

Abstract
Background
Zanthoxylum zanthoxyloides and Z. leprieurii fruits are commonly used in traditional system of medicine for diarrhea, pain, wound healing, etc. in Cameroon, Africa. Z. leprieurii fruits have been chemically studied for its bioactive compounds whereas the investigation on Z. zanthoxyloides fruits is lacking.
Results
After a detailed chemical analysis of the fruits of Z. leprieurii and Z. zanthoxyloides, a series of new acridone alkaloids, namely, 3-hydroxy-1,5,6-trimethoxy-9-acridone (1), 1,6-dihydroxy-3-methoxy-9-acridone (2), 3,4,5,7-tetrahydroxy-1-methoxy-10-methyl-9-acridone (3), 4-methoxyzanthacridone (8), 4-hydroxyzanthacridone (9), 4-hydroxyzanthacridone oxide (2,4’) (10) have been isolated. The known acridones which have been characterized are, helebelicine A (4), 1-hydroxy-3-methoxy-10-methyl-9-acridone (5), 1,3-dihydroxy-4-methoxy-10-methyl-9-acridone (6) and tegerrardin A (7). The in vitro antibacterial and cytotoxic screening of these acridones reveal that compound 3 has a moderate antibacterial activity (MIC 125 μg/mL) against Micrococcus luteus and Pseudomonas aeruginosa while compound 1 shows a moderate cytotoxic effect (IC50 of 86 μM) against WRL-68 (liver cancer cell line). Furthermore, the molecular modeling of these acridones predicted the structural basis for their mode of action and binding affinity for aromatase, quinone reductase and WAAG, a glycosyltransferase involved in bacterial lipopolysaccharide synthesis. Computational approaches, quantitative SAR and modeling studies predicted that acridones 1, 2, 3, 4, 9 and 10 were the inhibitors of glycosyltransferase while 1, 8, 9 and 10, the inhibitors of aromatase.
Conclusions
A total of 10 acridones have been isolated out of which 6 are new (1, 2, 3, 8, 9 and 10). Alkaloids 8, 9 and 10, having novel tetracyclic acridone structure with new carbon skeleton, have now been named as zanthacridone. The quantitative SAR and molecular modeling studies suggested that the compounds 1, 9 and 10 are inhibitors of both aromatase and glycosyltransferase.
Keywords: Rutaceae, Zanthoxylum zanthoxyloides, Z. leprieurii, Zanthacridones, Structure elucidation, Antibacterial, Cytotoxic activities, Molecular modeling, QSAR studies
Background
The pantropical genus Zanthoxylum (Rutaceae) is represented by 35 species in Africa [1]. Among them, Zanthoxylum zanthoxyloides (Lam.) Zepern. & Timler and Zanthoxylum leprieurii Guill. et Perr are found in Cameroon and are well reputed for their ethnomedicinal properties. Z. zanthoxyloides, formerly known as Fagara zanthoxyloides, a popular African medicinal plant, occurs abundantly in savanna and dry forest vegetations whereas Z. leprieurii, is a medium tree found in rain forests and savanna woodland, from sea-level up to the altitude of 2000 m [1,2]. In Cameroon, the leaves, stems and roots of these plants are used in the treatment of gonococci, urinary infections and dysentery [3]. In addition, the fruit infusion of Z. leprieurii is shown to relegate the symptoms of sickle cell anemia [2]. The aqueous ethanolic extracts of the leaves, roots and stem bark of Z. leprieurii have demonstrated moderate antifungal activity while the chloroform extract of the fruits showed moderate cytotoxicity with the brine-shrimp assay [4,5]. Recent reports on the dried fruits of Z. leprieurii from Cameroon described the isolation of acridone alkaloids that exhibited antiplasmodial activity and in vitro cytotoxicity [5,6]. Its roots and stem bark have yielded alkaloids (benzophenanthridines, acridone, aporphine), aliphatic amides and lignans [5,7,8]. Several studies on Z. zanthoxyloides have established its broad spectrum of biological activities, including sickle cell anemia [9-11]. The antisickling divanylloylquinic acids as well as the antifungal and antioxidant isobutylamide and benzophenanthridine alkaloids have been reported from the roots [12-14]. From its fruits, the components of essential oils were also characterized [15,16]. Besides the studies on its essential oil, no work has been carried out on the constituents of its non volatile extract. A recent study has revealed that the alcoholic extract of the fruits of this plant possessed in vitro cytotoxic activity against MCF-7, without identifying any bioactive constituents [17]. This prompted us to undertake a comparative study on the chemical constituents of the fruits of Z. zanthoxyloides and Z. leprieurii to identify their bioactive compounds.
In continuation of our work on the chemistry of medicinal and aromatic plants [18-22], we have investigated the fruits of two Zanthoxylum species collected from Cameroon, Africa and recently reported their bioactive non-alkaloidal phytoconstituents [23,24]. In the present paper, we describe the isolation and structure elucidation of several new acridone alkaloids, some of them with novel structures, from Z. zanthoxyloides and Z. leprieurii exhibiting antibacterial and cytotoxic activities. In order to explore the possible mechanism of these activities, we have also performed quantitative SAR of these acridones along with their molecular docking experiments to identify their putative biological targets.
Results and discussion
Isolation and identification of acridones
From the MeOH extract of the fruits of Z. zanthoxyloides, 6 new acridone alkaloids viz., 3-hydroxy-1,5,6-trimethoxy-9-acridone (1), 1,6-dihydroxy-3-methoxy-9-acridone (2), 3,4,5,7-tetrahydroxy-1-methoxy-10-methyl-9-acridone (3), 4-methoxyzanthacridone (8), 4-hydroxyzanthacridone (9), 4-hydroxyzanthacridone oxide (2,4’) (10) along with two known acridones, viz., helebelicine A (4) and 1-hydroxy-3-methoxy-10-methyl-9-acridone (5), have been isolated and identified (Figure 1). Compounds 8- 10 have a tetracyclic acridone carbon skeleton reported for the first time from Zanthoxylum spp. This novel acridone skeleton has, tentatively, been named as zanthacridone. From Z. leprieuri also, the new compound, 3-hydroxy-1,5,6-trimethoxy-9-acridone (1), was isolated along with three known acridones, namely, helebelicine A (4), 1,3-dihydroxy-4-methoxy-10-methyl-9-acridone (6) and tegerrardin A (7). All the compounds were Dragendorff positive and yellowish orange in color. The structures of the new compounds were elucidated mainly by UV, IR, NMR and MS spectroscopy (Additional file 1) and by comparison with the data already reported in the literature for acridone alkaloids [5,25-32].
Figure 1.

Structures of acridones 1- 10.
Compound 1 in its UV spectrum showed bands at 220, 233, 252 and 320 nm while in IR the bands appeared at 3448, 2929, 1620 cm-1 which suggested the presence of an acridone skeleton. Its molecular formula was determined by HRMS as C16H15O5N with [M]+ at m/z 301.0944. On the basis of the molecular formula and the assumption of an acridone skeleton through comparison with the literature data for similar compounds [5], compound 1 must be a trimethoxy-hydroxy-acridone. In the unsubstituted acridone, the signals of the peri-protons H-1 and H-8 are found at δ 8.25, while the neighbors of N-10 are found at approximately δ 7.5. It follows that the doublet at δ 7.99 ppm belongs to H-8; the coupling constant indicated a proton (H-7) in ortho-position. As the signal of H-7 did not show further couplings, the positions C-5 and C-6 must be oxygenated. It was further substantiated as in the 1H1H COSY spectrum H-7 showed correlation only with H-8 and in HMBC it showed correlation with C-7-OMe at δ 61.9. Its 1H NMR spectrum (Table 1) showed a downfield singlet at δ 9.02 for C-3-OH along with two meta coupled doublets (J = 3.0 Hz) at δ 7.01 and 7.55 indicating that C-1 and C-3 were substituted. The presence of three singlets at δ 4.40, 4.10 and 4.01 each of 3H in 1H NMR spectrum and δ 61.9, 59.3 and 57.5 in its 13C NMR spectrum indicated that it contained 3 OMe groups. The HMBC spectrum showed correlations as reported earlier [5] for 1,3 oxygenated acridones (Experimental). Its 13C NMR spectrum also exhibited four CH signals at 113.5, 104.8, 118.3 and 162.3 along with four C-O-R singlets at δ 158.0, 152.6, 145.1 and 143.3. These sets of signals clearly implied that in acridone skeleton, A and C rings are oxidized at 4 positions having 3 methoxy and a hydroxy functional group. The absence of the singlet of a chelated hydroxyl group in the region of δ 10-15 in its 1H NMR spectrum indicated that the peri positions C-1 and C-8 were not hydroxylated. This was further reinforced by the presence of the strongly downfielded signals at δ 4.40 in its 1H NMR spectrum and at δ 158.0 and 61.9 for C-1-OMe in its 13C NMR spectrum. The meta coupled doublets at δ 7.01 and 7.55 (J = 3.0 Hz) for H-2 and H-4 indicated that C-3 is also oxidized which was supported by the typical signal for C-3 in acridones at δ 152.6 in its 13C NMR spectrum. The C-3-OH signal at δ 9.02 correlated with C-3 at δ 152.6 in its HMBC spectrum, clearly indicating the placement of phenol in A ring. The downfield signal at δ 7.99 in 1H NMR spectrum which is typical for H-8 showed ortho coupling (J = 8.0 Hz) with H-7, correlation in 1H1H COSY and no meta coupling, suggesting that two other methoxy groups are present at C-5 and C-6. The relatively strong upfield shifted C-4 signal at δ 104.8 in 13C NMR spectrum for aromatic carbon fitted well with the earlier report [5]. The rest of the signals of 13C NMR spectrum, when compared with the literature data, [5,30,33,34] supported that 1 is a 3-hydroxy-1,5,6-trimethoxy-9-acridone.
