

Abstract
Background
In recent years, as the development of next-generation sequencing technology, a growing number of genes have been reported as being horizontally transferred from prokaryotes to eukaryotes, most of them involving arthropods. As a member of the phylum Arthropoda, the Pacific white shrimp Litopenaeus vannamei has to adapt to the complex water environments with various symbiotic or parasitic microorganisms, which provide a platform for horizontal gene transfer (HGT).
Results
In this study, we analyzed the genome-wide HGT events in L. vannamei. Through homology search and phylogenetic analysis, followed by experimental PCR confirmation, 14 genes with HGT event were identified: 12 of them were transferred from bacteria and two from fungi. Structure analysis of these genes showed that the introns of the two fungi-originated genes were substituted by shrimp DNA fragment, two genes transferred from bacteria had shrimp specific introns inserted in them. Furthermore, around other three bacteria-originated genes, there were three large DNA segments inserted into the shrimp genome. One segment was a transposon that fully transferred, and the other two segments contained only coding regions of bacteria. Functional prediction of these 14 genes showed that 6 of them might be related to energy metabolism, and 4 others related to defense of the organism.
Conclusions
HGT events from bacteria or fungi were happened in the genome of L. vannamei, and these horizontally transferred genes can be transcribed in shrimp. This is the first time to report the existence of horizontally transferred genes in shrimp. Importantly, most of these genes are exposed to a negative selection pressure and appeared to be functional.
Keywords: Horizontal gene transfer, Litopenaeus vannamei, Shrimp, Bacteria, Fungi
Background
Unlike vertical transfer, horizontal gene transfer (HGT) refers to the transfer of genes between organisms and allows recipients to acquire novel traits from the donors [1]. In most cases, horizontally transferred genes (HGT genes) are nonfunctional. However, functional HGTs often benefit the recipients in terms of adaptation to certain specialized niches and are thought to be an important evolutionary impetus [2]. Nowadays, most detected HGT events occur among bacteria inhabiting the same environment, and are a major source of genetic variation in bacteria [3,4]. However, HGT is also increasingly acknowledged to play an important role in animals [2]. Predominantly prokaryotes-to-animal HGT events have been described between prokaryotes and arthropods or nematodes, and an endosymbiont is the most probable donor for gene transfer because of the close and constant proximity of the cells from both organisms [1,5-9]. HGT in animals causes divergence in genetic materials and may result in physiological metabolism improvements and the gain of some other phenotypes, e.g. virulence gene transfer can cause emergence of new disease, while HGT genes in aphids can help it produce carotenoid [10-13]. Furthermore, functional HGT genes in animals have been identified play a vital role in the evolution of recipients [1,2].
The Pacific white shrimp, Litopenaeus vannamei, is an economically important marine aquaculture species worldwide. However, the development of the commercial culture of shrimps has generally been accompanied by increasing problems with diseases, which are mostly caused by opportunistic pathogens, such as viruses, bacteria and fungi [14]. Erosions of the cuticle, localized lesions and generalized septicemias are the three general symptoms seen in infected shrimps [14]. Generally, shrimps living in complex environments with various parasitic or symbiotic microorganisms, and the communications between them always make effects on their growth [15-18]. The close relationship between shrimps and their parasitic or symbiotic organisms may provide a platform for trans-kingdom horizontal transfer of genetic materials during evolutionary history [1,3]. However, no strong evidence for the HGT events in shrimp has been found.
The detection of animal HGT events has been based on complete genome sequences and the combination of homology searching and phylogenetic analysis [19]. Unfortunately, the whole genome and complete gene set of shrimp have not been published. However, recently, a large number of expressed sequences tags (ESTs) and high-throughput transcriptome sequencing data of L. vannamei were published [20,21]. These data provide good resources for the exhaustive HGTs detection in shrimp. In this study, the candidate HGT genes in the genome of L. vannamei were searched by sequence homology comparison, phylogenetic analysis, and experimental confirmation of the candidates HGT genes. By comparing the HGT genes-located shrimp genome contigs and corresponding donor genomes, the relatively large genomic segments, which included gene clusters, have been identified as being horizontally transferred from bacteria and integrated into the shrimp genome. Furthermore, their expression profiles at five developmental stages of shrimp were analyzed and their probable functions in shrimp were discussed.
Results
Fourteen HGT genes were detected in L. vannamei
In this study, an exhaustive detection method was used to identify HGT genes in L. vannamei (see Methods). Homologous BLAST analysis was used initially to detect HGT genes with a view for identifying homologous genes of non-mating species from shrimp (Figure 1). Initially, 65,582 gene segments were filtered out because there were no homologs detected among them. These sequences probably represent shrimp unique gene segments or non-coding DNA. In the second stage of homologous searching, more than 92% of the gene segments, which were most similar to other arthropod sequence, were excluded. Then, only 965 HGT candidates were left. During this procedure, two classes of sequences were filtered out: gene segments that only showed homology to arthropods and those having higher BLAST similarity scores with arthropods than any other species. However, several potential HGT events may be missed by this procedure, in that some truly HGT of Arthropoda may be considered as vertical inheritance. In the third homologous searching procedure, BLAST searches against the GenBank non-redundant protein database (nr) were implemented to extract sequences from an even larger spectrum of species. As small numbers of homologs are not sufficient for phylogenetic analysis and tree construction, we selected HGT candidates with more than 10 homologs in the nr database. Thus, after filtering genes in above procedures, only 722 HGT candidates remained for phylogenetic analysis (Figure 1).
Figure 1.

Flowchart for identification of HGT genes in L. vannamei. The eliminated and remaining numbers of gene segments in each step are listed on the figure.
Three kinds of phyloenetic analysis were performed on the remaining 722 HGT candidates after homology search (see Methods). Phylogenetic trees for each candidate HGT gene were constructed manually, and once a gene was nested within a donor clade and many other donor species formed basal branches, it was considered to be a HGT gene that had horizontally transferred from the donor to L. vannamei[8,22]. Most of the HGT candidates showed higher similarity with Arthropoda-related species and grouped within a single clade. Furthermore, several candidate HGTs showed a phylogenetic topology that did not support a HGT event. Thus, as for those 722 HGT candidates, 693 of them were eliminated by the phylogenetic analysis, leaving only 29 genes that displayed rational HGT phylogenetic topology (Figure 1). They were subjected to comparative phylogenetic analysis by constructing three kinds of phylogenetic trees. Ultimately, well-refined maximum likelihood (ML) trees were used to represent the HGT events.
Among the 29 candidate HGT genes above after phylogenetic selection, there were 12 genes showed significant similarity to the best-hit sequences (E-values ranged from 2.00E-82 to 5.00E-36, and identity values ranged from 95.83% to 100%). Considering that many candidate HGT genes might be contaminants derived from RNA extraction, library construction or sequencing, these sequences needed to be excluded from the 29 candidate HGT genes. Previously, we acquired 42× coverage of shotgun reads generated from whole genome sequencing of L. vannamei. A 1.9 Gb draft map of the contigs was assembled and used to identify genuine shrimp gene segments. Ultimately, only 14 candidate HGT genes were homologous to shrimp genomic contigs (E-values ranged from 0 to 9.00E-21, and identity values ranged from 88.22% to 100%). Thus, there are 15 genes were considered as non-shrimp genes and removed in this procedure. Some of HGT genes may be missed because of incompletely contigs assembly, but the 42× coverage of reads were full enough for identification of the existence of these gene segments. Generally, the sequenced reads tend to equally cover the assembly contigs, and eukaryotic contigs were expected to have different reads coverage to those contaminating contigs. Thus, with the help of SOAPaligner (http://soap.genomics.org.cn/soapaligner.html), we aligned all the reads to the 14 contigs which showed homologous with the 14 candidate HGT genes. The alignment results were compared with that of 1,000 randomly selected assembly contigs. It was found that the reads coverage of the 14 contigs (average coverage of 37.42) did not display any significant bias with that of 1,000 randomly selected contigs (average coverage of 38.25, Student’s t-test p < 0.05). Therefore, the 14 contigs seemed to possess eukaryotic characteristics rather than contaminates.
To avoid artificial mis-assembly of genomic contigs, PCR amplification was performed for these candidate HGT genes using shrimp genomic DNA as template. As expected, all 14 candidate HGT genes were successfully amplified. These amplified fragments were sequenced and showed nearly 100% sequence identity with correspondent genomic contigs. Furthermore, in order to verify the 14 HGT genes were integrated into the genome of L. vannamei, PCR amplification was also performed on both sides of the 14 genomic contigs on which the 14 HGT genes located. The two sides of the genome contigs, that did not show any similarity with the genome of the most probable donor, were most likely the eukaryotic sequences rather than HGT origin. BLAST results of the two sides of HGT fragments against nt database showed that most of them are similar to eukaryotic sequences (Additional file 1: Table S3). It indicates that the sides of these HGT fragments are eukaryotic portion, while the middle of the contigs is most probable HGT regions. Thus, we amplify the eukaryotic portion as long as possible to make sure the PCR products containing both eukaryotic and HGT regions. From the results, we can see that most of the edge products have been successfully amplified except for that of rpsN, which displayed a relatively weak amplification compared with the others. All the products were fully sequenced, and they were all identical to the correspondent regions of genome contigs. For the control of PCR amplification, all the products of positive control has been successfully amplified, while the products of negative control were amplified with nothing (see Methods section), which support for accuracy of the amplification of our target products. From the results above, it is reasonable to confirm that the 14 HGT genes were integrated into shrimp genome. In Table 1, the assigned names of the 14 HGT genes were based on their annotated functions or the gene name registered in NCBI (acsf, rpsF, rpsN, exbB, mopB, tnpA, stat, rpc2, dhfr, cata, sdrp, omtp, ankp and deha).
