

Abstract
Background
Streptococcus equi ssp. zooepidemicus (S. zooepidemicus) is an important pathogen causing swine streptococcosis in China. Pathogenicity islands (PAIs) of S. zooepidemicus have been transferred among bacteria through horizontal gene transfer (HGT) and play important roles in the adaptation and increased virulence of S. zooepidemicus. The present study used comparative genomics to examine the different pathogenicities of S. zooepidemicus.
Results
Genome of S. zooepidemicus ATCC35246 (Sz35246) comprises 2,167,264-bp of a single circular chromosome, with a GC content of 41.65%. Comparative genome analysis of Sz35246, S. zooepidemicus MGCS10565 (Sz10565), Streptococcus equi. ssp. equi. 4047 (Se4047) and S. zooepidemicus H70 (Sz70) identified 320 Sz35246-specific genes, clustered into three toxin-antitoxin (TA) systems PAIs and one restriction modification system (RM system) PAI. These four acquired PAIs encode proteins that may contribute to the overall pathogenic capacity and fitness of this bacterium to adapt to different hosts. Analysis of the in vivo and in vitro transcriptomes of this bacterium revealed differentially expressed PAI genes and non-PAI genes, suggesting that Sz35246 possess mechanisms for infecting animals and adapting to a wide range of host environments. Analysis of the genome identified potential Sz35246 virulence genes. Genes of the Fim III operon were presumed to be involved in breaking the host-restriction of Sz35246.
Conclusion
Genome wide comparisons of Sz35246 with three other strains and transcriptome analysis revealed novel genes related to bacterial virulence and breaking the host-restriction. Four specific PAIs, which were judged to have been transferred into Sz35246 genome through HGT, were identified for the first time. Further analysis of the TA and RM systems in the PAIs will improve our understanding of the pathogenicity of this bacterium and could lead to the development of diagnostics and vaccines.
Background
PAIs play important roles in the adaptation and increased virulence of pathogens. Bacterial PAI often encode both effector molecules responsible for disease and secretion systems that deliver these effectors to host cells [1,2]. PAIs are a distinct type of genomic island. PAIs contain mobile genetic elements (MGEs), which were acquired by the bacteria through HGT. Bacterial genomes contain various types of MGEs, such as transposons, plasmids, and bacteriophages. All of these elements may be acquired by HGT. Many MGEs serve as shuttles for genes that are beneficial to bacteria during their proliferation in a host environment. Several MGEs have been found in the genomes of pathogenic bacteria that contain genes conferring antibiotic resistance and genes encoding virulence factors, such as exotoxins, adhesins, and secretion systems [3].
Pathogenic bacteria often make use of suicide mechanisms, in which the death of individual cells benefits the survival of the population. This mechanism is regulated by the toxin-antitoxin system (TA system), which is related to DNA replication, mRNA stability, protein synthesis, cell-wall biosynthesis and ATP synthesis [4]. The ϵ antitoxin-ζ toxin system (ϵ/ζ system) is a type II TA system. It is distributed over plasmids and chromosomes of various pathogenic bacteria [5]. These systems benefit the stability of the genomic island in the bacterial genome.
S. zooepidemicus is the ancestor of Streptococcus equi ssp. equi (S. equi) and these two strains express many of the same proteins and virulence factors. However, unlike S. equi, which is host-restricted and only infects horses, S. zooepidemicus has no host preference. S. zooepidemicus is primarily an opportunistic pathogen infecting a wide variety of animal species, including important domestic species, which makes it a pathogen of veterinary concern. S. zooepidemicus causes mastitis in cows and mares, and is the most frequently isolated opportunistic pathogen of horses [6]. Occasionally, S. zooepidemicus can infect humans via zoonotic transmission from infected animals and causes invasive infections in humans such as septicemia and meningitis [7,8]. In 1975, Sichuan province experienced an S. zooepidemicus outbreak that resulted in the death of 300,000 pigs and great economic losses. S. zooepidemicus is an important pathogen of streptococcal diseases in swine [9,10] and it remains a great threat to Chinese swine breeding. In the present study, we used comparative genomic analyses between S. zooepidemicus ATCC35246 and other published S. zooepidemicus strains [11,12] to investigate the mechanisms underlying the differing pathogenicities of Streptococcus equi ssp. In particular, we tried to ascertain how S. zooepidemicus ATCC35246 is able to cause such a serious disease in pig. We determined the complete genome sequence of S. zooepidemicus ATCC35246 (Sz35246), a virulent strain isolated from a dead pig in China. The complete genome sequence not only permitted detailed analysis of the phylogenic relationship between species, but also provided insights into the biology and pathogenic capacity of this streptococcus.