Table 1.
1H NMR spectral data for 1-3, 8-10 (300 MHz, in CDCl3, δ in ppm)
| Proton | 1 | 2 | 3 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|
| 2 |
7.01(d,3.0) |
6.58(d,3.0) |
6.12(s) |
- |
- |
- |
| 4 |
7.55(d,3.0) |
6.44(d,3.0) |
- |
- |
- |
- |
| 5 |
- |
7.39(d,3.0) |
- |
7.40(dd,8.0,3.0) |
7.41(dd,8.0,3.0) |
7.34(dd,8.0,3.0) |
| 6 |
- |
- |
7.20(d,3.0) |
7.61(ddd,8.0,8.0,3.0) |
7.59(ddd,8.0,8.0,3.0) |
7.55(ddd,8.0,8.0,3.0) |
| 7 |
7.21(d,8.0) |
7.07(dd,8.0,3.0) |
- |
7.29(ddd,8.0,8.0,3.0) |
7.34(ddd,8.0,8.0,3.0) |
7.34(ddd,8.0,8.0,3.0) |
| 8 |
7.99(d,8.0) |
7.89(d,8.0) |
7.82(d,3.0) |
7.82(dd,8.0,3.0) |
8.40(dd, 8.0, 3.0) |
8.39(dd,8.0,3.0) |
| 1-OMe |
4.40(s) |
- |
4.44(s) |
- |
- |
- |
| 3-OMe |
- |
3.93(s) |
- |
- |
- |
- |
| 4-OMe |
- |
- |
- |
3.98(s) |
- |
- |
| 5-OMe |
4.10(s) |
- |
- |
- |
- |
- |
| 6-OMe |
4.01(s) |
- |
- |
- |
- |
- |
| N-Me |
- |
- |
3.90(s) |
3.77(s) |
3.69(s) |
3.70(s) |
| 2’-Me |
- |
- |
- |
1.30(s) |
1.33(s) |
1.32(s) |
| 1.30(s) |
1.42(s) |
1.28(s) |
||||
| 3’ |
- |
- |
- |
2.72(dd,13,10) |
2.68(dd,13,3.5) |
3.23(overlapping m) |
| 3.07(dd,13,3.5) |
3.00(dd,13,3.5) |
3.25(overlapping m) |
||||
| 4’ |
- |
- |
- |
3.50(dd,10,3.5) |
3.63(dd,10,3.5) |
4.85(dd,10.0,10.0) |
| 1-OH |
- |
12.12 |
- |
15.05 |
15.00 |
14.92 |
| 3-OH |
9.02 |
- |
9.05* |
- |
- |
- |
| 4-OH |
- |
- |
9.00* |
- |
9.05 |
9.10 |
| 5-OH |
- |
- |
9.32* |
- |
- |
- |
| 6-OH |
- |
8.90 |
- |
- |
- |
- |
| 7-OH | - | - | 8.92* | - | - | - |
*Assignments interchangeable.
Compound 2 exhibited UV and IR signals as in case of 1. Its HRMS showed [M]+ at m/z 257.0681 confirming C14H11O4N as its molecular formula. Its 1H NMR (Table 1) spectrum showed singlets at δ 12.12 and δ 8.90 for C-1-OH and C-6-OH, respectively, along with a relatively strong upfield singlet at δ 3.93 suggesting the presence of a methoxy group at C-3 in the acridone skeleton which was supported by the typical quartet at δ 56.4 in its 13C NMR spectrum. The 13C NMR spectrum (Table 2) further showed three downfield signals at δ 161.2, 162.4 and 144.0 suggesting 3 oxygenated carbons in A and C rings of acridone skeleton. The 1H NMR spectrum exhibited meta coupled doublets (J = 3.0 Hz) at δ 6.58 and 6.44 showing matching substitution in A ring as in case of compound 1. The typical downfield doublet at δ 7.89 (J = 8.0 Hz) for H-8 in 1H NMR spectrum showed correlation with a double doublet (J = 8.0, 3.0 Hz) for H-7 at δ 7.07 which in turn correlated with the doublet (J = 3.0 Hz) for H-5 at δ 7.39 in its 1H1H COSY. These data suggested that the second hydroxyl group is attached at C-6. The relatively upfield shift of C-4 at δ 90.7 in 13C NMR spectrum for aromatic carbon has been observed in earlier study [5]. The rest of 13C NMR values (Experimental) accorded well with the structure for 2 as 1,6-dihydroxy-3-methoxy-9-acridone.
Table 2.
13C NMR spectral data for 1-3, 8-10 (75 MHz, in CDCl3, δ in ppm)
| Carbon | 1 | 2 | 3 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|
| 1 |
158.0 (s) |
161.2 (s) |
160.0 (s) |
162.1 (s) |
160.0 (s) |
159.3(s) |
| 2 |
113.5 (d) |
94.3 (d) |
92.7 (d) |
118.2 (s) |
117.5 (s) |
160.2 (s) |
| 3 |
152.6 (s) |
162.4 (s) |
161.3 (s) |
166.5 (s) |
166.4 (s) |
166.5 (s) |
| 4 |
104.8 (d) |
90.7 (d) |
130.2 (s) |
123.0 (s) |
122.1 (s) |
131.2 (s) |
| 4a |
143.3 (s) |
140.7 (s) |
141.2 (s) |
139.3 (s) |
139.0 (s) |
124.8 (s) |
| 5a |
144.0 (s) |
140.7 (s) |
141.3 (s) |
139.3 (s) |
140.2 (s) |
133.2 (s) |
| 5 |
145.1 (s) |
104.4 (d) |
146.2 (s) |
115.0 (d) |
116.1 (d) |
114.5 (d) |
| 6 |
143.3 (s) |
144.0 (s) |
119.8 (d) |
131.2 (d) |
131.3 (d) |
131.4 (d) |
| 7 |
118.3 (d) |
117.2 (d) |
143.1 (s) |
123.0 (d) |
123.0 (d) |
123.6 (d) |
| 8 |
162.3 (d) |
126.9 (d) |
117.2 (d) |
124.0 (d) |
124.1 (d) |
126.8 (d) |
| 8a |
115.6 (s) |
120.4 (s) |
121.5 (s) |
121.5 (s) |
121.6 (s) |
122.1 (s) |
| 9 |
178.2 (s) |
180.8 (s) |
181.1 (s) |
177.2 (s) |
178.3 (s) |
181.6 (s) |
| 9a |
104.7 (s) |
108.2 (s) |
108.1 (s) |
114.9 (s) |
114.9 (s) |
116.2 (s) |
| OMe |
57.5 (q) |
56.4 (q) |
60.9 (q) |
62.8 (q) |
- |
- |
| 59.3 (q) |
- |
- |
- |
- |
- |
|
| 61.9 (q) |
- |
- |
- |
- |
- |
|
| N-Me |
- |
- |
34.3 (q) |
30.5 (q) |
31.5 (q) |
31.7 (q) |
| 2’ |
- |
- |
- |
73.4 (s) |
73.4 (s) |
72.3 (s) |
| 2’Me |
- |
- |
- |
24.4 (q) |
25.0 (q) |
24.7 (q) |
| 26.1 (q) |
26.2 (q) |
26.0 (q) |
||||
| 3’ |
- |
- |
- |
28.1 (t) |
28.2 (t) |
28.1 (t) |
| 4’ | - | - | - | 79.6 (d) | 78.6 (d) | 92.3 (d) |
*The multiplicity of the signals obtained by DEPT 135.