Table 1.
Fourteen predicted HGT genes in the L. vannamei genome
| Symbol | Length | Typesa | Contig position | GenBank ID | Function | Top hit speciesb | Phylum | Accession GI | E-value | Identity | Coverage | Figures |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
acsf |
672 |
B → L |
Contig124858619: 312-944 |
KC701594 |
acetyl-coenzyme A synthetase family protein |
Desulfotomaculum kuznetsovii |
Chloroflexi |
667983253 |
5.00E-48 |
66.87 |
72.77% |
Figure S1 |
|
rpsF |
395 |
B → L |
Contig124943021: 4082-4445 |
KC701595 |
30S ribosomal protein S6 |
Flavobacteriales bacterium |
Bacteroidetes |
1163788026 |
6.00E-50 |
96.61 |
44.81% |
Figure S2 |
|
rpsN |
593 |
B → L |
Contig5551: 3683-4206 |
KC701598 |
50S ribosomal protein L21 |
Gramella forsetii |
Bacteroidetes |
1160887756 |
2.00E-58 |
79.31 |
88.03% |
Figure S3 |
|
exbB |
522 |
B → L |
Contig124942315: 3448-3593 |
KC701596 |
biopolymer transport protein |
Robiginitalea biformata |
Bacteroidetes |
3320593070 |
1.00E-78 |
96.89 |
92.53% |
Figure S4 |
|
mopB |
760 |
B → L |
Contig124944249: 1863-1980 |
KC701597 |
molybdopterin oxidoreductase, iron-sulfur binding subunit |
Polaribacter irgensii |
Firmicutes |
3336447549 |
5.00E-123 |
83.27 |
99.08% |
Figure S5 |
|
tnpA |
352 |
B → L |
Contig5540: 2118-2468 |
KC701599 |
transposase |
Escherichia Coli |
Gama-proteobacteria |
269961834 |
1.00E-60 |
100 |
98.01% |
Figure S6 |
|
stat |
784 |
B → L |
Contig124277025: 193-313 |
KC701600 |
streptomycin 3″-adenylyltransferase |
Shigella flexneri |
Gama-proteobacteria |
3331678336 |
8.00E-78 |
100 |
56.25% |
Figure S7 |
|
rpc2 |
778 |
B → L |
Contig124717565: 157-356 |
KC701601 |
repressor protein C2 |
Escherichia Coli |
Gama-proteobacteria |
3333019710 |
6.00E-47 |
94.74 |
21.98% |
Figure S8 |
|
dhfr |
692 |
B → L |
Contig124922462: 426-1117 |
KC701602 |
dihydrofolate reductase |
Vibrio harveyi |
Gama-proteobacteria |
3323184048 |
2.00E-52 |
69.57 |
69.80% |
Figure S9 |
|
cata |
709 |
B → L |
Contig147675: 106-530 |
KC701603 |
chloramphenicol acetyltransferase |
Bacteroides uniformis |
Bacteroidetes |
1120437905 |
1.00E-81 |
100 |
59.24% |
Figure S10 |
|
sdrp |
286 |
B → L |
Contig124657743: 33-616 |
KC701604 |
short-chain dehydrogenase/reductase SDR |
Burkholderia phymatum |
Beta-proteobacteria |
1186471973 |
3.00E-29 |
67.37 |
99.99% |
Figure S11 |
|
omtp |
660 |
B → A |
Contig123317449: 1-217 |
KC701605 |
O-methyltransferase family protein |
Francisella sp. |
Gama-proteobacteria |
88802104 |
5.00E-23 |
44.65 |
72.27% |
Figure 2 |
|
ankp |
955 |
F → L |
Contig124712867: 86-719 |
KC701606 |
ankyrin repeat-containing protein |
Grosmannia clavigera |
Ascomycota |
2260060705 |
2.00E-25 |
46.39 |
30.47% |
Figure S12 |
| deha | 761 | F → L | Contig121079137: 1-172 | KC701607 | DEHA2A03014p | Debaryomyces hansenii | Ascomycota | 294654414 | 8.00E-22 | 37.23 | 54.01% | Figure S13 |
aThe HGT types of each HGT gene. B → L indicates HGT from Bacteria to L. vannamei or its ancestor; B → A indicates HGT from Bacteria-to-Arthropoda or its ancestor; F → L indicates HGT from Fungi-to-L. vannamei or its ancestor. bThe Arthropoda-related sequences were removed from the top hit BLAST species.
Bacteria and fungi were the two predominating donors of HGT genes
The phylogenetic trees can be used to indicate HGT events and their directions [22,23]. For 11 of the 14 HGT genes, the phylogenetic trees showed a phylogenetic topology of L. vannamei nesting with bacteria but far from other eukaryotes, which indicated a bacteria-to-L. vannamei HGT event (Table 1, Additional file 2: Figures S1, S2, S3, S4, S5, S6, S7, S8, S9, S10 and S11). The most probable donors of bacteria-to-L. vannamei HGT genes were Proteobacteria (five species) and Bacterioidetes (four species), and most of them were ecologically related to shrimp. Desulfotomaculum kuznetsovii, Flavobacteriales bacterium, Robiginitalea biformata, Polaribacter irgensii, Gramella forsetii, and Vibrio harveyi are marine bacteria, while Escherichia coli and Bacteroides uniformis are gut bacteria. These results supported the view that HGT events generally occur between species inhabiting the same environment [1,3]. The other two HGT genes were transferred from fungi to L. vannamei (ankp and deha, Table 1). Phylogenetic trees of ankp and deha showed that L. vannamei completely nested within the clade of fungi and far from bacteria and other eukaryotes, indicating a transfer of fungal origin (Additional file 2: Figures: S12, S13). The best-hit species of these two HGT genes belong to the Ascomycota, which is the largest phylum of fungi [24]. One of the two fungi donors, Grosmannia clavigera, was reported to be a symbiont of the mountain pine beetle [19]. Few fungi-to-higher eukaryote HGT events have been detected in previous studies, whereas, some reports of fungi-to-higher eukaryote HGT genes indicated that they greatly improved the host’s phenotypes and evolution [12,25]. Subsequent functional analysis indicated that these two fungi originated HGT genes may function in protein-protein interactions and electron transfer, respectively.
BLAST results of HGT genes against the nr database showed that the above 13 of the 14 HGT genes have no homologs among any other arthropods, which suggests that these HGT genes were transferred to L. vannamei or its ancestor after the speciation of Arthropoda [8]. One exception was a HGT gene, omtp, which showed high similarity with sequences of other Arthropoda-related species (arthropods and nematodes) that were all nested within the group of bacteria while being phylogenetically far from other invertebrates and vertebrates (Table 1, Figure 2). Previous phylogenetic analysis clustered arthropods and nematodes in a clade of molting animals termed the Ecdysozoa [26], which indicated they are evolved from the same ancestor. Besides, omtp was considered to encode a catechol-O-methyltransferase (COMT), a protein family which has ever been found to be horizontally transferred from bacteria to eukaryotes before the separation of animals and fungi [27,28]. This indicates that omtp was horizontally transferred earlier than the other HGT genes, and our phylogenetic tree of omtp suggested that it was transferred from a bacterial source to arthropods or its ancestors before the speciation of Ecdysozoa.
Figure 2.

Phylogenetic position of the shrimp HGT gene omtp. The phylogenetic tree was constructed using Phyml to perform ML analysis. The support values of ML, NJ and BI analysis are displayed beside each node.
Four HGT genes contain introns
Structure analysis of HGT genes was used to investigate whether there is structural evolvement of the HGT genes between the donor and recipient, which may provide evidence of the HGT events. Seven HGT genes were derived from single EST or transcriptome sequences: tnpA (GI: 41059456), stat (GI: 57504717), rpc2 (GI: 57504772), dhfr (GI: 171573934), cata (GI: 259510079), sdrp (GI: 349908938), deha (GI: 171560878). When comparing HGT gene segments to the shrimp genome contigs, we found that four HGT genes (acsf, sdrp, ankp and deha) contained introns in the shrimp genome. ankp and deha are the two HGT genes derived from fungi and their homologous genes in fungal genomes also contain introns, whereas acsf and sdrp were transferred from bacteria, which were free of introns. Surprisingly, it was found that the introns of genes from the donor fungi were different from those in the genome of shrimp, whereas the coding regions around the introns were highly conserved (E-values ranged from 8.00E-17 to 2.00E-13). Besides, by alignment of shrimp genome sequencing reads to this gene using SOAPaligner, we found that the intron regions were in highly coverage of nearly 88X, which is significantly higher than the sequencing depth of the whole genome (42X). Furthermore, these two HGT genes are evenly covered at 76X, while the random selected 100 genes is covered at 48X. Through using SOAPsnp v1.03 [29], three SNPs have been found on the contigs on which ankp located, and one of them has been detected locate on the intron of ankp. This indicated that the introns in two fungi-originated HGT genes may be highly repeated along with its surrounding sequences in shrimp genome. The shrimp acsf contains two introns (105 bp and 117 bp in length, respectively) and sdrp contains one intron (370 bp in length), but no introns were detected in the corresponding donor bacteria genome (D. kuznetsovii and B. phymatum). Therefore, it can be speculated that the introns might be integrated or changed in the HGT genes after horizontal transfer from the donor to L. vannamei.
