 3 μg/ml Mtbhsp60 for 15 min at 37 °C. Cells were lysed using Laemmli sample buffer as described earlier (38). Equal amounts of the extracts were separated by 12% SDS-PAGE, and following electrophoretic transfer, the nitrocellulose membranes were incubated with antibody to either phospho-p38 (BD Biosciences) or phospho-ERK1/2 (Cell Signaling Technology), total p38 (BD Biosciences), or total ERK1/2 (Cell Signaling Technology). Membranes were washed and incubated with appropriate secondary Ab conjugated to HRP (Sigma). Bound enzyme was detected by ECL following the manufacturer's protocol (GE Healthcare) as described earlier (38).
The expression levels of phosphorylated p38 and ERK1/2 in these macrophages were also determined by flow cytometry as described earlier (38, 39). For this, cells (1–2 × 106) were fixed with 1.5% paraformaldehyde (Sigma) for 10 min at 37 °C and chilled on ice for 1 min. After washing with staining buffer (1% BSA and 0.1% sodium azide in PBS), the cells were permeabilized using freshly prepared 90% ice-cold methanol for 30 min on ice. The cells were then washed three times in staining buffer and blocked in the same buffer for 10 min at 37 °C. Cells were then incubated with 100-fold diluted antibody to either phospho-p38 or phospho-ERK1/2 (Cell Signaling Technology) followed by incubation with anti-rabbit or anti-mouse IgG-FITC (Sigma) at 37 °C. Finally, the cells were washed and resuspended in PBS and analyzed in BD FACSVantage SE (BD Biosciences).
Electrophoretic Mobility Shift Assay (EMSA)
EMSA was performed following the protocol as described earlier (35). The nuclear extracts (10 μg) and 1 ng of γ-32P-end-labeled NF-κB consensus binding sequence 5′-AGTTGAGGGGACTTTCCCAGG-3′ were incubated for 30 min at room temperature in binding buffer (20 mm HEPES, 0.5 mm DTT, 1 mm MgCl2, 1 mm EDTA, and 5% glycerol) containing 2 μg of poly(dI-dC). The DNA-protein complex was resolved on 7% native gel in 1× TGE running buffer (25 mm Tris base, 190 mm glycine, 1 mm EDTA, pH 8.3). Specificity of the binding was confirmed by competition with 100-fold excess of unlabeled probe. The gel was dried at 80 °C for 1 h and exposed to imaging plate (Fuji Film) overnight. Visualization of the radioactive bands was carried out using a STARION image scanner (Fuji Film FLA-9000).
In Silico Analysis of Secondary Structures of Mtbhsp60 and Ecolihsp60
The Mtbhsp60 structure was determined by homology modeling using the Mtbhsp60.2 structure available in the Protein Data Bank (PDB) (chain A of PDB code 1SJP) as template. The sequence alignment of Mtbhsp60 and Mtbhsp60.2 was carried out using the Clustal W2 software. The protein sequence of Mtbhsp60 (43–527 amino acids) showed about 62% identity with that of 65-kDa HSP60.2 of M. tuberculosis, with an E-value of 2e-163 and a sequence coverage of 89%. The Modeler9 version 5 program (40, 41) was used for homology model building, and the structure was validated using the on-line server SAVES. The structural model with the best Verify3D profile was selected and further subjected to energy minimization (42, 43). The GROMACS program package 4.0.1 (44) was used for energy minimization by applying the GROMOS96 43a1 force field algorithms and the steepest descent methods.
The energy-minimized structure of Mtbhsp60 was used for structural comparison with Ecolihsp60 structure (crystal structure of Ecolihsp60 (chaperonin GroEL; chain A of PDB code 2EU1) retrieved from the PDB) (45). The degree of structural deviation between the Mtbhsp60 and Ecolihsp60 was calculated by determining the root mean square deviation by superimposing the two molecules using the Swiss PDB Viewer version 4.0.1 software (46). The root mean square deviation values suggest significant structural deviations between the two protein structures.
Statistical Analysis
Data were expressed as mean ± S.D. of at least three independent experiments performed with similar results. Student's t test was used to determine statistical differences between the groups. p < 0.05 was considered to be significant.
RESULTS
Mtbhsp60 Activates IL-10 Induction in Macrophages
Earlier, we found that Mtbhsp60 could inhibit PPD-induced pro-inflammatory response in macrophages (29). As IL-10 is known to inhibit production of pro-inflammatory cytokines (6, 30, 31), we speculated that Mtbhnsp60 can induce IL-10 production in macrophages. Therefore, to analyze the ability of Mtbhsp60 to activate IL-10 induction in macrophages, PMA-differentiated THP-1 macrophages were treated with either a fixed concentration (3 μg/ml) or titrating concentrations of Mtbhsp60, and the macrophages were either harvested after 1, 2, and 5 h post-treatment to analyze IL-10 mRNA expression by RT-PCR or cultured for a period of 48 h to measure the amount of IL-10 secreted in the culture supernatants by EIA. The recombinant Mtbhsp60 protein was found to activate IL-10 production in THP-1 macrophages in a dose-dependent manner (Fig. 1A). Mtbhsp60 could induce IL-10 transcript as early as 1 h post-treatment at 3 μg/ml concentration (Fig. 1B).
FIGURE 1.
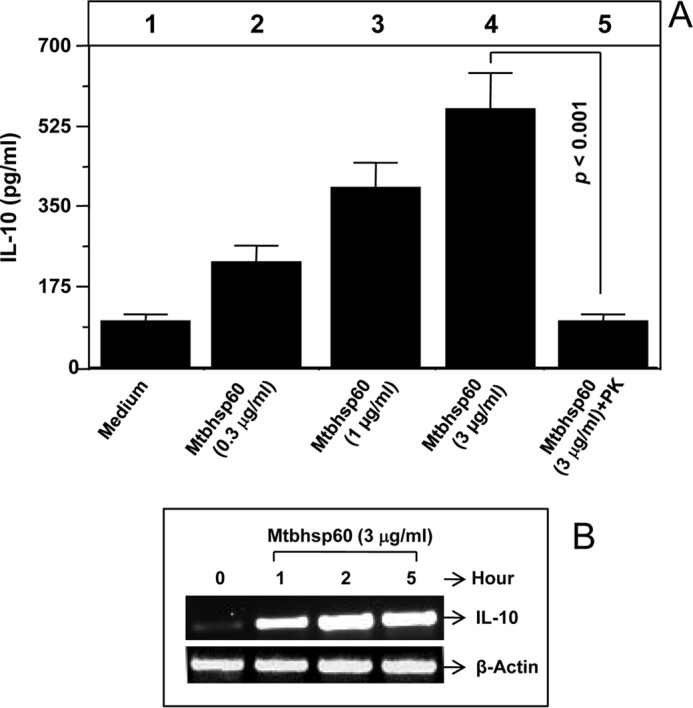
Mtbhsp60 induces IL-10 production in THP-1 macrophages. PMA-differentiated THP-1 macrophages were treated with either titrating concentrations of the purified Mtbhsp60 or a fixed concentration (3 μg/ml) of proteinase K-treated Mtbhsp60. A, IL-10 cytokine level was estimated by EIA in various culture supernatants harvested after 48 h of incubation. B, total RNA was extracted from various groups as described under “Experimental Procedures,” and IL-10 mRNA expression was measured at various time points by semi-quantitative RT-PCR. Results shown are representative of three to four different experiments.
To rule out the observed effects due to endotoxin contamination in the recombinant Mtbhsp60 preparation, the protein preparation was treated with polymyxin B, a specific inhibitor of bacterial lipopolysaccharide (LPS) (35), and the endotoxin content of the Mtbhsp60 protein preparation was found to be very low (less than 0.01 EU/ml) by Limulus amebocyte lysate assay. We found that treatment of Mtbhsp60 with proteinase K significantly abrogated its ability to induce IL-10, and the induction level was comparable with that of medium alone (Fig. 1A, compare lane 5 with lane 1) indicating that the observed effect is specific to Mtbhsp60. Mtbhsp60 was also found to induce IL-10 cytokine in human monocyte-derived macrophages (data not shown).
Interaction of Mtbhsp60 with TLR2 Receptors Is Important for Induction of IL-10 in Macrophages
Like many other HSPs (47, 48), we found that Mtbhsp60 can interact with both the TLR2 and TLR4 receptors. Time-dependent increase in the cell-bound Mtbhsp60-specific fluorescence could be detected in THP-1 cells preincubated with anti-TLR2 anti-TLR4 as well as isotype-matched control antibodies followed by treatment with Mtbhsp60 labeled with biotin and subsequent incubation with streptavidin-FITC conjugate (Fig. 2A). To determine the TLR receptor involved in Mtbhsp60-mediated IL-10 induction, specific TLR receptors were blocked by preincubating the THP-1 macrophages either with anti-TLR2, anti-TLR4 mAb, or isotype-matched control antibody and subsequently treated with Mtbhsp60, and the levels of IL-10 produced by these macrophages were measured by RT-PCR or EIA. It was found that compared with the group treated with medium alone, a significant increase in the IL-10 mRNA was observed upon exposure to Mtbhsp60 in the groups of those pretreated with either anti-TLR4 mAb or isotype-matched control antibody (Fig. 2B). However, in contrast, pretreatment with anti-TLR2 mAb had little effect on IL-10 gene expression in the presence of Mtbhsp60 (Fig. 2B). Similar results were also observed when IL-10 was measured in the culture supernatant by EIA (Fig. 2C, compare bars 5 and 7 with bar 3; p < 0.001 in both the cases). When cells were treated with both anti-TLR2 and anti-TLR4, the IL-10 level was comparable with that of medium alone (Fig. 2C, compare bar 9 with bar 1 and see supplemental Fig. 1). Comparable results were also observed in peritoneal macrophages from C57Bl/6 mice (Fig. 2D, compare bars 5 and 7 with bar 3; p < 0.001 in both the cases) suggesting that Mtbhsp60-mediated induction of IL-10 in macrophages is predominantly dependent on its interaction with TLR2. No significant differences were observed in the cell viability in antibody and Mtbhsp60 co-treated groups with that of the control group that was treated with medium alone (supplemental Fig. 2).