Results and discussion
Genomic features and basic transcriptomic structure
The 2,167,264-bp genome of Sz35246 comprises a single circular chromosome with a GC content of 41.65% (Additional file 1: Table S1 & Figure 1) and the genome information have been reported previously [11]. The GC content is similar to that of Streptococcus equi subsp. zooepidemicus MGCS10565 (Sz10565) [12], Streptococcus equi subsp. equi 4047 (Se4047) and Streptococcus equi subsp. zooepidemicus H70 (Sz70) [13]. The genome contains 2,087 protein-encoding genes, 57 tRNA genes, and five 5S-16S-23S rRNA operon gene clusters. Among the protein coding genes, 416 (19.93%) are predicted to encode conserved hypothetical proteins that are similar to proteins of unknown factions in other genomes, and 137 hypothetical genes (6.56%) have no matches in the nr protein database (Additional file 1: Table S1). The remaining 1534 genes were assigned putative functions. Eighty-one genes were identified as mobile elements, including those encoding a competence protein, a phage associated protein, a conjugation protein, a transposase and a site-specific recombinase, suggesting that these elements are used to take up and incorporate foreign DNA and are involved in reconstructing the genome architecture. Furthermore, global transcriptome analysis of Sz35246 using RNA-seq confirmed that 2048 of the 2,087 ORFs are expressed, but with different sequence coverages in vitro and in vivo (Additional file 1: Table S1). Comparative gene expression analysis reveals that 252 genes are upregulated and 142 genes are downregulated (Additional file 2: Table S2) by more than a 2-fold change in reads per kilo base per million (RPKM) values (p < 0.001) in vivo. The upregulated genes include 67 hypothetical protein coding genes and 28 response regulator, transcription regulator genes and chaperone protein encoding genes, suggesting that the differential expression of these ORFs plays an important role in survival of Sz35246 within the different host environments.
Figure 1.

Circular representation of the S. zoopedemicus ATCC35246 genome and comparative genome results. The ten circles (outer to inner) show the following. The outer three circles represent Sz35246 protein-encoding genes homologous to that of Sz10565, Sz70 and Se4047, respectively. The fourth circle represents the PAI in Sz35246 chromosome. The fifth circle shows the chromosome position scaled in kb from oriC. The sixth and seventh circles show the coding sequences on the plus and minus strands, respectively. All genes are color-coded based on the COG functional categories: cyan, information storage and processing; yellow, cellular processes and signaling; magenta, metabolism; and black, poorly characterized. The eighth circle shows rRNA in red and tRNA in blue. The ninth circle shows the GC content (in 1-kb windows).Values that are greater than and or less than the average (41.65%) are shown in green and red, respectively. The tenth circle shows the GC skew curve (10-kb window and 1-kb incremental shift). The values for plus and minus strands are shown in cobalt blue and purple, respectively.
Additionally, we found that some genes, including malA (SeseC_01626), malD (SeseC_01627), malE (SeseC_01633, SeseC_01622), malF (SeseC_01624, SeseC_01630), malG (SeseC_01625) and malQ (SeseC_01617) were upregulated when Sz35246 infected mice. These genes are related to maltose transport and metabolism and utilization of carbohydrates, which is essential for the ability of pathogenic bacteria to cause disease. Group A Streptococcus (GAS) strains express malE on their surface, and the transcript levels of the malE gene were significantly increased during growth in human saliva compared to common medium. MalE may contribute to the ability of GAS to colonize the oropharynx by utilizing maltose [14]. In addition, studies in S. pneumoniae have shown that deletions in carbon metabolism genes, including the maltose operon, lead to decreased production of known virulence factors, such as capsular polysaccharide and cholera toxin [15]. MalE of Sz35246 is a maltodextrin-binding protein, which also binds longer maltodextrins (e.g., maltotriose and maltotetraose). The upregulation of this protein and other maltose utilization-related proteins may contribute to the infection of Sz35246. Further investigation into these carbohydrate transport and metabolism pathways genes may yield novel insights into the pathogenesis of Sz35246. We also observed that certain known virulence factors were upregulated during Sz35246 infection, for example, streptokinase (SeseC_02411) and fibronectin-binding protein (sfs, SeseC_00464). The upregulation of bacteriocin (SeseC_02042) could help Sz35246 compete with other bacteria that colonize the host.