Three large exogenous segments integrated into L. vannamei genome
When comparing the donor genome with the shrimp genome contigs, three pairs of relatively large segments showed significant similarity around the position of HGT genes (Figure 3). HGT gene tnpA, which encodes a transposase, is derived from a transposon of E. coli that comprises three transposase-encoding genes (tnpX, tnpR, tnpA) and non-coding regions. In addition to the whole transposon, there was also a kch gene (encoding a voltage-gated potassium channel) downstream of the transposon, and the whole segment (6,913 bp) was completely integrated into the genome of L. vannamei. Transposons generally contribute little or nothing to the host’s phenotype, but some horizontally transferred transposons have been identified to benefit the recipient’s genomic evolution [30,31]. In addition to this large segment, there was one more gene, tonB, encoding a membrane spanning protein in the TonB-ExbB-ExbD complex [32], which was located downstream of the transposon in the donor genome. The other two genes (exbB and exbD, which encode ExbB and ExbD) were located far from tonB in the downstream. The exbB gene is one of the homologs of HGT gene exbB that was detected in this study (Table 1). It seems this horizontally transferred transposon might have served as a carrier for foreign genetic materials [31].
Figure 3.

Structures of three large horizontally transferred DNA fragments and their locations in both the donor and receptor genome. The conserved regions between shrimp genomic contigs and corresponding donor genomes are displayed.
HGT gene rpsF was predicted to encode 30S ribosomal protein S6, which is involved in protein synthesis (Table 1). When compared with the most probable donor genome of F. bacterium (NZ_ABI01000003), it was found that rpsF was almost in full-length like that on the donor genome (Figure 3b). Furthermore, it was also completely mapped with shrimp genome contigs without any integrated introns. The corresponding regions of rpsF on the shrimp and bacterial genomes were highly conserved (E value of 2.00E-59 and identity of 87.5%), which indicates that rpsF is an intron-less gene that may have transferred from bacteria to L. vannamei recently. Around rpsF, there is a large segment on the shrimp genome that is similar to the donor genome (E-values ranged from 2.00E-75 to 1.00E-11, identity values ranged from 78.01% to 92.31%). This large segment, 4,655 bp in length, contains six genes on a corresponding donor bacterial genome (Figure 3). Besides, it is surprising to found that non-coding regions on this segment were not conserved, and also a predicted gene hypo2 was not conserved as well. On donor bacterial genome, hypo2 is overlapped with both upstream hypo1 and downstream prsN. Homologous BLAST searching against the nt database detected a great many homologs (more than 100 matches) of this large segment, including hypo2. However, no evidence support hypo2 is actually a coding gene. If it is a gene prediction error, hypo2 may also a non-coding region. F. bacterium is just the best hit species of rpsF in GenBank database. It is known that there is a great deal of variation in genomes of bacteria. Therefore, the data of F. bacterium may be quite different from the true donor genome, which could lack hypo2. Thus, hypo2 may be missed in donor genome before the transfer of this large segment into shrimp genome. Whatever, the alignment results indicated that all these non-coding regions were absent from shrimp genome contigs. Similar results were also observed around mopB (Figure 3c).
The third horizontally transferred large DNA segment (4,118 bp) was around HGT gene mopB (Figure 3c). mopB is predicted to encode molybdopterin oxidoreductase and iron-sulfur binding subunit. Another molybdopterin oxidoreductase encoding gene, mopS, is located downstream of mopB and is also transferred to the shrimp genome. There is a region of low conservation (267 bp) upstream of mopB between the shrimp genome contig and the most probable donor genome. The alignment results indicated that most of the sequence (79.4%) from this region was absent in the bacterial genome. Eukaryotic gene prediction software packages including AUGEST [33], Glimmer-HMM [34] and FGENESH [35] were used to predict exons in this shrimp genome contig, and all the prediction results indicated that the low conserved region was part of a complete exon but not an intron that had inserted into the gene. This suggested that shrimp might have redirected this gene to integrate an additional coding sequence into the gene.
HGT genes under strong negative selection pressure
In order to identify the completeness of these HGT genes, we surveyed them from the genomic contigs with the help of gene prediction software packages AUGEST and FGENESH. Even though some of contigs are short in length, we still detect some HGT genes (acsf, rpsF, exbB, mopB, rpsN, tnpA and dhfr) have complete gene structure, including transcription start site, introns, extrons, polyA and stop codons. Moreover, comparing with the genes on the most probable donor genome, there are not any frameshifts among these HGT genes. Therefore, these HGT genes are relatively complete on shrimp genomes. Then, more efforts were made on the detection of whether HGT genes are exposed to selective pressure after the shrimps diverged from their ancestor. Through comparing HGT genes against the transcriptome unigenes of Fenneropenaeus chinensis and Penaeus monodon (two kinds of shrimps that phylogenetically close with L. vannamei), we collected 16 pairs of orthologous sequences (Table 2). Then, KaKs_Calculator [36] was used for calculating synonymous or nonsynonymous substitutions (dS, dN) among these pairs of genes. The statistical results showed that synonymous substitutions are relatively high in quantity (dS is around 0.7). Except stat, almost all the ratios of the nonsynonymous substitutions per site to synonymous substitutions per site (dN/dS) among these pairs of HGT genes are significantly lower than 1 (Table 2), which indicated that most of these HGT genes are under strong negative selection (especially for rpc2, sdrp, deha and omtp, dN/dS values lower than 0.1). Similar results have been found in HGT genes of aphid [37], the dN/dS ratios of which are more than 0.3, which is larger than that of L. vannamei. Previous researches indicated that genes subjected to negative selection tend to maintain their functions [38]. Therefore, most of the proteins encoded by these negative selected HGT genes appear to be functional.
Table 2.
The dN/dS values of HGT genes
| |
|
F. chinensis |
P. monodon |
||||
|---|---|---|---|---|---|---|---|
| HGT genes | Alignment length | dN/dS | P value | Alignment length | dN/dS | P value | |
| |
acsf |
618 |
0.4694 |
5.49E-03 |
140 |
0.0010 |
2.76E-04 |
|
stat |
199 |
0.5162 |
2.22E-01 |
199 |
0.3882 |
8.54E-02 |
|
|
rpc2 |
264 |
0.0527 |
1.54E-06 |
255 |
0.0516 |
6.28E-05 |
|
|
dhfr |
289 |
0.1057 |
3.42E-11 |
190 |
0.1123 |
2.77E-07 |
|
|
sdrp |
286 |
0.0752 |
5.22E-09 |
101 |
0.0488 |
8.54E-04 |
|
|
ankp |
384 |
0.4984 |
2.10E-04 |
148 |
0.3571 |
1.18E-01 |
|
|
deha |
574 |
0.0844 |
1.16E-17 |
317 |
0.1567 |
3.05E-05 |
|
| L. vannamei | omtp | 533 | 0.0850 | 5.60E-22 | 610 | 0.1263 | 1.13E-19 |
Most HGT genes are predicted to be associated with energy metabolism and defense mechanism
Both TCA cycle and the electron-transport chain are predominant pathways of energy metabolism in the matrix of the mitochondria [39,40]. The HGT gene acsf is predicted to Acetyl-CoA synthetase, which is involved in the metabolism of carbohydrate during the first step of energy generation in the tricarboxylic acid cycle (TCA cycle) [41]. Other four HGT genes (dhfr, sdrp, deha and mopB) are predicted as genes in electron transport (Figure 4). dhfr is an HGT gene annotated to encode dihydrofolate reductase which can use NADH as an electron donor to produce tetrahydrofolate for certain amino acids synthesis in 1-carbon transfer chemistry [42], and sdrp are predicted to encode NAD(H) or NADP(H) oxidoreductases. deha, predicted to be an HGT gene from fungi, encodes a cytochrome b5 reductase-like enzyme that catalyzes the reduction of cytochrome b5 using NADH as the electron donor [43]. mopB is predicted to encode molybdopterin oxidoreductase which participates in transferring electrons to cytochrome c [44]. Furthermore, a gene encoding cytochrome c, which contains a heme-binding domain for energy production and conversion, is located upstream of mopB and was transferred along with mopB (Figure 3). And at last, the energy-transducing protein ExbB, encoded by HGT gene exbB, is a part of TonB-ExbB-ExbD complex to form an energy transduction system [45-47]. Therefore, according to the information of predicted functions, all these six HGT genes may associate with energy metabolism.
