

Key Points
Gene expression in TCR-engineered cells resembles that of virus-reactive cells more than native tumor antigen-reactive cells.
Persisting TCR gene–engineered T cells are sensitive to PD-L1–PD-1 interaction but CD160-associated impairment is ligand-independent.
Abstract
Despite significant progress in the development of adoptive cell-transfer therapies (ACTs) using gene-engineered T cells, little is known about the fate of cells following infusion. To address that, we performed a comparative analysis of gene expression between T-cell receptor–engineered lymphocytes persisting in the circulation 1 month after administration and the product that was infused. We observed that 156 genes related to immune function were differentially expressed, including underexpression of stimulators of lymphocyte function and overexpression of inhibitory genes in postinfusion cells. Of genes overexpressed postinfusion, the product of programmed cell death 1 (PDCD1), coinhibitory receptor PD-1, was expressed at a higher percentage in postinfusion lymphocytes than in the infusion product. This was associated with a higher sensitivity to inhibition of cytokine production by interaction with its ligand PD-L1. Coinhibitory receptor CD160 was also overexpressed in persisting cells, and its expression was associated with decreased reactivity, which surprisingly was found to be ligand-independent. These results contribute to a deeper understanding of the properties of transgenic lymphocytes used to treat human malignancies and may provide a rationale for the development of combination therapies as a method to improve ACT. This trial was registered at www.clinicaltrials.gov as #NCT00509288, #NCT00923195, and #NCT01273181.
Introduction
Clinical protocols based on administration of ex vivo–stimulated tumor-infiltrating lymphocytes (TILs) demonstrated that cell-based immunotherapy is a safe and efficacious option for the treatment of otherwise incurable malignancies.1 More recently, advances in gene transfer technologies facilitated the development of alternative approaches that involved the delivery of transgenes encoding antitumor antigen receptors into readily available peripheral blood lymphocytes (PBLs). Antibody-based chimeric antigen receptors (CARs), or natural T-cell receptor (TCR)–engineered T cells from peripheral blood have mediated cancer regression in both hematological and solid malignancies,2-5 but the biology of these genetically modified cells has not been thoroughly characterized in vivo.
Previous reports have shown that circulating lymphocytes derived from the adoptively transferred gene-modified PBLs displayed different phenotypic and molecular traits than the cellular product administered to patients.4,6 While the study of the biology of genetically engineered PBLs in patients is limited, we previously reported that expression of introduced TCR genes was decreased in postinfusion cells, which compromised the antitumor reactivity of these cells.7
In the present study, we conducted a detailed analysis of immune-related gene expression using platforms that allowed for the direct quantitation of gene expression in order to study the molecular mechanisms governing the fate of these engineered cells in vivo. These studies used murine-derived TCRs as a unique cell-surface tag to directly isolate engineered T cells from clinical samples taken from 10 patients undergoing adoptive cell-transfer therapies (ACTs). We focused our attention on genes involved in T-cell reactivity, and observed overexpression of several inhibitors of T-cell function in persisting cells, including programmed cell death 1 (PDCD1). Expression of the gene product, PD-1, was confirmed in persisting lymphocytes and was associated with reduced production of interferon (IFN)γ upon in vitro stimulation. Moreover, we studied the potential detrimental effects of surface expression of CD160, a molecule previously described as a coinhibitory receptor for T cells, and demonstrated the novel finding that, in the context of ACT, CD160 expression is associated with decreased reactivity of TCR-engineered CD8+ lymphocytes in a ligand-independent manner.
Materials and methods
Clinical samples
PBLs used in this study were obtained from melanoma patients enrolled in ACT protocols at the Surgery Branch, National Cancer Institute, National Institutes of Health (NCT00509288, NCT00923195, NCT01273181). All patients were treated under protocols reviewed and approved by the National Institutes of Health institutional biosafety committee, the National Institutes of Health recombinant DNA advisory committee, the National Cancer Institute institutional review board, and the Food and Drug Administration (all Bethesda, MD). All patients gave written informed consent for protocol enrollment in accordance with the Declaration of Helsinki. Details on retroviral vector transductions were previously reported.4,8
Flow cytometry and magnetic separation
Analytical flow cytometry analyses were performed using a FACSCanto I or FACSCanto II flow cytometer with FACSDiva software (BD Biosciences), and analyzed using FlowJo software (Tree Star). Preparative flow cytometry was performed using a FACSAria cell sorter (BD Biosciences). Details on antibodies can be found in supplemental Methods (available on the Blood website). Negative selection of T cells was performed using the Pan T cell isolation kit II (Miltenyi Biotec).
Gene-expression analysis
Total RNA from T cells was isolated using an RNeasy kit (Qiagen) according to the manufacturer’s instructions. The nCounter Analysis System9 (NanoString Technologies) was used to screen for the expression of 511 genes involved in human immunology using the nCounter GX Human Immunology Kit (NanoString Technologies) as instructed by the manufacturer. One hundred nanograms of total RNA was used. Counts were normalized to internal controls and reference genes (HPRT1, G6PD, and ABCF1) using nSolve software. Analysis of changes in gene expression was performed using Partek Genomic Suite (Partek Incorporated). For real-time polymerase chain reaction (PCR) analysis of gene expression, we used pathway-focused T Cell Anergy and Tolerance RT2 Profiler PCR array (PAHS-074C; SABiosciences). Data were analyzed using RT2 Profiler PCR Array Data Analysis software, provided by the manufacturer, normalizing gene expression to that of 5 reference genes (ACTB, HPRT1, B2M, GAPDH, and RPL13A). The complete lists of genes analyzed by NanoString and by PCR arrays are shown in supplemental Tables 4 and 5. CD160 and ACTB expression was measured using commercially available TaqMan probe/primer sets (Applied Biosystems) according to the manufacturer’s instructions.
Results
Murine TCRs permit direct isolation of gene-marked T cells from patient blood
Nine metastatic melanoma patients, and 1 synovial cell sarcoma patient, enrolled in clinical trials involving ACTs of lymphocytes engineered with murine TCRs were analyzed for persistence of TCR gene expression. Seven patients received HLA-A*0201–restricted TCRs specific for premelanosome protein gp100,4 2 of which also received anti-MART1 TCR-transduced cells in combination with total body irradiation (Clinical Trial #NCT00923195), and 3 patients received anti-MAGE-A3 TCR-engineered T cells.8 All patients received nonmyeloablative lymphodepleting chemotherapy prior to cell infusion and high-dose interleukin (IL)-2 postinfusion. Five of the patients included in this study were nonresponders, 3 were partial responders, and 2 were complete responders based on RECIST10 criteria (Table 1). We were able to accurately quantify the presence of transgenic lymphocytes in the patient circulation 1 month postinfusion by staining for the murine TCR β chain. Persistence at 1 month, determined by flow cytometry, ranged from 14% to 86% of the peripheral blood CD3+ cell and did not correlate with infused cell dose or with clinical response (Table 1).
Table 1.