Comparative genomic analysis and pathogenicity islands (PAIs)
Comparative analysis of Sz35246 genome with three other genomes revealed that the evolution of Sz35246 has been driven by genomic rearrangements and HGT. X-alignment analysis of Sz35246 versus Sz10565 [12], Se4047 and Sz70 [13] revealed that small and large scale chromosome inversions have occurred during replication termination between Sz35246 and Se4047 and between Sz35246 and Sz70 (Figure 2). These genome rearrangements may influence the transcription of surrounding genes after the HGT process, which has contributed to the shaping of the Sz35246 genome.
Figure 2.

Synteny between the Sz35246 genome and the Sz10565, Sz70 and Se4047 genomes, respectively. The x-axis shows the position on Sz35246 genome; the y-axis shows the position on the Sz10565 genome (A), the position on the Sz70 genome (B) and the position on the Se4047 genome (C).
The comparative analysis of the Sz35246 genome with the three other genomes identified 1,397 orthologous genes that are shared by all four strains (Figure 3). In addition, 191, 184 and 93 genes are shared between Sz35246 and Sz10565, Sz70 and Se4047, respectively, suggesting that Sz35246 and Sz10565 are more closely related than the other strains. Furthermore, X-alignment analysis of Sz35246 versus Sz10565 [12], Se4047 and Sz70 [13] also suggested that Sz35246 is closer to Sz10565 than to the other two species. Phylogenetic trees of the four strains were constructed based on the sequences of the 1,397 orthologous genes using minimum evolution and neighbor joining phylogenic reconstruction methods available in the MEGA package (Figure 4). The phylogenic trees also indicated that Sz35246 is much closer to Sz10565 than to the other two species, which is consistent with the r genome-scale alignment analysis.
Figure 3.
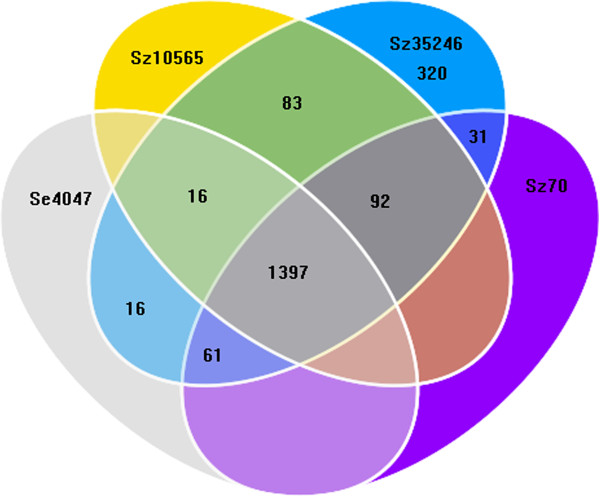
The whole genome comparison among Sz35246 and Sz10565, Sz70, Se4047 genome. Venn diagram representing unique and shared gene numbers among Sz35246, Sz10565, Sz70 and Se4047.
Figure 4.

Phylogenetic trees were inferred using neighbour joining (A), maximum parsimony (B) and minimum evolution (C) among the four genomes based on concatenated alignments of 1397 orthologous proteins.
Further comparative analysis showed that 320 genes are specific to Sz35246, which include 197 (61%) that were annotated as “hypothetical protein”, among which 149 encode small proteins with lengths of no more than 100 amino acids (Additional file 3: Table S3). These small proteins are annotated as hypothetical proteins; however, certain highly conserved hypothetical proteins may play important roles in response to specific environmental stresses and host adaptation. For example, these small proteins have been reported to have evolved in response to specific environmental stress and to participate in the suppression of the type III secretion system [16]. The remaining functional genes encode 40 virulence proteins, 14 phage-associated proteins, eight transposases, five site-specific recombinases, a conjugation protein, a phage integrase, a phage recombinase, an IS transposase and a relaxase. These results suggest that the Sz35246 genome acquired these virulence genes through HGT, either by transduction with phages or by conjugation with plasmids or chromosomal fragments.