Figure 4.

Nine HGT genes participate in the TCA cycle and electron transport chain of the shrimp mitochondrion. The majority of the functions of these HGT genes are predicted relate to energy metabolism. The HGT genes are shown in gray oblongs and the HGT genes differentially expressed during five development stages are marked with additional red borders.
As for the six energy metabolism related HGT genes, it is of interest to find whether they are transferred from mitochondria in the distant past. It is well known that nuclear mitochondrial DNA (NUMTs) is commonly found to be transferred into nuclear genomes in various species [48]. However, in this study, none of six energy metabolism related HGT genes have been identified to show any similarity to the whole mitochondrial genome of L. vannamei (NCBI accession number of NC_009626.1). Therefore, unlike NUMTs, these HGT genes are not transferred from mitochondria in recent past, but transferred from non-mating organisms of shrimp in the long evolutionary history.
In Figure 4, the other three HGT genes are predicted to function in protein synthesis and interaction. rpsF and rpsN are predicted to encode two ribosomal protein subunits that are used for protein synthesis. ankp encodes a protein containing ankyrin repeats, which are used for mediating protein-protein interactions in diverse families of proteins [49]. Interestingly, the spread of ankyrin repeats has been suggested to have occurred by HGT, and their occurrence in yeast excludes exon shuffling [49]. This supported the HGT events of ankp, which maintained the stability of its gene structure.
According to the annotated functions of four of the HGT genes, they are associated with defense mechanisms. There are two antibiotic resistance genes (stat and cata) that are predicted to encode streptomycin 3′-adenylyltransferase and chloramphenicol acetyltransferase, respectively (Table 1). Both transferases are responsible for the streptomycin and chloramphenicol resistance and prevent them from binding to ribosomes [50,51]. Accidently, streptomycin and chloramphenicol are specifically bind to 30S and 50S ribosomal subunit, respectively, which are predicted encoded by two other HGT genes, rpsF and rpsN (Table 1). Thus stat and cata may be involved in protection of protein synthesis. As the only bacteria-to-Arthropoda HGT gene in L. vannamei (Figure 2), omtp was annotated to encode O-methyltransferase, which is ubiquitously dispersed in various organisms and plays an important role in animal growth, development and defense [52,53]. rpc2 is annotated to encode repressor protein C2, which is a gene involved in the SOS response [54,55]. Generally, the SOS response is induced by various antibiotics, such as chloramphenicol, trimethoprim and streptomycin. The SOS response can promote HGT events of antibiotic resistance genes among bacteria [54]. Above all, the proteins encoded by these four HGT genes are predicted as defense-related.
Gene expression of the HGT genes in differential developmental stages of shrimp
To detect whether HGT genes have effects on shrimp growth and development, we performed differential gene expression (DGE) analysis on these HGT genes, based on the transcriptomes of five developmental stages of shrimp larvae (see Methods). The number of reads aligned to the transcripts were calculated (Additional file 1: Table S4), and the expression level of each transcript was measured by FPKM values (fragments per kilobase of exon per million fragments mapped). High coverage of reads successfully supported for the following DGE analysis. Nine HGT genes showed differential expression among five stages (Figure 5, Additional file 1: Table S5). stat displayed the highest expression level (FPKM values ranged from 1018.57 to 2910.86) at all five stages, while rpsF was expressed at the lowest (FPKM values ranged from 0 to 1.43). rpsF seems only expressed at the stage of zoea and mysis. Similar to rpsF, the other two fundamental DNA and protein synthesis related HGT genes (dhfr and ankp) displayed an expression pattern that gene expression level at the stage of zoea and mysis was generously higher than the other stages (Figure 5, Additional file 1: Table S4). In addition to providing fundamental nucleic acid precursors and certain amino acids, dhfr also participates in energy metabolism. Three other energy metabolism-related HGT genes showed DGE, and two of them (acsf and sdrp) displayed similar expression pattern with dhfr. deha showed the reverse expression pattern, in that it showed lower expression at stages of zoea and mysis than at other stages (Figure 5). In addition, three defense-related HGT genes (rpc2, omtp and stat) were universally more highly expressed than other HGT genes (Figure 5, Additional file 1: Table S5), indicating that they may play an important role in early shrimp development.
Figure 5.
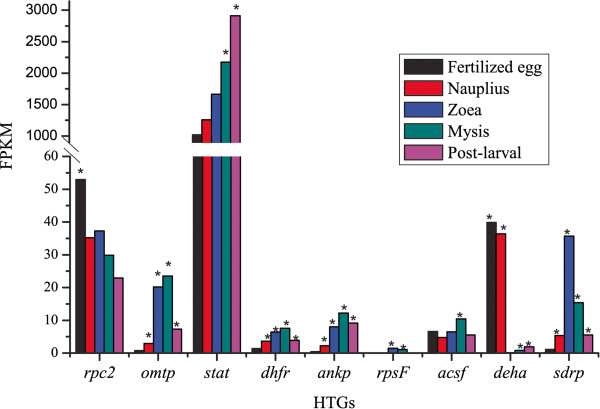
Differential expression of eight HGT genes at five developmental stages of shrimp. Gene expression level was evaluated by FPKM value. * indicates the HGT genes were significantly differentially expressed when compared with the lowest expressed development stage.
Discussion
Structure alteration of HGT genes indicated the mechanisms of HGT
Previous research on HGT events suggested that some of the HGT genes transferred from bacteria acquired eukaryotic features, such as signal peptides and spliceosomal-type introns [13,37]. Similar occurrences were observed in the present work: five spliceosomal-type introns are integrated into four HGT genes in the shrimp genome. Two HGT genes originating from fungi (ankp and deha) seem to have differential introns to that of the most probable donor, while the other two HGT genes (acsf and sdrp) originated from bacteria and new introns were directly inserted into the HGT genes. As for ankp and deha, the introns on it may exchanged between donor and recipient genome, otherwise, it may originated from an un-sequenced fungus, of which the intron regions are also similar to that of shrimp genome. Whereas, the introns on ankp and deha are predicted to be highly repeated in shrimp genome, which indicated these introns maybe arose from the shrimp. Spliceosomal introns in eukaryotic genes have been identified as a possible barrier to HGT from fungi. However, it would still possible for fungi-originated genes transfer through mediation of spliced RNA intermediate [1]. Thus, the introns on the two fungi-originated genes (ankp and deha) are most probably exchanged with shrimp introns rather than originated from un-sequenced fungi. Although introns are generally thought to be mildly deleterious elements for mature mRNA generation, there are still indications that some introns are essential for eukaryote genomes, e.g. alternative splicing, RNA editing and enhancing mRNA transcription [56]. Thus, the introns integrated into these HGT genes may play essential roles in gene expression and regulation. In addition to introns, other non-coding DNA and even coding regions were changed in the HGT genes. The horizontally transferred large segment around rpsF was observed to exclude non-coding regions in the shrimp genome (Figure 3). By contrast, an inserted coding region was detected at the 5′ end of mopB in the shrimp genome. Although there is no evidence that this inserted coding sequence enhances the protein expression level or function, it may undergo an adaptive fitness in shrimp [57].
There are several mechanisms to explain HGT events in eukaryotes. The mechanism of bacteria-to-animal HGT most probably arises from endosymbiosis [1,58]. The close and constant proximity of cells provides a platform for the transfer of genetic materials. Generally, the genetic materials that horizontally transferred are mostly complete or fragments of genes, but not large DNA fragments. However, horizontally transferred large DNA fragments were detected in previous studies and in the present study [59-62]. In the case of single HGT genes, spliceosomal-type introns were found, which indicated that these genes acquired the introns for their function after they were transferred to the host genome. In a previous study [63], several conserved introns in both recipient and donor taxa were found in HGT genes, which indicated that those HGT genes are the products of direct eukaryote-to-eukaryote transfer. By contrast, in this study, the introns in two of the fungi-to-L. vannamei HGT genes seem to be exchanged, which suggested the introns may have not have transferred into host genomes along with the exons. The single HGT genes, including intron-free HGT genes, were most probably acquired from mature RNA after gene transcription in the donor cell. It is also possible that these genes were acquired from raw DNA segments and shrimp could recognize the splice sites accurately and substitute with its own introns, which seems mechanistically unlikely. Operons have been reported playing important role in transfer of large DNA segments which include three or more genes [64]. Through operon prediction by DOOR (Database of prOkaryotic OpeRons) [65], we found that this large horizontally transferred segment around rpsF was predicted as an operon. Furthermore, there is a gene priA, located upstream of this operon, encoded primosomal protein that are responsible for creating RNA primers during DNA replication [66]. Primosomal proteins are essential for operon HGT events, and actually, primosomal operon have ever been detected in previous researches [67]. Therefore, it is reasonable to think that the large segment around rpsF is completely transferred. As for the large horizontally transferred segment around mopB, operon prediction indicated that mopB and mopS are also involved in an operon, which implies that they are horizontally transferred together (Figure 3c). Although these large segments are transferred together, non-coding regions were removed from them (Figure 3, the large segment around tnpA was an exception because it was transferred as a single transposon), which suggested intergenic regions may not transferred into the shrimp genome. This supports the hypothesis that mature RNA was the main substrate for HGT.