Compound 3 had the UV and IR bands for acridone alkaloids as described in the case of 1. Its HRMS showed [M]+ at m/z 303.0734 corresponding to C15H13O6N. Its 1H NMR spectrum showed four downfield singlets at δ 9.05, 9.32, 9.00 and 8.92 for phenolic protons. The downfield meta coupled doublet (J = 3.0 Hz) at δ 7.82 typical for H-8 in its 1H NMR spectrum, clearly suggested that the doublet (J = 3.0 Hz) at δ 7.20 corresponds to H-6. Its 1H NMR spectrum showed a typical downfield singlet at δ 4.44 for OMe at C-1 and δ 3.90 for N-Me which was supported by the quartets in 13C NMR spectrum at δ 60.9 and 34.3, respectively. A singlet at δ 6.12 in the 1H NMR spectrum further suggested that rest of the 3 carbons in A ring along with 2 in C ring are oxygenated which was strengthened by the presence of a total of five C-O-R signals in its 13C NMR spectrum at δ 160.0, 161.3, 130.2, 146.2 and 143.1 for C-1, C-3, C-4, C-5 and C-7, respectively. The relatively strong upfield shift of C-4 at δ 130.2 for oxygenated carbon with N-10 methylated acridones fits well with the published data [5]. These data (Tables 1 and 2) established that 3 is 3,4,5, 7-tetrahydroxy-1-methoxy-10-methyl-9-acridone.
Compound 8 showed UV and IR spectral values for acridone skeleton as described in the case of 1. Its HRMS showed [M]+ at m/z 355.1416 for C20H21O5N. An OH (H-bonded) singlet appeared at δ 15.05 in its 1H NMR spectrum (Table 1) indicating the presence of a phenol at C-1. In its 1H NMR spectrum, two sets of double doublets (J = 8.0 Hz and 3.0 Hz) at δ 7.82 and 7.40 for H-8 and H-5 along with two sets of triple doublets (J = 8.0, 8.0, 3.0 Hz) at δ 7.61 and 7.29 for H-6 and H-7, were available. These signals showed respective correlations from H-5 to H-8 in the 1H1H COSY spectrum (Experimental) revealing that C ring is unsubstituted at C-5 to C-8 positions. This fact was justified by the doublets at δ 115.0, 131.2, 123.0 and 124.0 available in its 13C NMR spectrum (Table 2). Since no signals, other than these four for C ring, were seen in the typically unsubstituted aromatic region in 1H NMR spectrum of 8, it was concluded that all the four free carbons in A ring were substituted. The downfield signals at δ 162.1, 166.5 and 123.0 in its 13C NMR spectrum implied that 3 carbons in A ring are oxygenated while 4th carbon is C-substituted. The relatively strong upfield shift of C-4 at δ 123.0 for oxygenated carbon with N-10 methylated acridones concurred well with previous report [5]. A set of additional signals in its 1H NMR spectrum at δ 3.50 (dd, J = 10, 3.5 Hz), δ 2.72 and 3.07 (dd each, J = 13.0, 10.0 Hz) and two methyl singlets (6H) at δ 1.30 designated the presence of a three carbon chain attached with A ring. The 1H1H COSY experiment showed a cyclic ring of -CH(OH)CH2C(O)(CH3)2- attached at C-2 and C-3. Although the oxidation at C-3 is biogenetically preferred in this class of compounds, the HMBC experiment established that C-2 is C-substituted while C-3 is O-substituted. In HMBC spectrum, the signal for 4’-H at δ 3.50 showed strong correlation with C-2 at δ 118.2. This type of cyclization is justified by the 13C NMR values at δ 79.6 (d), 28.1 (t), 73.4 (s) as well as 24.4 and 26.1 (q) along with C-2 and C-3 singlets at δ 118.2 and 166.5, respectively. In the NOESY spectrum of 8, the C-4’-H at δ 3.50 showed a weak correlation with phenolic proton at C-1 at δ 15.05 and no correlation with C-2’-Me, suggesting that OH at C-4’ is β-oriented. The molecular model of 8 further reinforced this statement. Its HMQC and HMBC spectra also supported these observations (Experimental). The 3, 4-cyclization is normally preferred for a more stable linear structure of acridones. Recently, the linear tetracyclic acridones were reported from Glycosmis parva by Chansriniyom, et al. [25] having a double bond in pyran ring. Moreover, similar cyclization product was earlier reported from the synthesis of 1-hydroxy-1,2-dihydroisoacronycine by Magiatis et al. [35] but without oxidation at C-4. Acetylation of 8 yielded 8a showing the downfield shifting of CH(OH) from δ 3.50 to δ 4.34 along with an additional signal at δ 2.10 for OAc in its 1H NMR spectrum. This observation clearly supported the presence of a CHOH group in this ring. Since the OMe appeared slightly upfield at δ 3.98 in 1H NMR, it was placed at C-4 and not at C-1, obviously indicating that C-1 is substituted by a hydroxy group. The typical signals at δ 3.77 (s) in 1H NMR and δ 30.5 (quartet) in 13C NMR spectra justified the presence of N-Me. The present tetracyclic skeleton was, tentatively, named as zanthacridone and the data described as above supported the structure of 8 as 4-methoxyzanthacridone.
Compound 9 had almost similar spectral data except the methoxy signal in 1H NMR and 13C NMR spectra (Tables 1 and 2) which clearly suggested that C-4 is now substituted with a hydroxy group rather than a methoxy, as was in case of compound 8. In its HRMS, [M]+ appeared at m/z 341.1257 for C19H19O5N. Therefore, the structure of 9 was assigned as 4-hydroxyzanthacridone.
Compound 10 showed almost similar spectral data as 9 with the following exceptions: In its 1H NMR spectrum, the CHOH appeared downfield at δ 4.85, as compared to 9 at δ 3.63; the -CH(OH)- also appeared more downfield at δ 92.3 in 10 than at δ 78.6 in compound 9 in its 13C NMR spectrum. Moreover, a downfield shift was observed for C-2 from 117.5 in 9 to 160.2 in 10 in the 13C NMR spectrum. The relatively strong upfield shift of C-4 at δ 131.2 for oxygenated carbon with N-10 methylated acridones, was also in accordance with earlier studies [5]. Acetylation of 10 gave a downfield shift of CHOH from δ 4.85 to δ 5.35 with an additional singlet for OAc at δ 2.11 in 1H NMR spectrum. In the NOESY spectrum, one of the C-2’-Me at δ 1.32 correlated with C-4OH at δ 9.10 evidencing that there is no change in the position of the additional ring and happens to be quite similar as in case of 8 and 9. Since H-4’ did not correlate with C-1 in its HMBC spectrum (as was in cases of 8 and 9), it was evident that there is an extra bond formation by an oxide between C-2 and C-4’. This observation fits well with the value of C-2 at δ 160.2 in its 13C NMR spectrum. The C-4’-OH had a β-orientation as in its NOESY spectrum H-4’ showed correlation with C-1-OH (Experimental) similar to 8 and 9. In its HRMS, it showed [M]+ at m/z 357.1210 for C19H19O6N. These data clearly supported the structure of 10 as 4-hydroxyzanthacridone oxide (2,4’).
Biological evaluation
In vitro antibacterial evaluation
The results of the antibacterial activity of the isolated major acridones (1, 3, 4, 7, 8 and 10) against three Gram-positive and one Gram-negative bacteria are given in Table 3. Compound 3 exhibited moderate activity (MIC 125 μg/mL) when tested against Gram-positive Micrococcus luteus and Gram-negative Pseudomonas aeruginosa whereas 1, 8 and 10 showed activity against P. aeruginosa with MIC values of 500, 250 and 250 μg/mL, respectively. Compound 7 had MIC values of 500 and 250 μg/mL against Staphylococcus aureus and Bacillus subtilis, respectively (Table 3) while 4 displayed a MIC of 250 μg/mL against both bacteria. Against M. luteus, compounds 4 and 7 demonstrated a moderate activity with MIC 125 and 500 μg/mL, respectively but failed to show appreciable activity against P. aeruginosa. The present study related the antimicrobial activity with acridone alkaloids from the genus Zanthoxylum, although the acridones from the genus Boronea have exhibited this activity, earlier [36].
Table 3.
In vitro antibacterial activity of extracts of Z. leprieurii, Z. zanthoxyloides and acridone alkaloids
| |
MIC expressed in μg/mL |
|||
|---|---|---|---|---|
| Compounds | BS | ML | PA | SA |
|
1 |
>500 |
>500 |
500 |
>500 |
|
3 |
nt |
125 |
125 |
nt |
|
4 |
250 |
125 |
>500 |
250 |
|
7 |
250 |
500 |
>500 |
500 |
|
8 |
nt |
>500 |
250 |
nt |
|
10 |
nt |
>500 |
250 |
nt |
| Kanamycin |
0.53 |
4.16 |
62.5 |
6.94 |
| Extracts |
|
|
|
|
|
Z. leprieurii |
>1000 |
250 |
1000 |
>1000 |
| Z. zanthoxyloides | >1000 | 500 | >1000 | >1000 |
Gram-positive bacteria: BS = Bacillus subtilis MTCC121, ML = Micrococcus luteus MTCC2470 and SA = Staphylococcus aureus MTCC96, Gram-negative bacteria: PA = Pseudomonas aeruginosa MTCC741, nt = not tested. Kanamycin is the standard antibiotic.