Furthermore, these Sz35246-specific genes are tightly clustered into four regions, varying in length from about 10 kb to 50 kb, which were as termed PAIs (SeseCisland_1~4) (Figure 1). An orthologous genes analysis between Sz35246 and Sz10565, Sz70, Se4047 confirmed these genomic islands are present in the Sz35246 genome only (Figure 1). The genes located in these four PAIs might be involved in Sz35246’s pathogenesis in causing swine streptococcosis and its strong virulence. These islands were further confirmed the annotation information and the co-linearity comparison of the Sz35246 genome with those of the three other genomes.
Significantly, sequence and annotation analyses of these islands revealed that SeseCisland_1, SeseCisland_2 and SeseCisland_3 contain the same type of virulence genes involved in the bacterial TA systems that have been reported to play subtle roles in the survival of bacteria under harsh natural environments [4,17]. Based on previous analyses of TA systems in Escherichia coli K12 [4], Mycobacterium tuberculosis[17] and Mycobacterium smegmatis[18], we speculated that acquired-TA systems might play a positive role in survival of Sz35246 under different host environments. The RM system is used by bacteria to protect themselves from foreign DNA, such as bacteriophages and other viruses. Genes encoding RM system proteins, which include a restriction endonuclease and a restriction endonuclease control protein, were identified in a cluster in SeseCisland_4. Based on these results, we speculated that the acquired RM system might be involved in defense against infection by foreign DNA such as prophages and viruses. Thus, the PAIs may allow Sz35246 to adapt to various host stress conditions and to defend itself against infection by prophages, other bacteria and viruses. The expression and potential impact of these islands on the physiology, pathogenesis and host adaptation of Sz35246 are discussed below.
I. SeseCisland_1: Phd/Doc TA system
SeseCisland_1 contains 54 genes (53,095 bp), 42 of which are Sz35246-specific genes, including mobile elements resembling the IS200 family transposase (SeseC_00874), a prophage site-specific recombinase resolvase family protein (SeseC_00919), a putative conjugal transfer protein (SeseC_00927), a conjugation protein (SeseC_00935) and a tnpX site-specific recombinase family protein (SeseC_00939), suggesting that this island is an integrative conjugative element (Additional file 4: Table S4 & Figure 5A). SeseCisland_1 contains 20 structural phage loci, indicating that MGEs, such as phages, are also implicated in HGT in Streptococcus species. Further analysis showed that the island has an abnormal GC skew and that the island-located genes have an average G+C content of 39.42% (Additional file 4: Table S4), which is significantly different from the mean value for the genome (41.65%) (p=0.002).
Figure 5.

Schematic representation of the putative PAIs and their expression in S. zoopedemicus ATCC35246 (A), SeseCisland_1; (B), SeseCisland_2; (C), SeseCisland_3; (D), SeseCisland_4. The expression levels of in vitro and in vivo conditions are shown at single-nt resolution in red and green, respectively. All genes are color-coded based on the annotation information as follows: yellow, toxin-Antitoxin system (TA system); blue, phage associated protein; green, other virulence protein; red, virB4 components; cyan, mobile elements; grey, hypothetical protein.
A major feature within SeseCisland_1 is the presence of two genes (SeseC_00898 and SeseC_00899) encoding proteins homologous to addiction module antitoxin Phd protein and killer Doc protein, respectively. The Phd-Doc TA system is the Type II TA system that first identified in bacteriophage P1 [19]. Phylogenetic trees based on Phd/Doc proteins also suggested that Phd/Doc proteins are highly homologous to those of Streptococcus phage phi-m461 and phi-SsuD1 (Figure 6). The Phd/Doc mRNAs are co-expressed from the same promoter and both are translated into proteins. These proteins form a stable TA complex to block the functions of the Doc toxin. Doc is a toxin that kills plasmid-free segregants, and Phd is an unstable antidote that neutralizes the toxin. Doc inhibits translation elongation by association with the 30S ribosomal subunit [20]. Under stress conditions or host change, Phd is degraded by ATP-dependent serine proteases, such as ClpXP protease [21], resulting in freeing of the toxin from the TA system and inducing cell growth inhibition and cell death [22,23]. Interestingly, a gene (SeseC_00903) encoding a protein homologous to the E. coli Clpxp protein is present in this island. Doc toxins are expressed in two conditions (in vivo and in vitro) (Figure 5A), which agrees with previous reports that Doc causes cell growth and death by inhibiting translation without affecting transcription and replication. The observations and results reported here support the hypothesis that SeseCisland_1 helps Sz35246 to adapt to environmental and host changes.