FIGURE 2.

IL-10 activation by Mtbhsp60 is TLR2-dependent. A, PMA-differentiated THP-1 macrophages were pretreated with either 10 μg/ml of anti-TLR2 mAb or anti-TLR4 mAb or isotype-matched control antibody for 1 h and then incubated with 10 μg/ml of biotin-labeled Mtbhsp60 at 4 °C for 5 and 30 min followed by incubation with streptavidin-FITC. The fluorescence was measured by flow cytometry. B, PMA-differentiated THP-1 macrophages were pretreated with 10 μg/ml of anti-TLR2 mAb or anti-TLR4 mAb or isotype-matched control antibody for 1 h and cultured for 2 h in the presence of 3 μg/ml of Mtbhsp60. Total RNA was extracted, and IL-10 levels were measured by semi-quantitative RT-PCR, and quantification of the IL-10 mRNA was performed by densitometric analysis using AlphaEaseFC software and the Spot Denso tool (version 7.0.1; Alpha Innotech, San Leandro, CA). Data are expressed as mean ± S.D. of three independent experiments. C, PMA-differentiated THP-1 macrophages were pretreated with neutralizing mAb to either TLR2 or TLR4, isotype-matched control antibody, or with both anti-TLR2 mAb and anti-TLR4 mAb in the absence or presence of Mtbhsp60 (3 μg/ml). After 48 h, IL-10 cytokine levels in culture supernatants from various groups were measured by EIA. D, experiments were also set to measure IL-10 levels by EIA in C57Bl/6 peritoneal macrophages treated with 10 μg/ml of anti-TLR2 Ab, anti-TLR4 Ab, or isotype control antibody in the absence or presence of Mtbhsp60 (3 μg/ml). Results shown are representative of three different experiments.
To further confirm the role of TLR2 in Mtbhsp60-mediated IL-10 induction, we determined the amount of IL-10 produced in macrophages that were deficient in either TLR2 or TLR4. Therefore, siRNA-mediated gene silencing was carried out using TLR-specific siRNAs in THP-1 macrophages, and the consequent reductions in the levels of surface expression of TLR2 or TLR4 were confirmed by flow cytometry (supplemental Fig. 3). Consistent with our previous observations with neutralizing antibodies (Fig. 2), we found that Mtbhsp60-mediated IL-10 induction was higher in TLR4-deficient cells when compared with that of TLR2-deficient cells (Fig. 3, compare bar 6 with bar 5; p < 0.001). Collectively these data suggest that IL-10 induction by Mtbhsp60 is predominantly mediated through TLR2-induced signaling.
FIGURE 3.
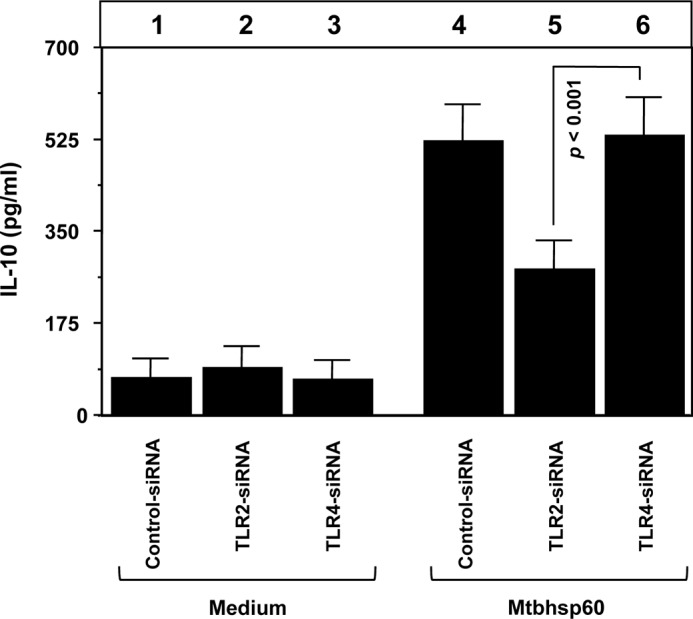
Silencing of TLR2 expression by siRNA down-regulates Mtbhsp60-induced IL-10 in THP-1 macrophages. PMA-differentiated THP-1 macrophages were transfected with negative control siRNA or TLR2-specific siRNA or TLR4-specific siRNA. After 24 h post-transfection, cells were incubated with Mtbhsp60 (3 μg/ml) for 48 h, and levels of IL-10 were measured by EIA in the culture supernatants. Results shown are representative of three independent experiments.
IL-10 Activation by Mtbhsp60 Is Dependent on the Endocytosis of Mtbhsp60 via TLR2 Receptors
We next investigated the probable mechanisms involved in the TLR2-dependent activation of IL-10 by Mtbhsp60. Earlier studies on human and chlamydial HSP60s suggest that activation of macrophage innate-effector responses by heat shock proteins is dependent to a great extent on the TLR-mediated, clathrin-dependent endocytosis (48), and clathrin-dependent endocytosis is known to be a major pathway for internalization of transmembrane receptors (49). Therefore, we investigated whether receptor-mediated endocytosis of Mtbhsp60 is a prerequisite for activation of IL-10. PMA-differentiated THP-1 macrophages were incubated with FITC-labeled Mtbhsp60 (Mtbhsp60-FITC) in the absence or presence of MDC, a selective cytoskeleton microtubule inhibitor that is known to block clathrin-dependent endocytosis (50), and we examined the cellular distribution of Mtbhsp60-associated fluorescence using confocal microcopy. It was observed that THP-1 macrophages treated with MDC had poorly internalized Mtbhsp60-FITC (Fig. 4A) resulting in increased cell surface accumulation of Mtbhsp60 (supplemental Fig. 4). After incubation of Mtbhsp60 with macrophages, the protein was found to be co-localized with early endosome antigen 1 (EEA1), an early endosome-specific marker (supplemental Fig. 5) suggesting receptor-mediated endocytosis of the protein. Concurrently, Mtbhsp60-mediated IL-10 induction was also significantly compromised in THP-1 macrophages treated with MDC as compared with the macrophages treated with medium alone (Fig. 4B, compare bar 4 and bar 5 with bar 3; p < 0.01 and p < 0.001, respectively). MDC had no significant effect on cell viability at both 50 and 100 μm concentrations used in these experiments (supplemental Fig. 6). These results suggest that receptor-mediated clathrin-dependent endocytosis of Mtbhsp60 is required for IL-10 induction in macrophages.
FIGURE 4.
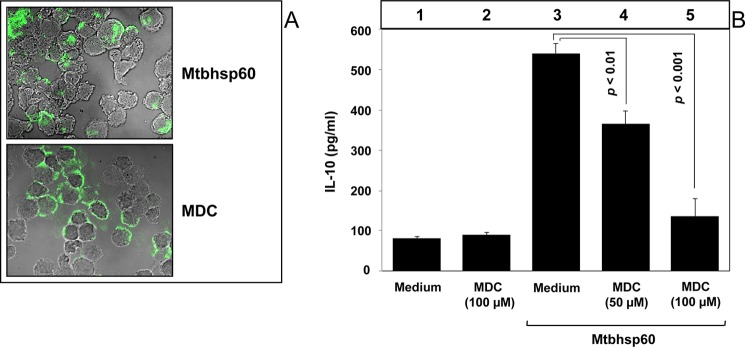
MDC inhibits Mtbhsp60-mediated induction of IL-10 in THP-1 macrophages. A, PMA-differentiated THP-1 macrophages in a chamber slide were preincubated for 30 min without or with MDC (100 μm) followed by incubation with 10 μg/ml of Mtbhsp60-FITC at 37 °C for 15 min. Cells were fixed and washed, and the cell bound fluorescence was analyzed by confocal laser scanning microscopy. B, PMA-differentiated THP-1 macrophages were either left untreated or pretreated with 50 or 100 μm MDC and subsequently incubated with 3 μg/ml of Mtbhsp60. IL-10 cytokine level was estimated by EIA in various culture supernatants harvested after 48 h of incubation. Results shown are representative of three different experiments.
Because in previous experiments we observed a direct role of TLR2 in the Mtbhsp60-mediated activation of IL-10 (Figs. 2 and 3), we next examined whether internalization of Mtbhsp60 was actually mediated through the TLR2 receptor. Therefore, PMA-differentiated THP-1 macrophages were pretreated with 10 μg/ml of anti-TLR2 or anti-TLR4 mAb to block the TLR2 or TLR4 receptors, respectively, and then incubated with Mtbhsp60-FITC (10 μg/ml) for 15 min at 37 °C, and we examined the intracellular distribution of Mtbhsp60. The control group received isotype-matched antibody. We found that the macrophages pretreated with anti-TLR2 mAb showed higher cell surface-associated fluorescence as compared with the macrophages pretreated with either anti-TLR4 mAb or isotype control antibody (Fig. 5A) indicating impaired endocytosis in the anti-TLR2 antibody-treated group. These results suggest that Mtbhsp60 is internalized upon interaction with TLR2, and its interaction with TLR4 stalls this protein predominantly at the cell surface (Fig. 5A). Upon internalization, the Mtbhsp60 was also found to be colocalized along with TLR2 (supplemental Fig. 7). In the next experiments, we pretreated THP-1 macrophages with anti-TLR4 antibodies to allow Mtbhsp60 to interact predominantly with TLR2, and we blocked the TLR2-mediated endocytosis by MDC and measured the levels of IL-10 in these cells. As expected, IL-10 induction was found to be strongly inhibited when endocytosis of Mtbhsp60 through TLR2 was inhibited (Fig. 5B, compare bar 4 with bar 3; p < 0.01). Similar results were obtained using peritoneal macrophages from C57Bl/6 mice (Fig. 5C, compare bar 4 with bar 3; p < 0.001). These data suggest that endocytosis of Mtbhsp60 is required to induce IL-10 post-binding to the TLR2 receptors.