Some HGT genes appeared to be functional in L. vannamei
Given that shrimp cells are frequently in proximity to prokaryotic cells, it is not surprising that HGT can be detected between them. The type of prokaryote-to-eukaryote HGT is generally of interest because it could potentially providing novel functions to eukaryotes, allow adaptation to novel niches, and affect their evolution [1]. However, finding prokaryotic DNA in eukaryotic genome is not sufficient to imply that this DNA is functional in the host genome. In this study, several evidences suggest the functionality of these HGT genes for shrimp. Firstly, most of HGT genes had complete gene structure, and none of frameshift mutations could be found among them, which indicated they are not pseudogenes. Secondly, selection pressure analysis of these HGT genes showed that there was a strong negative selection for most of them (dN/dS values significantly lower than 1), indicating deleterious effects for most mutations at the protein levels [38], thus, these HGT genes in shrimp seemed to be effectively functional after the transfer. Lastly, these HGT sequences were derived from EST or transcriptome sequences, which imply they could transcribe in shrimp cells. Furthermore, most of the HGT genes were found to be transcribed at the early development stages of shrimp larvae, and they displayed significant DGE at five development stages. This indicated that these HGT genes were tightly associated with the development of shrimp. Therefore, these genes transferred from bacteria or fungi appeared to be functional in shrimp cells. According to the predicted functions of these HGT genes, six of them were annotated as energy metabolism related, and four of them were predicted to be associated with defense of shrimp. Among them, one of defense-related HGT gene, omtp, was reported to be involved in defense response against bacterial infection in Chinese shrimp, F. chinensis[28]. In a previous report [68], there was strong evidence to show that invertebrates could obtain defensive complex polyketides from bacterial symbionts via O-methyltransferase (encoded by omtp) methylation of the marine compounds. Therefore, omtp was assumed to be an effective functional defense gene transferred from bacteria. Given the unlikely transfer and transcription of genes that in the donor are related to energy metabolism and defense, we speculate that these HGT genes may have the same role in the recipient. This is consistent with what is known about the demands of shrimp where molting is energy intensive and current aquaculture methods stressful [14,16,69]. However, the precise roles of these proteins need to be demonstrated through further studies.
Conclusions
Through sequence homology comparison, phylogenetic analysis and experimental verification, fourteen bacteria or fungi originated HGT genes were detected in L. vannamei. Spliceosomal-type introns were found to be inserted in four HGT genes, while non-coding regions of two large horizontally transferred segments were lacked on the shrimp genome. These structure alterations provide evidence that mature RNA may be the substrate for HGT events. Among 14 HGT genes, most of them were detected to be exposed to negative selection pressure and they appeared to be functional. Functions prediction annotated them to be associated with energy metabolism and defense of shrimp. Further studies should be taken to demonstrate the precise roles of these HGT genes.
Methods
Sources of genome and transcriptome dataset of L. vannamei
A total of 1.9 Gb shrimp genome contigs, which cover approximately 76% of the whole genome, were assembled from 42-fold coverage of whole genome shotgun reads of L. vannamei in our laboratory. The genome contigs were used for mapping of the candidate HGT genes on the shrimp genome. Information on the genome contigs to which candidate HGT genes mapped were submitted to NCBI (http://www.ncbi.nlm.nih.gov, KC701594 - KC701607).
The gene set of L. vannamei was predominantly composed of two online datasets from NCBI: 161,796 ESTs that were generated from multiple tissues [20,70], and 74,111 transcriptome sequences that were generated from the whole body of L. vannamei larvae [21]. The ESTs were assembled into 32,550 unigenes using CAP3 with default parameters [71]. The software Minimus2 (http://sourceforge.net/apps/mediawiki/amos/index.php?title=Minimus2) was then used to merging the two sequence sets and remove duplicated, redundant sequences. Ultimately, 82,579 gene segments were generated and used for subsequent HGT genes detection.
Local protein database construction
There are 6,529,500 protein sequences collected from the complete proteomes of 4,080 species on the NCBI ftp site (ftp.ncbi.nlm.nih.gov), and these were used for HGT detection. The constructed local database included seven arthropods, 32 fungi, seven plants, 31 other eukaryotes, 1,607 bacteria and 2,387 viruses (Additional file 1: Table S1).
BLASTx-based HGT search and detection
BLASTx-based [72] homologous sequence searching was the first procedure for HGT detection, and there were three steps of similar sequences identification in the detection pipeline (Figure 1). First, each candidate L. vannamei gene segment was compared with the total protein sequences in the local database with cutoff thresholds of E value ≤ 1E-20, identity value ≥ 25% and overlap value ≥ 25 [72]. The remaining 13,388 highest similarity sequences were extracted for further analysis. The second step was the similarity comparison between arthropods and other species. By comparing the BLAST results, candidate genes with higher BLASTx similarity scores to Arthropoda than other species were excluded because these genes were most probably the Arthropoda unique genes. In step three, using the same cutoff thresholds as the first step, the remaining sequences were used to search against the nr database. If there were less than 10 homologs in one phylogeny, the corresponding gene segments were not considered for subsequent phylogenetic analysis.
Phylogenetic analysis
Homologous sequences of the remaining candidate HGT genes were extracted from the nr database, and the candidate HGT genes were translated from nucleotides to amino acid sequences. The combination of the candidate HGT genes sequences and their homologs from the nr database were used to construct phylogenetic trees, which were used to assess standard HGT or non-HGT events [22,23]. Three types of phylogenetic tree were constructed based on different methods and were compared with each other to generate the best phylogenetic topology.
The first type of tree was generated by complement alignment of the sequences using ClustalX [73], and computing the pairwise distance-matrix of the aligned sequences with PHYLIP software package [74]. A neighbor-joining tree was then constructed with the distance-matrix and 1000 bootstrap replicates were performed. The alignment method of the other two phylogenetic analyses was implemented on MUSCLE 3.6 [75] and the conserved region of each alignment was trimmed using Gblocks [76], which allows less strict flanking positions and gap positions within the final blocks, but does not allow many contiguous nonconserved positions. Using Phyml [77], we performed an approximate likelihood-ratio test (aLRT, Minimum of SH-like and Chi2-based parametric) on the conserved aligned sequences [78], and constructed the phylogenetic tree with the substitution model of JTT. The maximum likelihood (ML) phylogeny was the last type of analysis, and the ML trees are shown in the figures. ML trees were constructed by Phyml using a WAG + gamma +Inv model, and 100% bootstraps were performed to gain the branch support values [8,22]. The phylogenetic trees were used to judge HGT events and HGT directions, based on previous studies that considered that HGT genes tend to form a monophyletic branch with a set of far-related species, which are considered as the donors [22,23]. The alignment datasets and phylogenetic tree-files of the HGT genes from analyses have been deposited in TreeBASE as study S14162 (http://www.treebase.org/treebase/). To test the support for contentious topology of the extracted HGT genes, Bayesian phylogenetic inference (BI) was performed on them with the help of the program Mrbayes 3.2.1 [79]. In the BI analysis, two independent runs, each with four chains, were analyzed for millions of generations until the standard deviation of split frequencies converged towards zero. A burn-in of 25% samples is used for summarizing the parameter values and trees.
Verification of HGT genes by PCR
ESTs and transcriptome sequences may be contaminated by other organisms in the procedures of RNA extraction, library construction and sequencing; therefore, the candidate HGT genes were verified to confirm whether they were part of the genome of L. vannamei. Each candidate sequence that showed significant HGT events was compared against the draft genome contigs using BLASTn with a cutoff threshold of E value ≤ 1E-40, identity value ≥ 90% [72]. To avoid the effects of genome mis-assembly, a DNA-based PCR was implemented to amplify the genomic segments containing HGT genes. DNA was isolated from the muscle tissue of the adult shrimps. The information of the genome contigs including HGT genes were extracted for primer design and the information of the primer pairs for each HGT gene are listed in Additional file 1: Table S2. Besides, PCR amplification was also performed on the edge of HGT fragments on genome contigs for the attempts of amplify both HGT region and eukaryotic portion of the genome. The edges of the HGT fragments were determined based on the BLAST (E value ≤ 1E-5) of the genome contigs against the most probable donor genome. The edges were the initial sites of two sides which showed homologous with the most probable donor genome. The primers were designed to amplify the products covering both HGT and eukaryotic portions as long as possible on both sides of the HGT region. The edge information and the primer pairs are listed in Additional file 1: Table S3. The PCR parameters were 94°C for 3 min; 35 cycles of 94°C for 30 s, annealing temperature (It is different for each product and are listed in Table S2-S3) for 30 s, and 72°C for 45 s; and a final 72°C for 7 min. The PCRs were conducted using ABI GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA, USA). Besides, negative controls were set using Escherichia coli genomic DNA or distilled water as template for PCR amplification, and a positive control were performed using primers designed in previous study, which has been successfully amplified a specific shrimp genomic sequence (GI:124633758) [80]. All the PCR products were sequenced to test their integrity and accuracy using ABI 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA). The sequences generated from sequencer were compared with the original genomic contigs.