Patient characteristics and treatment
| Patient (previous identifier) | Age/sex | Diagnosis | Sites of metastases | TCR | Culture conditions* | No. of IL-2 doses | No. of cells received, ×109 | % mTCRb+/CD3+ at 1 mo | Tumor response (duration, mo) |
|---|---|---|---|---|---|---|---|---|---|
| 1 (7)† | 60/M | Melanoma | In, bo, li, lu, sc | gp100 | S1d7 | 4 | 1.8 | 56 | NR |
| 2 (12)† | 62/M | Melanoma | lu, ln | gp100 | R2d13 | 7 | 47 | 38 | NR |
| 3 | 58/M | Melanoma | iliac ln | gp100 | R2d11 | 8 | 56 | 14 | NR |
| 4 | 56/F | Melanoma | lu | gp100‡ | R2d12 | 4 | 34‡ | 29 | NR |
| 5 (6)§ | 44/M | Melanoma | Right thigh, bilat inguinal and iliac nodes | MAGE A3 | R2d12 | 4 | 53 | 61 | NR |
| 6 (14)† | 51/F | Melanoma | lu, li | gp100 | R2d12 | 6 | 110 | 24 | PR (4) |
| 7 (1)§ | 59/M | Melanoma | lu | MAGE A3 | R2d12 | 6 | 28 | 33 | CR (23+) |
| 8 (11)† | 50/M | Melanoma | retroperit ln, iliac ln; br | gp100 | R2d12 | 8 | 69 | 81 | CR (48+) |
| 9 | 52/M | Melanoma | sc, lu, ln | gp100|| | R2d12 | 6 | 24|| | 44 | PR (3) |
| 10 (4)§ | 21/F | Synovial cell sarcoma | lu | MAGE A3 | R2d12 | 1 | 41 | 86 | PR (5) |
bilat, bilateral; bo, bone; br, brain; CR, complete response; li, liver; ln, lymph node; lu, lung; mTCRb+, murine-TCR β-chain positive; NR, no response; PR, partial response; retroperit, retroperitoneal; sc, subcutaneous; TCR, T-cell receptor.
S1d7 indicates initial stimulation with OKT3 and IL-2 during 7 days. R2d11, R2d12, and R2d13 indicate second stimulation with OKT3, IL-2, and feeder cells for 11, 12, and 13 days, respectively, using the Rapid Expansion Protocol.11
Patient identifying number as reported in Johnson et al.4
Also received 0.5e9 MART1 TCR-transduced cells.
Patient identifying number as reported in Morgan et al.8
Also received 23.5e9 MART1 TCR-transduced cells.
We isolated 3 cell populations from each patient for comparative gene-expression analysis: postinfusion, infusion, and preinfusion samples (Figure 1A). Transgenic T cells persisting 1 month postinfusion were fluorescence-activated cell sorter (FACS)–sorted based on dual staining with anti-CD3 and anti-mouse TCR β-chain (mTCRb) antibodies to high purity (Figure 1B-C). Infusion samples consisted of the infusion preparations administered to patients (>97% CD3+ cells). Nine of 10 patient cell preparations (patients 2-10, Table 1) were grown using the Rapid Expansion Protocol (REP),11 which included stimulation with anti-CD3 monoclonal antibody (mAb), IL-2, and addition of irradiated allogeneic peripheral blood mononuclear cell (PBMC) feeder cells (patient 1 received short-term cultured cells grown without feeders). A detailed description of the production of infusion cells is available in the supplemental Methods. Preinfusion samples consisted of CD3+ cells isolated by negative selection from patients’ peripheral blood before any treatment was initiated. In all cases, cryopreserved samples were thawed and immediately processed without further culture in vitro. Cells were kept on ice during the procedures to minimize manipulation-induced changes in gene expression.
Figure 1.

Isolation of TCR-engineered lymphocytes from peripheral blood. (A) Timeline representing the outline of TCR-transfer clinical protocols and the experimental design for the isolation of cells used in gene-expression analysis. PBMCs are isolated ∼24 days before infusion by apheresis and subsequently stimulated with OKT3 in presence of IL-2 (Stim1). Retroviral transductions take place 23 and 22 days before infusion and a REP is started 2 weeks before administration of the cells. Culture media are supplemented with IL-2 during the ex vivo culture. The resulting cellular product is administered on day 0, and IL-2 is given for the first few days (1-3 days) to support engraftment. Approximately 1 month after infusion PBMCs are isolated by apheresis. Samples included in this study were prepared as follows: preinfusion samples are CD3+ cells isolated by negative selection, using magnetic bead sorting, from PBMCs collected at ∼day −24. Infusion samples are aliquots of the final cellular product, isolated from the infusion bag. Postinfusion samples are CD3+ mTCRb+ cells isolated from PBMC samples 1 month after infusion by FACS sorting. (B-C) FACS sorting of engineered T cells from PBMCs at 1 month postinfusion. (B) Presort staining of CD3 and mTCRb. (C) Postsort analysis showing enrichment of T cells expressing murine TCRs. Gated on lymphoid, single, PI− cells.
Analysis of differentially expressed genes shows major differences between postinfusion and infusion samples, but fewer differences between postinfusion and preinfusion T cells
Expression of 511 genes involved in immune function was analyzed using a direct, solution-hybridization–based methodology (Nanostring) that permits unbiased quantitation of mRNA levels. Gene-expression analysis was performed on FACS-sorted postinfusion transgenic T cells, as well as in matched infusion preparations and untransduced preinfusion T cells from patients. Unsupervised exploration of gene expression by principal component analysis (PCA) showed a separation of samples in three groups corresponding to preinfusion, infusion, and postinfusion samples, with a principal component accounting for 23.5% of the variability (Figure 2A). This analysis, however, failed to group samples based on clinical response (supplemental Figure 1A).
Figure 2.

Gene-expression analysis. (A) PCA of 8 sets of preinfusion (blue), infusion (green), and postinfusion (red) samples analyzed by NanoString. (B) Venn diagram showing the number of differentially expressed genes identified in each of the 3 comparisons, among a code set of 511 targets. (C) Volcano plot for the 511 genes, comparing matched postinfusion and infusion engineered T cells. Horizontal line at y = 1.31 represents the threshold of statistical significance (P = .05) and vertical lines at x = +/−2 represent the threshold of FC set as cutoff values for the definition of differentially expressed genes.
A total of 156 genes were identified as differentially expressed (fold change [FC], absolute value > 2, P < .05) between postinfusion and infusion samples, and 170 genes between infusion and preinfusion samples (Figure 2B). In contrast, only 84 genes were differentially expressed between postinfusion and preinfusion cells (Figure 2B). Eighteen genes were differentially expressed in all 3 comparisons, in that their expression levels in persisting transgenic lymphocytes were intermediate between expression levels found in preinfusion and in infusion cells (Figures 2C and 3A). Genes with differential expression between postinfusion and preinfusion cells that did not follow this pattern (45 in total) included markers of differentiation such as GNLY (granulysin) and PRF1 (perforin), both overexpressed postinfusion, and chemokine receptor CCR7, downregulated postinfusion) (Figure 3B). The complete sets of differentially expressed genes for the comparisons between postinfusion and infusion cells, infusion and preinfusion, and between postinfusion and preinfusion lymphocytes are listed in Table 2, supplemental Table 1, and supplemental Table 2, respectively.