FIGURE 5.

IL-10 activation by Mtbhsp60 is dependent on TLR2-mediated endocytosis of Mtbhsp60. A, PMA-differentiated THP-1 macrophages were pretreated with 10 μg/ml of anti-TLR2 mAb or anti-TLR4 mAb or isotype-matched control antibody for 1 h and further incubated for 15 min in the presence of Mtbhsp60-FITC (10 μg/ml) at 37 °C. Cells were fixed and washed, and the endocytosis was examined by confocal laser scanning microscopy. PMA-differentiated THP-1 macrophages (B) or peritoneal macrophages (C) from C57Bl/6 mice were pretreated with 10 μg/ml of anti-TLR4 Ab or isotype control Ab for 1 h and then incubated with 3 μg/ml of Mtbhsp60 in the absence or presence of 100 μm MDC. After 48 h of incubation, IL-10 cytokine level was measured by EIA in various culture supernatants. Results shown are representative of at least three different experiments.
Mtbhsp60-induced TNF-α Production Is Largely Dependent on the Signaling Generated by Surface-bound TLR-Mtbhsp60 Immune Complex
It is believed that Mtbhsp60 is also responsible for TNF-α induction as M. tuberculosis mutants lacking the groEL homologue Cpn60.1 (Mtbhsp60) failed to induce an inflammatory response and was unable to induce formation of granuloma in the murine experimental model (51). TNF-α is known to be one of the critical factors involved in the granuloma formation (3, 52–54). Interestingly, in human endothelial cells, induction of TNF-α by some other mycobacterial heat shock proteins like HSP65 and HSP70 was found to be TLR4-dependent (28). Because our previous experiments suggest that Mtbhsp60 interacts with both the TLR2 and TLR4 receptors and TLR2 is involved in Mtbhsp60-mediated IL-10 induction, we speculated a role of TLR4 in inducing TNF-α production. Therefore, we blocked either the TLR2 or the TLR4 or both the TLR2 and TLR4 receptors using respective blocking antibodies and then treated the cells with 3 μg/ml Mtbhsp60. It was observed that inhibition of TLR4-Mtbhsp60 interaction with anti-TLR4 antibody resulted in significantly poorer TNF-α production in THP-1 macrophages in response to Mtbhsp60 when compared with that of anti-TLR2- or isotype-matched control antibody-treated cells (Fig. 6, compare bar 4 with bar 6 and bar 2; p < 0.01 in both the cases). These results indicate that Mtbhsp60 probably targets the TLR4 to trigger induction of the pro-inflammatory cytokines like TNF-α. Earlier we found that when Mtbhsp60 interacted with TLR2 it undergoes receptor-mediated endocytosis to induce IL-10, whereas its interaction with TLR4 left the protein mostly surface-bound (Fig. 5A). These observations tempted us to speculate that TNF-α induction is triggered by signals generated predominantly by the surface-bound Mtbhsp60-TLR4 complexes, whereas induction of IL-10 is driven by the signals generated by Mtbhsp60 localized in the endosome upon receptor-mediated endocytosis through TLR2. To test this hypothesis, we next blocked the TLR2-mediated endocytosis of Mtbhsp60 by MDC and examined whether surface-bound TLR2-Mtbhsp60 complexes can also induce production of TNF-α. The macrophages were therefore pretreated either with isotype control antibody or anti-TLR4 antibody and then treated with Mtbhsp60 in the absence or presence of MDC. True to our expectations, we found that blockage of receptor-mediated endocytosis in the presence of MDC could significantly increase the production of TNF-α in isotype control antibody-treated cells (Fig. 6, compare bar 3 with bar 2; p < 0.05). When TLR2-mediated endocytosis of Mtbhsp60 was blocked in the presence MDC, production of TNF-α was also significantly increased (Fig. 6, compare bar 5 with bar 4; p < 0.01). Interestingly, in such situations, production of IL-10 was found to be significantly reduced in macrophages when TLR2-mediated endocytosis of Mtbhsp60 was inhibited in the presence of MDC (Fig. 5, compare bar 4 with bar 3). These data suggest that Mtbhsp60 can induce starkly opposite cytokine responses in macrophages depending on its cellular localization post-binding to TLRs. Because blockage of TLR2 and TLR4 receptors together with the respective antibodies had no significant increase in the Mtbhsp60-mediated TNF-α production (Fig. 6, lane 7) or IL-10 (Fig. 2C, lane 9; supplemental Fig. 1) over medium control, it appears that the Mtbhsp60 primarily targets the TLR2 and the TLR4 receptors to influence cytokine signaling in macrophages.
FIGURE 6.

Interaction of Mtbhsp60 with TLR4 or TLR2 in the presence of MDC triggers TNF-α production. PMA-differentiated THP-1 macrophages were pretreated with 10 μg/ml of neutralizing mAb to either TLR2 or TLR4, or isotype-matched control antibody, or with both anti-TLR2 mAb and anti-TLR4 mAb for 1 h and subsequently incubated with Mtbhsp60 (3 μg/ml) in the absence or presence of MDC (100 μm), and TNF-α cytokine levels were quantified after 48 h in different culture supernatants by EIA. Results shown are representative of three different experiments.
To further corroborate our findings, we also used macrophages harvested from TLR2 KO and TLR4 KO mice. The thioglycolate-elicited peritoneal macrophages from C57Bl/6 WT, TLR2 KO, and TLR4 KO mice were left untreated or treated with Mtbhsp60 at a concentration of 3 μg/ml, and the levels of IL-10 and TNF-α produced by these macrophages were measured by semi-quantitative RT-PCR. The results shown in Fig. 7 indicate that Mtbhsp60 could increase IL-10 mRNA expression in macrophages harvested from either wild-type or TLR4 KO mice; however, a near control level of IL-10 mRNA expression in response to Mtbhsp60 was observed in macrophages harvested from TLR2 KO mice (Fig. 7, A and B). In contrast, a strong TNF-α induction by Mtbhsp60 was observed predominantly in macrophages harvested from either wild-type or TLR2 KO mice, whereas Mtbhsp60 failed to trigger TNF-α mRNA expression in macrophages harvested from TLR4 KO mice, and the mRNA level was almost similar to the medium-treated control (Fig. 7, A and C). These results clearly demonstrate that Mtbhsp60 primarily targets the TLR2 to induce IL-10 and the TLR4 to activate TNF-α cytokine in macrophages.
FIGURE 7.

Mtbhsp60 targets the TLR2 to induce IL-10 and the TLR4 to activate production of TNF-α in macrophages. Thioglycolate-elicited peritoneal macrophages from C57Bl/6 WT, TLR2 KO, and TLR4 KO mice were either left untreated or treated with Mtbhsp60 (3 μg/ml) for 1 h. A, total RNA was extracted, and IL-10 and TNF-α levels were measured by semi-quantitative RT-PCR. Densitometric analyses were performed for IL-10 (B) and TNF-α (C) using the AlphaEaseFC software and the Spot Denso tool, normalized for GAPDH, and reported as arbitrary densitometric units. Values are means ± S.D. of the densitometric analysis of three independent experiments.
E. coli Heat Shock Protein 60 (Ecolihsp60) Is Retained Mainly on the Macrophage Surface upon Interaction with Either TLR2 or TLR4 and Triggers Induction of TNF-α
The E. coli homologue of Mtbhsp60, Ecolihsp60 (groEL), is known to induce predominantly a pro-inflammatory cytokine response in macrophages and human monocytes (25, 55, 56). We found that Ecolihsp60 can bind to both the TLR2 and TLR4 receptors on THP-1 macrophages (supplemental Fig. 8). Interestingly, interaction of Ecolihsp60 with either TLR2 or TLR4 left the protein mostly stranded on the cell surface. When THP-1 macrophages were pretreated with isotype-matched control or anti-TLR2 or anti-TLR4 Ab and subsequently incubated with Ecolihsp60-FITC and allowed it to endocytose at 37 °C, we observed very strong cell surface-bound fluorescence using confocal microscopy due to poor TLR-mediated endocytosis of Ecolihsp60 (Fig. 8A). As earlier we observed triggering of TNF-α production by surface-bound Mtbhsp60, we speculated that interaction of Ecolihsp60 with either TLR2 or TLR4 would trigger TNF-α production due to its inherent inability to undergo receptor-mediated endocytosis unlike interaction of Mtbhsp60 with TLR2. We found that the blockage of either TLR2 or TLR4 receptors using respective blocking antibodies led to production of similar levels of TNF-α (Fig. 8B, compare bar 3 with bar 4). Interestingly, in the presence of isotype-matched Ab, where both the TLR2 and TLR4 receptors are free to interact with the Ecolihsp60, the levels of TNF-α were found to be significantly higher as compared with that of TLR2 or TLR4 alone and almost summed up the levels of TNF-α produced together by these receptors (Fig. 8B, compare bar 2 with bars 3 and 4).
FIGURE 8.