Figure 6.
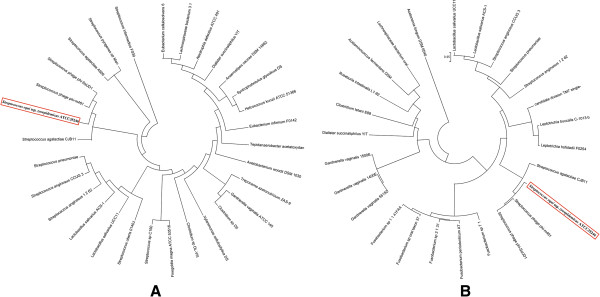
Phylogeny of antitoxin Phd and addiction module killer Doc protein sequences. Thirty phd toxin sequences (A) and twenty-six anti-toxin sequences (B) from loci of bacterial chromosomes, plasmids and phages were aligned. The sequences were aligned with clustalW and the genetic relationships inferred using the unweighted pair group method with arithmetic mean (UPGMA) implanted in MEGA 4.0 software.
II. SeseCisland_2: Fic/Doc TA system
SeseCisland_2 contains an important open reading frame (ORF), SeseC_01334, which encodes a protein with a filamentation induced by cAMP (Fic) domain (Additional file 5: Table S5 and Figure 5B). The Fic domain is classified together with a second family of sequences, doc (death on curing), in the Pfam protein families database [24]. The Fic/Doc family protein sequences are aligned against this protein present inside other bacteria. Interestingly, phylogenetic analysis revealed that the Fic/Doc protein is homologous to that of Fusobacterium nucleatum subsp fusiforme (Figure 7). Fic/Doc family proteins are known as members of a TA system, the functional sites are common to both families [25]. The Fic protein has been reported to be involved in cell division and synthesis of folate, indicating that the Fic protein and cAMP are involved in a regulatory mechanism of cell division via folate metabolism [26,27]. Fic family virulence proteins may be important in many bacterial pathogens. For example, the immunoglobulin-binding protein A (IbpA) of Histophilus somni contains a direct repeat of two Fic domains, and mutation of IbpA or just the fic domain of IbpA decreased the virulence of this bacteria. The Fic domain has been shown to covalently modify host Rho GTPases with AMP, which may explain how the Fic domain influences bacterial virulence [28]. Thus, the Fic family protein in SeseCisland_2 may be involved in the pathogenicity of Sz35246.
Figure 7.
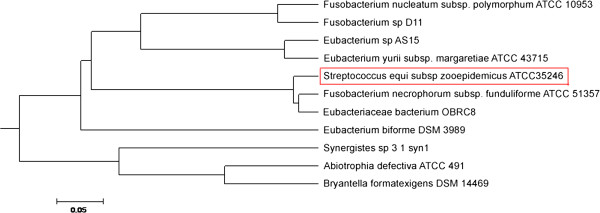
Phylogeny of Fic/DOC domain protein sequences. Twelve protein sequences of bacteria were aligned using clustalW and the genetic relationship trees were constructed with MEGA4.0 software as well as Figure 6.
SeseCisland_2 encodes 16 genes (17,293 bp), 10 of which are Sz35246-specific. SeseCisland_2 also contains certain mobile elements, including an endonuclease relaxase (SeseC_01323), a bacterial mobilization protein (SeseC_01324) integrase/recombinase (SeseC_01328) and transposase protein (SeseC_01332). Thus, we speculate that this region also plays important roles in bacterial adaptation, virulence and physiology.
III. SeseCisland_3: ϵ/ζ TA system
SeseCisland_3 contains 21 Sz35246-specific genes (Additional file 6: Table S6 and Figure 5C), the most notable of which are two genes annotated as Type II TA system genes encoding a ζ toxin protein (SeseC_01875) and an ϵ antitoxin protein (SeseC_01876). VirB4/VirB6/VirD4 components (SeseC_01908, SeseC_01912 SeseC_01914 and SeseC_01916) from the type IV secretion system (T4SS) are also present. Additionally, virulence-associated factors, such as glucan-binding protein and abortive infection protein, are also encoded by this region. All the virulence genes are expressed under in vitro and in vivo conditions. Several MGEs such as site-specific recombinases (SeseC_01863, SeseC_01864 and SeseC_01865) and transposases (SeseC_01867&SeseC_001869) were also identified in this island. The bioinformatics analysis showed that a Type II TA system, a type IV secretion system and other virulence genes were present in this island, which may contribute directly to the bacterium’s pathogenicity and host adaption.
