

Abstract
Background
Qualitative alterations of lipoproteins underlie the high incidence of atherosclerosis in diabetes. The objective of this study was to assess the impact of low-density lipoprotein (LDL) subfraction phenotype on the qualitative characteristics of LDL and high-density lipoprotein (HDL) in patients with type 2 diabetes.
Methods
One hundred twenty two patients with type 2 diabetes in poor glycemic control and 54 healthy subjects were included in the study. Patients were classified according to their LDL subfraction phenotype. Seventy-seven patients presented phenotype A whereas 45 had phenotype B. All control subjects showed phenotype A. Several forms of modified LDL, HDL composition and the activity and distribution of lipoprotein-associated phospholipase A2 (Lp-PLA2) were analyzed.
Results
Oxidized LDL, glycated LDL and electronegative LDL were increased in both groups of patients compared with the control group. Patients with phenotype B had increased oxidized LDL and glycated LDL concentration than patients with phenotype A. HDL composition was abnormal in patients with diabetes, being these abnormalities more marked in patients with phenotype B. Total Lp-PLA2 activity was higher in phenotype B than in phenotype A or in control subjects. The distribution of Lp-PLA2 between HDL and apoB-containing lipoproteins differed in patients with phenotype A and phenotype B, with higher activity associated to apoB-containing lipoproteins in the latter.
Conclusions
The presence of LDL subfraction phenotype B is associated with increased oxidized LDL, glycated LDL and Lp-PLA2 activity associated to apoB-containing lipoproteins, as well as with abnormal HDL composition.
Keywords: Low-density lipoprotein, High-density lipoprotein, Lipoprotein-associated phospholipase A2, Modified LDL, Type 2 diabetes, LDL subfraction phenotype
Background
Atherosclerosis-related pathologies are the main cause of mortality and morbidity in type 2 diabetes. Plasma lipid profile, including hypertriglyceridemia, low high-density lipoprotein (HDL) cholesterol levels and hyperapolipoprotein B, is frequently abnormal in these subjects and is a cause of the high prevalence of macrovascular and microvascular complications [1,2]. However, these quantitative abnormalities per se cannot explain all the increase in cardiovascular risk (CVR) in diabetes, and it is currently accepted that alterations in the qualitative properties of low-density lipoprotein (LDL) and HDL underlie the development of atherosclerosis [1]. The concurrence of hypertriglyceridemia and low HDL cholesterol usually promote the formation of small dense LDL (sdLDL) particles. This set of features, known as diabetic dyslipidemia, is partly for environmental reasons but also has a significant genetic component [3]. The predominance of sdLDL is known as LDL subfraction phenotype B, in contrast to phenotype A, which is characterized by large buoyant LDL particles. Phenotype A is predominant in normolipemic subjects with low CVR, whereas phenotype B is associated with pathological situations with increased CVR, including type 2 diabetes [4].
Other pathological features, such as hyperglycemia, lipoperoxidation and systemic inflammation are exacerbated in diabetes. These processes alter the lipoprotein function [5], favoring the formation of modified forms of LDL, such as glycosylated LDL (glLDL), oxidized LDL (oxLDL) and electronegative LDL (LDL(−)). A common feature shared by glLDL and oxLDL is the increased negative electric charge. LDL(−), therefore, includes oxLDL and glLDL, but a major fraction of LDL(−) particles is generated by alternative mechanisms, such as lipolysis and non-esterified fatty acid (NEFA)-loading [6]. This heterogeneity adds value to the measurement of LDL(−), which can be considered as a pool of modified LDL particles in plasma. The proportion of oxLDL, glLDL and LDL(−) is increased in subjects with high CVR, including diabetes [7]. These modified LDLs have a high content of lipid metabolites with high inflammatory potential, including lysophospholipids, whose concentration is increased in diabetes [8].
Diabetes also alters normal HDL function. The antiatherogenic properties of HDL, including its key role in reverse cholesterol transport and its antioxidant properties, are disturbed in these patients [9]. Both functions are modulated by changes in the relative composition of lipids and proteins in HDL, and also by enzymatic activities associated to HDL, such as paraoxonase 1 (PON1) and lipoprotein-associated phospholipase A2 ((Lp-PLA2), also known as platelet-activating factor acetylhydrolase). PON1 is exclusively bound to HDL and it has been suggested that it has an antioxidant function by deactivating lipid peroxides [10]. In contrast to PON1, Lp-PLA2 can associate with HDL, LDL and VLDL. Controversy exists as to whether Lp-PLA2 has a pro- or anti-inflammatory action. It has been recently suggested that the relative distribution of Lp-PLA2 between LDL and HDL determines its pro- or anti-inflammatory action. According to this assumption, Lp-PLA2 in HDL is anti-inflammatory whereas Lp-PLA2 associated to apoB-containing lipoproteins is pro-inflammatory [11]. For this reason, the relative distribution of Lp-PLA2, rather than its total activity, could be a better CVR marker.
In vitro studies have shown that sdLDL is prone to modification and it is also known that HDL from patients with type 2 diabetes has an impaired function. However, to our knowledge, no previous study has compared diabetic patients with phenotype A and phenotype B. This would allow specifically assessing the impact of LDL subfraction phenotype on the distribution of Lp-PLA2 and other qualitative characteristics of HDL and LDL. The aim of the current work was to quantify modified forms of LDL, to study the composition of HDL and to determine the distribution of Lp-PLA2 in patients with poorly-controlled type 2 diabetes who were classified according to their LDL subfraction phenotype.