Figure 3.

Differentially expressed genes. (A) RNA expression levels for 18 genes that were differentially expressed in all three comparisons. Each line represents an individual patient. (B) Expression patterns of genes differentially expressed between postinfusion and preinfusion samples, which did not return to levels similar to preinfusion after engraftment.
Table 2.
Differentially expressed genes (postinfusion vs infusion)
| Gene | Description | FC | P |
|---|---|---|---|
| CYBB | Cytochrome b-245, β polypeptide | 27.08 | .03 |
| CSF3R | Colony stimulating factor 3 receptor (granulocyte) | 21.76 | .02 |
| TNFAIP3 | Tumor necrosis factor, α-induced protein 3 | 19.03 | .004 |
| CD14 | CD14 molecule | 18.81 | .02 |
| LILRA2 | Leukocyte immunoglobulin-like receptor, subfamily A (with TM domain), member 2 | 18.27 | .02 |
| CD36 | CD36 molecule (thrombospondin receptor) | 14.10 | .04 |
| FCGR2C | Fc fragment of IgG, low affinity IIc, receptor for (CD32) | 12.36 | .02 |
| CR1 | Complement component (3b/4b) receptor 1 | 11.65 | .01 |
| CX3CR1 | Chemokine (C-X3-C motif) receptor 1 | 11.29 | .009 |
| FCGR3A | Fc fragment of IgG, low affinity IIIa, receptor (CD16a) | 10.30 | .002 |
| IL7R | Interleukin 7 receptor | 9.31 (12.80) | .02 |
| TGFBI | Transforming growth factor, β-induced, 68kDa | 9.03 | .03 |
| TLR8 | Toll-like receptor 8 | 7.73 | .02 |
| NFKBIA | Nuclear factor of κ light polypeptide gene enhancer in B-cells inhibitor, α | 7.57 | .006 |
| KLRD1 | Killer cell lectin-like receptor subfamily D, member 1 | 7.35 | .001 |
| LILRA3 | Leukocyte immunoglobulin-like receptor, subfamily A (without TM domain), member 3 | 7.17 | .007 |
| FCGR2A | Fc fragment of IgG, low affinity IIa, receptor (CD32) | 6.99 | .03 |
| IL1RN | Interleukin 1 receptor antagonist | 6.94 | .03 |
| LILRB2 | Leukocyte immunoglobulin-like receptor, subfamily B (with TM and ITIM domains), member 2 | 6.68 | .02 |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase) | 6.32 (14.02) | .03 |
| PPBP | Pro-platelet basic protein (chemokine (C-X-C motif) ligand 7) | 6.26 | .04 |
| LTBR | Lymphotoxin B receptor | 6.14 | .04 |
| SELL | Selectin L | 6.10 (11.62) | .02 |
| IFIT2 | Interferon-induced protein with tetratricopeptide repeats 2 | 5.61 | .003 |
| LILRB4 | Leukocyte immunoglobulin-like receptor, subfamily B (with TM and ITIM domains), member 4 | 5.57 | .03 |
| BST1 | Bone marrow stromal cell antigen 1 | 5.49 | .02 |
| TCF7 | Transcription factor 7 (T-cell specific, HMG-box) | 5.41 | .01 |
| FCGRT | Fc fragment of IgG, receptor, transporter, α” | 5.38 | .02 |
| MSR1 | Macrophage scavenger receptor 1 | 5.17 | .0009 |
| CXCR4 | Chemokine (C-X-C motif) receptor 4 | 5.09 | .003 |
| LILRA1 | Leukocyte immunoglobulin-like receptor, subfamily A (with TM domain), member 1 | 5.08 | .01 |
| TNFSF13B | Tumor necrosis factor (ligand) superfamily, member 13b | 5.00 | .008 |
| CD1D | CD1d molecule | 4.99 | .003 |
| LITAF | Lipopolysaccharide-induced TNF factor | 4.98 | .001 |
| KIR3DS1 | Killer cell immunoglobulin-like receptor, three domains, short cytoplasmic tail, 1 | 4.98 | .03 |
| FCER1A | Fc fragment of IgE, high affinity I, receptor for; α polypeptide | 4.87 | .01 |
| KIR2DL2 | Killer cell immunoglobulin-like receptor, two domains, long cytoplasmic tail, 2 | 4.79 | .03 |
| CFD | Complement factor D (adipsin) | 4.74 | .004 |
| TLR2 | Toll-like receptor 2 | 4.68 | .03 |
| SOCS3 | Suppressor of cytokine signaling 3 | 4.61 | .01 |
| LILRB1 | Leukocyte immunoglobulin-like receptor, subfamily B (with TM and ITIM domains), member 1 | 4.59 | .001 |
| CLEC4E | C-type lectin domain family 4, member E | 4.52 | .02 |
| NCR1 | Natural cytotoxicity triggering receptor 1 | 4.35 | .002 |
| BTK | Bruton agammaglobulinemia tyrosine kinase | 4.18 | <.05 |
| PDCD1 | Programmed cell death 1 | 4.06 | .009 |
| SERPING1 | Serpin peptidase inhibitor, clade G (C1 inhibitor), member 1 | 3.98 | .002 |
| CLEC4A | C-type lectin domain family 4, member A | 3.94 | .02 |
| ITGA6 | Integrin, α 6 | 3.92 | .001 |
| LILRB3 | Leukocyte immunoglobulin-like receptor, subfamily B (with TM and ITIM domains), member 3 | 3.84 | .005 |
| NOS2 | Nitric oxide synthase 2, inducible | 3.52 | .01 |
| IL6R | Interleukin 6 receptor | 3.50 | .03 |
| CXCR2 | Chemokine (C-X-C motif) receptor 2 | 3.40 | .01 |
| FYN | FYN oncogene related to SRC, FGR, YES | 3.31 | .00006 |
| PTAFR | Platelet-activating factor receptor | 3.26 | .01 |
| IRAK3 | Interleukin-1 receptor-associated kinase 3 | 3.25 | .02 |
| FCGR2B | Fc fragment of IgG, low affinity IIb, receptor (CD32) | 3.25 | .0003 |
| CFP | Complement factor properdin | 3.23 | .01 |
| TAGAP | T-cell activation RhoGTPase activating protein | 3.05 | .0009 |
| IL18 | Interleukin 18 (interferon-γ-inducing factor) | 3.03 | .003 |
| CSF1R | Colony stimulating factor 1 receptor | 3.00 | .01 |
| IL10 | Interleukin 10 | 2.99 (20.78) | .02 |
| RARRES3 | Retinoic acid receptor responder (tazarotene induced) 3 | 2.99 | .001 |
| IL19 | Interleukin 19 | 2.97 | .009 |
| ITGAM | Integrin, α M (complement component 3 receptor 3 subunit)” | 2.94 | .01 |
| TLR3 | Toll-like receptor 3 | 2.94 | .0009 |
| TRAF6 | TNF receptor-associated factor 6, E3 ubiquitin protein ligase” | 2.90 | .002 |
| C2 | Complement component 2 | 2.89 | .0003 |
| PTGER4 | Prostaglandin E receptor 4 (subtype EP4) | 2.89 | .001 |
| CCL11 | Chemokine (C-C motif) ligand 11 | 2.83 | .01 |
| IL23A | Interleukin 23, α subunit p19 | 2.83 | .01 |
| IL12A | Interleukin 12A (natural killer cell stimulatory factor 1, p35) | 2.80 | .003 |