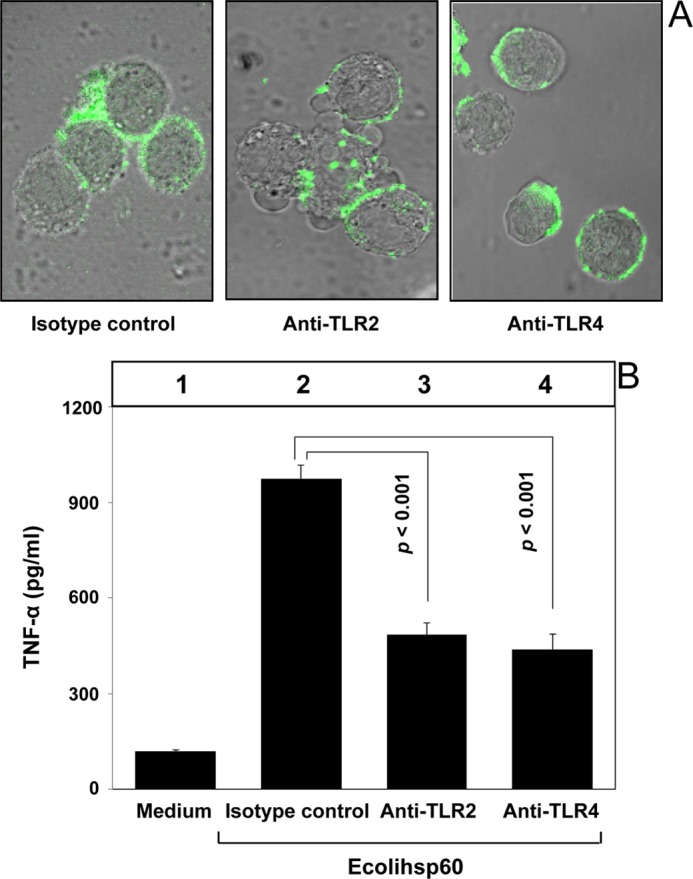
Interaction of Ecolihsp60 either with TLR2 or TLR4 results in induction of TNF-α. A, PMA-differentiated THP-1 macrophages were pretreated with 10 μg/ml of anti-TLR2 mAb or anti-TLR4 mAb or isotype-matched control antibody for 1 h, followed by incubation with Ecolihsp60-FITC (10 μg/ml) at 37 °C for 15 min. Cells were fixed and washed, and endocytosis of the protein was assessed by confocal laser scanning microscopy. B, PMA-differentiated THP-1 macrophages were pretreated with 10 μg/ml of anti-TLR2 mAb or anti-TLR4 mAb or isotype-matched control antibody for 1 h and then incubated with 3 μg/ml Ecolihsp60. TNF-α levels were quantified after 48 h in different culture supernatants by EIA. Data are representative of mean ± S.D. of three different experiments.
Although the heat shock proteins are highly conserved, the biochemical features of Mtbhsp60 are known to be deviated significantly from its E. coli homologue, Ecolihsp60. Mtbhsp60 appears to exist in a lower oligomeric state as compared with its E. coli counterpart due to substitutions in some crucial interface residues required to stabilize its inter-subunit interactions (33). It is likely that there are also significant deviations at the structural level of the monomers that might explain some of the observed functional differences between these two homologous proteins. Therefore, we determined the three-dimensional structure of Mtbhsp60 using homology modeling (Fig. 9A) and superimposed with that of Ecolihsp60 (Fig. 9B). The overall root mean square deviation of the Mtbhsp60 (green)/Ecolihsp60 (blue) (Fig. 9C) was found to be 4.89 Å, indicating a significant deviation exists between these two proteins at the conformational level.
FIGURE 9.
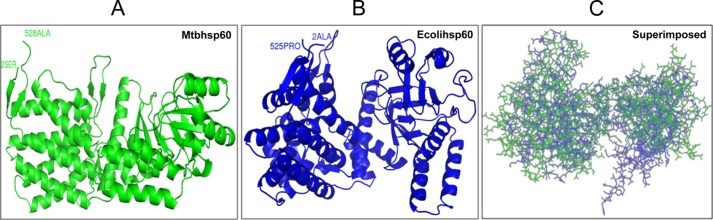
Secondary structure comparison of Mtbhsp60 with Ecolihsp60. A, secondary structure of the Mtbhsp60 protein model predicted based on homology modeling with Modeler software is shown (green) after its energy minimization. B, solved crystal structure of Ecolihsp60 obtained from Protein Data bank (chain A of PDB code 2EU1) is displayed (blue). C, superimposed structures of Mtbhsp60 and Ecolihsp60.
p38 MAPK and ERK1/2 Signaling Play Important Roles in the TLR-dependent Induction of IL-10 and TNF-α by Mtbhsp60
A number of TLR ligands are known to activate various MAPKs, including p38 MAPK, ERK1/2, and JNK 1/2 (57, 58), and MAPKs play crucial roles in regulating the innate cytokine production in macrophages (59). We and others have shown that p38 MAPK and ERK1/2 signalings are crucial in regulating the anti-inflammatory and pro-inflammatory cytokines in macrophages by mycobacterial components (38, 60–67). It has been reported that activation of p38 MAPK plays a critical role for IL-10 production (62, 66), whereas TNF-α secretion is predominantly dependent on ERK1/2 activation in macrophages (11, 61). Therefore, we compared the phosphorylation status of p38 MAPK and ERK1/2 in situations where Mtbhsp60 is endocytosed by interacting with TLR2 and thereby resulting in dominant production of IL-10 to situations where it is predominantly sequestered on the cell surface, like interactions with TLR4 or with TLR2 in the presence of MDC, resulting in increased production of TNF-α. Therefore, THP-1 macrophages were treated either with anti-TLR2 antibody to allow binding of Mtbhsp60 to TLR4 or with anti-TLR4 antibody to allow binding of Mtbhsp60 to TLR2 receptors, respectively, and we examined the phosphorylation status of p38 MAPK and ERK1/2 by flow cytometry and Western blotting. It was observed that interaction of Mtbhsp60 with TLR2 leads to phosphorylation of p38 MAPK predominantly as early as 10 min and to a lesser extent ERK1/2 (Fig. 10, A and B). However, engagement of Mtbhsp60 with TLR4 predominantly phosphorylated the ERK1/2 at the same time point (Fig. 10, A and B). Conversely, blocking of TLR2-mediated endocytosis of Mtbhsp60 by MDC had little effect on p38 MAPK phosphorylation status, but ERK1/2 phosphorylation was found to be significantly increased (Fig. 10C). Interestingly, activation of p38 MAPK has been reported to be crucial for triggering early endocytic membrane traffic (68, 69). Taken together, these data suggest that endocytosis of Mtbhsp60 through TLR2 predominantly activates p38 MAPK leading to IL-10 induction, whereas surface-bound Mtbhsp60 activates ERK1/2 causing higher induction of TNF-α. Therefore, it appears that the cellular localization of Mtbhsp60 following interaction with TLRs dictates the type of MAPKs to be activated and subsequently the kind of cytokine responses to be produced in the macrophages.
FIGURE 10.

Comparison of phosphorylation status of p38 MAPK and ERK1/2 mediated by Mtbhsp60 between the TLR2 receptor that undergoes endocytosis against TLR2 or TLR4 that does not undergo endocytosis. A, PMA-differentiated THP-1 macrophages were pretreated with 10 μg/ml of anti-TLR2 mAb or anti-TLR4 mAb or isotype-matched control Ab for 1 h and then treated with Mtbhsp60 (3 μg/ml) for 15 min. After permeabilization, macrophages were incubated with antibody to either phospho-p38 or phospho-ERK1/2 followed by incubation with anti-rabbit IgG-FITC or anti-mouse IgG-FITC, and fluorescence was analyzed by flow cytometry. B, in another experiment, THP-1 macrophages were pretreated with 10 μg/ml of either anti-TLR2 mAb or anti-TLR4 mAb or isotype-matched control Ab followed by incubation with Mtbhsp60 (3 μg/ml). Cells were lysed, and the levels of phosphorylated and total p38 MAPK as well as phosphorylated and total ERK1/2 levels were measured by Western blotting. C, PMA-differentiated THP-1 macrophages were pretreated with neutralizing mAb to TLR4 (10 μg/ml) for 1 h and then treated with Mtbhsp60 for 15 min in the absence or presence of MDC (100 μm). The phosphorylation status of p38 MAPK and ERK1/2 was analyzed by flow cytometry. Results are representative of three experiments.
Because TNF-α is shown to be dominantly regulated by the NF-κB transcription factors (70), and becauseTLR4 is known to activate NF-κB (71, 72), it is expected that interaction of TLR4 with Mtbhsp60 could activate the NF-κB transcription factor more dominantly as compared with the condition where Mtbhsp60 was allowed to interact with TLR2. Therefore, THP-1 macrophages were treated with Mtbhsp60 in the absence or presence of neutralizing Ab to either TLR2 or TLR4 or isotype-control Ab, and nuclear extracts were used to check the specific DNA binding activity of the NF-κB complex by EMSA using NF-κB consensus oligonucleotide probe (73) labeled with [γ-32P]ATP. The EMSA result indeed indicates a stronger activation of NF-κB when Mtbhsp60 interacted with TLR4 (Fig. 11A). Blocking of NF-κB activity in the anti-TLR2 Ab-treated macrophages by specific NF-κB inhibitors like BAY 11-7082 (74, 75) as well as pyrrolidine dithiocarbamate (35, 76) could inhibit TNF-α induction in these cells (Fig. 11B) indicating a role of NF-κB downstream of TLR4 in the regulation of pro-inflammatory cytokines by Mtbhsp60.
FIGURE 11.