Methods
Study population
One hundred twenty two consecutive poor controlled type 2 diabetes subjects who attended the outpatient diabetic clinic during 2007–2008 to optimize glycemic control were recruited. Previous hypoglycemic treatment consisted of diet (22%), oral agents (21.1%), insulin plus oral agents (29.4%) and insulin (27.5%). Regarding hypolipidemic drugs, 25.4% of patients were treated with statins or statin plus ezetimibe, 4.1% with fibrate and 2.5% with statin plus fibrate. Patients with acute or chronic infections, active inflammatory diseases or treatment with anti-inflammatory drugs were excluded. High sensibility C-reactive protein (hsCRP, Roche Diagnostics) was routinely measured and subjects with values higher than 20 mg/L were excluded from the study. Hypertension was defined as subjects with a systolic blood pressure of ≥140 mm Hg, with a diastolic blood pressure of ≥90 mm Hg, or those who were receiving antihypertensive therapy at the time of examination. Coronary Heart Disease (CHD) was defined as documented diagnosis of CHD, self-reported positive history of CHD or electrocardiogram (ECG) positive for CHD. Fifty four normoglycemic and normolipemic healthy subjects were included as control group. The study was conducted in accordance with the principles of the Declaration of Helsinki and was approved by the Hospital Ethics Committee. All patients and control subjects gave informed consent.
Laboratory determinations
Lipid profile, modified LDLs, HDL composition and Lp-PLA2 activity distribution were determined from plasma obtained in EDTA-containing Vacutainer tubes. PON1 activity was determined from sera obtained in non-additive Vacutainer tubes. HbA1c was measured by ion-exchange high-performance liquid chromatography (HPLC; Variant, Bio-Rad).
Lipid profile included total cholesterol (Roche) and triglyceride (Roche), very-low density lipoprotein (VLDL), LDL and HDL cholesterol, total non-esterified fatty acids (NEFA) (Wako), apoB (Roche), apoA-I (Roche) and apoA-II (Kamiya Biomedical). Cholesterol of lipoprotein fractions was measured using a direct method to quantify HDL cholesterol (HDL-C plus, Roche) or by ultracentrifugation when the triglyceride concentration was higher than 3 mmol/L, according to NCEP recommendations [12]. All determinations were performed in a Hitachi 917 autoanalyzer.
LDL size was determined by non-denaturing polyacrylamide gradient (2–16%) gel electrophoresis (GGE), as described [13]. LDL subfraction phenotype B was defined by a predominant LDL diameter lower than 25.5 nm, whereas phenotype A subjects had a LDL diameter higher than 25.5 nm.
oxLDL (Mercodia) and glLDL (Exocell/Glycacor) were quantified by commercial ELISA. For LDL(−) quantification, total LDL was previously isolated by sequential ultracentrifugation (density range 1.019-1.050 g/mL). LDL(−) proportion was determined from total LDL by anion-exchange chromatography in a MonoQ HR 5/5 column (GE Healthcare) using a NaCl stepwise gradient [14].
HDL was isolated by sequential ultracentrifugation (density range 1.063-1.210 g/mL), using KBr gradients. All steps were performed at 4°C and all KBr solutions contained 1 mmol/L EDTA and 2 μmol/L BHT to prevent lipoperoxidation during isolation procedures. HDL composition was determined by measuring the content of cholesterol (Roche), triglyceride (Roche), phospholipid (Wako), NEFA (Wako), apoA-I (Roche) and apoA-II (Kamiya), in a Hitachi 917 autoanalyzer.
PON-1 activity in serum was measured using phenylacetate as substrate, as described [15]. Lp-PLA2 activity was measured using 2-thio-PAF (Cayman) as substrate, according to the manufacturer’s instructions [16]. To determine the distribution of Lp-PLA2 between lipoprotein fractions, apoB-containing lipoproteins were precipitated from serum using dextran sulfate, as described [17]. Briefly, 200 μL of serum were mixed with 50 μL of dextran sulfate reagent, incubated for 5 min at room temperature, and centrifuged for 10 min at 10,000 g. The supernatant (depleted of apoB-containing lipoproteins) was collected and assayed for HDL-associated Lp-PLA2 activity.
Statistical analysis
Statistical analysis was performed using SPSS 19 (SPSS Inc). Before statistical analysis, normal distribution and homogeneity of the variances were tested using Kolmogorov-Smirnov and Levène tests, respectively. Data that were not normally distributed were logarithmically transformed before analysis. Analysis of variables was performed by one-way analysis of variance (ANOVA). ANCOVA test was used to assess if the differences seen in ANOVA persisted after adjustment for triglycerides and HbA1c. Differences between two groups were compared using the Student’s T test for unpaired data (variables with normal distribution) or the Mann–Whitney U test (variables with non-normal distribution). Correlations between parameters were analyzed using the Pearson R test for variables with normal distribution and the Spearman test for variables with non-normal distribution. Data are expressed as mean ± SD. P < 0.05 was considered significant.
Results
Table 1 shows anthropometrics, clinical characteristics and lipid profile of patients and controls. One third (n = 45) of patients with type 2 diabetes presented phenotype B (LDL size < 25.5 nm), in contrast to the control group in which all subjects (n = 54) had phenotype A. Table 1 shows the lipid profile of patients with type 2 diabetes and control subjects. Both groups of patients had higher plasma levels of triglyceride, VLDL cholesterol, apoB and NEFA and lower concentration of HDL cholesterol and apoA-I than control subjects. The lipid profile was worse in patients with phenotype B. The differences observed in the two groups of patients compared to controls were larger in phenotype B patients, who also presented higher total cholesterol and LDL cholesterol than controls. The differences between phenotype A and phenotype B patients included higher total cholesterol, triglyceride, VLDLc, LDLc, apoB and lower HDLc and apoA-I in patients with phenotype B than in patients with phenotype A (Table 1).
Table 1.