ϵ/ζ systems ensure stable plasmid inheritance by inducing death in plasmid-deprived offspring cells. Members of the ϵ/ζ systems have been found on resistance plasmids in major human pathogens [29,30]. By contrast, chromosomally encoded ϵ/ζ systems were reported to contribute to virulence of pathogenic bacteria. Brown et al. compared clinical serotype 3 isolates with ζ toxin gene knockout strains in mixed systemic and respiratory infections of mice, and thus connected the ζ toxin with virulence in Streptococcus pneumonia[31]. The ϵ/ζ system also exists in the 89 k pathogenicity island of Streptococcus suis serotype 2. This bacterium is an important zoonotic pathogen, causing more than 200 cases of severe human infection worldwide [32]. The ζ toxin is inhibited by its cognate antitoxin, ϵ. The structure of the complex of ζ toxin inactivated by ϵ antitoxin (ϵ2ζ2) was solved by X-ray crystallography [33]. Upon degradation of ϵ, the ζ toxin is released, allowing this enzyme to inhibit bacterial cell wall synthesis, which eventually triggers autolysis [34]. The toxic effect of the ζ toxin has also been demonstrated in a diverse array of organisms, including Saccharomyces cerevisiae[35].
Phylogenetic analysis of ζ proteins and ϵ antitoxin proteins showed that the proteins from Sz35246 are highly homologous to those of Streptococcus urinalis 2285–97, Streptococcus intermedius F0395 and Streptococcus vestibularis F0396 and widely distributed in many bacteria (Figure 8). This broad distribution has been reported that the zeta toxin family on plasmids [21,36,37], bacterial chromosomes [23,38] and in Streptococcus pneumonia and Streptococcus suis serotype 2 PAIs. The broad distribution of this system within the bacterial kingdom suggests that it uses a ubiquitous bacteriotoxic mechanism to overcome host defenses and environmental changes. On the other hand, we hypothesized that horizontal transfer of this island may occur through T4SS-mediated conjugation process, because four genes products display similarities to Streptococcus T4SS components.
Figure 8.
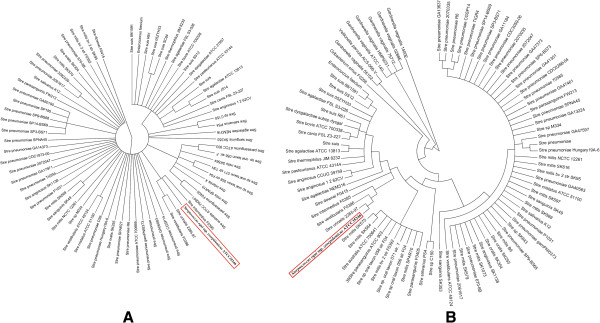
Phylogeny of zeta toxin (PezT) (A) and epsilon antitoxin (PezA) (B) protein sequences. Eighty-eight zeta toxin protein sequences (A) and seventy-two epsilon antitoxin protein sequences (B) from Streptococcus, Enterococcus, Oribacterium, Veillonella and Gardnerella bacterial species were aligned. The genetic relationships were determined as detailed in Figure 6.
IV SeseCisland_4: RM system and virulence island
SeseCisland_4 contains eight Sz35246-specific genes (from a total of 10 genes), the two mobile elements (SeseC_02358, SeseC_02362) are transposase IS1167 and phage integrase (Additional file 7: Table S7 and Figure 5D), suggesting that this island has been acquired by HGT from another microorganism. The major feature of this island is three strain specific genes (SeseC_02360, SeseC_02361 and SeseC_02362) that were annotated as RM system proteins, which protect bacteria from foreign DNA, such as bacteriophages. The RM system is strategy that permits bacteria to live in difference environments [39], allowing bacteria erect a barrier to gene transfer and making them resistant to phage infection [40]. Taken together, these data suggest that the RM systems is a remarkable characteristic of Sz35246 and is probably involved in the adaptation of these bacteria to different environmental conditions.