Mtbhsp60 targets TLR4 to activate NF-κB transcription factors. A, PMA-differentiated THP-1 macrophages were pretreated with 10 μg/ml of either anti-TLR2 mAb or anti-TLR4 mAb or isotype-matched control Ab for 60 min and then incubated with 3 μg/ml of Mtbhsp60 for another 60 min. Cells were harvested, and nuclear extracts were prepared. The DNA-binding activity of NF-κB complex was measured by EMSA. B, in another experiment, PMA-differentiated THP-1 macrophages were pretreated with 10 μg/ml of either isotype-control Ab or anti-TLR2 Ab and further incubated with 3 μg/ml of Mtbhsp60 in the absence or presence of BAY 11-7082 (10 μm) or pyrrolidine dithiocarbamate (3 μm). After 48 h, culture supernatants were harvested, and TNF-α levels were quantified by EIA. Results shown are representative of three different experiments.
DISCUSSION
Activation of macrophages via distinct pathogen recognition receptors like the TLRs results in stimulation of specific cytokines in the macrophages. Several studies have highlighted the distinct roles of TLR2 and TLR4 agonists to modulate T cell immune responses by regulating expression of anti-inflammatory and pro-inflammatory cytokines (11, 16, 19, 21, 77, 78).
In addition to their classical chaperone functions, several HSPs were implicated to play a role in modulating the innate effector functions of macrophages (22). Several microbial heat shock proteins, including M. tuberculosis, are known to activate macrophage-derived cytokine production (23–28). Among the M. tuberculosis heat shock proteins, the Mtbhsp60 (Cpn60.1, Rv3417c) is known to be a very potent stimulator of cytokine production in macrophages and is believed to be an important virulence factor for M. tuberculosis (26). Up-regulation of Rv3417c expression inside the macrophages suggests that Mtbhsp60 might offer a survival advantage to the bacteria within the host (79).
The Mtbhsp60 is known to induce both pro-inflammatory cytokines like TNF-α, IL-12, and anti-inflammatory cytokine IL-10 (26); however, the mechanisms for such opposing cytokine responses were not well understood. Previously, we found that Mtbhsp60 could differentially modulate PPD-induced IL-12 p40 production in macrophages in a TLR-dependent fashion. Blockage of Mtbhsp60-TLR4 interaction using anti-TLR4 antibody resulted in significant reduction in IL-12 p40 production, whereas blocking its interaction with TLR2 using anti-TLR2 antibody could significantly augment PPD-induced IL-12 p40 production (29). These data indicated that interaction of Mtbhsp60 with TLR2 predominantly induced anti-inflammatory signaling. Herein, we found that interaction of Mtbhsp60 with TLR2 led to predominantly anti-inflammatory IL-10 production as opposed to its interaction with TLR4 that induced very little IL-10 but triggered predominantly production of pro-inflammatory cytokine like TNF-α. Interestingly, a complete inhibition of IL-10 was observed in primary macrophages but not in PMA-differentiated THP-1 macrophages using 10 μg/ml of anti-TLR2 blocking Ab. However, a combination of both the TLR2 and TLR4 Abs at the same concentration was able to completely inhibit the production of IL-10 in these cells. This could possibly be due to the fact that THP-1 is a monocyte/macrophage tumor cell line known to respond differently when compared with the primary macrophages (80, 81). Therefore, it is possible that in the PMA-differentiated THP-1 macrophages, downstream TLR signaling pathways may not be as tightly regulated as in the primary macrophages. However, anti-TLR2 Ab was able to inhibit almost 70% of the IL-10 produced by the control group treated with isotype control Ab. This indicates a predominant role of the TLR2 receptors in triggering IL-10 production by Mtbhsp60 even in a surrogate cell line like THP-1, which was used frequently by several groups (29, 82, 83). The data obtained by using macrophages from the TLR2 as well as TLR4 knock-out mice once again support our findings that Mtbhsp60 predominantly targets the TLR2 receptors to induce IL-10 cytokine. Similarly, the anti-TLR4 Ab alone was probably not sufficient to completely block the TLR4-specific signaling cascades crucial for induction of TNF-α in THP-1 macrophages.
The TLR2 and TLR4 receptors are known to regulate the effector functions differently in response to various mycobacterial ligands by producing different levels of these counteractive anti-inflammatory and pro-inflammatory cytokines (11, 16–21). TLR2-triggered signaling cascades can swing toward either the anti-inflammatory or the pro-inflammatory response phenotype depending on the ligand (38, 84, 85). For example, in macrophages interaction of a synthetic lipopeptide Pam3CSK4 with TLR2 induced a pro-inflammatory response (86), whereas PPE18, a mycobacterial TLR2 ligand, induced production of anti-inflammatory IL-10 response (38). Interestingly, signaling through TLR4 predominantly triggers production of pro-inflammatory cytokines that favor Th1 lineage commitment (87, 88). Similar to the Mtbhsp60, other mammalian and microbial HSP60s were also found to require functional TLR4 on macrophages and dendritic cells for pro-inflammatory activity (27, 48, 89, 90).
Although in this study we found that Mtbhsp60 interacts with both the TLR2 and TLR4 receptors, at this moment it is unclear whether Mtbhsp60 preferentially interacts with any one of these receptors. It appears that it is the cellular location of Mtbhsp60 after its receptor interaction that dictates the kind of cytokine to be produced because blockage of its endocytosis (using MDC), even when it interacts with TLR2, leads to production of TNF-α just like its interaction with TLR4. Interestingly, we demonstrated earlier that Mtbhsp60 can skew the TLR2/TLR4 ratio on the macrophage surface (29) to favor a pronounced Th2-type response. Therefore, we speculate that it is the relative proportion of TLR2 and TLR4 available to interact with Mtbhsp60 on the macrophage surface that determines whether the outcome will be predominantly a Th1- or Th2-type cytokine rather than the receptor preference of Mtbhsp60 itself.
Upon interaction with TLR2, Mtbhsp60 was found to be colocalized with the early endosome-specific marker EEA1. The confocal data demonstrate that Mtbhsp60 was colocalized along with TLR2. Interestingly, TLR2-mediated endocytosis of Mtbhsp60 was found to be required for induction of IL-10. In contrast, inhibition of TLR2-mediated endocytosis by MDC resulted in an increase in the cell surface accumulation of Mtbhsp60 and severely compromised its ability to induce IL-10. Interestingly, in such a situation, we also observed an increase in the TNF-α production indicating that retention of TLR2-bound Mtbhsp60 on the cell surface is sufficient to trigger higher expression of TNF-α. However, upon interaction with TLR4, the Mtbhsp60 predominantly remained stranded on the cell surface and induced a dominant pro-inflammatory-type response characterized by higher TNF-α production. The Mtbhsp60 homologue of E. coli, Ecolihsp60, is known to induce predominantly a pro-inflammatory-type signaling (25, 55, 56). We found that Ecolihsp60 interacts with both the TLR2 and TLR4 to induce production of TNF-α. However, Ecolihsp60 has remained predominantly cell surface-bound even when it interacted with TLR2, unlike Mtbhsp60. These observations led us to speculate that the cellular localization of Mtbhsp60 post-binding to TLRs activates different signaling cascades that finally dictate the type of inflammatory response to be produced in macrophages.
Although the protein sequence of Ecolihsp60 is significantly similar to that of Mtbhsp60, biochemical features of Mtbhsp60 deviate significantly from the characteristic properties of the Ecolihsp60 (33, 91). The Mtbhsp60 exists in a lower oligomeric state as compared with its E. coli counterpart due to substitutions in some crucial interface residues required to stabilize its inter-subunit interactions, and it also lacks ATPase activity (33). We also found that at the structural level Mtbhsp60 also significantly deviates from Ecolihsp60, which may be responsible for the differential amenability of these two proteins to undergo endocytosis post-binding to TLR2. However, both these proteins are endocytosed poorly when interacted through TLR4. The exact reasons for such receptor-dependent preferences for internalization of Mtbhsp60 are not clear. Moreover, it appears from our data that the decisions made at the level of receptor-ligand interactions have important consequences in dictating the subsequent immune responses in macrophages. It is likely that interaction of Mtbhsp60 with the ectodomains of TLR2 and TLR4 can induce different structural plasticity resulting in differential localization of the protein.
Interestingly, the human and chlamydial HSP60s were found to engage both the TLR2 and TLR4 receptors to induce production of TNF-α in macrophages that required clathrin-dependent endocytosis and were dependent on MyD88, an adaptor protein that passes the post-receptor signaling cues downstream of many TLRs (48). However, IL-6 inducing ability of Helicobacter pylori HSP60 was found to be independent of TLR2, TLR4, or MyD88 (23). In contrast, we found that TLR2-mediated clathrin-dependent endocytosis was required to induce an anti-inflammatory response characterized by an increase in the p38 phosphorylation. Therefore, although the HSP60 proteins are highly conserved across different species, it appears that the mechanisms of their immunoregulatory functions vary considerably.