| C7 | Complement component 7 | 2.77 | .02 |
| IL15 | Interleukin 15 | 2.71 (3.73) | .02 |
| SIGIRR | Single immunoglobulin and toll-interleukin 1 receptor (TIR) domain | 2.67 | .0003 |
| LILRA6 | Leukocyte immunoglobulin-like receptor, subfamily A (with TM domain), member 6 | 2.67 | .01 |
| C1S | Complement component 1, s subcomponent | 2.66 | .01 |
| MASP2 | Mannan-binding lectin serine peptidase 2 | 2.65 | .006 |
| CEBPB | CCAAT/enhancer binding protein (C/EBP), β” | 2.64 | .03 |
| PRDM1 | PR domain containing 1, with ZNF domain | 2.64 | .0009 |
| RAG2 | Recombination activating gene 2 | 2.58 | <.05 |
| TLR1 | Toll-like receptor 1 | 2.58 | .001 |
| FKBP5 | FK506 binding protein 5 | 2.52 | .01 |
| TGFB1 | Transforming growth factor, β 1 | 2.49 (2.83) | .002 |
| NFKBIZ | Nuclear factor of κ light polypeptide gene enhancer in B-cells inhibitor, ζ | 2.48 | .004 |
| IRF1 | Interferon regulatory factor 1 | 2.48 | .00002 |
| DEFB1 | Defensin, β 1” | 2.46 | .003 |
| CLU | Clusterin | 2.45 | .02 |
| FCGR3B | Fc fragment of IgG, low affinity IIIb, receptor (CD16b) | 2.44 | .003 |
| ITGB1 | Integrin, β 1 (fibronectin receptor, β polypeptide, antigen CD29 includes MDF2, MSK12) | 2.44 | .01 |
| IRF7 | Interferon regulatory factor 7 | 2.43 | .0005 |
| MARCO | Macrophage receptor with collagenous structure | 2.41 | .01 |
| IRGM | Immunity-related GTPase family, M | 2.38 | .02 |
| IL6ST | Interleukin 6 signal transducer (gp130, oncostatin M receptor) | 2.37 | .002 |
| CASP8 | Caspase 8, apoptosis-related cysteine peptidase | 2.36 | .001 |
| ETS1 | v-ets erythroblastosis virus E26 oncogene homolog 1 (avian) | 2.35 | .0002 |
| IL1RL2 | Interleukin 1 receptor-like 2 | 2.33 | .04 |
| LTB4R | Leukotriene B4 receptor | 2.33 | .003 |
| LTF | Lactotransferrin | 2.32 | .006 |
| MX1 | Myxovirus (influenza virus) resistance 1, interferon-inducible protein p78 | 2.32 | .03 |
| ITGA2B | Integrin, α 2b (platelet glycoprotein IIb of IIb/IIIa complex, antigen CD41) | 2.29 | .02 |
| GNLY | Granulysin | 2.29 | .03 |
| IFIH1 | Interferon induced with helicase C domain 1 | 2.29 | .0006 |
| CXCL10 | Chemokine (C-X-C motif) ligand 10 | 2.28 | .04 |
| LTB4R2 | Leukotriene B4 receptor 2 | 2.26 | .0007 |
| PDCD2 | Programmed cell death 2 | 2.26 | .002 |
| CD209 | CD209 molecule | 2.24 | .004 |
| XCR1 | Chemokine (C motif) receptor 1 | 2.23 | .03 |
| TLR5 | Toll-like receptor 5 | 2.22 | .0005 |
| MBP | Myelin basic protein | 2.22 | .007 |
| ITGA5 | Integrin, α 5 (fibronectin receptor, α polypeptide) | 2.21 | .0006 |
| IKZF3 | IKAROS family zinc finger 3 (Aiolos) | 2.21 | .00008 |
| C1QB | Complement component 1, q subcomponent, B chain | 2.21 | .002 |
| C8G | Complement component 8, γ polypeptide | 2.18 | .005 |
| JAK1 | Janus kinase 1 | 2.18 | .0001 |
| TBX21 | T-box 21 | 2.18 (2.03) | .02 |
| STAT5B | Signal transducer and activator of transcription 5B | 2.15 | .004 |
| ATG16L1 | Autophagy related 16-like 1 (S. cerevisiae) | 2.13 | .002 |
| C4B | Complement component 4B (Chido blood group) | 2.12 | .008 |
| IL1R2 | Interleukin 1 receptor, type II | 2.12 | .04 |
| IRF8 | Interferon regulatory factor 8 | 2.09 | <.05 |
| HLA-C | Major histocompatibility complex, class I, C | 2.09 | .0002 |
| PLA2G2E | Phospholipase A2, group IIE | 2.09 | <.05 |
| ZAP70 | ζ-chain (TCR) associated protein kinase 70kDa | 2.09 | .0002 |
| ATM | Ataxia telangiectasia mutated | 2.08 | .01 |
| PTPRC | Protein tyrosine phosphatase, receptor type, C | 2.08 | .0006 |
| BCL6 | B-cell CLL/lymphoma 6 | 2.07 | .04 |
| CD44 | CD44 molecule (Indian blood group) | 2.04 | .01 |
| RAG1 | Recombination activating gene 1 | 2.03 | .04 |
| PML | Promyelocytic leukemia | 2.02 | .006 |
| CASP1 | Caspase 1, apoptosis-related cysteine peptidase | 2.01 | .0006 |
| MYD88 | Myeloid differentiation primary response 88 | 2.01 | .0001 |
| IL21R | Interleukin 21 receptor | 2.01 | <.05 |
| BCL2 | B-cell CLL/lymphoma 2 | −2.03 | .004 |
| CEACAM1 | Carcinoembryonic antigen-related cell adhesion molecule 1 (biliary glycoprotein) | −2.06 | .02 |
| CCR5 | Chemokine (C-C motif) receptor 5 | −2.09 | .003 |
| SOCS1 | Suppressor of cytokine signaling 1 | −2.29 | .002 |
| HLA-DRB1 | Major histocompatibility complex, class II, DR β 1 | −2.38 | .01 |
| IRF4 | Interferon regulatory factor 4 | −2.49 | .0005 |
| GZMA | Granzyme A (granzyme 1, cytotoxic T-lymphocyte-associated serine esterase 3) | −2.55 | .01 |
| CCR2 | Chemokine (C-C motif) receptor 2 | −2.60 | .0002 |
| CSF1 | Colony stimulating factor 1 (macrophage) | −2.84 (−2.26) | .0002 |
| DPP4 | Dipeptidyl-peptidase 4 | −2.97 | .0002 |
| CCL3 | Chemokine (C-C motif) ligand 3 | −3.10 | .03 |
| CSF2RB | Colony stimulating factor 2 receptor, β, low-affinity (granulocyte-macrophage) | −3.24 | .02 |
| CXCR6 | Chemokine (C-X-C motif) receptor 6 | −3.41 | .01 |
| GPI | Glucose-6-phosphate isomerase | −3.72 | .008 |
| CD40LG | CD40 ligand | −3.87 (−3.83) | .005 |
| CSF2 | Colony stimulating factor 2 (granulocyte-macrophage) | −4.66 (−55.17) | .0002 |
| CCR8 | Chemokine (C-C motif) receptor 8 | −4.67 | .02 |
| FOXP3 | Forkhead box P3 | −4.82 (−4.88) | .02 |
| HLA-DQA1 | Major histocompatibility complex, class II, DQ α 1 | −4.95 | .03 |
| LTA | Lymphotoxin α | −5.34 (−5.49) | .0002 |
| IL2RA | Interleukin 2 receptor, α | −7.40 (−5.18) | .009 |
| CISH | Cytokine inducible SH2-containing protein | −8.31 | .00002 |
| TNFRSF8 | Tumor necrosis factor receptor superfamily, member 8 | −11.14 (−10.32) | .007 |
| IL13 | Interleukin 13 | −11.44 (−61.60) | .02 |
NanoString data. RT-PCR data in parentheses.