Relationship between PAIs and Sz35246 virulence
To prove that the genes located within the PAIs affect the virulence of Sz35246, we deleted part of SeseCisland_3 from SeseC_01869 to SeseC_01898. PCR was used to confirm the deletion (Figure 9A and Additional file 8: Table S8), sequencing results showed that exactly 28,606 bp of SeseCisland_3 was deleted, including the genes belong to the ϵ /ζ TA system (Figure 9B). The deleted region started with Tn5252 transposon gene (SeseC_01869), and two repeat sequences, including transposase genes (SeseC_01867 and SeseC_01901), were located at the flank of the deleted region. These two repeat sequences and the Tn5252 transposon gene formed the structural basis for deleting such a long fragment. The mutant strain ∆Island3-Sz35246 and wild-type Sz35246, were used to infect ICR mice to evaluate the influence of partial PAI deletion on bacterial virulence. The percent survival significantly increased in ∆Island3-Sz35246 infected mice (Figure 10), 5 days post-infection, only one of the ten mice was dead; however, of the mice infected with wild-type Sz35246, only one was alive. The survival curve indicated that partial deletion of a PAI did affect the virulence of Sz35246, and that some of these genes in the PAI are important for bacterial pathogenicity. Genes located in the other three PAIs require further study to determine their role in bacterial virulence.
Figure 9.

PCR detection and schematic diagram of partial SeseCisland_3 deletion. (A) PCR detection of genes located in SeseCisland_3 deleted region (markers are 2000 bp, 1000 bp, 750 bp, 500 bp, 250 bp and 100 bp), the number of genes corresponds to Additional file 8: Table S8. (B) Genomic organization of the partial SeseCisland_3 deletion locus and its flanking repeat sequences in Sz35246. The fragment from 1551608 to 1580213 was knocked out, including the zeta toxin (PezT) and epsilon antitoxin (PezA) genes. The deleted region started with a Tn5252 transposon protein gene and is flanked by two repeated regions. After the reciprocal recombination, only one repeat region remained.
Figure 10.

Survival curves for ICR mice infected with the wild-type Sz35246 and ∆Island3-Sz35246. Two groups of eight-week-old ICR mice were inoculated i.p. with 2.5×105 CFU bacteria, and mouse survival was monitored over a 5-day period. Data are expressed as the mean percentage of live animals in each group (n = 10). The virulences of these two strains were significantly different (P<0.05).
Other potential virulence genes dispersed in the Sz35246 genome
Strain Sz70 was isolated from a nasal swab taken from a healthy thoroughbred racehorse in Newmarket, England, in 2000 [41]. A genome wide comparison of Sz35246 with Sz70 identified Sz35246-specific genes, some of which may be involved in the virulence of Sz35246 and may provide clues as to why Sz35246 causes such a serious swine streptococcosis but other strains do not. Virulence-associated protein E (vapE, SeseC_01325), which was originally identified in Dichelobacter nodosus[42], is part of a vap region of D. nodosus that is associated with virulence [43]. The mechanism by which VapE affects virulence has not been determined yet, but the presence of an integrase gene XerC (SeseC_01328) immediately upstream of vapE, suggested a role for bacteriophages in the evolution and transfer of these bacterial virulence determinants; i.e., it is possible that exchange of this putative virulence factor with other bacteriophages could take place [44]. Moreover, a vapE-like gene has also been identified in a pathogenicity island of Staphylococcus aureus[45]. The pathogens of a footrot outbreak in a Debre Zeit swine farm were identified as Staphylococcus aureus and Dichelobacter nodosus, both bacteria contain the vapE gene. VapE has not been identified in other strains of S. zooepidemicus, but only in Sz35246. This gene may be related to Sz35246 pathogenicity towards pig. The role of the vapE gene in the virulence of Sz35246 remains to be clarified.
Adherence is an essential requirement for invasion of cells by bacterial pathogens. Long extracellular structures resembling fimbriae mediate adhesion to components of the host extracellular matrix, such as collagen and fibronectin. We identified seven Sz35247 unique proteins that contain an LPXTG motif (found in cell wall anchor domains), including collagen-like protein SclZ.1 (SeseC_00092), fibrinogen- and Ig-binding protein precursor (SeseC_00180), cell surface protein (SeseC_00619), T-antigen-like fimbrial structural subunit protein (SeseC_02472), putative cell surface protein (SeseC_02304), InlA-like domain containing cell surfaced-anchored protein (SeseC_01462) and collagen-like surface-anchored protein SclE (SeseC_00246). All of these proteins are anchored on the bacterial surface and may be involved in bacterial adhesion and invasion.