Pharmacophore modeling and molecular docking studies against Glycosyltransferase (WAAG)
Since compounds 3 and 4 showed a MIC of 125 μg/mL against M. luteus, we carried out a quantitative SAR and molecular docking to explore the possible mechanism of action for the observed biological activities targeting glycosyltransferase. Results of pharmacophore modeling showed that compounds 1, 2, 3, 4, 9 and 10 have significant activity against M. luteus. Molecular docking study of these compounds against WAAG protein, a glycosyltransferase involved in lipopolysaccharide biosynthesis (PDB ID: 2IW1) [37], revealed active conformation and interacting binding site amino acid residues (Figure 2). For validation of molecular docking co-crystallised uridine derivative was re-docked on bacterial protein WAAG (PDB: 2IW1) (Table 4). Compounds 3 and 4 were experimentally validated to be significantly active against M. luteus. Quantitative SAR studies showed that compounds 2 and 9 seem to be significantly active against M. luteus. However, experimentally non-active compounds 7 and 8 also predicted inactive against M. luteus. Moreover, compounds 1 and 10 were also predicted to be inactive against M. luteus (Table 5).
Figure 2.

Molecular docking of active acridone alkaloids against (A) anticancer target protein aromatase in human (compounds 1, 8, 9 and 10) and (B) antibacterial WAAG protein of bacteria (compounds 1, 2, 3, 4, 9 and 10).
Table 4.
Results of molecular docking experiments targeting antibacterial protein WAAG
| Molecule | H-bonds | Bound amino acid residues of WAAG | Binding affinity (kcal/mol) |
|---|---|---|---|
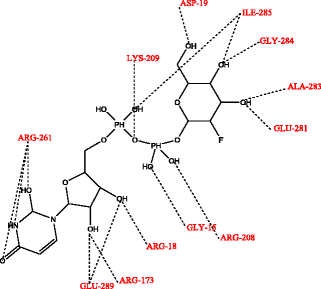 |
16 |
ASP-19, ILE-285, GLY-284, LYS-209, ALA-283, GLU-281, ARG-261, GLY-15, ARG-208, ARG-18, ARG-173, GLU-289 |
-10.7 |
| Uridine-5’-diphosphate-2-deoxy-2-fluoro-alpha- d-glucose (Control inhibitor) | |||
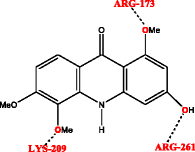 |
03 |
ARG-173, LYS-209, ARG-261 |
-8.6 |
| Compound 1 | |||
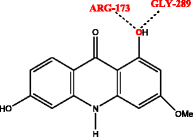 |
02 |
ARG-173, GLY-289 |
-8.5 |
| Compound 2 | |||
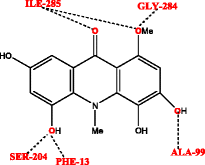 |
06 |
PHE-13, ALA-99, SER-204, GLY-284, ILE-285, |
-7.6 |
| Compound 3 | |||
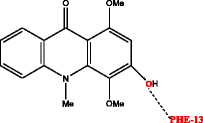 |
01 |
PHE-13 |
-7.0 |
| Compound 4 | |||
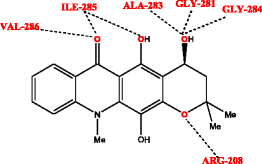 |
07 |
ARG-208, VAL-286, GLY-281, ALA-283, GLY-284, ILE-285, |
-8.6 |
| Compound 9 | |||
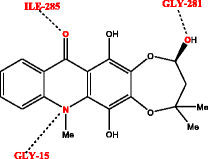 |
03 | GLY-15, GLY-281, ILE-285 | -8.1 |
| Compound 10 |
Table 5.
Derived pharmacophore model for antibacterial activity of acridones against M. luteus
| Acridones | Score obtained | Categorizing through threshold (S = 4.1466) | Predicted activity | Experimental activity (MIC in μg/mL) | Experimental response |
|---|---|---|---|---|---|
|
1. |
4.668387 |
>4.1466 |
significantly active |
>500 |
Non active |
|
2. |
4.655514 |
>4.1466 |
significantly active |
Not tested |
Not tested |
|
3. |
4.409138 |
>4.1466 |
significantly active |
125 |
Active |
|
4. |
5.006913 |
>4.1466 |
significantly active |
125 |
Active |
|
5. |
3.892896 |
<4.1466 |
Non-significant activity |
Not tested |
Not tested |
|
6. |
4.049106 |
<4.1466 |
Non-significant activity |
Not tested |
Not tested |
|
7. |
2.944231 |
<4.1466 |
Non-significant activity |
500 |
Less active |
|
8. |
4.008224 |
<4.1466 |
Non-significant activity |
>500 |
Non active |
|
9. |
4.189883 |
>4.1466 |
significantly active |
Not tested |
Not tested |
| 10. | 4.479721 | >4.1466 | significantly active | >500 | Not active |
The threshold score (S) of 4.1466 has been selected for the screening of lead compounds.
In vitro cytotoxicity evaluation
We screened compounds 1, 4, 7 and 8 for their in vitro cytotoxicity (Table 6) against liver (WRL-68), colon (CaCO2), breast (MCF-7) and prostate (PC-3) cancer cell lines using the MTT assay. We found that compounds 4 and 7 failed to produce a significant activity against the tested human cancer cell lines. However, a previous study [5] has reported 7 having a moderate antiproliferative activity against lung carcinoma and colorectal adenocarcinoma cell lines indicating that 7 possesses a selective cytotoxic activity. Compound 1 showed a moderate activity (IC50 of 86 μM) against WRL-68; both 1 and 8 exhibited a poor activity against MCF-7 with IC50 229 and 205 μM, respectively.
Table 6.
In vitro cytotoxicity of acridone 1, 4, 7 and 8
| |
IC50 expressed in μM |
|||
|---|---|---|---|---|
| Compounds | WRL-68 | CaCO2 | MCF-7 | PC-3 |
|
1 |
86 |
>300 |
229 |
272 |
|
4 |
>300 |
>300 |
>300 |
>300 |
|
7 |
>300 |
>300 |
>300 |
>300 |
|
8 |
>300 |
>300 |
205 |
>300 |
| Doxorubicin | 1.4 | 6.0 | 3.6 | 8.6 |
WRL-68 = Liver cancer cell lines, CaCO2 = colon cancer, MCF-7 = breast cancer, PC-3 = prostate cancer. Doxorubicin Sigma D-1515 is the standard used.
Pharmacophore modeling and molecular docking studies against aromatase and quinone Reductase
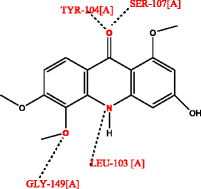
Results of pharmacophore modeling showed that compounds 1, 8, 9 and 10 have significant activity against human anticancer target aromatase [38]. Molecular docking on human aromatase, quinone reductase, not on helicase [39], was preferred because it is the popular enzyme in vertebrates known to catalyse the biosynthesis of all oestrogens from androgens and is therefore considered as a frontline therapy for oestrogen-dependent breast cancer [38]. Molecular docking study of studied compounds (1, 8, 9, 10) against anticancer targets aromatase (PDB: 3EQM), quinone reductase (PDB: 3G5M) showed active conformations comparable to control molecule. During docking experiments interacting binding site amino acids were namely, LEU-103, TYR-104, SER-107, GLY-149 for compound 1 with docking energy -6.1 kcal/mol and four H-bonds. Similarly, THR-148, TYR-155, SER-164 residues were identified for compound 8 with docking energy -6.7 kcal/mol and four H-bonds. Likewise residues PHE-106, THR-147, THR-148, TYR-155, SER-164 were identified in the binding site for compound 9 with docking energy -7.3 kcal/mol and showed six H-bonds. Compound 10 showed interaction with LEU-103, PHE-106, THR-147 with docking energy -7.7 kcal/mol and three H-bonds (Table 7, Figure 2). All compounds (1, 8, 9, 10) showed strong binding affinity with aromatase when compared with control molecule. Control experiment showed binding site residues namely, PHE-17, THR-147 with docking energy -5.8 kcal/mol and two H-bonds. For validation of docking experiment, co-crystallised inhibitor 6-methoxy-9-methyl[1,3]dioxolo[4,5-h]quinolin-8(9 h)-one was used as control molecule targeting anticancer protein aromatase (PDB: 3EQM), quinone reductase (PDB: 3G5M) (Table 7). Compounds 1 and 8 were experimentally validated to be significantly active against MCF-7 human breast cancer cell line. Results showed that out of ten compounds four showed significant anticancer activity against MCF-7 (Table 8). Besides, all studied compounds showed compliance with Lipinski's rule of five parameters for drug likeness and oral bioavailability (Table 9).
Table 7.