Structure analysis of HGT genes
The structures of HGT segments and their surrounding sequences on both the shrimp and donor genome were analyzed. The tBLASTx program was used to compare the HGT segments and shrimp genome contigs against the complete donor genomes with an E value cutoff of 1.00E-10 in consideration of a longer match length. The up- and downstream genes of the HGT genes positions on the donor genome were identified and analyzed to determine whether they were co-transferred to shrimp genome. Using ClustalX, the conservation between shrimp genome contigs and donor genomes was displayed along the alignment sequences.
Calculation of dN/dS ratios
We used KaKs_Calculator to calculate synonymous and nonsynonymous substitutions between orthologous pairs to identify whether there is natural selection subjected to these genes [36]. The 46,676 transcriptome unigenes of F. chinensis[81] and 7.8 Gb transcriptome reads of P. monodon were downloaded from NCBI (SRX110652, SRX110651 and SRX110649). The transcriptome reads of P. monodon were assembled into 182,648 unigenes through using the Trinity program [82]. BLASTn-based homologous sequence searching was performed for predicted HGT genes of L. vannamei against the transcriptome unigenes of F. chinensis and P. monodon. The homologous sequences of each HGT gene were aligned using ClustalX and the open reading frames were determined, then, we calculated the dN/dS for each orthologous pair using YN algorithm with KaKs_Calculator.
DGE profiles of HGT genes
Recently, five transcriptomes of L. vannamei were sequenced at different developmental stages in our laboratory: fertilized egg; nauplius; zoea; mysis and post-larval [16]. More than 23.7 Gb of high quality clean data were generated and each library contained a mean number of 52,660,075 reads. All the reads were then assembled into 66,815 unigenes using the Trinity program [82]. The HGT related transcriptome reads have been submitted to NCBI SRA database with the accession number of SRR839222, SRR839236, SRR842572, SRR842625 and SRR842627. The ESTs, transcriptome sequences of L. vannamei on NCBI, and our transcriptome sequences were generated from different individuals; therefore, the DGE analysis will be affected when analyzing the HGT gene segments directly. Therefore, the subsequent DGE analysis of HGT genes was represented by that of their extremely homologous unigenes in five transcriptomes. For each transcriptome, all reads were processed and aligned to all unigenes using SOAPaligner v2.21 which allow two base mismatches, and coverage of every unigene was analyzed. The transcript abundance was measured by FPKM values:
Set FPKM to be the expression of unigene A, and C to be number of fragments that uniquely aligned to unigene A, N to be total number of fragments that uniquely aligned to all unigenes, and L to be the base number in the CDS of unigene A. The FPKM method is able to eliminate the influence of different gene length and sequencing level on the calculation of gene expression. As the data came from five developmental stages for DGE analysis, with the help of the edgeR package [83], we performed 10 pairwise comparisons of the expression between any two developmental stages of shrimp. The DGE ratios, which were calculated using the ratio of the normalized FPKM values from any two samples, were used to test the statistical significance, as described recently [84,85]. The p-values were analyzed using the edgeR package for significant differences, and q-values were calculated by applying a false discovery rate (FDR) adjustment for multiple testing [86,87]. The criteria for DGE evaluation are DGE ratio ≥ 2, p value ≤ 0.001 and FDR ≤ 0.001.
Abbreviations
HGT: Horizontal gene transfer; ESTs: Expression sequence tags; DGE: Differential gene expression.
Competing interests
The authors have declared that no competing interest exists.
Authors’ contributions
JHX designed the study. JBY carried out the analysis, drafted and revised the manuscript. XJZ revised the manuscript. JKW carried out partial analysis. XJZ, CZL, FHL and JHX supervised the study. All authors read and approved the final manuscript.
Supplementary Material
Local genome database for HGT identification. Table S2. Primers of the 14 HGT genes for PCR. Table S3. The PCR primers of the edge products of 14 HGT genes. Table S4. The FPKMs of each HGT genes in different development stage. Table S5. Differential expression of the 14 HGT genes at five development stages of shrimp larvae.
Phylogenetic tree of acsf and its homologs. Figure S2. Phylogenetic tree of rpsF and its homologs. Figure S3. Phylogenetic tree of rpsN and its homologs. Figure S4. Phylogenetic tree of exbB and its homologs. Figure S5. Phylogenetic tree of mopB and its homologs. Figure S6. Phylogenetic tree of tnpA and its homologs. Figure S7. Phylogenetic tree of stat and its homologs. Figure S8. Phylogenetic tree of rpc2 and its homologs. Figure S9. Phylogenetic tree of dhfr and its homologs. Figure S10. Phylogenetic tree of cata and its homologs. Figure S11. Phylogenetic tree of sdrp and its homologs. Figure S12. Phylogenetic tree of ankp and its homologs. Figure S13. Phylogenetic tree of deha and its homologs.
Contributor Information
Jian-Bo Yuan, Email: jianbone@163.com.
Xiao-Jun Zhang, Email: xjzhang@qdio.ac.cn.
Cheng-Zhang Liu, Email: liu@qdio.ac.cn.
Jian-Kai Wei, Email: weijiankai10@mails.gucas.ac.cn.
Fu-Hua Li, Email: fhli@qdio.ac.cn.
Jian-Hai Xiang, Email: jhxiang@qdio.ac.cn.
Acknowledgements
I would like to thank Dr. Feng Jiang for his thoughtful discussions on this work, and also thank Dr. Elayaraja Sivaramasamy for his help on editing the manuscript. This work was supported by a grant from National High Tech Research and Development Program (2012AA092205, 2012AA10A404) and National Program on Key Basic Research Project (973 Program, Grant No. 2012CB114403).
References
- Dunning Hotopp JC. Horizontal gene transfer between bacteria and animals. Trends Genet. 2011;27(4):157–163. doi: 10.1016/j.tig.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling PJ, Palmer JD. Horizontal gene transfer in eukaryotic evolution. Nature reviews Genetics. 2008;9(8):605–618. doi: 10.1038/nrg2386. [DOI] [PubMed] [Google Scholar]
- Beiko RG, Harlow TJ, Ragan MA. Highways of gene sharing in prokaryotes. Proc Natl Acad Sci USA. 2005;102(40):14332–14337. doi: 10.1073/pnas.0504068102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogarten JP, Doolittle WF, Lawrence JG. Prokaryotic evolution in light of gene transfer. Mol Biol Evol. 2002;19(12):2226–2238. doi: 10.1093/oxfordjournals.molbev.a004046. [DOI] [PubMed] [Google Scholar]
- Nikoh N, Tanaka K, Shibata F, Kondo N, Hizume M, Shimada M, Fukatsu T. Wolbachia genome integrated in an insect chromosome: evolution and fate of laterally transferred endosymbiont genes. Genome research. 2008;18(2):272–280. doi: 10.1101/gr.7144908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty SN, Foster JM, Mitreva M, Dunning Hotopp JC, Martin J, Fischer K, Wu B, Davis PJ, Kumar S, Brattig NW. et al. Endosymbiont DNA in endobacteria-free filarial nematodes indicates ancient horizontal genetic transfer. PloS one. 2010;5(6):e11029. doi: 10.1371/journal.pone.0011029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JA, Kirkness EF, Simakov O, Hampson SE, Mitros T, Weinmaier T, Rattei T, Balasubramanian PG, Borman J, Busam D. et al. The dynamic genome of Hydra. Nature. 2010;464(7288):592–596. doi: 10.1038/nature08830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Lou MM, Xie GL, Zhang GQ, Zhou XP, Li B, Jin GL. Horizontal gene transfer in silkworm. Bombyx mori. BMC genomics. 2011;12:248. doi: 10.1186/1471-2164-12-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CX, Bhattacharya D, Reyes-Prieto A. Endosymbiotic and horizontal gene transfer in microbial eukaryotes: impacts on cell evolution and the tree of life. Mobile genetic elements. 2012;2(2):101–105. doi: 10.4161/mge.20110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen TL, Stukenbrock EH, Liu Z, Meinhardt S, Ling H, Faris JD, Rasmussen JB, Solomon PS, McDonald BA, Oliver RP. Emergence of a new disease as a result of interspecific virulence gene transfer. Nature genetics. 2006;38(8):953–956. doi: 10.1038/ng1839. [DOI] [PubMed] [Google Scholar]
- Rumpho ME, Worful JM, Lee J, Kannan K, Tyler MS, Bhattacharya D, Moustafa A, Manhart JR. Horizontal gene transfer of the algal nuclear gene psbO to the photosynthetic sea slug Elysia chlorotica. Proc Natl Acad Sci U S A. 2008;105(46):17867–17871. doi: 10.1073/pnas.0804968105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA, Jarvik T. Lateral transfer of genes from fungi underlies carotenoid production in aphids. Science. 2010;328(5978):624–627. doi: 10.1126/science.1187113. [DOI] [PubMed] [Google Scholar]