Anthropometrics, clinical characteristics and lipid profile of patients with type 2 diabetes and control subjects
| Type 2 diabetes phenotype A (n = 77) | Type 2 diabetes phenotype B (n = 45) |
Control group |
ANOVA |
|
|---|---|---|---|---|
| (n = 54) | p-value | |||
| Age (years) |
60 ± 11 |
56 ± 11 a |
56 ± 14 a |
0.108 |
| Male (%) |
65.9 |
64.6 |
57.4 a, b |
<0.001 |
| BMI (kg/m2) |
29.6 ± 6.2 |
31.1 ± 4 a |
25.9 ± 3.6 a, b |
<0.001 |
| Waist (cm) |
104 ± 15 |
108 ± 13 a |
92 ± 11 a, b |
<0.001 |
| Diabetes duration (years) |
12 ± 11 |
9 ± 8 a |
- |
|
| Insulin use (%) |
58 |
52 a |
- |
|
| Lipid lowering drugs (%) |
30 |
28 |
- |
|
| HbA1c (%) |
9.1 ± 2.1 |
9.6 ± 2.3 a |
5.3 ± 0.6 a, b |
<0.001 |
| Hypertension (%) 1 |
69.4 |
68.2 |
- |
|
| Retinopathy (%) |
29.6 |
29.5 |
- |
|
| Albuminuria (%) |
24 |
29.6 a |
- |
|
| Coronary heart disease (%) 2 |
5.6 |
15.9 a |
- |
|
| Total cholesterol (mmol/L) |
4.78 ± 1.09 |
5.64 ± 1.53 a |
5.00 ± 0.79 b |
0.004 |
| Triglycerides (mmol/L) |
1.46 ± 0.79 |
3.89 ± 3.30 a |
0.87 ± 0.37 a,b |
<0.001 |
| VLDLc (mmol/L) |
0.65 ± 0.30 |
1.43 ± 1.12 a |
0.40 ± 0.17 a,b |
<0.001 |
| LDLc (mmol/L) |
2.86 ± 0.95 |
3.10 ± 0.96 a |
2.94 ± 0.66 b |
0.281 |
| HDLc (mmol/L) |
1.28 ± 0.33 |
1.02 ± 0.27 a |
1.64 ± 0.37 a,b |
<0.001 |
| NEFA (mmol/L) |
0.64 ± 0.29 |
0.90 ± 0.48 |
0.41 ± 0.19 a,b |
<0.001 |
| apoB (g/L) |
0.93 ± 0.32 |
1.09 ± 0.28 a |
0.80 ± 0.19 a,b |
<0.001 |
| apoA-I (g/L) |
1.46 ± 0.24 |
1.37 ± 0.25 a |
1.57 ± 0.31 a,b |
0.001 |
| apoA-II (g/L) |
0.33 ± 0.07 |
0.34 ± 0.06 |
0.35 ± 0.05 |
0.081 |
| LDL size (nm) | 26.0 ± 0.4 | 25.0 ± 0.4 a | 26.3 ± 0.1 b | <0.001 |
a, P < 0.05 vs phenotype A patients; b, P < 0.05 vs phenotype B patients. 1 Hypertension was defined as a systolic blood pressure of ≥140 mm Hg, a diastolic blood pressure of ≥90 mm Hg or those who were receiving antihypertensive therapy at the time of examination. 2 Coronary Heart Disease (CHD) was defined as documented diagnosis of CHD, self-reported positive history of CHD or ECG positive for CHD.
Regarding modified LDLs, both groups of patients had higher plasma concentration of oxLDL, glLDL and LDL(−) than control subjects (Table 2). oxLDL and glLDL concentrations were higher in patients with phenotype B than in patients with phenotype A. In contrast, the proportion of LDL(−) was similar between both groups of patients.
Table 2.
Modified LDLs, HDL composition and Lp-PLA2 activity in patients with type 2 diabetes and control subjects
| Type 2 diabetes phenotype A (n = 77) | Type 2 diabetes phenotype B (n = 45) |
Control group |
ANOVA |
|
|---|---|---|---|---|
| (n = 54) | p-value | |||
| Modified LDL | ||||
| Oxidized LDL (U/L) |
61.4 ± 20.0 |
78.6 ± 26.7 a |
51.2 ± 19.3 a,b |
0.005 |
| Glycated LDL (mg/dL) |
2.17 ± 0.94 |
2.77 ± 1.41 a |
1.85 ± 0.73 a,b |
0.030 |
| LDL(−) (%) |
7.6 ± 3.5 |
7.7 ± 3.1 |
6.1 ± 2.0 a,b |
0.017 |
| HDL composition | ||||
| Cholesterol (%)1 |
17.1 ± 2.3 |
15.6 ± 3.0 a |
17.3 ± 2.3 b |
0.204 |
| Trigycerides (%) |
4.3 ± 1.7 |
6.7 ± 1.9 a |
3.1 ± 1.0 a,b |
<0.001 |
| Phospholipids (%) |
33.5 ± 4.3 |
31.3 ± 3.3 a |
32.8 ± 3.7 |
0.076 |
| apoA-I (%) |
31.9 ± 5.8 |
32.8 ± 6.3 |
34.3 ± 5.7 a,b |
0.193 |
| apoA-II (%) |
13.2 ± 3.4 |
13.6 ± 2.6 |
12.5 ± 2.9 a,b |
0.265 |
| NEFA(mol/mol apoA-I) |
1.1 ± 0.6 |
1.0 ± 0.6 |
1.0 ± 0.9 |
0.907 |
| Ratio lipid/protein |
1.20 ± 0.28 |
1.19 ± 0.27 |
1.16 ± 0.27 a,b |
0.539 |
| Ratio A-I/A-II |
2.61 ± 0.92 |
2.54 ± 0.81 |
2.98 ± 1.12 a,b |
0.112 |
| Ratio chol/trigl |
4.65 ± 2.19 |
2.57 ± 0.91 |
6.32 ± 2.68 a,b |
<0.001 |
| PON1 activity |
78.5 ± 25.5 |
82.6 ± 25.5 |
82.5 ± 23.1 |
0.152 |
| (μmol/min/mL) | ||||
| Lp-PLA2 activity(μmol/min/mL) | ||||
| Total Lp-PLA2 |
20.1 ± 6.6 |
23.0 ± 7.2 a |
19.1 ± 6.2 b |
0.627 |
| HDL-Lp-PLA2 |
6.8 ± 2.7 |
7.1 ± 3.5 |
6.2 ± 2.6 |
0.995 |
| apoB-associated-Lp-PLA2 |
13.3 ± 4.8 |
15.9 ± 4.6 a |
12.9 ± 3.3 b |
0.539 |
| % HDL-Lp-PLA2 |
33.9 ± 10.5 |
30.9 ± 11.4 a |
32.4 ± 10.6 |
0.510 |
| % apoB-associated-Lp-PLA2 | 66.1 ± 19.4 | 69.1 ± 21.2 a | 67.6 ± 19.8 | 0.510 |
a, P < 0.05 vs phenotype A patients; b, P < 0.05 vs phenotype B patients.