Results of molecular docking experiments targeting anticancer protein uinine reductase (PDB: 3G5M) and aromatase (PDB: 3EQM)
| Molecule | H-bonds | Bound amino acid residues | Binding affinity (kcal/mol) |
|---|---|---|---|
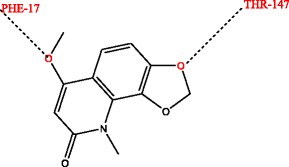 |
2 |
ASP-309, MET-374 |
-9.5 |
| Androstenedione (control inhibitor) | |||
 |
02 |
PHE-17, THR-147 |
-5.8 |
| 6-methoxy-9-methyl[1,3]dioxolo[4,5-h]quinolin- 8(9 h)-one (Control inhibitor) | |||
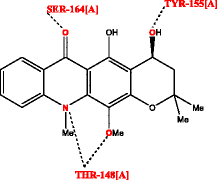 |
04 |
LEU-103, TYR-104, SER-107, GLY-149 |
-6.1 |
| Compound 1 | |||
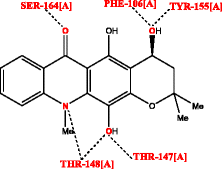 |
04 |
THR-148, TYR-155, SER-164 |
-6.7 |
| Compound 8 | |||
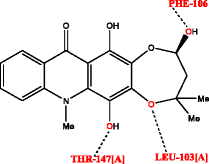 |
06 |
PHE-106, THR-147, THR-148, TYR-155, SER-164 |
-7.3 |
| Compound 9 | |||
 |
03 | LEU-103, PHE-106, THR-147 | -7.7 |
| Compound 10 |
Table 8.
Derived pharmacophore model for anticancer activity of acridones active against human cancer cell line MCF-7
| Acridones | Score obtained | Categorizing through threshold (S = 4.1274) | Predicted activity | Experimental activity (IC50 in μM) | Experimental response |
|---|---|---|---|---|---|
| 1. |
4.2712 |
> 4.1274 |
significantly active |
229 |
Poor activity |
| 2. |
3.5826 |
< 4.1274 |
Non-significant activity |
Not tested |
Not tested |
| 3. |
3.6316 |
< 4.1274 |
Non-significant activity |
Not tested |
Not tested |
| 4. |
3.6796 |
< 4.1274 |
Non-significant activity |
>300 |
Non active |
| 5. |
3.3015 |
< 4.1274 |
Non-significant activity |
>300 |
Non active |
| 6. |
2.9402 |
< 4.1274 |
Non-significant activity |
Not tested |
Not tested |
| 7. |
2.4587 |
< 4.1274 |
Non-significant activity |
Not tested |
Not tested |
| 8. |
5.1457 |
> 4.1274 |
Significantly active |
205 |
Poor activity |
| 9. |
4.9482 |
> 4.1274 |
Significantly active |
Not tested |
Not tested |
| 10. | 5.5092 | > 4.1274 | Significantly active | Not tested | Not tested |
The threshold score (S) of 4.1274 has been selected for the screening of lead compounds.
Table 9.
Compliance of acridone alkaloids with standard parameters of Lipinski’s rule of five for drug likeness and oral bioavailability
| Acridone alkaloids | miLOGP | MW | nON | nOHNH | nviolations |
|---|---|---|---|---|---|
|
1 |
1.926 |
301.298 |
6 |
2 |
0 |
|
2 |
1.862 |
257.245 |
5 |
3 |
0 |
|
3 |
1.119 |
303.27 |
7 |
4 |
0 |
|
4 |
2.189 |
285.299 |
5 |
1 |
0 |
|
5 |
2.433 |
255.273 |
4 |
1 |
0 |
|
6 |
1.913 |
271.272 |
5 |
2 |
0 |
|
7 |
2.173 |
255.273 |
4 |
1 |
0 |
|
8 |
2.476 |
355.39 |
6 |
2 |
0 |
|
9 |
2.2 |
341.363 |
6 |
3 |
0 |
| 10 | 2.411 | 357.362 | 7 | 3 | 0 |
miLogP = octanol/water partition coefficient, MW = molecular weight, nON = number of hydrogen acceptor, nOHNH = number of hydrogen donor, nviolations = violations from Lipinski’s rule.
Experimental
General experimental procedures
1H and 13C NMR spectra were obtained with a FT-NMR 300 MHz equipped with a 5 mm 1H and 13C (ATP) probe operating at 300 and 75 MHz, respectively, with TMS as internal standard. Chemical shifts were reported in δ (ppm) and coupling constants (J) were measured in Hz. QTOF-HRMS spectra were recorded on Agilent 6520-QTOF LC/MS having an ESI source in positive mode. Dry Nitrogen was used with 11 LPM flow rate for ionization at 350 Dc. The ESIMS was recorded on API-3000 LC-MS/MS ABSCIEX in 1.0 ppm solution through MS grade acetonitrile-water with injection mode: infusion through motor driven hardware syringe. UV analysis was carried out on a Spectronic Genesys 2 spectrophotometer and IR was recorded with FT-IR Perkin Elmer instrument. Optical rotations were taken with a Horiba SEPA-300 polarimeter. Melting points were determined on a Fisher-Johns melting point apparatus and were uncorrected. Precoated aluminium sheets silica gel 60 F254 TLC plates were used to check the purity of compounds and preparative TLC was performed on glass plates of silica gel 60 (20×20 cm, Merck). Flash chromatography was performed with a Buchi Pump manager C-615 flash model operating with two pump modules C-605. Spots were viewed under UV lamp (254 nm and 365 nm) or sprayed with Anisaldehyde- sulfuric acid and Draggendorff reagents. All reagents used, were of analytical grades.
Plant material
The dried fruits of Z. leprieurii and Z. zanthoxyloides were collected at Douala, Littoral Province of Cameroon in December 2009. The plants were identified at the University of Douala (TF) and the samples were compared with the voucher specimen number 2713/SRFK/CAM and 21793/SFR/CAM for Z. leprieurii and Z. zanthoxyloides, respectively at the National Herbarium of Cameroon, Yaounde.
Extraction and isolation
Extraction and isolation from Z. zanthoxyloides
The dried and ground fruits (400 g) were successively extracted under percolation at room temperature overnight with n-hexane three times. Thereafter, they were extracted by methanol at room temperature overnight which gave 16 g of crude extracts. A portion of this extract (10 g) was dissolved in CHCl3 (30 mL) and extracted three times with 5 N HCL. The aqueous layer was basified with 25% ammonium hydroxide and the basic alkaloidal part was extracted with CHCl3 (3×100 mL). The extract was dried over anhydrous sodium sulfate and solvent was evaporated. The extract (240 mg) obtained as above, after silica gel column chromatography (58 g, 60-120 mesh, 2.3 × 93 cm) with the mixtures of n-hexane, EtOAc and MeOH in the increasing order of polarity in basic medium (2 drops ammonium hydroxide/100 mL solvent), afforded 16 fractions. From fraction 8, after preparative TLC, 4 (5 mg) was obtained. While fraction 9 (n-hexane-EtOAc, 1:4) was left to crystallize at room temperature to obtain orange crystals of 3 (3.5 mg, TLC, EtOAc- CHCl3- MeOH, 97:1:2, Rf 0.24), fraction 10 eluted with n-hexane-EtOAc (1:4 and 1:9) was further purified through preparative TLC to afford 2 (6 mg, TLC, MeOH + 1% NH4OH- C6H6- CH2Cl2, 1:2:2, Rf 0.64). Fractions (11-13) which were complex mixtures were pooled together and purified through flash chromatography (20 g, 230-400 mesh, 15 × 100 mm glass column C-690 Sepacore Buchi fitted with a precolumn of ID 15 mm) with EtOAc as solvent A and MeOH plus 2 drops ammonium hydroxide/100 mL as solvent B. The flow rate was set up to 2.5 mL/min. A total of 52 fractions (each of 12 mL) were collected and pooled according to their TLC pattern into four fractions. Fractions 1 and 2 gave yellowish crystals of 1 (32 mg, TLC, EtOAc- MeOH + 1% NH4OH, 9:1, Rf 0.71), 8 (19.2 mg, TLC, EtOAc- MeOH+ 1% NH4OH, 9:1, Rf 0.67) and 5. Fraction 3 afforded 9 (10.3 mg, TLC, EtOAc- MeOH + 1% NH4OH, 9:1, Rf 0.53) and fraction 4 yielded 10 (22 mg, TLC, EtOAc- MeOH + 1% NH4OH 9:1, Rf 0.49). All the isolated alkaloids gave yellowish orange colored crystals or crystalline yellow mass and positive test with Dragendorff reagent.
Extraction and isolation from Z. leprieurii
The dried fruits (380 g) after extraction as described above, yielded 48 g of methanol extract from which 40 g was dissolved in CHCl3 (110 mL) and the alkaloids extracted as detailed above. The silica gel cc (120 g, 60-120 mesh, 2.5 × 100 cm) yielded complex mixtures (340 mg) which after flash chromatography (116 g, 230-400 mesh, 36 × 230 mm glass column C-690 Sepacore Buchi) using n-Hexane as solvent A and EtOAc plus 2 drops ammonium hydroxide/100 mL as solvent B with the flow rate stated as above, afforded eight fractions. Recrystallisation of fractions 3, 5 and 6 in MeOH- CHCl3 afforded 7 (10 mg), 1 (3 mg) and 4 (40 mg), respectively. Compound 6 (5 mg) was obtained from fraction 7, after recrystallization in MeOH- CHCl3.