- Nikoh N, McCutcheon JP, Kudo T, Miyagishima SY, Moran NA, Nakabachi A. Bacterial genes in the aphid genome: absence of functional gene transfer from Buchnera to its host. PLoS genetics. 2010;6(2):e1000827. doi: 10.1371/journal.pgen.1000827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightner DV. A review of the diseases of cultured penaeid shrimps and prawns with emphasis on recent discoveries and developments. Iloilo City, Philippines: Proceedings of the First International Conference on the Culture of Penaeid Prawns/Shrimps, 4–7 December 1984; 1985. pp. 79–103. [Google Scholar]
- Klaoudatos SD, Klaoudatos DS. Phylogeny biology and ecology of Crustaceans(Phylum Arthropoda; Subphylum Crusacea). Reproductive Biology of Crustaceans. New Hampshire, USA: Case studies of Decapod Crustaceans Science Publishers; 2008. [Google Scholar]
- Center GSL. The brine shrimp life cycle. Genetics: Learn; 2012. [Google Scholar]
- Gil-Turnes MS, Hay ME, Fenical W. Symbiotic marine bacteria chemically defend crustacean embryos from a pathogenic fungus. Science. 1989;246(4926):116–118. doi: 10.1126/science.2781297. [DOI] [PubMed] [Google Scholar]
- Overstreet RM. Parasites of some penaeid shrimps with emphasis on reared hosts. Aquaculture. 1973;2:105–140. [Google Scholar]
- DiGuistini S, Wang Y, Liao NY, Taylor G, Tanguay P, Feau N, Henrissat B, Chan SK, Hesse-Orce U, Alamouti SM. et al. Genome and transcriptome analyses of the mountain pine beetle-fungal symbiont Grosmannia clavigera, a lodgepole pine pathogen. Proc Natl Acad Sci USA. 2011;108(6):2504–2509. doi: 10.1073/pnas.1011289108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary NA, Trent HF 3rd, Robalino J, Peck ME, McKillen DJ, Gross PS. Analysis of multiple tissue-specific cDNA libraries from the Pacific whiteleg shrimp, Litopenaeus vannamei. Integrative and comparative biology. 2006;46(6):931–939. doi: 10.1093/icb/icl006. [DOI] [PubMed] [Google Scholar]
- Li C, Weng S, Chen Y, Yu X, Lu L, Zhang H, He J, Xu X. Analysis of Litopenaeus vannamei transcriptome using the next-generation DNA sequencing technique. PloS one. 2012;7(10):e47442. doi: 10.1371/journal.pone.0047442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZW, Shen YH, Xiang ZH, Zhang Z. Pathogen-origin horizontally transferred genes contribute to the evolution of Lepidopteran insects. BMC evolutionary biology. 2011;11:356. doi: 10.1186/1471-2148-11-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanhope MJ, Lupas A, Italia MJ, Koretke KK, Volker C, Brown JR. Phylogenetic analyses do not support horizontal gene transfers from bacteria to vertebrates. Nature. 2001;411(6840):940–944. doi: 10.1038/35082058. [DOI] [PubMed] [Google Scholar]
- Sutton BC. Mitosporic fungi (Deuteromycetes) in the fictionary-of-the-fungi. 1993. pp. 27–55. (Fungal Holomorph : Mitotic, Meiotic and Pleomorphic Speciation in Fungal Systematics). http://apps.webofknowledge.com/InboundService.do?SID=T2mU3W69zf7xznsDuJS&product=WOS&UT=A1993BY77H00003&SrcApp=EndNote&DestFail=http%3A%2F%2Fwww.webofknowledge.com&Init=Yes&action=retrieve&Func=Frame&customersID=ResearchSoft&SrcAuth=ResearchSoft&IsProductCode=Yes&mode=FullRecord.
- Altincicek B, Kovacs JL, Gerardo NM. Horizontally transferred fungal carotenoid genes in the two-spotted spider mite Tetranychus urticae. Biol Lett. 2012;8(2):253–257. doi: 10.1098/rsbl.2011.0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguinaldo AM, Turbeville JM, Linford LS, Rivera MC, Garey JR, Raff RA, Lake JA. Evidence for a clade of nematodes, arthropods and other moulting animals. Nature. 1997;387(6632):489–493. doi: 10.1038/387489a0. [DOI] [PubMed] [Google Scholar]
- Iyer LM, Aravind L, Coon SL, Klein DC, Koonin EV. Evolution of cell-cell signaling in animals: did late horizontal gene transfer from bacteria have a role? Trends in genetics : TIG. 2004;20(7):292–299. doi: 10.1016/j.tig.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Li DX, Du XJ, Zhao XF, Wang JX. Cloning and expression analysis of an o-methyltransferase (OMT) gene from Chinese shrimp, Fenneropenaeus chinensis. Fish & shellfish immunol. 2006;21(3):284–292. doi: 10.1016/j.fsi.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K. SNP detection for massively parallel whole-genome resequencing. Genome research. 2009;19(6):1124–1132. doi: 10.1101/gr.088013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert C, Schaack S, Pace JK 2nd, Brindley PJ, Feschotte C. A role for host-parasite interactions in the horizontal transfer of transposons across phyla. Nature. 2010;464(7293):1347–1350. doi: 10.1038/nature08939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodelsperger C, Sommer RJ. Computational archaeology of the Pristionchus pacificus genome reveals evidence of horizontal gene transfers from insects. BMC evolutionary biology. 2011;11:239. doi: 10.1186/1471-2148-11-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeck GS, Coulton JW. TonB-dependent iron acquisition: mechanisms of siderophore-mediated active transport. Mol Microbiol. 1998;28(4):675–681. doi: 10.1046/j.1365-2958.1998.00817.x. [DOI] [PubMed] [Google Scholar]
- Stanke M, Morgenstern B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005;33:W465–W467. doi: 10.1093/nar/gki458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20(16):2878–2879. doi: 10.1093/bioinformatics/bth315. [DOI] [PubMed] [Google Scholar]
- Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268(1):78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Li J, Zhao XQ, Wang J, Wong GK, Yu J. KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics, proteomics & bioinformatics. 2006;4(4):259–263. doi: 10.1016/S1672-0229(07)60007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoh N, Nakabachi A. Aphids acquired symbiotic genes via lateral gene transfer. BMC biology. 2009;7:12. doi: 10.1186/1741-7007-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters AE, Bavishi A, Cho H, Choudhary M. Evolutionary constraints and expression analysis of gene duplications in Rhodobacter sphaeroides 2.4.1. BMC research notes. 2012;5:192. doi: 10.1186/1756-0500-5-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg HL. Krebs citric-acid cycle - half a century and still turning - introductory. Biochem Soc Symp. 1987;54:1–2. [Google Scholar]
- Karp G. Cell and Molecular Biology. John Wiley & Sons; 2012. http://as.wiley.com/WileyCDA/WileyTitle/productCd-EHEP002533.html. [Google Scholar]
- Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci USA. 2006;103(27):10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell JR, Dyson HJ, Wright PE. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu Rev Bioph Biom. 2004;33:119–140. doi: 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- Giordani R, Buc J. Evidence for two different electron transfer pathways in the same enzyme, nitrate reductase A from Escherichia coli. Eur J Biochem/FEBS. 2004;271(12):2400–2407. doi: 10.1111/j.1432-1033.2004.04159.x. [DOI] [PubMed] [Google Scholar]
- Li XZ, Luo QW, Wofford NQ, Keller KL, McInerney MJ, Wall JD, Krumholz LR. A molybdopterin oxidoreductase is involved in H-2 oxidation in desulfovibrio desulfuricans G20. J Bacteriol. 2009;191(8):2675–2682. doi: 10.1128/JB.01814-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs PI, Larsen RA, Postle K. Quantification of known components of the Escherichia coli TonB energy transduction system: TonB, ExbB, ExbD and FepA. Mol Microbiol. 2002;44(1):271–281. doi: 10.1046/j.1365-2958.2002.02880.x. [DOI] [PubMed] [Google Scholar]
- Eick-Helmerich K, Braun V. Import of biopolymers into Escherichia coli: nucleotide sequences of the exbB and exbD genes are homologous to those of the tolQ and tolR genes, respectively. J Bacteriol. 1989;171(9):5117–5126. doi: 10.1128/jb.171.9.5117-5126.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letain TE, Postle K. TonB protein appears to transduce energy by shuttling between the cytoplasmic membrane and the outer membrane in Escherichia coli. Mol Microbiol. 1997;24(2):271–283. doi: 10.1046/j.1365-2958.1997.3331703.x. [DOI] [PubMed] [Google Scholar]
- Tsuji J, Frith MC, Tomii K, Horton P. Mammalian NUMT insertion is non-random. Nucleic acids research. 2012;40(18):9073–9088. doi: 10.1093/nar/gks424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork P. Hundreds of ankyrin-like repeats in functionally diverse proteins - mobile modules that cross phyla horizontally. Proteins. 1993;17(4):363–374. doi: 10.1002/prot.340170405. [DOI] [PubMed] [Google Scholar]