Emerging evidence suggests that IL-10 and TNF-α cytokines in macrophages are regulated upstream mainly by the p38 MAPK and ERK1/2 (11, 62, 63, 92) signaling cascades. Although ERK1/2 activation is mainly required for TNF-α production (63, 66), IL-10 induction is predominantly dependent on p38 MAPK signaling (60, 65). Accordingly, we have observed that internalization of Mtbhsp60 through TLR2 is associated with an increased phosphorylation of p38 MAPK, whereas engagement of this protein with TLR4 induced predominantly ERK1/2 phosphorylation. Again, blocking the TLR2-mediated endocytosis of Mtbhsp60 by MDC resulted in the enhancement of ERK1/2 phosphorylation without any significant change in the p38 MAPK phosphorylation status. Therefore, the dichotomous nature of such opposing signal transduction appears to be due to the divergent MAPK signaling triggered by the Mtbhsp60 localized in the endosome and the cell membrane. We further indicated a role of NF-κB downstream of TLR4 in the regulation of pro-inflammatory cytokines by Mtbhsp60. We observed a higher NF-κB activity when Mtbhsp60 interacted with TLR4 as compared with the TLR2. The predominant induction of NF-κB during its interaction with TLR4 could be due to the increased activation of ERK1/2 signaling as indicated elsewhere (93, 94). Although many studies have indicated that hsp60 can trigger NF-κB and pro-inflammatory responses (48), our studies for the first time indicated that specific interaction of hsp60 of M. tuberculosis with TLR4 resulted in stronger NF-κB activity leading to production of pro-inflammatory cytokines like TNF-α and IL-12 (29). Interestingly, p65 NF-κB is also known to be required for optimal IL-10 gene expression (95). However, the level of nuclear p65 NF-κB was found to be significantly low in the TLR4 Ab-treated group as compared with the group treated either with TLR2 Ab or isotype control Ab (supplemental Fig. 9) indicating that the p65 NF-κB probably does not play a dominant role in the production of IL-10 by Mtbhsp60 through its interaction with TLR2. This could be due to different TLRs utilized to induce IL-10. Saraiva et al. (95) used LPS, a TLR4 ligand, as a stimulator unlike the Mtbhsp60 used in this study, which induces IL-10 upon interaction with TLR2.
In summary, our study highlights the possible mechanisms by which Mtbhsp60 can potentially endow an anti-inflammatory milieu by undergoing TLR2-mediated endocytosis to induce production of IL-10 in the macrophages leading to a pronounced Th2 response favoring survival of M. tuberculosis inside the host. However, it can also interact with TLR4 to induce a host protective pro-inflammatory response, and its ability to increase the surface expression of TLR2 (28) can mitigate the TLR4-mediated protective effects. Our study hints to the possible mechanisms by which Mtbhsp60 may play a role in the virulence of M. tuberculosis.
Acknowledgments
We thank Dr. Nooruddin Khan for critical suggestions. We also thank Dr. K. H. Bhat and Vedprakash Dwivedi for help in animal experiments and EMSA.
This work was supported in part by grants from the Indian Council of Medical Research, Government of India Grant Imm./18/11/12/2008-ECD-1, the Department of Biotechnology, Government of India Grant BT/PR12854/BRB/10/730/2009, and a core grant from the Department of Biotechnology (to Centre for DNA Fingerprinting and Diagnostics).

This article contains supplemental Figs. 1–9.
- Th2
- T-helper 2
- Mtbhsp60
- M. tuberculosis heat shock protein 60
- Ecolihsp60
- E. coli heat shock protein 60
- MDC
- monodansylcadaverine
- EIA
- enzyme immune assay
- TLR
- Toll-like receptor, NF-κB, nuclear factor-κB
- Ab
- antibody
- PPD
- purified protein derivative
- PDB
- Protein Data Bank
- PMA
- phorbol 12-myristate 13-acetate
- HSP
- heat shock protein.
REFERENCES
- 1. Schluger N. W., Rom W. N. (1998) The host immune response to tuberculosis. Am. J. Respir. Crit. Care Med. 157, 679–691 [DOI] [PubMed] [Google Scholar]
- 2. Rook G. A., Dheda K., Zumla A. (2005) Immune responses to tuberculosis in developing countries: implications for new vaccines. Nat. Rev. Immunol. 5, 661–667 [DOI] [PubMed] [Google Scholar]
- 3. Flynn J. L., Goldstein M. M., Chan J., Triebold K. J., Pfeffer K., Lowenstein C. J., Schreiber R., Mak T. W., Bloom B. R. (1995) Tumor necrosis factor-α is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 2, 561–572 [DOI] [PubMed] [Google Scholar]
- 4. Flynn J. L. (2004) Immunology of tuberculosis and implications in vaccine development. Tuberculosis 84, 93–101 [DOI] [PubMed] [Google Scholar]
- 5. Quesniaux V. F., Jacobs M., Allie N., Grivennikov S., Nedospasov S. A., Garcia I., Olleros M. L., Shebzukhov Y., Kuprash D., Vasseur V., Rose S., Court N., Vacher R., Ryffel B. (2010) TNF in host resistance to tuberculosis infection. Curr. Dir. Autoimmun. 11, 157–179 [DOI] [PubMed] [Google Scholar]
- 6. Turner J., Gonzalez-Juarrero M., Ellis D. L., Basaraba R. J., Kipnis A., Orme I. M., Cooper A. M. (2002) In vivo IL-10 production reactivates chronic pulmonary tuberculosis in C57BL/6 mice. J. Immunol. 169, 6343–6351 [DOI] [PubMed] [Google Scholar]
- 7. Goldfeld A., Ellner J. J. (2007) Pathogenesis and management of HIV/TB co-infection in Asia. Tuberculosis 87, S26-S30 [DOI] [PubMed] [Google Scholar]
- 8. Mukhopadhyay S., Nair S., Hasnain S. E. (2007) Nitric oxide: friendly rivalry in tuberculosis. Curr. Signal. Transduct. Ther. 2, 121–128 [Google Scholar]
- 9. Rook G. A. (2007) Th2 cytokines in susceptibility to tuberculosis. Curr. Mol. Med. 7, 327–337 [DOI] [PubMed] [Google Scholar]
- 10. Higgins D. M., Sanchez-Campillo J., Rosas-Taraco A. G., Lee E. J., Orme I. M., Gonzalez-Juarrero M. (2009) Lack of IL-10 alters inflammatory and immune responses during pulmonary Mycobacterium tuberculosis infection. Tuberculosis 89, 149–157 [DOI] [PubMed] [Google Scholar]
- 11. Jo E. K., Yang C. S., Choi C. H., Harding C. V. (2007) Intracellular signalling cascades regulating innate immune responses to Mycobacteria: branching out from Toll-like receptors. Cell. Microbiol. 9, 1087–1098 [DOI] [PubMed] [Google Scholar]
- 12. Barton G. M., Medzhitov R. (2004) Toll signaling: RIPping off the TNF pathway. Nat. Immunol. 5, 472–474 [DOI] [PubMed] [Google Scholar]
- 13. Ryffel B., Fremond C., Jacobs M., Parida S., Botha T., Schnyder B., Quesniaux V. (2005) Innate immunity to mycobacterial infection in mice: critical role for toll-like receptors. Tuberculosis 85, 395–405 [DOI] [PubMed] [Google Scholar]
- 14. Takeda K., Akira S. (2005) Toll-like receptors in innate immunity. Int. Immunol. 17, 1–14 [DOI] [PubMed] [Google Scholar]
- 15. Collins H. L., Kaufmann S. H. (2001) The many faces of host responses to tuberculosis. Immunology 103, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Re F., Strominger J. L. (2004) IL-10 released by concomitant TLR2 stimulation blocks the induction of a subset of Th1 cytokines that are specifically induced by TLR4 or TLR3 in human dendritic cells. J. Immunol. 173, 7548–7555 [DOI] [PubMed] [Google Scholar]
- 17. Underhill D. M., Ozinsky A., Smith K. D., Aderem A. (1999) Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc. Natl. Acad. Sci. U.S.A. 96, 14459–14463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jones B. W., Heldwein K. A., Means T. K., Saukkonen J. J., Fenton M. J. (2001) Differential roles of Toll-like receptors in the elicitation of proinflammatory responses by macrophages. Ann. Rheum. Dis. 60, Suppl. 3, 6–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Agrawal S., Agrawal A., Doughty B., Gerwitz A., Blenis J., Van Dyke T., Pulendran B. (2003) Cutting edge: different Toll-like receptor agonists instruct dendritic cells to induce distinct Th responses via differential modulation of extracellular signal-regulated kinase-mitogen-activated protein kinase and c-Fos. J. Immunol. 171, 4984–4989 [DOI] [PubMed] [Google Scholar]
- 20. Quesniaux V., Fremond C., Jacobs M., Parida S., Nicolle D., Yeremeev V., Bihl F., Erard F., Botha T., Drennan M., Soler M. N., Le Bert M., Schnyder B., Ryffel B. (2004) Toll-like receptor pathways in the immune responses to mycobacteria. Microbes Infect. 6, 946–959 [DOI] [PubMed] [Google Scholar]
- 21. Netea M. G., Van der Meer J. W., Sutmuller R. P., Adema G. J., Kullberg B. J. (2005) From the Th1/Th2 paradigm toward a Toll-like receptor/T-helper bias. Antimicrob. Agents Chemother. 49, 3991–3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Henderson B., Henderson S. (2009) Unfolding the relationship between secreted molecular chaperones and macrophage activation states. Cell Stress Chaperones 14, 329–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gobert A. P., Bambou J. C., Werts C., Balloy V., Chignard M., Moran A. P., Ferrero R. L. (2004) Helicobacter pylori heat shock protein 60 mediates interleukin-6 production by macrophages via a toll-like receptor (TLR)-2-, TLR-4-, and myeloid differentiation factor 88-independent mechanism. J. Biol. Chem. 279, 245–250 [DOI] [PubMed] [Google Scholar]