1 Composition is expressed as the percentage of the mass of cholesterol, triglycerides, phospholipids, apoA-I and apoA-II.
The composition analysis of isolated HDL particles showed that both groups of patients had an abnormal composition compared to control samples (Table 2), with some of these alterations being stronger in patients with phenotype B. Differences between patients with phenotype B and controls included a lower content of cholesterol and apoA-I, and a higher content of triglycerides and apoA-II. Similar differences were observed when controls were compared with patients with phenotype A, except that cholesterol content was similar in both groups. As a consequence, the apoA-I/apoA-II ratio was lower and the lipid/protein ratio was higher in HDL from both groups of patients than in control subjects. Both ratios were similar in patients with phenotype A and phenotype B. These observations suggest that HDL in both groups of patients with diabetes has lower density and larger size than HDL in control subjects. Regarding PON1 activity, no difference was observed.
Total and apoB-associated Lp-PLA2 activity was higher in patients with phenotype B than in control subjects and in patients with phenotype A, and was similar in the two latter groups (Table 2, Figure 1A). No statistical difference was observed in the HDL-associated Lp-PLA2 activity. Consequently, the distribution of Lp-PLA2 between HDL and apoB-containing lipoproteins (Table 2, Figure 1B) was different between patients with phenotype A and those with phenotype B. The latter had lower proportion of Lp-PLA2 in HDL and higher proportion in apoB-containing lipoproteins than in patients with phenotype A.
Figure 1.
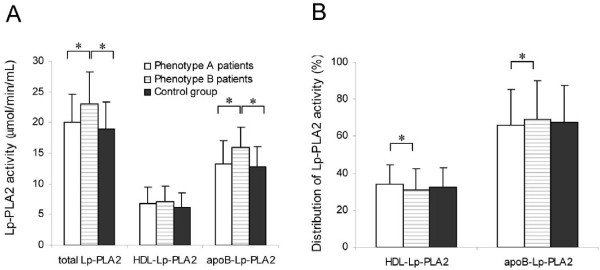
Lp-PLA2 activity. Enzymatic activity (A) and relative distribution between HDL and apoB-containing lipoproteins (B) of Lp-PLA2 in control subjects and patients with type 2 diabetes classified according their LDL subfraction phenotype. * indicates P < 0.05.
Covariance analysis (ANCOVA) showed that the differences seen in apoB and glLDL disappeared after adjusting for TG. Differences observed in LDL(−) and oxLDL lost their significance after adjusting for HbA1c and TG (data not shown). These associations are expected and underline the known relation of LDL(−) and oxLDL with the presence of diabetes and hypertriglyceridemia, and of apoB and glLDL with hypertriglyceridemia. Table 3 shows the correlation analysis of LDL size with lipid profile, modified LDLs, HDL composition and Lp-PLA2 activity. In diabetic patients, significant correlations with the parameters of lipid profile were similar to those previously reported: LDL size correlated negatively with triglycerides, cholesterol, VLDLc, apoB and NEFA, and positively with HDLc and apoA-I. LDL size also correlated negatively with oxLDL and glLDL. Regarding HDL composition, LDL size correlated positively with the content of phospholipids, cholesterol and Lp-PLA2, and negatively with the content of triglycerides and apoA-I. The correlation analysis of the control group showed some differences compared to diabetic patients. Most of correlations observed between LDL size and lipid profile were similar to that in DM2 patients, except the positive correlation with HDLc and the negative correlation with plasma NEFA, which were lost in the control group. The negative correlation with glLDL as well as the correlations with some HDL components also were lost in control subjects. These observations suggest that the influence of LDL size on HDL composition and glLDL is stronger in diabetic patients than in control subjects.
Table 3.