Spectroscopic data of new compounds
3-Hydroxy-1,5,6-trimethoxy-9-acridone (1): Mp: 165°C; UV (CHCl3) λmax (log ϵ) nm: 233 (0.781), 252 (2.511), 320 (0.426); IR (KBr) νmax cm-1: 3448, 2929, 1620, 1579, 1507; 1H and 13C NMR see Tables 1 and 2; 1H1H COSY: H-8 at δ 7.99 ― H-7 at δ 7.21; HMBC: C-3-OH → C-3, C-2, C-4; C-5-OMe → C-5; C-6-OMe → C-6; ESIMS m/z (rel. int.): 301 (5) [M]+, 282 (100), 260 (82), 227 (30), 202 (42); HRMS: 301.0944 (Calc. for C16H15O5N 301.0945).
1,6-Dihydroxy-3-methoxy-9-acridone (2): UV (CHCl3) λmax (log ϵ) nm: 251 (2.011), 321 (0.406); IR (KBr) νmax cm-1: 3438, 2929, 1620, 1579; 1H and 13C NMR see Tables 1 and 2; 1H1H COSY: H-8 at δ 7.89 ― H-7 at δ 7.07; HMBC: C-3-OMe → C-3; ESIMS m/z (rel. int.): 257 (15) [M]+, 245 (60), 214 (100); HRMS: 257.0681 (Calc. for C14H11O4N 257.0683).
3,4,5,7-Tetrahydroxy-1-methoxy-10-methyl-9-acridone (3): Mp > 250°C; UV (CHCl3) λmax (log ϵ) nm: 262 (2.511), 320 (0.426); IR (KBr) νmax cm-1: 3430, 2929, 1620, 1580; 1H and 13C NMR see Tables 1 and 2; HMBC: C-1-OMe → C-1; H-2 → C-2, C-1, C-3; ESIMS m/z (rel. int.): 303 (5) [M+], 298 (8), 286 (22), 282 (100); HRMS: 303.0734 (Calc. for C15H13O6N 303.0737).
4-Methoxyzanthacridone (8): [α]D22: -3.69o (CHCl3, c = 0.22); UV (CHCl3) λmax (log ϵ) nm: 243 (1.898), 275 (1.204), 327 (1.005); IR (KBr) νmax cm-1: 3368, 2929, 1654, 1620, 1579; 1H and 13C NMR see Tables 1 and 2; 1H1H COSY: H-8 at δ 7.82 ― H-7 at δ 7.29 ― H-6 at δ 7.61 ― H-5 at δ 7.40; H-4’ at δ 3.50 ― H-3a’ at δ 2.72 ― H-3b’ at δ 3.07; HMBC: H-4’ à C-2, C-1, C-2’, C-3’; H-3a’ and H-3b’à C-2, C-4’, C-2’; C-2’Me àC-2’, C-3’; NOESY: H4’― C-1-OH, H-3’a, H-3’b; ESIMS m/z (rel. int.): 355 (2) [M]+, 330 (18), 314 (100), 202 (8); HRMS: 355.1416 (Calc. for C20H21O5N 355.1414).
Acetylation of 4-methoxyzanthacridone (8)
Using 1 mL of acetic anhydride and one drop of pyridine, 8 (2 mg) was acetylated and left for reflux at ambient temperature for 6 hrs. The reaction mixture after solvent evaporation under rotavapor yielded compound 8a. 1H NMR (CDCl3, δ): 4.34 (dd, J = 10.0, 3.5 Hz) H4’, 2.10, s, OAc.
4-Hydroxyzanthacridone (9): [α]D22: -4.11o (CHCl3, c = 0.11); UV (CHCl3) λmax (log ϵ) nm: 243 (1.826), 318 (0.618), 329 (0.562); IR (KBr) νmax cm-1: 3360, 2931, 1652, 1620, 1581; 1H and 13C NMR see Tables 1 and 2; 1H1H COSY: H-8 at δ 8.40 ― H-7 at δ 7.34 ― H-6 at δ 7.59 ― H-5 at δ 7.41; H-4’ at δ 3.63 ― H-3a’ at δ 2.68 ― H-3b’ ― at δ 3.00; HMBC: H-4’ → C-2, C-1, C-2’, C-3’; H-3a’and H-3b’ → C-2, C-4’, C-2’; C-2’Me →C-2’, C-3’; NOESY: H-4’—C-1-OH, H-3’a, H-3’b; ESIMS m/z (rel. int.): 341 (2) [M]+, 296 (32), 282 (100), 260 (35), 224 (40); HRMS: 341.1257 (Calc. for C19H19O5N 341.1258).
Acetylation of 4-hydroxyzanthacridone (9)
Using 1 mL of acetic anhydride and one drop of pyridine, 9 (2 mg) was acetylated and left for reflux at ambient temperature for 6 hrs. The reaction mixture after solvent evaporation under rotavapor yielded compound 9a. 1H NMR (CDCl3, δ): 4.22 (dd, J = 10.0, 3.5 Hz) H-4’, 2.10, s, OAc.
4-Hydroxyzanthacridone oxide (2,4’) (10): [α]D22: -1.32o (CHCl3, c = 0.25); UV (CHCl3) λmax (log ϵ) nm: 242 (1.247), 312 (0.507); IR (KBr) νmax cm-1: 3423, 1654, 1623, 1582; 1H and 13C NMR see Tables 1 and 2; 1H1H COSY: H-8 at δ 8.39 ― H-7 at δ 7.34 ― H-6 at δ 7.55 ― H-5 at δ 7.34; H-4’ at δ 4.85 ― H-3a’ at δ 3.23 and H-3b’ at δ 3.25; HMBC: H4’ → C-2, C-2’, C-3’; H-3a’and H-3b’ → C-4’, C-2’; C-2’Me → C-2’, C-3’; NOESY: H-4’― C-1-OH, H-3’a, H-3’b; C-2’Me— C-4OH; ESIMS m/z (rel. int.): 357 (2) [M]+, 316 (28), 296 (72), 282 (100), 260 (48); HRMS: 357.1210 (Calc. for C19H19O6N 357.1207).
Acetylation of 4-hydroxyzanthacridone oxide (2,4’) (10)
Acetylation was done on 2 mg of 10 using 1 mL of acetic anhydride and one drop of pyridine and left for reflux at ambient temperature for 6 hrs. The reaction mixture after solvent evaporation under rotavapor yielded compound 10a. 1H NMR (CDCl3, δ): 5.35 (dd, J = 10.0, 3.5 Hz) H-4’, 2.11, s, OAc.
Biological assays
Antibacterial assay
The four bacterial strains viz., Micrococcus luteus MTCC2470, Staphylococcus aureus MTCC96, Bacillus subtilis MTCC121 and Pseudomonas aeruginosa MTCC741 used for the assay, were acquired from Microbial Type Culture Collection (MTCC), Chandigarh, India. MIC values of the tested compounds 1, 3, 4, 7, 8 and 10 against pathogens, were determined by microdilution method [40]. The inocula of microorganisms were prepared from a 17 h broth culture and the suspensions were adjusted to 0.5 Mc Farland turbidity. The tested samples were first dissolved in dimethyl sulfoxide (DMSO), then in Mueller Hinton Broth to the highest dilution of 500 μg/mL. Then, serial twofold dilutions were made in a series of concentration with 0.0078 μg/mL being the smallest concentration in the 96 wells microplate. Ampicillin and Kanamycin diluted prior in water were used as reference antibiotics. Negative control was made with DMSO.
Cytotoxicity assay
IC50 values for compounds 1, 4, 7 and 8 were determined as described previously [41]. 1.5-2.0X103 cells/well were incubated in the 5% CO2 incubator for 24 h to enable them to adhere properly to the 96-well microplates (Greiner, Germany). Test compounds dissolved in DMSO were added at varying concentrations and left for 4 h. After 48 h, 10 μL MTT (Sigma M-2128) was added and the plates were incubated for an additional 4 h. The absorbance was read on a Fluostar Omega Microplate reader (BMG LABTECH, Australia) at 570 nm within 1 h of DMSO addition and the respective IC50 were calculated. The experiments were done in triplicate.
Molecular modeling and docking studies
A pharmacophore model has been derived to predict the cytotoxic and antibacterial activities of acridone alkaloids against human cancer cell line MCF-7 and M. luteus, respectively. Derived QSAR models have been validated using standard method [42]. For QSAR model development against MCF-7, a total of 27 known compounds were used in the training data set. These compounds showed >70% structural similarity with acridone alkaloid skeleton. For statistical relationship eight chemical descriptors namely, AlogP, BCUTw-1 l, Eccentric connectivity index, LipoaffinityIndex, McGowan volume, Petitigen number, TPSA and Zagreb index were calculated for QSAR model building. The similar approach has been used for the development of QSAR model for antibacterial activity. On the other hand, molecular docking simulation study was performed by AutoDock-Vina software [43]. Aromatase (PDB:3EQM), quinone reductase (PDB: 3G5M) and glycosyltransferase (WAAG) (PDB: 2IW1) target proteins have been used during docking experiments to reveal the binding affinity against anticancer and antibacterial targets, respectively [37,44]. Drug likeness and oral bioavailability of studied compounds were evaluated through Lipinski’s rule of five.