We next used a real-time PCR-based assay to analyze the expression of a gene set that partially overlapped with that analyzed by Nanostring (supplemental Figure 2). This additional analysis confirmed the changes in gene expression identified by Nanostring (shown in parenthesis in Table 2). Of the genes analyzed by PCR array, only 4 were differentially expressed between postinfusion and preinfusion samples. FOXP1, FOXP3, and CCR4 were underexpressed, while CD70 was overexpressed. FOXP1 expression, which was differentially expressed in all 3 comparisons by real-time PCR, was confirmed by western blot (supplemental Figure 3).
Among the differentially regulated genes, a series of inhibitory receptors were overexpressed postinfusion, including PDCD1 (PD-1) and a group of MHC-binding negative receptors known to modulate T-cell activation by competing with TCR for binding of HLA molecules and by signaling through immunoreceptor tyrosine-based inhibition motifs (ITIMs): LILRA1, LILRA6, LILRB1, LILRB2, LILRB3, and LILRB4 (FC 6.69-2.67; Table 2). KIR2DL2, a gene encoding for another MHC-binding negative receptor previously described as overexpressed in CD8 T cells of melanoma patients,12 was overexpressed in persisting lymphocytes compared with both infusion and preinfusion cells (FC 4.79, P = .03; Table 2; supplemental Table 2). In the analysis of 2 other well-studied genes, mRNA encoding for inhibitory cytokine TGFB1 was higher in postinfusion samples than in infusion cells (Table 2; Figure 3A) whereas mRNA encoding for antiapoptotic gene BCL2 was less abundant in postinfusion cells (Table 2). Anergy-related genes13 DGKA, NFATC2, CBLB, and ITCH were also overexpressed postinfusion (supplemental Table 3).
Postinfusion lymphocytes express PD-1 and are sensitive to PD-L1 expressed in target cells
PDCD1, which encodes for inhibitory receptor PD-1, was significantly overexpressed in postinfusion cells (FC= 4.06, P = .009; Table 2). To verify PD-1 surface expression, flow cytometry was performed on infusion and postinfusion CD8+ T cells, which showed a significant increase in PD-1 staining on postinfusion cells compared with infusion counterparts (Figure 4A). To test for the biological relevance of increased PD-1 expression, we performed coculture experiments of patients’ postinfusion lymphocytes against target cells overexpressing PD-1 ligand, PD-L1 (supplemental Figure 4). As shown in Figure 4B, postinfusion CD8+ cells produced less IFNγ when cocultured with PD-L1 overexpressing targets, suggesting that persisting TCR-engineered lymphocytes were sensitive to PD-1/PD-L1 inhibition.
Figure 4.

PD-1 and CD160 are overexpressed in persisting transgenic lymphocytes. (A) Percentages of PD-1 expression in infusion and postinfusion CD8 T cells. (B) Reduced IFNγ production in CD8 T cells cocultured in vitro with relevant targets engineered to expressed PD-L1. (C) LAG-3, 2B4, and CD160 exhaustion markers surface expression in CD3+CD8+mTCRb+ infusion and postinfusion lymphocytes. Cells wre gated on lymphoid, PI−, CD3+, CD8+, mTCRb+ cells. (D) HVEM surface expression in melanoma tumor cells present in tumor digests of 4 melanoma patients. (E) Histograms showing surface staining of CD160 and costimulatory receptor LIGHT in persisting engineered lymphocytes of 5 patients. In all samples analyzed, LIGHT expression was lower than that of CD160.
CD160 is overexpressed postinfusion and its expression is associated with lower IFNγ production
PD-1 upregulation has been associated with exhaustion due to sustained antigen stimulation. To determine whether PD-1 expression in transgenic T cells might be an indicator of exhaustion in the context of ACT, we analyzed, by FACS, the expression of other exhaustion markers such as LAG-3, 2B4 (CD244), and CD160. While LAG-3 was underexpressed on the surface of postinfusion T cells compared with infusion cells, 2B4 was expressed at high levels in both cell types (Figure 4C). CD160 was expressed at higher levels in postinfusion cells than in infusion counterparts, both at RNA (supplemental Figure 5) and protein levels (Figure 4C).
The natural ligand for CD160 is herpes virus entry mediator (HVEM) (encoded by TNFRSF14), and CD160/HVEM interaction can inhibit CD4+ cell activation.14 However, ligation of HVEM by the alternate receptor LIGHT (TNFSF14) acts as a costimulatory signal for T cells.15,16 Consistent with previous reports,17 we found that HVEM is expressed in human melanoma tumors (Figure 4D). Since LIGHT was significantly underexpressed in postinfusion samples according to RT-PCR data (supplemental Table 3; FC −3.02, P = .003), we analyzed the balance of surface expression of LIGHT and CD160 in patient’s postinfusion cells. While coinhibitory receptor CD160 was consistently expressed, the costimulatory receptor LIGHT was present at very low or undetectable levels (Figure 4E), suggesting that binding to HVEM would result in an inhibitory effect on T cell activation.
To test the relevance of this pathway for the functionality of TCR-engineered T cells, we performed coculture experiments using both infusion and postinfusion patient samples with melanoma lines engineered to overexpress HVEM (supplemental Figure 4), and monitored IFNγ production by intracellular staining. Figure 5A depicts a representative coculture experiment, where mTCRb+ CD8+ cells were gated separately according to CD160 expression and IFNγ expression was determined. Strikingly, CD160+ cells expressed less IFNγ than CD160− regardless of the levels of HVEM expression in the target cells. Results from postinfusion samples of 8 different patients and infusion samples of 10 patients are summarized in Figure 5B. Postinfusion CD160+ lymphocytes had a significantly lower percentage of IFNγ-producing cells than CD160− counterparts when cocultured with either melanoma line 624 (mean: 30% vs 5%, respectively, P = .0003) or 624-HVEM (mean: 31% vs 4%, respectively, P = .0002). No statistically significant difference was found for CD160+ postinfusion lymphocytes between those cocultured with 624 and with 624-HVEM (mean 5% vs 4%, P = .4), or for CD160− cocultured with 624 and with 624-HVEM (mean: 30% vs 31%, P = 1). A similar pattern was observed for infusion cells, where CD160+ lymphocytes displayed a lower percentage of IFNγ+ cells than CD160−, when cocultured with either 624 (mean: 22% vs 45%, P = .002) or with 624-HVEM (mean: 17% vs 38%, P = .003). Consistent with the observations on postinfusion cells, no statistically significant difference was observed between the percentages of IFNγ+ lymphocytes when infusion cells were cocultured with 624 and with 624-HVEM (means: 45% vs 38%, P = .4 for CD160− cells; 22% vs 17%, P = .09 for CD160+ cells). Similar results were obtained using an HLA-A*0201–expressing variant of MAGE A3-expressing lung cancer cell line, H1299 (data not shown), indicating that the observation of reduced reactivity in CD160+ T cells was not specific for either the target cell or the introduced TCR.