Correlation analysis of LDL size with lipid profile, modified LDLs, HDL composition and Lp-PLA2 activity in patients with type 2 diabetes and control subjects
| DM2 patients | Control subjects | |||
|---|---|---|---|---|
| |
R |
P |
R |
P |
| HbA1c |
−0.160 |
0.071 |
−0.103 |
0.601 |
| Insulin |
−0.076 |
0.418 |
0.008 |
0.937 |
| Total cholesterol |
−0.185 |
0.035 |
−0.326 |
0.016 |
| Triglycerides |
−0.553 |
< 0.001 |
−0.427 |
0.001 |
| VLDLc |
−0.513 |
< 0.001 |
−0.423 |
0.001 |
| LDLc |
−0.152 |
0.083 |
−0.333 |
0.014 |
| HDLc |
0.407 |
< 0.001 |
0.096 |
0.491 |
| NEFA |
−0.226 |
0.004 |
−0.042 |
0.765 |
| apoB |
−0.246 |
0.005 |
−0.458 |
< 0.001 |
| apoA-I |
0.264 |
0.002 |
−0.033 |
0.811 |
| apoA-II |
−0.068 |
0.472 |
−0.099 |
0.476 |
| Oxidized LDL |
−0.390 |
< 0.001 |
−0.472 |
< 0.001 |
| Glycated LDL |
−0.273 |
0.003 |
−0.151 |
0.307 |
| LDL(-) |
−0.020 |
0.828 |
0.030 |
0.849 |
| Cholesterol in HDL |
0.321 |
0.002 |
0.208 |
0.244 |
| Trigycerides in HDL |
−0.398 |
< 0.001 |
−0.150 |
0.405 |
| Phospholipids in HDL |
0.437 |
< 0.001 |
0.067 |
0.712 |
| apoA-I in HDL |
−0.205 |
0.047 |
0.086 |
0.635 |
| apoA-II in HDL |
−0.169 |
0.103 |
−0.069 |
0.739 |
| NEFA in HDL |
−0.010 |
0.925 |
0.086 |
0.635 |
| PON1 |
0.016 |
0.867 |
0.024 |
0.872 |
| Total Lp-PLA2 activity |
−0.123 |
0.172 |
0.025 |
0.862 |
| HDL-Lp-PLA2 activity |
0.030 |
0.745 |
0.061 |
0.680 |
| % of HDL-Lp-PLA2 | 0.191 | 0.034 | 0.064 | 0.662 |
Bold type indicates the loss of statistical significance in the control group compared to diabetic patients.
Discussion
Several authors have reported differences in the qualitative characteristics of HDL and LDL between diabetic and healthy subjects. However, most of these studies analyzed separately LDL and HDL and, despite the high prevalence of LDL phenotype B in type 2 diabetes, these characteristics have not been usually compared classifying diabetic subjects according to their LDL subfraction phenotype. Regarding Lp-PLA2, its distribution has been analyzed comparing LDL phenotypes only in one study [18], but not in patients with diabetes. The novelty of our study is to analyze simultaneously oxLDL, glLDL, LDL(−), HDL composition and distribution of Lp-PLA2 in patients with type 2 diabetes classified according to LDL phenotype.
Results in the current study concur with previous findings reporting that oxLDL, glLDL and LDL(−) are increased in diabetes [7,19,20]. Our data show that oxLDL and glLDL are increased in patients with phenotype A compared to control subjects, but this situation is more marked in patients with phenotype B. The high concentration of oxLDL and glLDL observed in patients with phenotype B could be attributed to several mechanisms. It is known that sdLDL has an impaired plasma clearance, which could lead to increased residence time in blood and favor further modifications. This could be related to the high susceptibility to oxidation and to non-enzymatic glycosylation that has been demonstrated both “in vitro” and “in vivo” in sdLDL particles [5]. In this context, it is worthy mentioning the work by Younis et al., who recently reported that sdLDL concentration is an even stronger determinant of LDL glycation than hyperglycemia [21]. However, differences in oxLDL and glLDL between patients with phenotype A and control subjects still remained significant. Therefore, factors other than LDL size could account for these differences. First, hyperglycemia and increased oxidative stress, which are hallmarks of diabetes, affect circulating LDL, even in patients with phenotype A. Another possible explanation is related to the altered composition of HDL observed in both groups of patients with type 2 diabetes. Our results indicate that HDL composition was abnormal in both groups of patients, although alterations were stronger in phenotype B patients. These differences included apoA-I/apoA-II and lipid/protein ratios. A decrease of apoA-I in HDL with a concomitant increase of apoA-II produces particles with lower antioxidant capacity [15]. Likewise, higher lipid/protein ratio suggests an impairment of the antioxidant properties of HDL, since the HDL3 subfraction (with lower lipid/protein ratio) has a stronger antioxidant capacity than the HDL2 subfraction (with higher lipid/protein ratio) [22].
The alterations in the composition of HDL from patients with diabetes also point to a decreased ability to promote reverse cholesterol transport. An increased triglyceride and decreased cholesterol content in HDL has been related with impaired cholesterol efflux from adipose and hepatic cells [23]. This alteration was present in HDL isolated from our patients and could be indicative of such impairment. This would be pronounced in diabetic subjects with phenotype B in which the ratio cholesterol/triglyceride in HDL is 2.5-fold lower than in control subjects.
Although the concentration of oxLDL and glLDL was increased in patients with phenotype B, the relative content of LDL(−) was not affected by the presence of sdLDL particles, because there was no difference between phenotype A and phenotype B patients. This observation suggests that although oxidation and glycosylation could be partially involved in the generation of LDL(−), these would not be the major mechanisms in diabetes. Other mechanism involved in the formation of LDL(−) in diabetes is NEFA-loading. [14]. As a consequence, the increased plasma level of NEFA is probably a major cause of increased LDL(−) in diabetes.