- Shaw WV, Packman LC, Burleigh BD, Dell A, Morris HR, Hartley BS. Primary structure of a chloramphenicol acetyltransferase specified by R plasmids. Nature. 1979;282(5741):870–872. doi: 10.1038/282870a0. [DOI] [PubMed] [Google Scholar]
- Harwood JH, Smith DH. Resistance factor-mediated streptomycin resistance. J Bacteriol. 1969;97(3):1262–1271. doi: 10.1128/jb.97.3.1262-1271.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahlbrock K, Scheel D. Physiology and molecular-biology of phenylpropanoid metabolism. Annu Rev Plant Physiol Plant Mol Biol. 1989;40:347–369. doi: 10.1146/annurev.pp.40.060189.002023. [DOI] [Google Scholar]
- Chiron H, Drouet A, Claudot AC, Eckerskorn C, Trost M, Heller W, Ernst D, Sandermann H. Molecular cloning and functional expression of a stress-induced multifunctional O-methyltransferase with pinosylvin methyltransferase activity from Scots pine (Pinus sylvestris L.) Plant Mol Biol. 2000;44(6):733–745. doi: 10.1023/A:1026507707186. [DOI] [PubMed] [Google Scholar]
- Beaber JW, Hochhut B, Waldor MK. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature. 2004;427(6969):72–74. doi: 10.1038/nature02241. [DOI] [PubMed] [Google Scholar]
- Little JW, Mount DW, Yanischperron CR. Purified lexa protein is a repressor of the reca and lexa genes. Proc Natl Acad Sci U S A. 1981;78(7):4199–4203. doi: 10.1073/pnas.78.7.4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffares DC, Mourier T, Penny D. The biology of intron gain and loss. Trends in genetics: TIG. 2006;22(1):16–22. doi: 10.1016/j.tig.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A, Keightley PD. The distribution of fitness effects of new mutations. Nat Rev Genet. 2007;8(8):610–618. doi: 10.1038/nrg2146. [DOI] [PubMed] [Google Scholar]
- Dyall SD, Brown MT, Johnson PJ. Ancient invasions: from endosymbionts to organelles. Science. 2004;304(5668):253–257. doi: 10.1126/science.1094884. [DOI] [PubMed] [Google Scholar]
- Kondo N, Nikoh N, Ijichi N, Shimada M, Fukatsu T. Genome fragment of Wolbachia endosymbiont transferred to X chromosome of host insect. Proc Natl Acad Sci USA. 2002;99(22):14280–14285. doi: 10.1073/pnas.222228199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aikawa T, Anbutsu H, Nikoh N, Kikuchi T, Shibata F, Fukatsu T. Longicorn beetle that vectors pinewood nematode carries many Wolbachia genes on an autosome. P R Soc B. 2009;276(1674):3791–3798. doi: 10.1098/rspb.2009.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudoumis V, Alam U, Aksoy E, Abd-Alla AMM, Tsiamis G, Brelsfoard C, Aksoy S, Bourtzis K. Tsetse-Wolbachia symbiosis: comes of age and has great potential for pest and disease control. J Invertebr Pathol. 2013;112:S94–S103. doi: 10.1016/j.jip.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotopp JCD, Clark ME, Oliveira DCSG, Foster JM, Fischer P, Torres MC, Giebel JD, Kumar N, Ishmael N, Wang SL. et al. Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science. 2007;317(5845):1753–1756. doi: 10.1126/science.1142490. [DOI] [PubMed] [Google Scholar]
- Richards TA, Soanes DM, Jones MD, Vasieva O, Leonard G, Paszkiewicz K, Foster PG, Hall N, Talbot NJ. Horizontal gene transfer facilitated the evolution of plant parasitic mechanisms in the oomycetes. Proc Natl Acad Sci USA. 2011;108(37):15258–15263. doi: 10.1073/pnas.1105100108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Rogozin IB, Kondrashov AS, Koonin EV. Genome alignment, evolution of prokaryotic genome organization, and prediction of gene function using genomic context. Genome research. 2001;11(3):356–372. doi: 10.1101/gr.GR-1619R. [DOI] [PubMed] [Google Scholar]
- Dam P, Olman V, Harris K, Su ZC, Xu Y. Operon prediction using both genome-specific and general genomic information. Nucleic acids research. 2007;35(1):288–298. doi: 10.1093/nar/gkl1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen GC, Kornberg A. Assembly of the Primosome of DNA-Replication in Escherichia-Coli. J Biol Chem. 1993;268(26):19204–19209. [PubMed] [Google Scholar]
- Masai H, Arai K. Operon structure of dnaT and dnaC genes essential for normal and stable DNA replication of Escherichia coli chromosome. J Biol Chem. 1988;263(29):15083–15093. [PubMed] [Google Scholar]
- Zimmermann K, Engeser M, Blunt JW, Munro MH, Piel J. Pederin-type pathways of uncultivated bacterial symbionts: analysis of o-methyltransferases and generation of a biosynthetic hybrid. J Am Chem Soc. 2009;131(8):2780–2781. doi: 10.1021/ja808889k. [DOI] [PubMed] [Google Scholar]
- Sanchez-Paz A, Garcia-Carreno F, Hernandez-Lopez J, Muhlia-Almazan A, Yepiz-Plascencia G. Effect of short-term starvation on hepatopancreas and plasma energy reserves of the Pacific white shrimp (Litopenaeus vannamei) J Exp Mar Biol Ecol. 2007;340(2):184–193. doi: 10.1016/j.jembe.2006.09.006. [DOI] [Google Scholar]
- Robalino J, Almeida JS, McKillen D, Colglazier J, Trent HF 3rd, Chen YA, Peck ME, Browdy CL, Chapman RW, Warr GW. et al. Insights into the immune transcriptome of the shrimp Litopenaeus vannamei: tissue-specific expression profiles and transcriptomic responses to immune challenge. Physiological genomics. 2007;29(1):44–56. doi: 10.1152/physiolgenomics.00165.2006. [DOI] [PubMed] [Google Scholar]
- Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome research. 1999;9(9):868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins DG, Thompson JD, Gibson TJ. Using CLUSTAL for multiple sequence alignments. Methods in enzymology. 1996;266:383–402. doi: 10.1016/s0076-6879(96)66024-8. [DOI] [PubMed] [Google Scholar]
- Ropelewski AJ, Nicholas HB, Gonzalez Mendez RR. MPI-PHYLIP: parallelizing computationally intensive phylogenetic analysis routines for the analysis of large protein families. PloS one. 2010;5(11):e13999. doi: 10.1371/journal.pone.0013999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular biology and evolution. 2000;17(4):540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic biology. 2003;52(5):696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Systematic biology. 2006;55(4):539–552. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19(12):1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Li Q, Liu QH, Huang J. FATP synthase b-chain of Litopenaeus vannamei involved in White Spot Syndrome Virus infection. Virus genes. 2013. http://dx.doi.org/10.1007/s11262-013-0907-1. http://link.springer.com/article/10.1007%2Fs11262-013-0907-1. [DOI] [PubMed]
- Li S, Zhang X, Sun Z, Li F, Xiang J. Transcriptome analysis on Chinese shrimp fenneropenaeus chinensis during WSSV acute infection. PloS one. 2013;8(3):e58627. doi: 10.1371/journal.pone.0058627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng QD. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–U130. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twine NA, Janitz K, Wilkins MR, Janitz M. Whole transcriptome sequencing reveals gene expression and splicing differences in brain regions affected by Alzheimer’s disease. PloS one. 2011;6(1):e16266. doi: 10.1371/journal.pone.0016266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate - apractical and powerful approach to multiple testing. J Roy Stat Soc B Met. 1995;57(1):289–300. [Google Scholar]
- Winbush A, Reed D, Chang PL, Nuzhdin SV, Lyons LC, Arbeitman MN. Identification of gene expression changes associated with long-term memory of courtship rejection in Drosophila males. G3 (Bethesda) 2012;2(11):1437–1445. doi: 10.1534/g3.112.004119. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Local genome database for HGT identification. Table S2. Primers of the 14 HGT genes for PCR. Table S3. The PCR primers of the edge products of 14 HGT genes. Table S4. The FPKMs of each HGT genes in different development stage. Table S5. Differential expression of the 14 HGT genes at five development stages of shrimp larvae.
Phylogenetic tree of acsf and its homologs. Figure S2. Phylogenetic tree of rpsF and its homologs. Figure S3. Phylogenetic tree of rpsN and its homologs. Figure S4. Phylogenetic tree of exbB and its homologs. Figure S5. Phylogenetic tree of mopB and its homologs. Figure S6. Phylogenetic tree of tnpA and its homologs. Figure S7. Phylogenetic tree of stat and its homologs. Figure S8. Phylogenetic tree of rpc2 and its homologs. Figure S9. Phylogenetic tree of dhfr and its homologs. Figure S10. Phylogenetic tree of cata and its homologs. Figure S11. Phylogenetic tree of sdrp and its homologs. Figure S12. Phylogenetic tree of ankp and its homologs. Figure S13. Phylogenetic tree of deha and its homologs.