Figure 5.
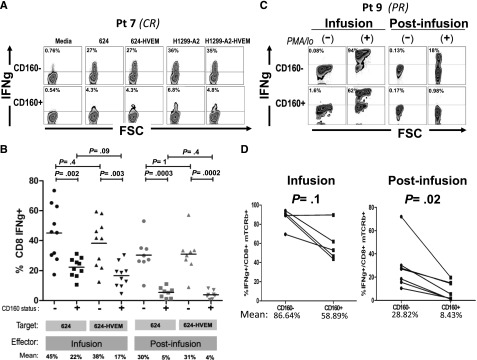
CD160 is a marker of T cells with reduced IFNγ-secretion capacity. (A) Representative example of IFNγ intracellular staining in 1-month postinfusion MAGE A3 TCR+ CD8 lymphocytes (gated separately based on CD160 expression) cocultured against MAGE A3+ 624 and H1299-A2, or HVEM-transduced derivatives. (B) Summary of results from 10 infusion samples and 8 postinfusion samples cocultured with 624 or 624-HVEM. (C) Intracellular staining of IFNγ in infusion and postinfusion lymphocytes of patient 9 stimulated with phorbol 12-myristate 13-acetate (PMA)/Ionomicyn during 4 hours or nonstimulated controls. (D) Summary of 6 patients.
CD160-expressing T cells show decreased IFNγ production independently of the presence of ligands in target cells
To test whether CD160-associated impairment in IFNγ production was an intrinsic property of postinfusion cells, or if it was rather mediated by binding to a different ligand, we analyzed IFNγ production in CD160+ and CD160− cells after a strong, nonspecific stimulation with phorbol 12-myristate 13-acetate/ionomicyn. Cells were seeded sparsely (5 × 105 cells per well, in 6-multiwell plates) in order to minimize lymphocyte-to-lymphocyte contact because HVEM is expressed in some postinfusion T cells (data not shown). IFNγ production was measured after 4 hours of stimulation and, consistent with results from coculture experiments, a lower percentage of IFNγ+ cells was observed in the CD160+ cells than in CD160− counterparts, an example of which is shown in Figure 5C. This trend was observed in both infusion and postinfusion samples, but the difference was statistically significant only in postinfusion samples. The mean percentage of IFNγ+ cells was 86.64% vs 58.89% for CD160− and CD160+ infusion cells, respectively (P = .1), whereas the mean percentage of IFNγ+ cells was 28.82% vs 8.43% for CD160− and CD160+ postinfusion cells, respectively (P = .02) (Figure 5D).
Discussion
Advanced therapies based on adoptive transfer of tumor targeted T lymphocytes have proven efficacious in treating human malignancies, even in the context of disseminated, heavily pretreated disease.1 Long-term persistence of transferred cells has been documented by us3,4,6 and others18 and, moreover, a correlation between persistence of adoptively transferred TILs and clinical response was found.1 These observations suggest that the antitumor effect of adoptively transferred cells might rely on a sustained immune reaction against tumor cells. Long-term persistence of these cells might also constitute an attractive immune-surveillance system potentially preventing recurrences in patients undergoing clinical responses.
Although the dynamics of cellular immunity have been extensively studied in the context of antiviral and, to some extent, antitumor responses,19 those findings may not necessarily apply to the biology of adoptively transferred genetically engineered cells. Unlike naturally occurring effector T cells, adoptively transferred lymphocytes have undergone extensive ex vivo manipulation including, but not limited to, strong polyclonal stimulation, in vitro culture in presence of supraphysiological concentrations of growth factors, and in vivo expansion within lymphodepleted hosts. In the case of CAR/TCR-transduced PBLs, manipulations also include the introduction of transgenes by means of viral infection, and expression of a high-affinity immune receptor in addition to the endogenous TCR. Thus, the fate of transferred cells in vivo might substantially differ from the well-characterized processes leading to establishment of natural immune memory or, alternatively, tolerance and/or exhaustion.20
We previously reported that persisting TCR-transduced T cells displayed reduced expression levels of the exogenous TCR compared with the infusion sample, which might negatively impact on the antitumor activity of persisting cells.7 In the present study, we sought to further clarify aspects of the biology of gene-engineered T cells, by exploring the differences in expression of immune-related genes between persisting engineered cells and the cellular product that was initially administered to the patients. Using technologies that directly quantitate RNA levels (Nanostring and Q-RT-PCR arrays) we observed a high proportion of genes with differential expression between the persisting and infusion cell populations. Expression patterns of differentiation markers such as IL-7R and CD62L (SELL) were consistent with previous observations demonstrating an increase postinfusion.6 Interestingly, by analyzing preinfusion T cells, we observed that most differences in gene expression between preinfusion and infusion cells were the mirror image of those found when comparing postinfusion vs infusion cells, suggesting that persisting cells either acquire or are selected for gene-expression patterns that are similar to those present in their parental nonmanipulated peripheral blood T cells.
Ma et al analyzed the pattern of cytokine expression in 1-month postinfusion transgenic lymphocytes of a single patient who had received ACT of MART-1 TCR-engineered T cells, compared with peripheral blood CD8+ cells from healthy donors. They reported a higher percentage of polyfunctional cytotoxic T lymphocytes within the MART-1–specific lymphocyte population in the melanoma patient than in CD8+ cells of healthy donors, proposing that this polyfunctionality might be associated with the clinical outcome.21 Recently, the same authors reported the single cell-based multiplexed analysis of secreted molecules in 3 ACT patients, confirming the presence of polyfunctional transgenic lymphocytes and hypothesizing on the impact of adoptively transferred cells on the endogenous antitumor response.22 The present study is, to our knowledge, the first multipatient multi–time-point gene-expression analysis of adoptively transferred, gene-engineered human lymphocytes. The changes in gene expression described in our study were likely associated to a general process occurring both in patients undergoing clinical responses and in those with progressive disease because minimal differences in gene expression were found between responders and nonresponders. A larger number of samples would likely be needed to identify gene-expression patterns associated with clinical outcomes. Of note, several negative regulators of cell function were overexpressed in persisting lymphocytes, including FOXP1,23 CBL-B,24-26 DGKA,27,28 DGKZ,29 ITCH,30 TGFB1,31 LILRB1 and LILRB3,32 SOCS3,33 CD160, PDCD1,34 and KIR2DL2 among others. Of these, we focused on 2 extracellular inhibitory receptors, CD160 and PD-1. Blocking inhibitory signals mediated by ligation of HVEM and PD-L1 within the tumor microenvironment might serve as a potential approach to increase ACT efficacy.