The role of Lp-PLA2 in atherogenesis is controversial. Lp-PLA2 is thought to play a role in the prevention of oxidative modifications [24], but it has been positively associated to coronary heart disease. Most epidemiological studies have found a relationship between Lp-PLA2 and coronary events or ischemic stroke [25]. However, Lp-PLA2 shows a strong association with total and LDL cholesterol levels, and there is no definitive agreement regarding whether this biomarker is independent from LDL concentration [26]. Few studies have been performed in patients with type 2 diabetes and results are controversial. Nelson et al. recently reported that Lp-PLA2 predicts future risk of incident type 2 diabetes [27], but the Rotterdam Study did not find an association between the presence of diabetes and total Lp-PLA2 activity [28]. Regarding the association of Lp-PLA2 with cardiovascular disease, Hatoum et al. described that Lp-PLA2 activity was associated with incident coronary heart disease in patients with type 2 diabetes [29], but Nelson et al. reported that the Lp-PLA2 activity did not explain the excess of cardiovascular risk in diabetes [30]. To further complicate the matter, Kizer et al. found divergent associations between Lp-PLA2 activity and Lp-PLA2 mass and the risk of cardiovascular disease in a population with high prevalence of diabetes [31]. These authors suggest that a possible explanation for these paradoxical findings could be related to the distribution of Lp-PLA2 among lipoprotein classes. However, only three studies have previously analyzed the distribution of Lp-PLA2 between lipoproteins in diabetes. Kujiraoka et al. reported that, although no difference was observed in total Lp-PLA2 activity between patients with diabetes and control subjects, Lp-PLA2 distribution was altered in the former, with a lower proportion bound to HDL [32]. In a recent study, Mitsutake et al. obtained similar results in diabetic patients and also found that Lp-PLA2 in HDL was lower in subjects with increased coronary artery calcification [33]. Finally, Onat et al., which measured Lp-PLA2 mass, agreed with these authors and also found decreased content of this enzyme in HDL from diabetic patients [34]. However, none of these studies took into account the presence of phenotype B. Our data indicate that increased total Lp-PLA2 activity and abnormal distribution of Lp-PLA2 is strongly dependent on the presence of LDL subfraction phenotype B. The higher content of Lp-PLA2 in apoB-containing lipoproteins from patients with phenotype B is probably due to the higher affinity of this enzyme for the binding to sdLDL particles than to large, buoyant LDL particles [18]. The observation that Lp-PLA2 has a lower content in HDL from patients with phenotype B than from those with phenotype A concurs with the concept that this enzyme has an antioxidant role when is bound to HDL [35]. This could account for the increased concentration of oxLDL in diabetic patients with phenotype B.
Conclusions
Our data agree with previous reports of alterations in the qualitative characteristics of LDL and HDL in patients with type 2 diabetes and demonstrate that some, but not all, of these abnormalities, are closely related to the presence of LDL subfraction phenotype B. Specifically, a high concentration of oxLDL and glLDL and a higher content of Lp-PLA2 in apoB-containing lipoproteins are favored by the presence of sdLDL particles. In addition, alterations observed in the composition of HDL from patients with phenotype B were stronger than in patients with phenotype A. The relevance of these results resides in the high prevalence of phenotype B among patients with poorly-controlled type 2 diabetes. Therapies focused on changing the LDL subfraction phenotype, such as fibrates [36] or glycemic optimization [37], would improve some qualitative pro-atherogenic characteristics of LDL and help to lower the increase in CVR present in type 2 diabetes.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
J.L.S-Q. conceived of the study, wrote the manuscript and researched data. I.V. selected patients and reviewed the manuscript. E.J-F. researched data and reviewed the manuscript. J.S-H performed statistical analysis and reviewed the manuscript. R.B. researched data. F.B-V participated in the design of the study, contributed to the discussion and reviewed/edited the manuscript. J.O-L. participated in the design of the study, contributed to the discussion and reviewed/edited the manuscript. A.P. conceived of the study, selected patients and wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Jose Luis Sánchez-Quesada, Email: jsanchezq@santpau.cat.
Irene Vinagre, Email: ivinagre@clinic.es.
Elena De Juan-Franco, Email: elenitadejuan@yahoo.es.
Juan Sánchez-Hernández, Email: jsanchezh@santpau.cat.
Rosa Bonet-Marques, Email: rbonetm@santpau.cat.
Francisco Blanco-Vaca, Email: fblancova@santpau.cat.
Jordi Ordóñez-Llanos, Email: jordonez@santpau.cat.
Antonio Pérez, Email: aperez@santpau.cat.
Acknowledgements
This work was supported by grants from Spanish Ministry of Health (ISCIII/FIS) PI05/0500, PI05/2099 and PI10/00265. JLS-Q is supported by ISCIII/FIS CP06/0220. JLS-Q, FB-V and JO-L are members of the Quality Research Group 2009-SGR-1205 from Generalitat de Catalunya. J.S-H, F.B-V and A.P. are members of CIBER de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM) from ISCIII. The authors are grateful to Carolyn Newey (IIB Sant Pau) for editorial assistance.