Conclusions
Although acridone class of alkaloids are uncommon in nature but they have been found to be present in several Genus of Family Rutaceae. Only one specy of Zanthoxylum, i.e. Z. leprieurii, has recently been investigated for acridone alkaloids, in detail. Now, we have investigated the fruits of medicinally important species of Cameroonian Z. zanthoxyloides and Z. leprieurii, for their acridone alkaloids and confirmed their antibacterial and cytotoxic activities. The prediction of possible mechanism of action has been done through molecular modeling of the isolated compounds. Moreover, the present work of isolation of novel acridone alkaloids from Z. zanthoxyloides emphasizes their uniqueness as taxonomic markers among the African species of this genus. We have isolated and identified 10 acridones out of which 6 were new (1, 2, 3, 8, 9 and 10). Acridones 8, 9 and 10 are described with an unusual tetracyclic acridone which has now been named as zanthacridone because of their new carbon skeleton. The quantitative SAR and molecular modeling studies suggested that the compounds 1, 9 and 10 are inhibitors of aromatase, quinone reductase and glycosyltransferase. Therefore, these compounds hold the promise for further studies to use them as chemopreventive agents and LPS inhibitors.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LM and WNAV conceived the work; WNAV collected the fruits for onward extraction and purification of compounds, the antibacterial assay and drafted the manuscript. LM identified the compounds after purification, finalized the manuscript and supervised the bench work. SK performed the in vitro cytotoxic activity. OP and FK did the QSAR and modeling studies. TF identified the plant material and helped in the collections. VK supervised the work and improved the manuscript. All the authors read and approved the manuscript.
Supplementary Material
Figure 1a: 1H NMR of compound 1. b: 13C NMR of compound 1. c: DEPT of compound 1. d: COSY of compound 1. e: Mass of compound 1. f: IR of compound 1. Figure 2a: 1H NMR of compound 2. b: Mass of compound 2. Figure 3a: 1H NMR of compound 3. b: 13C NMR of compound 3. c: DEPT of compound 3. d: Mass of compound 3. e: IR of compound 3. Figure 4a: 1H NMR of compound 8. b: 13C NMR of compound 8. c: DEPT of compound 8. d: COSY of compound 8. e: NOESY of compound 8. f: Mass of compound 8. g: IR of compound 8. Figure 5a: 1H NMR of compound 9. b: 13C NMR of compound 9. c: 13C NMR of compound 9. d: DEPT of compound 9. e: DEPT of compound 9. f: Mass of compound 9. Figure 6a: 1H NMR of compound 10. b: 13C NMR of compound 10. c: DEPT of compound 10. d: COSY of compound 10. e: Mass of compound 10. f: IR of compound 10.
Contributor Information
Vyry NA Wouatsa, Email: alives1@yahoo.fr.
Laxminarain Misra, Email: laxmisra@hotmail.com.
Shiv Kumar, Email: ars198922@gmail.com.
Om Prakash, Email: mailbox4op@gmail.com.
Feroz Khan, Email: f.khan@cimap.res.in.
Francois Tchoumbougnang, Email: tchoumbougnang@yahoo.fr.
R Kumar Venkatesh, Email: drvenkateshkumar@yahoo.com.
Acknowledgements
Ms Wouatsa N. A. Vyry is grateful to TWAS and CSIR for the award of a fellowship and Director, CSIR-CIMAP is thanked for his encouragements and providing research facilities.
References
- Matu EN. In: PROTA (Plant Resources of Tropical Africa/Ressources végétales de l’Afrique tropicale) Schmelzer GH, Gurib-Fakim A, editor. Wageningen: Internet record from Protabase; 2011. Zanthoxylum zanthoxyloides (Lam.) Zepern. & Timler. [Google Scholar]
- Tabuti JRS. In: PROTA (Plant Resources of Tropical Africa/Ressources végétales de l’Afrique tropicale) Schmelzer GH, Gurib-Fakim A, editor. Wageningen; 2011. Zanthoxylum leprieurii Guill. & Perr. (Internet record from Protabase). [Google Scholar]
- Noumi E. Les plantes à épices, à condiments et à aromates du Cameroun. Université de Yaoundé; 1984. (Thèse de Doctorat). [Google Scholar]
- Ngane AN, Biyiti L, Zollo PHA, Bouchet P. Evaluation of antifungal activity of extracts of two Cameroonian Rutaceae: Zanthoxylum leprieurii Guill. et Perr. and Zanthoxylum xanthoxyloides Waterm. J Ethnopharmacol. 2000;70:335–342. doi: 10.1016/S0378-8741(99)00188-9. [DOI] [PubMed] [Google Scholar]
- Ngoumfo RM, Jouda J-B, Mouafo FT, Komguem J, Mbazoa CD, Shiao TC, Choudhary MI, Laatsch H, Legault J, Pichette A, Roy A. In vitro cytotoxic activity of isolated acridone alkaloids from Zanthoxylum leprieurii Guill. & Perr. Bioorg Med Chem. 2010;18:3601–3605. doi: 10.1016/j.bmc.2010.03.040. [DOI] [PubMed] [Google Scholar]
- Tchinda AT, Fuendjiep V, Sajjad A, Matchawe C, Wafo P, Khan S, Tane P, Choudhary MI. Bioactive compounds from the fruits of Zanthoxylum leprieurii. Pharmacologyonline. 2009;1:406–415. [Google Scholar]
- Adesina SK. Further new constituents of Zanthoxylum leprieurii. Fitoterapia. 1987;58:123–126. [Google Scholar]
- Adesina SK. The Nigerian Zanthoxylum; chemical and biological values. Afr J Trad CAM. 2005;2:282–301. [Google Scholar]
- Gansané A, Sanon S, Ouattara LP, Traoré A, Hutter S, Olivier E, Azas N, Sirima SB, Nebié I. Antiplasmodial activity and toxicity of crude extracts from alternative parts of plants widely used for the treatment of malaria in Burkina Faso: contribution to their preservation. Parasitol Res. 2010;106:335–340. doi: 10.1007/s00436-009-1663-y. [DOI] [PubMed] [Google Scholar]
- Udo IO. Potentials of Zanthoxylum zanthoxyloides (Lam.) for the control of stored product insect pests. J Stored Prod Postharvest Res. 2011;2:40–44. [Google Scholar]
- Ynalvez RA, Cardenas C, Addo JK, Adukpo GE, Dadson BA, Addo-Mensah A. Evaluation of the antimicrobial activity of Zanthoxylum zanthoxyloides root bark extracts. Res J Med Pl. 2012;6:149–159. [Google Scholar]
- Ouattara B, Jansen O, Angenot L, Guissou IP, Frederich M, Fondu P, Tits M. Anti-sickling properties of divanilloylquinic acids isolated from Fagara zanthoxyloides Lam. (Rutaceae) Phytomedicine. 2009;16:125–129. doi: 10.1016/j.phymed.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Chaaib F, Quieroz EF, Ndjoko K, Diallo D, Hostettmann K. Antifungal and antioxidant compounds from the root bark of Fagara zanthoxyloides. Planta Med. 2003;69:316–320. doi: 10.1055/s-2003-38877. [DOI] [PubMed] [Google Scholar]
- Queiros EF, Hay A-E, Chaaib F, van Diemen D, Diallo D, Hostettmann K. New and bioactive aromatic compounds from Zanthoxylum zanthoxyloides. Planta Med. 2006;72:746–750. doi: 10.1055/s-2006-941504. [DOI] [PubMed] [Google Scholar]
- Adesina SK. Further novel constituents of Zanthoxylum zanthoxyloides root and pericarp. J Nat Prod. 1986;49:715–716. doi: 10.1021/np50046a035. [DOI] [Google Scholar]
- Fogang HPD, Tapondjou LA, Womeni HM, Quassinti L, Bramucci M, Vitali LA, Petrelli D, Lupidi G, Maggi F, Papa F, Vittoric S, Barbonid L. Characterization and biological activity of essential oils from fruits of Zanthoxylum xanthoxyloides Lam. and Z. leprieurii Guill. & Perr., two culinary plants from Cameroon. Flav and Fragr J. 2012;27:171–179. doi: 10.1002/ffj.3083. [DOI] [Google Scholar]
- Choumessi AT, Loureiro R, Silva AM, Moreira AC, Pieme AC, Tazoacha A, Oliveira PJ, Penlap VB. Toxicity evaluation of some traditional African spices on breast cancer cells and isolated rat hepatic mitochondria. Food Chem Toxicol. 2012;50:4199–4208. doi: 10.1016/j.fct.2012.08.008. [DOI] [PubMed] [Google Scholar]