- 24. Bulut Y., Faure E., Thomas L., Karahashi H., Michelsen K. S., Equils O., Morrison S. G., Morrison R. P., Arditi M. (2002) Chlamydial heat shock protein 60 activates macrophages and endothelial cells through Toll-like receptor 4 and MD2 in a MyD88-dependent pathway. J. Immunol. 168, 1435–1440 [DOI] [PubMed] [Google Scholar]
- 25. Tabona P., Reddi K., Khan S., Nair S. P., Crean S. J., Meghji S., Wilson M., Preuss M., Miller A. D., Poole S., Carne S., Henderson B. (1998) Homogeneous Escherichia coli chaperonin 60 induces IL-1β and IL-6 gene expression in human monocytes by a mechanism independent of protein conformation. J. Immunol. 161, 1414–1421 [PubMed] [Google Scholar]
- 26. Lewthwaite J. C., Coates A. R., Tormay P., Singh M., Mascagni P., Poole S., Roberts M., Sharp L., Henderson B. (2001) Mycobacterium tuberculosis chaperonin 60.1 is a more potent cytokine stimulator than chaperonin 60.2 (Hsp 65) and contains a CD14-binding domain. Infect. Immun. 69, 7349–7355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Habich C., Baumgart K., Kolb H., Burkart V. (2002) The receptor for heat shock protein 60 on macrophages is saturable, specific, and distinct from receptors for other heat shock proteins. J. Immunol. 168, 569–576 [DOI] [PubMed] [Google Scholar]
- 28. Bulut Y., Michelsen K. S., Hayrapetian L., Naiki Y., Spallek R., Singh M., Arditi M. (2005) Mycobacterium tuberculosis heat shock proteins use diverse toll-like receptor pathways to activate pro-inflammatory signals. J. Biol. Chem. 280, 20961–20967 [DOI] [PubMed] [Google Scholar]
- 29. Khan N., Alam K., Mande S. C., Valluri V. L., Hasnain S. E., Mukhopadhyay S. (2008) Mycobacterium tuberculosis heat shock protein 60 modulates immune response to PPD by manipulating the surface expression of TLR2 on macrophages. Cell. Microbiol. 10, 1711–1722 [DOI] [PubMed] [Google Scholar]
- 30. Moore K. W., de Waal Malefyt R., Coffman R. L., O'Garra A. (2001) Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 19, 683–765 [DOI] [PubMed] [Google Scholar]
- 31. O'Garra A., Robinson D. (2004) Development and function of T helper 1 cells. Adv. Immunol. 83, 133–162 [DOI] [PubMed] [Google Scholar]
- 32. Rahim S. S., Khan N., Boddupalli C. S., Hasnain S. E., Mukhopadhyay S. (2005) Interleukin-10 (IL-10)-mediated suppression of IL-12 production in RAW 264.7 cells also involves c-rel transcription factor. Immunology 114, 313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qamra R., Srinivas V., Mande S. C. (2004) Mycobacterium tuberculosis GroEL homologues unusually exist as lower oligomers and retain the ability to suppress aggregation of substrate proteins. J. Mol. Biol. 342, 605–617 [DOI] [PubMed] [Google Scholar]
- 34. Clark A. C., Ramanathan R., Frieden C. (1998) Purification of GroEL with low fluorescence background. Methods Enzymol. 290, 100–118 [DOI] [PubMed] [Google Scholar]
- 35. Khan N., Ghousunnissa S., Jegadeeswaran S. M., Thiagarajan D., Hasnain S. E., Mukhopadhyay S. (2007) Anti-B7–1/B7–2 antibody elicits innate-effector responses in macrophages through NF-κB-dependent pathway. Int. Immunol. 19, 477–486 [DOI] [PubMed] [Google Scholar]
- 36. Mukhopadhyay S., Mohanty M., Mangla A., George A., Bal V., Rath S., Ravindran B. (2002) Macrophage effector functions controlled by Bruton's tyrosine kinase are more crucial than the cytokine balance of T cell responses for microfilarial clearance. J. Immunol. 168, 2914–2921 [DOI] [PubMed] [Google Scholar]
- 37. Mukhopadhyay S., Srivastava V. M., Murthy P. K., Hasnain S. E. (2004) Poorer NF-κB signaling by microfilariae in macrophages from BALB/c mice affects their ability to produce cytotoxic levels of nitric oxide to kill microfilariae. FEBS Lett. 567, 275–280 [DOI] [PubMed] [Google Scholar]
- 38. Nair S., Ramaswamy P. A., Ghosh S., Joshi D. C., Pathak N., Siddiqui I., Sharma P., Hasnain S. E., Mande S. C., Mukhopadhyay S. (2009) The PPE18 of Mycobacterium tuberculosis interacts with TLR2 and activates IL-10 induction in macrophage. J. Immunol. 183, 6269–6281 [DOI] [PubMed] [Google Scholar]
- 39. Krutzik P. O., Nolan G. P. (2003) Intracellular phospho-protein staining techniques for flow cytometry: monitoring single cell signaling events. Cytometry A 55, 61–70 [DOI] [PubMed] [Google Scholar]
- 40. Martí-Renom M. A., Stuart A. C., Fiser A., Sánchez R., Melo F., Sali A. (2000) Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291–325 [DOI] [PubMed] [Google Scholar]
- 41. Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., Sali A. (2007) Comparative protein structure modeling using Modeller. Curr. Protoc. Protein Sci. Chapter 2, Unit 2.9 [DOI] [PubMed] [Google Scholar]
- 42. Bowie J. U., Lüthy R., Eisenberg D. (1991) A method to identify protein sequences that fold into a known three-dimensional structure. Science 253, 164–170 [DOI] [PubMed] [Google Scholar]
- 43. Lüthy R., Bowie J. U., Eisenberg D. (1992) Assessment of protein models with three-dimensional profiles. Nature 356, 83–85 [DOI] [PubMed] [Google Scholar]
- 44. Lindahl E., Hess B., van der Spoel D. (2001) GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 7, 306–317 [Google Scholar]
- 45. Cabo-Bilbao A., Spinelli S., Sot B., Agirre J., Mechaly A. E., Muga A., Guérin D. M. (2006) Crystal structure of the temperature-sensitive and allosteric-defective chaperonin GroELE461K. J. Struct. Biol. 155, 482–492 [DOI] [PubMed] [Google Scholar]
- 46. Guex N. (1996) Swiss-Pdb Viewer: a new fast and easy to use PDB viewer for the Macintosh. Experientia 52, A26 [Google Scholar]
- 47. Ohashi K., Burkart V., Flohé S., Kolb H. (2000) Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J. Immunol. 164, 558–561 [DOI] [PubMed] [Google Scholar]
- 48. Vabulas R. M., Ahmad-Nejad P., da Costa C., Miethke T., Kirschning C. J., Häcker H., Wagner H. (2001) Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J. Biol. Chem. 276, 31332–31339 [DOI] [PubMed] [Google Scholar]
- 49. Schmid S. L. (1997) Clathrin-coated vesicle formation and protein sorting: an integrated process. Annu. Rev. Biochem. 66, 511–548 [DOI] [PubMed] [Google Scholar]
- 50. Davies P. J., Davies D. R., Levitzki A., Maxfield F. R., Milhaud P., Willingham M. C., Pastan I. H. (1980) Transglutaminase is essential in receptor-mediated endocytosis of α2-macroglobulin and polypeptide hormones. Nature 283, 162–167 [DOI] [PubMed] [Google Scholar]
- 51. Hu Y., Henderson B., Lund P. A., Tormay P., Ahmed M. T., Gurcha S. S., Besra G. S., Coates A. R. (2008) A Mycobacterium tuberculosis mutant lacking the groEL homologue cpn60.1 is viable but fails to induce an inflammatory response in animal models of infection. Infect. Immun. 76, 1535–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kindler V., Sappino A. P., Grau G. E., Piguet P. F., Vassalli P. (1989) The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell 56, 731–740 [DOI] [PubMed] [Google Scholar]
- 53. Roach D. R., Bean A. G., Demangel C., France M. P., Briscoe H., Britton W. J. (2002) TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J. Immunol. 168, 4620–4627 [DOI] [PubMed] [Google Scholar]
- 54. Algood H. M., Lin P. L., Flynn J. L. (2005) Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin. Infect. Dis. 41, S189–S193 [DOI] [PubMed] [Google Scholar]
- 55. Retzlaff C., Yamamoto Y., Hoffman P. S., Friedman H., Klein T. W. (1994) Bacterial heat shock proteins directly induce cytokine mRNA and interleukin-1 secretion in macrophage cultures. Infect. Immun. 62, 5689–5693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Marcatili A., Cipollaro de l'Ero G., Galdiero M., Folgore A., Petrillo G. (1997) TNF-α, IL-lα, IL-6 and lCAM-l expression inhuman keratinocytes stimulated in vitro with Escherichia coli heat-shock proteins. Microbiology 143, 45–53 [DOI] [PubMed] [Google Scholar]
- 57. O'Neill L. A. (2006) How Toll-like receptors signal: what we know and what we don't know. Curr. Opin. Immunol. 18, 3–9 [DOI] [PubMed] [Google Scholar]
- 58. O'Neill L. A., Bowie A. G. (2007) The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 7, 353–364 [DOI] [PubMed] [Google Scholar]
- 59. Nakahara T., Moroi Y., Uchi H., Furue M. (2006) Differential role of MAPK signaling in human dendritic cell maturation and Th1/Th2 engagement. J. Dermatol. Sci. 42, 1–11 [DOI] [PubMed] [Google Scholar]
- 60. Blumenthal A., Ehlers S., Ernst M., Flad H. D., Reiling N. (2002) Control of mycobacterial replication in human macrophages: roles of extracellular signal-regulated kinases 1 and 2 and p38 mitogen-activated protein kinase pathways. Infect. Immun. 70, 4961–4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tse H. M., Josephy S. I., Chan E. D., Fouts D., Cooper A. M. (2002) Activation of the mitogen-activated protein kinase signaling pathway is instrumental in determining the ability of Mycobacterium avium to grow in murine macrophages. J. Immunol. 168, 825–833 [DOI] [PubMed] [Google Scholar]