References
- Witztum JL. Role of modified lipoproteins in diabetic macroangiopathy. Diabetes. 1997;46(Suppl 2):S112–S114. doi: 10.2337/diab.46.2.s112. [DOI] [PubMed] [Google Scholar]
- Toth PP, Simko RJ, Palli SR, Koselleck D, Quimbo RA, Cziraky MJ. The impact of serum lipids on risk for microangiopathy in patients with type 2 diabetes mellitus. Cardiovasc Diabetol. 2012;11:109. doi: 10.1186/1475-2840-11-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollin TI, Isakova T, Jablonski KA, De Bakker PI, Taylor A, McAteer J, Pan Q, Horton ES, Delahanty LM, Altshuler D. et al. Genetic modulation of lipid profiles following lifestyle modification or metformin treatment: the Diabetes Prevention Program. PLoS Genet. 2012;8(8):e1002895. doi: 10.1371/journal.pgen.1002895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss RM. Lipids and lipoproteins in patients with type 2 diabetes. Diabetes Care. 2004;27(6):1496–1504. doi: 10.2337/diacare.27.6.1496. [DOI] [PubMed] [Google Scholar]
- Younis N, Sharma R, Soran H, Charlton-Menys V, Elseweidy M, Durrington PN. Glycation as an atherogenic modification of LDL. Curr Opin Lipidol. 2008;19(4):378–384. doi: 10.1097/MOL.0b013e328306a057. [DOI] [PubMed] [Google Scholar]
- Sanchez-Quesada JL, Benitez S, Ordonez-Llanos J. Electronegative low-density lipoprotein. Curr Opin Lipidol. 2004;15(3):329–335. doi: 10.1097/00041433-200406000-00014. [DOI] [PubMed] [Google Scholar]
- Sanchez-Quesada JL, Perez A, Caixas A, Rigla M, Payes A, Benitez S, Ordonez-Llanos J. Effect of glycemic optimization on electronegative low-density lipoprotein in diabetes: relation to nonenzymatic glycosylation and oxidative modification. J Clin Endocrinol Metab. 2001;86(7):3243–3249. doi: 10.1210/jc.86.7.3243. [DOI] [PubMed] [Google Scholar]
- Ha CY, Kim JY, Paik JK, Kim OY, Paik YH, Lee EJ, Lee JH. The association of specific metabolites of lipid metabolism with markers of oxidative stress, inflammation and arterial stiffness in men with newly diagnosed type 2 diabetes. Clin Endocrinol (Oxf) 2012;76(5):674–682. doi: 10.1111/j.1365-2265.2011.04244.x. [DOI] [PubMed] [Google Scholar]
- Kontush A, Chapman MJ. Antiatherogenic function of HDL particle subpopulations: focus on antioxidative activities. Curr Opin Lipidol. 2010;21(4):312–318. doi: 10.1097/MOL.0b013e32833bcdc1. [DOI] [PubMed] [Google Scholar]
- Mackness MI, Durrington PN, Mackness B. The role of paraoxonase 1 activity in cardiovascular disease: potential for therapeutic intervention. Am J Cardiovasc Drugs. 2004;4(4):211–217. doi: 10.2165/00129784-200404040-00002. [DOI] [PubMed] [Google Scholar]
- Rosenson RS, Stafforini DM. Modulation of oxidative stress, inflammation, and atherosclerosis by lipoprotein-associated phospholipase A2. J Lipid Res. 2012;53(9):1767–1782. doi: 10.1194/jlr.R024190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AM, Perez A, Zapico E, Ordonez-Llanos J. Non-HDL cholesterol and apolipoprotein B in the dyslipidemic classification of type 2 diabetic patients. Diabetes Care. 2003;26(7):2048–2051. doi: 10.2337/diacare.26.7.2048. [DOI] [PubMed] [Google Scholar]
- Sanchez-Quesada JL, Benitez S, Otal C, Franco M, Blanco-Vaca F, Ordonez-Llanos J. Density distribution of electronegative LDL in normolipemic and hyperlipemic subjects. J Lipid Res. 2002;43(5):699–705. [PubMed] [Google Scholar]
- Benitez S, Sanchez-Quesada JL, Lucero L, Arcelus R, Ribas V, Jorba O, Castellvi A, Alonso E, Blanco-Vaca F, Ordonez-Llanos J. Changes in low-density lipoprotein electronegativity and oxidizability after aerobic exercise are related to the increase in associated non-esterified fatty acids. Atherosclerosis. 2002;160(1):223–232. doi: 10.1016/S0021-9150(01)00565-2. [DOI] [PubMed] [Google Scholar]
- Ribas V, Sanchez-Quesada JL, Anton R, Camacho M, Julve J, Escola-Gil JC, Vila L, Ordonez-Llanos J, Blanco-Vaca F. Human apolipoprotein A-II enrichment displaces paraoxonase from HDL and impairs its antioxidant properties: a new mechanism linking HDL protein composition and antiatherogenic potential. Circ Res. 2004;95(8):789–797. doi: 10.1161/01.RES.0000146031.94850.5f. [DOI] [PubMed] [Google Scholar]
- Benitez S, Sanchez-Quesada JL, Ribas V, Jorba O, Blanco-Vaca F, Gonzalez-Sastre F, Ordonez-Llanos J. Platelet-activating factor acetylhydrolase is mainly associated with electronegative low-density lipoprotein subfraction. Circulation. 2003;108(1):92–96. doi: 10.1161/01.CIR.0000072791.40232.8F. [DOI] [PubMed] [Google Scholar]
- De Juan-Franco E, Pérez A, Ribas V, Sanchez- Hernandez JA, Blanco-Vaca F, Ordonez-Llanos J, Sanchez-Quesada JL. Standarization of a method to evaluate the antioxidant capacity of high-density lipoproteins. Int J Biomed Sci. 2009;5(4):402–410. [PMC free article] [PubMed] [Google Scholar]
- Gazi I, Lourida ES, Filippatos T, Tsimihodimos V, Elisaf M, Tselepis AD. Lipoprotein-associated phospholipase A2 activity is a marker of small, dense LDL particles in human plasma. Clin Chem. 2005;51(12):2264–2273. doi: 10.1373/clinchem.2005.058404. [DOI] [PubMed] [Google Scholar]
- Hoogeveen RC, Ballantyne CM, Bang H, Heiss G, Duncan BB, Folsom AR, Pankow JS. Circulating oxidised low-density lipoprotein and intercellular adhesion molecule-1 and risk of type 2 diabetes mellitus: the Atherosclerosis Risk in Communities Study. Diabetologia. 2007;50(1):36–42. doi: 10.1007/s00125-006-0533-8. [DOI] [PubMed] [Google Scholar]