- Misra LN, Lal P, Chaurasia ND, Sangwan RS, Sinha S, Tuli R. Selective reactivity of 2-mercaptoethanol with 5,6β-epoxide in steroids from Withania somnifera. Steroids. 2008;73:245–251. doi: 10.1016/j.steroids.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Misra LN, Mishra P, Pandey A, Sangwan RS, Sangwan NS, Tuli R. Withanolides from Withania somnifera roots. Phytochemistry. 2008;69:1000–1004. doi: 10.1016/j.phytochem.2007.10.024. [DOI] [PubMed] [Google Scholar]
- Misra LN, Mishra P, Pandey A, Sangwan RS, Sangwan NS. 1,4-Dioxane and ergosterol derivatives from Withania somnifera roots. J Asian Nat Prod Res. 2012;14:39–45. doi: 10.1080/10286020.2011.622719. [DOI] [PubMed] [Google Scholar]
- Misra LN, Saikia AK. Chemotypic variation in Indian Lantana camara essential oil. J Essential Oil Res. 2011;23:1–5. [Google Scholar]
- Rout PK, Sahoo D, Misra LN. Comparison of extraction methods of Mimusops elengi L. Flowers. Ind Crops and Prod. 2010;32:678–680. doi: 10.1016/j.indcrop.2010.05.019. [DOI] [Google Scholar]
- Misra LN, Wouatsa NAV, Kumar S, Kumar VR, Tchoumbougnang F. Antibacterial, cytotoxic activities and chemical composition of fruits of two Cameroonian Zanthoxylum species. J Ethnopharmacol. 2013;148:74–80. doi: 10.1016/j.jep.2013.03.069. [DOI] [PubMed] [Google Scholar]
- Wouatsa NAV, Misra LN, Kumar RV, Darokar MP, Tchoumbougnang F. Zantholic acid, a new monoterpenoid from Zanthoxylum zanthoxyloides. Nat Prod Res. 2013. in press. [DOI] [PubMed]
- Chansriniyom C, Ruangrungsi N, Lipipun V, Kumamoto T, Ishikawa T. Isolation of acridone alkaloids and N-[(4-monoterpenyloxy)phenylethyl]- substituted sulfur-containing propanamide derivatives from Glycosmis parva and their anti-herpes simplex virus activity. Chem Pharm Bull. 2009;57:1246–1250. doi: 10.1248/cpb.57.1246. [DOI] [PubMed] [Google Scholar]
- Costes N, Le Deit H, Michel S, Tillequin F, Koch M, Pfeiffer B, Renard P, Leonce S, Guilbaud N, Kraus-Berthier L, Pierre A, Atassi G. Synthesis and cytotoxic and antitumor activity of Benzo[b]pyrano[3,2-h]acridin-7-one analogues of acronycine. J Med Chem. 2000;43:2395–2402. doi: 10.1021/jm990972l. [DOI] [PubMed] [Google Scholar]
- Elomri A, Mitaku S, Michel S, Skaltsounis A-L, Tillequin F, Koch M, Pierre A, Guilbaud N, Leonce S, Kraus-Berthier L, Rolland Y, Atassi G. Synthesis and cytotoxic and antitumor activity of esters in the 1, 2-dihydroxy-1,2-dihydroacronycine series. J Med Chem. 1996;39:4762–4766. doi: 10.1021/jm9602975. [DOI] [PubMed] [Google Scholar]
- Ito C, Kondo Y, Wu T-S, Furukawa H. Structures of four new acridones and three new quinolone alkaloids. Chem Pharm Bull. 2000;48:65–70. doi: 10.1248/cpb.48.65. [DOI] [PubMed] [Google Scholar]
- Magiatis P, Mitaku S, Michel S, Skaltsounis A-L, Tillequin F, Pierre A, Atassi G. Ring expansion reactions of acronycine to fused 1,4-oxazepine and 1,4-dioxepin systems. Nat Prod Lett. 2000;14:183–190. doi: 10.1080/10575630008041229. [DOI] [Google Scholar]
- Naidoo D, Coombes PH, Mulholland DA, Crouch NR, van den Bergh AJJ. Substituted acridone alkaloids from Toddaliopsis bremekampii (Rutaceae: Toddalioideae) of South-central Africa. Phytochemistry. 2005;66:1724–1728. doi: 10.1016/j.phytochem.2005.04.036. [DOI] [PubMed] [Google Scholar]
- Wu T-S, Chen C-M. Acridone alkaloids from the root bark of Severinia buxifolia in Hainan. Chem Pharm Bull. 2000;48:85–90. doi: 10.1248/cpb.48.85. [DOI] [PubMed] [Google Scholar]
- Yang Z-L, Xie X-H, Jiang X-J, Huang Y-B, Liu J-K. A new acridone alkaloid from Micromelum integerrimum. Chem Pharm Bull. 2009;57:734–735. doi: 10.1248/cpb.57.734. [DOI] [PubMed] [Google Scholar]
- Ahsan M, Gray AI, Leach G, Waterman PG. Quinolone and acridone alkaloids from Boronia lanceolata. Phytochemistry. 1993;33:1507–1510. doi: 10.1016/0031-9422(93)85122-8. [DOI] [Google Scholar]
- Bergenthal D, Mester I, Rozsa Z, Reisch J. 13C-NMR-Spektren einiger acridon-alkaloide. Phytochemistry. 1979;18:161–163. doi: 10.1016/S0031-9422(00)90937-3. [DOI] [Google Scholar]
- Magiatis P, Mitaku S, Michel S, Pierre A, Atassi G. Synthesis and cytotoxic activitiy of isoacronycine and its derivatives. Heterocycles. 2002;57:341–351. doi: 10.3987/COM-01-9402. [DOI] [Google Scholar]
- Nazrul Islam SK, Gray AI, Waterman PG, Ahasan M. Screening of eight alkaloids and ten flavonoids isolated from four species of the genus Boronia (Rutaceae) for antimicrobial activities against seventeen clinical microbial strains. Phytother Res. 2002;16:672–674. doi: 10.1002/ptr.999. [DOI] [PubMed] [Google Scholar]
- Perumal D, Sakharkar KR, Tang TH, Chow VTK, Lim CS, Samal A, Sugiura N, Sakharkar MK. Cloning and targeted disruption of two lipopolysaccharide biosynthesis genes, kdsA and waaG, of Pseudomonas aeruginosa PAO1 by site-directed mutagenesis. J Mol Microbiol Biotechnol. 2010;19:169–179. doi: 10.1159/000322157. [DOI] [PubMed] [Google Scholar]
- Ghosh D, Griswold J, Erman M, Pangborn W. Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature. 2009;457:219–223. doi: 10.1038/nature07614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz-Drogon A, Dorner B, Erker T, Boguszewska-Chachulska AM. Synthesis of new acridone derivatives, inhibitors of NS3 helicase, which efficiently and specifically inhibit subgenomic HCV replication. J Med Chem. 2010;53:3117–3126. doi: 10.1021/jm901741p. [DOI] [PubMed] [Google Scholar]
- Zgoda JR, Porter JR. A convenient microdilution method for screening natural products against bacteria and fungi. Pharmaceutical Biol. 2001;39:221–225. doi: 10.1076/phbi.39.3.221.5934. [DOI] [Google Scholar]
- Saxena M, Faridi U, Mishra R, Gupta MM, Darokar MP, Srivastava SK, Singh D, Luqman S, Khanuja SPS. Cytotoxic agents from Terminalia arjuna. Planta Med. 2007;73:1486–1490. doi: 10.1055/s-2007-990258. [DOI] [PubMed] [Google Scholar]
- Prakash O, Khan F, Sangwan RS, Misra LN. ANN-QSAR model for virtual screening of Androstenedione C-Skeleton containing phytomolecules and analogues for cytotoxic activity against Human Breast cancer cell line MCF-7. Comb Chem High Throughput Screen. 2013;16:57–72. doi: 10.2174/1386207311316010008. [DOI] [PubMed] [Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comp Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balunas MJ, Su B, Brueggemeier RW, Kinghorn AD. Natural products as aromatase inhibitors. Anticancer Agents Med Chem. 2008;8:646–682. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1a: 1H NMR of compound 1. b: 13C NMR of compound 1. c: DEPT of compound 1. d: COSY of compound 1. e: Mass of compound 1. f: IR of compound 1. Figure 2a: 1H NMR of compound 2. b: Mass of compound 2. Figure 3a: 1H NMR of compound 3. b: 13C NMR of compound 3. c: DEPT of compound 3. d: Mass of compound 3. e: IR of compound 3. Figure 4a: 1H NMR of compound 8. b: 13C NMR of compound 8. c: DEPT of compound 8. d: COSY of compound 8. e: NOESY of compound 8. f: Mass of compound 8. g: IR of compound 8. Figure 5a: 1H NMR of compound 9. b: 13C NMR of compound 9. c: 13C NMR of compound 9. d: DEPT of compound 9. e: DEPT of compound 9. f: Mass of compound 9. Figure 6a: 1H NMR of compound 10. b: 13C NMR of compound 10. c: DEPT of compound 10. d: COSY of compound 10. e: Mass of compound 10. f: IR of compound 10.