- 62. Song C. H., Lee J. S., Lee S. H., Lim K., Kim H. J., Park J. K., Paik T. H., Jo E. K. (2003) Role of mitogen-activated protein kinase pathways in the production of tumor necrosis factor-α, interleukin-10, and monocyte chemotactic protein-1 by Mycobacterium tuberculosis H37Rv-infected human monocytes. J. Clin. Immunol. 23, 194–201 [DOI] [PubMed] [Google Scholar]
- 63. Yadav M., Roach S. K., Schorey J. S. (2004) Increased mitogen-activated protein kinase activity and TNF-α production associated with Mycobacterium smegmatis- but not Mycobacterium avium-infected macrophages requires prolonged stimulation of the calmodulin/calmodulin kinase and cyclic AMP/protein kinase A pathways. J. Immunol. 172, 5588–5597 [DOI] [PubMed] [Google Scholar]
- 64. Pathak S. K., Basu S., Bhattacharyya A., Pathak S., Kundu M., Basu J. (2005) Mycobacterium tuberculosis lipoarabinomannan-mediated IRAK-M induction negatively regulates Toll-like receptor-dependent interleukin-12 p40 production in macrophages. J. Biol. Chem. 280, 42794–42800 [DOI] [PubMed] [Google Scholar]
- 65. Chi H., Barry S. P., Roth R. J., Wu J. J., Jones E. A., Bennett A. M., Flavell R. A. (2006) Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc. Natl. Acad. Sci. U.S.A. 103, 2274–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Souza C. D., Evanson O. A., Weiss D. J. (2006) Mitogen-activated protein kinase p38 pathway is an important component of the anti-inflammatory response in Mycobacterium avium subsp. paratuberculosis-infected bovine monocytes. Microb. Pathog. 41, 59–66 [DOI] [PubMed] [Google Scholar]
- 67. Yang C. S., Shin D. M., Lee H. M., Son J. W., Lee S. J., Akira S., Gougerot-Pocidalo M. A., El-Benna J., Ichijo H., Jo E. K. (2008) ASK1-p38 MAPK-p47phox activation is essential for inflammatory responses during tuberculosis via TLR2-ROS signalling. Cell. Microbiol. 10, 741–754 [DOI] [PubMed] [Google Scholar]
- 68. Cavalli V., Corti M., Gruenberg J. (2001) Endocytosis and signaling cascades: a close encounter. FEBS Lett. 498, 190–196 [DOI] [PubMed] [Google Scholar]
- 69. Zwang Y., Yarden Y. (2006) p38 MAP kinase mediates stress-induced internalization of EGFR: implications for cancer chemotherapy. EMBO J. 25, 4195–4206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Baldwin A. S., Jr. (1996) The NF-κB and IκB proteins: new discoveries and insights. Annu. Rev. Immunol. 14, 649–683 [DOI] [PubMed] [Google Scholar]
- 71. Fitzgerald K. A., Rowe D. C., Barnes B. J., Caffrey D. R., Visintin A., Latz E., Monks B., Pitha P. M., Golenbock D. T. (2003) LPS-TLR4 signaling to IRF-3/7 and NF-κB involves the toll adapters TRAM and TRIF. J. Exp. Med. 198, 1043–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang G., Ghosh S. (2001) Toll-like receptor-mediated NF-κB activation: a phylogenetically conserved paradigm in innate immunity. J. Clin. Invest. 107, 13–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wagner D. H., Jr., Vaitaitis G., Sanderson R., Poulin M., Dobbs C., Haskins K. (2002) Expression of CD40 identifies a unique pathogenic T cell population in type 1 diabetes. Proc. Natl. Acad. Sci. U.S.A. 99, 3782–3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lee J., Rhee M. H., Kim E., Cho J. Y. (2012) BAY 11-7082 is a broad-spectrum inhibitor with anti-inflammatory activity against multiple targets. Mediators Inflamm. 2012, 416036–416047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mori N., Yamada Y., Ikeda S., Yamasaki Y., Tsukasaki K., Tanaka Y., Tomonaga M., Yamamoto N., Fujii M. (2002) Bay 11-7082 inhibits transcription factor NF-κB and induces apoptosis of HTLV-I-infected T-cell lines and primary adult T-cell leukemia cells. Blood 100, 1828–1834 [DOI] [PubMed] [Google Scholar]
- 76. Bhat K. H., Chaitanya C. K., Parveen N., Varman R., Ghosh S., Mukhopadhyay S. (2012) Proline-proline-glutamic acid (PPE) protein Rv1168c of Mycobacterium tuberculosis augments transcription from HIV-1 long terminal repeat promoter. J. Biol. Chem. 287, 16930–16946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hirschfeld M., Weis J. J., Toshchakov V., Salkowski C. A., Cody M. J., Ward D. C., Qureshi N., Michalek S. M., Vogel S. N. (2001) Signaling by toll-like receptor 2 and 4 agonists results in differential gene expression in murine macrophages. Infect. Immun. 69, 1477–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Qian C., Jiang X., An H., Yu Y., Guo Z., Liu S., Xu H., Cao X. (2006) TLR agonists promote ERK-mediated preferential IL-10 production of regulatory dendritic cells (diffDCs), leading to NK-cell activation. Blood 108, 2307–2315 [DOI] [PubMed] [Google Scholar]
- 79. Monahan I. M., Betts J., Banerjee D. K., Butcher P. D. (2001) Differential expression of mycobacterial proteins following phagocytosis by macrophages. Microbiology 147, 459–471 [DOI] [PubMed] [Google Scholar]
- 80. Daigneault M., Preston J. A., Marriott H. M., Whyte M. K., Dockrell D. H. (2010) The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One 5, e8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Carta S., Tassi S., Pettinati I., Delfino L., Dinarello C. A., Rubartelli A. (2011) The rate of interleukin-1β secretion in different myeloid cells varies with the extent of redox response to toll-like receptor triggering. J. Biol. Chem. 286, 27069–27080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yokota S., Minota S., Fujii N. (2006) Anti-HSP autoantibodies enhance HSP-induced pro-inflammatory cytokine production in human monocytic cells via toll-like receptors. Int. Immunol. 8, 573–580 [DOI] [PubMed] [Google Scholar]
- 83. Tsuei A. C., Martinus R. D. (2012) Metformin induced expression of Hsp60 in human THP-1 monocyte cells. Cell Stress Chaperones 17, 23–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Quesniaux V. J., Nicolle D. M., Torres D., Kremer L., Guérardel Y., Nigou J., Puzo G., Erard F., Ryffel B. (2004) Toll-like receptor 2 (TLR2)-dependent-positive and TLR2-independent-negative regulation of proinflammatory cytokines by mycobacterial lipomannans. J. Immunol. 172, 4425–4434 [DOI] [PubMed] [Google Scholar]
- 85. Doz E., Rose S., Nigou J., Gilleron M., Puzo G., Erard F., Ryffel B., Quesniaux V. F. (2007) Acylation determines the toll-like receptor (TLR)-dependent positive versus TLR2-, mannose receptor-, and SIGNR1-independent negative regulation of pro-inflammatory cytokines by mycobacterial lipomannan. J. Biol. Chem. 282, 26014–26025 [DOI] [PubMed] [Google Scholar]
- 86. Ozinsky A., Underhill D. M., Fontenot J. D., Hajjar A. M., Smith K. D., Wilson C. B., Schroeder L., Aderem A. (2000) The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc. Natl. Acad. Sci. U.S.A. 97, 13766–13771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Loures F. V., Pina A., Felonato M., Araújo E. F., Leite K. R., Calich V. L. (2010) Toll-like receptor 4 signaling leads to severe fungal infection associated with enhanced proinflammatory immunity and impaired expansion of regulatory T cells. Infect. Immun. 78, 1078–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Józefowski S., Sobota A., Kwiatkowska K. (2008) How Mycobacterium tuberculosis subverts host immune responses. BioEssays 30, 943–954 [DOI] [PubMed] [Google Scholar]
- 89. Sasu S., LaVerda D., Qureshi N., Golenbock D. T., Beasley D. (2001) Chlamydia pneumoniae and chlamydial heat shock protein 60 stimulate proliferation of human vascular smooth muscle cells via toll-like receptor 4 and p44/p42 mitogen-activated protein kinase activation. Circ. Res. 89, 244–250 [DOI] [PubMed] [Google Scholar]
- 90. Flohé S. B., Brüggemann J., Lendemans S., Nikulina M., Meierhoff G., Flohé S., Kolb H. (2003) Human heat shock protein 60 induces maturation of dendritic cells versus a Th1-promoting phenotype. J. Immunol. 170, 2340–2348 [DOI] [PubMed] [Google Scholar]
- 91. Gaston J. S. (2002) Heat shock proteins and innate immunity. Clin. Exp. Immunol. 127, 1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Reiling N., Blumenthal A., Flad H. D., Ernst M., Ehlers S. (2001) Mycobacteria-induced TNF-α and IL-10 formation by human macrophages is differentially regulated at the level of mitogen-activated protein kinase activity. J. Immunol. 167, 3339–3345 [DOI] [PubMed] [Google Scholar]
- 93. Hayashi T., Kishiwada M., Fujii K., Yuasa H., Nishioka J., Ido M., Gabazza E. C., Suzuki K. (2006) Lipopolysaccharide-induced decreased protein S expression in liver cells is mediated by MEK/ERK signaling and NFκB activation involvement of membrane-bound CD14 and toll like receptor-4. J. Thromb. Haemost. 4, 1763–1773 [DOI] [PubMed] [Google Scholar]
- 94. Jiang B., Xu S., Hou X., Pimentel D. R., Brecher P., Cohen R. A. (2004) Temporal control of NF-κB activation by ERK differentially regulates interleukin-1β-induced gene expression. J. Biol. Chem. 279, 1323–1329 [DOI] [PubMed] [Google Scholar]
- 95. Saraiva M., Christensen J. R., Tsytsykova A. V., Goldfeld A. E., Ley S. C., Kioussis D., O'Garra A. (2005) Identification of a macrophage-specific chromatin signature in the IL-10 locus. J. Immunol. 175, 1041–1046 [DOI] [PubMed] [Google Scholar]