CD160 is an MHC class I–binding glycophosphatidylinositol-anchored glycoprotein expressed in natural killer (NK) cells, activated CD4+ T cells and in subsets of CD8+ T cells.14 Unlike other receptors, CD160 lacks an intracellular signaling domain and the details of its ultimate molecular mechanism of action remain elusive. Its inhibitory effect on proliferation and cytokine secretion by CD4+ T cells was shown to be dependent on HVEM binding.14 HVEM can be a ligand for inhibitory or stimulatory receptors expressed in T cells15,16 and has been described as a mechanism of inhibition induced by regulatory T cells.35 Its role as a ligand is particularly relevant in the immune response against melanoma tumors, which express HVEM at high frequency.17 We conducted a series of experiments aimed at clarifying the functional relevance of CD160 overexpression, and we did it in parallel to PD-1, a well-characterized coinhibitory receptor that was also overexpressed in postinfusion cells. Functional experiments conducted ex vivo revealed that CD160+ persisting CD8+ lymphocytes were impaired in their ability to produce IFNγ in response to TCR stimulation, but this did not appear to be dependent on the presence of HVEM on cellular targets. PD-1, in contrast, may play an active role in T-cell inhibition since IFNγ production by CD8+ T cells was sensitive to the presence of PD-L1 in target cells. In light of the extensive body of evidence showing that PD-1/PD-L1 signaling is an important modulator of immune response against virus36,37 and most importantly against cancer,38-40 we suggest that treatments involving disruption of PD-1/PD-L1 interaction by means of blocking antibodies might impact positively on the clinical outcome of ACT using gene-engineered T cells. Moreover, it has been reported that PD-L1 is induced by IFNγ in antigen-presenting cells41 and melanoma cells,42 and that PD-L1 expression in melanoma cells is higher in areas of the tumor presenting a higher T-cell infiltration,43 suggesting that PD-L1 expression in the tumor microenvironment might be increased after ACT.
The relevance of coinhibitory signaling in the context of immunotherapy of cancer has been highlighted by findings from several preclinical studies17,44 and clinical trials38-40,45,46 where disruption of immune checkpoint signaling pathways resulted in enhanced antitumor immunity.45 The detrimental effects of coinhibitory signaling might not be restricted to an acute inhibition of T-cell function because it has been postulated that sustained signaling can lead to cellular exhaustion.46 The induction of exhaustion in T cells specific for tumor antigens has been explored based on the similarities between chronic viral infections and cancer vis-a-vis the sustained exposure of T cells to cognate antigens in presence of inflammation. In this regard, Baitsch et al characterized a population of MART-1–specific T CD8+ cells present in melanoma metastatic lesions of patients vaccinated with MART-1 peptide, finding that TILs presented a transcriptomic profile enriched in markers of exhaustion.47 Interestingly, they found that although CD160 was expressed by T cells specific for latent viral infections, it was not expressed by MART-1–specific T cells present either in peripheral blood or within metastatic lesions. This is in contrast with our results on CD160 expression in peripheral blood gp100- and MAGE A3-specific TCR-engineered T cells and may reflect the inherent differences between the biology of endogenous antigen-specific lymphocytes arising from vaccination (or via priming by tumor cells) and that of adoptively transferred gene-engineered T cells.
The pattern PD1+2B4+CD160+LAG3− of persisting TCR-transduced cells was similar to that described for exhausted HIV-specific CD8+ lymphocytes48 suggesting that some aspects of T-cell immunity based on adoptively transferred TCR gene-engineered T cells may be more similar to an antiviral response than to a naturally occurring antitumor response. A recent report by Peretz et al documented the coexpression of PD-1 and CD160 in dysfunctional HIV-specific CD8 T cells, where PD-1+ CD160+ cells presented higher levels of expression of KIR2DL2 than activated PD-1+ CD160− counterparts.49 In our dataset, KIR2DL2 mRNA was overexpressed in postinfusion compared with infusion cells, and was the only inhibitory receptor of its kind that was not significantly expressed in preinfusion cells (Figure 3B). It is plausible that the observed association between CD160 expression and functional impairment is indicative of an exhaustion-like process occurring in adoptively transferred gene-engineered T cells.
In summary, our results provide new insight into the biology of TCR-transgenic human T cells across different steps of adoptive cell transfer therapy protocols, showing the dynamics of mRNA expression of genes involved in immune function. These studies were made possible by the use of a unique reagent, murine TCRs, which permit the highly specific isolation of gene-marked T cells directly from patient blood. The knowledge of gene-expression dynamics may serve as a basis for the development of refined gene therapies in which the expression of therapeutic genes might be regulated by coupling them to the transcriptional control of genes or microRNAs that are specifically expressed in infusion or postinfusion samples. Finally, the expression of PD-1 in 1-month postinfusion lymphocytes and the sensitivity of these cells to PD-L1–induced inhibition, may represent an opportunity for therapeutic intervention. Combination immunotherapy involving blocking antibodies, or PDCD1 gene inactivation using zinc finger nucleases, might lead to the development of more efficacious ACT treatments for cancer.
Supplementary Material
Acknowledgments
The authors would like to thank Arnold Mixon and Shawn Farid for technical support, and the TIL laboratory at the Surgery Branch for handling of clinical samples. Nanostring data were acquired by Xiaolin Wu and Nina Bubunenko of the Laboratory of Molecular Technology, SAIC-Frederick, Inc (Frederick).
This work was supported by the intramural program of the National Cancer Institute, Center for Cancer Research, National Institutes of Health (Bethesda, MD).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: D.A.-D., J.L.D., and K.-i.H. performed experiments; and D.A.-D., J.L.D., K.-i.H., J.C.Y., R.A.M., and S.A.R. discussed data, designed experiments, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Richard A. Morgan, National Cancer Institute/Surgery Branch, Building 10, CRC, Room 3W-5940, 10 Center Dr, Bethesda, MD 20892; e-mail: rmorgan@mail.nih.gov.