- Cohen MP, Lautenslager G, Shea E. Glycated LDL concentrations in non-diabetic and diabetic subjects measured with monoclonal antibodies reactive with glycated apolipoprotein B epitopes. Eur J Clin Chem Clin Biochem. 1993;31(11):707–713. doi: 10.1515/cclm.1993.31.11.707. [DOI] [PubMed] [Google Scholar]
- Younis NN, Soran H, Sharma R, Charlton-Menys V, Greenstein A, Elseweidy MM, Durrington PN. Small-dense LDL and LDL glycation in metabolic syndrome and in statin-treated and non-statin-treated type 2 diabetes. Diab Vasc Dis Res. 2010;7(4):289–295. doi: 10.1177/1479164110383063. [DOI] [PubMed] [Google Scholar]
- Kontush A, Chantepie S, Chapman MJ. Small, dense HDL particles exert potent protection of atherogenic LDL against oxidative stress. Arterioscler Thromb Vasc Biol. 2003;23(10):1881–1888. doi: 10.1161/01.ATV.0000091338.93223.E8. [DOI] [PubMed] [Google Scholar]
- Camont L, Chapman MJ, Kontush A. Biological activities of HDL subpopulations and their relevance to cardiovascular disease. Trends Mol Med. 2011;17(10):594–603. doi: 10.1016/j.molmed.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Zalewski A, Macphee C. Role of lipoprotein-associated phospholipase A2 in atherosclerosis: biology, epidemiology, and possible therapeutic target. Arterioscler Thromb Vasc Biol. 2005;25(5):923–931. doi: 10.1161/01.ATV.0000160551.21962.a7. [DOI] [PubMed] [Google Scholar]
- Koenig W, Khuseyinova N. Lipoprotein-associated and secretory phospholipase A2 in cardiovascular disease: the epidemiological evidence. Cardiovasc Drugs Ther. 2009;23(1):85–92. doi: 10.1007/s10557-008-6135-6. [DOI] [PubMed] [Google Scholar]
- Bhatti S, Hakeem A, Cilingiroglu M. Lp-PLA(2) as a marker of cardiovascular diseases. Curr Atheroscler Rep. 2010;12(2):140–144. doi: 10.1007/s11883-010-0095-6. [DOI] [PubMed] [Google Scholar]
- Nelson TL, Biggs ML, Kizer JR, Cushman M, Hokanson JE, Furberg CD, Mukamal KJ. Lipoprotein-associated phospholipase A2 (Lp-PLA2) and future risk of type 2 diabetes: results from the Cardiovascular Health Study. J Clin Endocrinol Metab. 2012;97(5):1695–1701. doi: 10.1210/jc.2011-3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oei HH, Van Der Meer IM, Hofman A, Koudstaal PJ, Stijnen T, Breteler MM, Witteman JC. Lipoprotein-associated phospholipase A2 activity is associated with risk of coronary heart disease and ischemic stroke: the Rotterdam Study. Circulation. 2005;111(5):570–575. doi: 10.1161/01.CIR.0000154553.12214.CD. [DOI] [PubMed] [Google Scholar]
- Hatoum IJ, Hu FB, Nelson JJ, Rimm EB. Lipoprotein-associated phospholipase A2 activity and incident coronary heart disease among men and women with type 2 diabetes. Diabetes. 2010;59(5):1239–1243. doi: 10.2337/db09-0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson TL, Kamineni A, Psaty B, Cushman M, Jenny NS, Hokanson J, Furberg C, Mukamal KJ. Lipoprotein-associated phospholipase A(2) and future risk of subclinical disease and cardiovascular events in individuals with type 2 diabetes: the Cardiovascular Health Study. Diabetologia. 2011;54(2):329–333. doi: 10.1007/s00125-010-1969-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizer JR, Umans JG, Zhu J, Devereux RB, Wolfert RL, Lee ET, Howard BV. Lipoprotein-associated phospholipase A(2) mass and activity and risk of cardiovascular disease in a population with high prevalences of obesity and diabetes: the Strong Heart Study. Diabetes Care. 2012;35(4):840–847. doi: 10.2337/dc11-1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujiraoka T, Iwasaki T, Ishihara M, Ito M, Nagano M, Kawaguchi A, Takahashi S, Ishi J, Tsuji M, Egashira T. et al. Altered distribution of plasma PAF-AH between HDLs and other lipoproteins in hyperlipidemia and diabetes mellitus. J Lipid Res. 2003;44(10):2006–2014. doi: 10.1194/jlr.D300021-JLR200. [DOI] [PubMed] [Google Scholar]
- Mitsutake R, Miura S, Zhang B, Saku K. HDL-associated factors provide additional prognostic information for coronary artery disease as determined by multi-detector row computed tomography. Int J Cardiol. 2010;143(1):72–78. doi: 10.1016/j.ijcard.2009.01.058. [DOI] [PubMed] [Google Scholar]
- Onat A, Hergenc G, Can G, Ugur M, Nartop F. Dual activity of serum lipoprotein-associated phospholipase A(2) yielding positive and inverse associations with cardiometabolic risk. Clin Chem Lab Med. 2011;49(8):1349–1357. doi: 10.1515/CCLM.2011.110. [DOI] [PubMed] [Google Scholar]
- Rallidis LS, Tellis CC, Lekakis J, Rizos I, Varounis C, Charalampopoulos A, Zolindaki M, Dagres N, Anastasiou-Nana M, Tselepis AD. Lipoprotein-associated phospholipase A(2) bound on high-density lipoprotein is associated with lower risk for cardiac death in stable coronary artery disease patients: a 3-year follow-up. J Am Coll Cardiol. 2012;60(20):2053–2060. doi: 10.1016/j.jacc.2012.06.057. [DOI] [PubMed] [Google Scholar]
- Wagner AM, Jorba O, Bonet R, Ordonez-Llanos J, Perez A. Efficacy of atorvastatin and gemfibrozil, alone and in low dose combination, in the treatment of diabetic dyslipidemia. J Clin Endocrinol Metab. 2003;88(7):3212–3217. doi: 10.1210/jc.2003-030153. [DOI] [PubMed] [Google Scholar]
- Sanchez-Quesada JL, Vinagre I, De Juan-Franco E, Sanchez-Hernandez J, Blanco-Vaca F, Ordonez-Llanos J, Perez A. Effect of improving glycemic control in patients with type 2 diabetes mellitus on low-density lipoprotein size, electronegative low-density lipoprotein and lipoprotein-associated phospholipase A2 distribution. Am J Cardiol. 2012;110(1):67–71. doi: 10.1016/j.amjcard.2012.02.051. [DOI] [PubMed] [Google Scholar]