References
- 1.Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114(3):535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Powell DJ, Jr, Dudley ME, Robbins PF, Rosenberg SA. Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood. 2005;105(1):241–250. doi: 10.1182/blood-2004-06-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burns WR, Zheng Z, Rosenberg SA, Morgan RA. Lack of specific gamma-retroviral vector long terminal repeat promoter silencing in patients receiving genetically engineered lymphocytes and activation upon lymphocyte restimulation. Blood. 2009;114(14):2888–2899. doi: 10.1182/blood-2009-01-199216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan RA, Chinnasamy N, Abate-Daga D, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36(2):133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geiss GK, Bumgarner RE, Birditt B, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26(3):317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 10.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92(3):205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 11.Riddell SR, Greenberg PD. The use of anti-CD3 and anti-CD28 monoclonal antibodies to clone and expand human antigen-specific T cells. J Immunol Methods. 1990;128(2):189–201. doi: 10.1016/0022-1759(90)90210-m. [DOI] [PubMed] [Google Scholar]
- 12.Casado JG, Soto R, DelaRosa O, et al. CD8 T cells expressing NK associated receptors are increased in melanoma patients and display an effector phenotype. Cancer Immunol Immunother. 2005;54(12):1162–1171. doi: 10.1007/s00262-005-0682-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baine I, Abe BT, Macian F. Regulation of T-cell tolerance by calcium/NFAT signaling. Immunol Rev. 2009;231(1):225–240. doi: 10.1111/j.1600-065X.2009.00817.x. [DOI] [PubMed] [Google Scholar]
- 14.Cai G, Anumanthan A, Brown JA, Greenfield EA, Zhu B, Freeman GJ. CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat Immunol. 2008;9(2):176–185. doi: 10.1038/ni1554. [DOI] [PubMed] [Google Scholar]
- 15.Steinberg MW, Cheung TC, Ware CF. The signaling networks of the herpesvirus entry mediator (TNFRSF14) in immune regulation. Immunol Rev. 2011;244(1):169–187. doi: 10.1111/j.1600-065X.2011.01064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pasero C, Speiser DE, Derré L, Olive D. The HVEM network: new directions in targeting novel costimulatory/co-inhibitory molecules for cancer therapy. Curr Opin Pharmacol. 2012;12(4):478–485. doi: 10.1016/j.coph.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 17.Derré L, Rivals JP, Jandus C, et al. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J Clin Invest. 2010;120(1):157–167. doi: 10.1172/JCI40070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. 2006;211:214–224. doi: 10.1111/j.0105-2896.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 21.Ma C, Fan R, Ahmad H, et al. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat Med. 2011;17(6):738–743. doi: 10.1038/nm.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma C, Cheung AF, Chodon T, et al. Multifunctional T-cell analyses to study response and progression in adoptive cell transfer immunotherapy. Cancer Discov. 2013;3(4):418–429. doi: 10.1158/2159-8290.CD-12-0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng X, Wang H, Takata H, Day TJ, Willen J, Hu H. Transcription factor Foxp1 exerts essential cell-intrinsic regulation of the quiescence of naive T cells. Nat Immunol. 2011;12(6):544–550. doi: 10.1038/ni.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paolino M, Thien CB, Gruber T, et al. Essential role of E3 ubiquitin ligase activity in Cbl-b-regulated T cell functions. J Immunol. 2011;186(4):2138–2147. doi: 10.4049/jimmunol.1003390. [DOI] [PubMed] [Google Scholar]
- 25.Karwacz K, Bricogne C, MacDonald D, et al. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol Med. 2011;3(10):581–592. doi: 10.1002/emmm.201100165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stromnes IM, Blattman JN, Tan X, Jeevanjee S, Gu H, Greenberg PD. Abrogating Cbl-b in effector CD8(+) T cells improves the efficacy of adoptive therapy of leukemia in mice. J Clin Invest. 2010;120(10):3722–3734. doi: 10.1172/JCI41991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olenchock BA, Guo R, Carpenter JH, et al. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat Immunol. 2006;7(11):1174–1181. doi: 10.1038/ni1400. [DOI] [PubMed] [Google Scholar]
- 28.Zha Y, Marks R, Ho AW, et al. T cell anergy is reversed by active Ras and is regulated by diacylglycerol kinase-alpha. Nat Immunol. 2006;7(11):1166–1173. doi: 10.1038/ni1394. [DOI] [PubMed] [Google Scholar]
- 29.Zhong XP, Hainey EA, Olenchock BA, et al. Enhanced T cell responses due to diacylglycerol kinase zeta deficiency. Nat Immunol. 2003;4(9):882–890. doi: 10.1038/ni958. [DOI] [PubMed] [Google Scholar]
- 30.Shembade N, Harhaj NS, Parvatiyar K, et al. The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat Immunol. 2008;9(3):254–262. doi: 10.1038/ni1563. [DOI] [PubMed] [Google Scholar]
- 31.Margherita G, Loreto G, Elena R. TGF-beta: a master switch in tumor immunity. Curr Pharm Des. 2012;18(27):4126–4134. doi: 10.2174/138161212802430378. [DOI] [PubMed] [Google Scholar]
- 32.Vivier E, Anfossi N. Inhibitory NK-cell receptors on T cells: witness of the past, actors of the future. Nat Rev Immunol. 2004;4(3):190–198. doi: 10.1038/nri1306. [DOI] [PubMed] [Google Scholar]
- 33.Palmer DC, Restifo NP. Suppressors of cytokine signaling (SOCS) in T cell differentiation, maturation, and function. Trends Immunol. 2009;30(12):592–602. doi: 10.1016/j.it.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Odorizzi PM, Wherry EJ. Inhibitory receptors on lymphocytes: insights from infections. J Immunol. 2012;188(7):2957–2965. doi: 10.4049/jimmunol.1100038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tao R, Wang L, Murphy KM, Fraser CC, Hancock WW. Regulatory T cell expression of herpesvirus entry mediator suppresses the function of B and T lymphocyte attenuator-positive effector T cells. J Immunol. 2008;180(10):6649–6655. doi: 10.4049/jimmunol.180.10.6649. [DOI] [PubMed] [Google Scholar]
- 36.Bengsch B, Seigel B, Ruhl M, et al. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog. 2010;6(6):e1000947. doi: 10.1371/journal.ppat.1000947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Macatangay BJ, Rinaldo CR. PD-1 blockade: A promising immunotherapy for HIV? Cellscience. 2009;5(4):61–65. [PMC free article] [PubMed] [Google Scholar]
- 38.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inozume T, Hanada K, Wang QJ, et al. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J Immunother. 2010;33(9):956–964. doi: 10.1097/CJI.0b013e3181fad2b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taube JM, Anders RA, Young GD, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127):127ra137. [DOI] [PMC free article] [PubMed]
- 44.Lasaro MO, Sazanovich M, Giles-Davis W, et al. Active immunotherapy combined with blockade of a coinhibitory pathway achieves regression of large tumor masses in cancer-prone mice. Mol Ther. 2011;19(9):1727–1736. doi: 10.1038/mt.2011.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Norde WJ, Hobo W, van der Voort R, Dolstra H. Coinhibitory molecules in hematologic malignancies: targets for therapeutic intervention. Blood. 2012;120(4):728–736. doi: 10.1182/blood-2012-02-412510. [DOI] [PubMed] [Google Scholar]
- 47.Baitsch L, Baumgaertner P, Devêvre E, et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J Clin Invest. 2011;121(6):2350–2360. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamamoto T, Price DA, Casazza JP, et al. Surface expression patterns of negative regulatory molecules identify determinants of virus-specific CD8+ T-cell exhaustion in HIV infection. Blood. 2011;117(18):4805–4815. doi: 10.1182/blood-2010-11-317297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peretz Y, He Z, Shi Y, et al. CD160 and PD-1 co-expression on HIV-specific CD8 T cells defines a subset with advanced dysfunction. PLoS Pathog. 2012;8(8):e1002840. doi: 10.1371/journal.ppat.1002840. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.