Fibronectin (Fn)-binding proteins have been reported to mediate the invasion of host cells without the need for other bacterial factors [46]. Fn, which has received much attention as a target of bacterial adhesins, it is a glycoprotein found in the extracellular matrix and body fluids of vertebrates. Fn-binding proteins are found in Streptococcus pyogenes (SfbI/F1), Staphylococcus aureus (FnBPA and FnBPB), Streptococcus dysgalactiae (FnBA and FnBB), and other bacterial species [47]. In previous research, an fnz gene was found in S. zooepidemicus and a sfs gene was only found in S. equi, both of which genes encode a cell surface protein that binds Fn [48]. The sfs gene (SeseC_00464) was identified in Sz35246 for the first time. The transcriptome data showed that the sfs gene was upregulated infection of a host by Sz35246 (in vivo). Presumably, the expression of this gene promoted bacterial pathogenicity by inhibiting the binding between collagen and Fn.
The Sz35246 and Sz10565 genomes both have the Fim III operon (type II fimbriae) (SeseC_02471-02473 and SeseC_02475). The structural proteins of type III fimbriae have an amino-terminal secretion signal and a carboxy-terminal sorting signal, and their assembly into fimbriae is dependent on the adjacently encoded dedicated sortases [49]. Sz70 contains two loci that encode genes putatively required for pilus expression, but lacks this putative pilus locus. Recent studies of Salmonella enterica revealed that the presence of fimbriae increases the ability of host-restricted bacteria to invade normally restrictive cells [50]. Thus, we hypothesize that the presence of the Fim III operon might be associated with breaking host-restriction by S. zooepidemicus.
Conclusions
The genome and expression analysis of Sz35246 provided fundamental information on the physiology and potential pathogenic capacity of this bacterium. The comparison of the genomes of Sz35246, Se4047, Sz10565 and Sz70 identified gens that are specific to Sz35246. These genes may be related to the bacterium’s pathogenic function, including causing swine streptococcosis and breaking host-restriction. We identified novel MGEs, which may have been involved in the evolution of Sz35246. The presence of the elements and the phylogenetic analysis indicated that this genome has been shaped by chromosomal inversion, recombination and HGT events. Sz35246 probably acquired its PAIs and certain specific genes through HGT. The presence of TA systems exists in three of genomic islands of Sz35246 may be related to this strains pathogenicity. Study of these systems will form the basis of our future research. The availability of the complete Sz35246 genome sequence will facilitate further studies of this pathogen and the development of diagnostics and vaccines.
Methods
Strain and growth conditions
S. zooepidemicus ATCC35246 was isolated from a dead pig in Sichuan, China.. To prepare total cellular DNA from S. zooepidemicus ATCC35246, bacteria were grown in Bacto™ Todd-Hewitt Broth at 37°C, in a 10% CO2 atmosphere. Total cellular DNA was isolated from the mid-exponential (OD600= 0.6) phase culture using a Genomic Purification System (Promega).
Preparation of RNA for transcriptome analysis
From pure culture
cultures for preparing RNA samples were grown overnight at 37°C under aerobic conditions in liquid medium with shaking. Overnight pre-cultures were diluted in liquid medium and incubated at 37°C under aerobic conditions with shaking. Exponentially growing cells (OD600= 0.6) were harvested by centrifugation for 10 min at 10,000 rpm at 4°C. Total RNA was extracted as previously described [51]. RNA quality was assessed by determining the OD260/280 ratio with a Nanodrop 2000 (Thermo) and by visualization following agarose gel electrophoresis.
From infected mice organs
specific pathogen-free female BALB/c mice were intravenously infected with S. zooepidemicus ATCC35246 [52]. At 24 h post-infection, the mice were sacrificed and dissected. The livers and spleens were harvested and immediately frozen in liquid nitrogen. The organs were stored at −80°C. Before RNA isolation, the organs were thawed on ice and homogenized in 20 ml of an ice-cold solution composed of 0.2 M sucrose/0.01% SDS. The homogenate was gently centrifuged for 20 min at 300 rpm and filtered to remove large tissue debris. The tissue suspension was centrifuged for 20 min at 8000 rpm to pellet the bacteria. Centrifugations were performed at 4°C. Bacterial RNA extraction was performed as previously described [51]. RNA quality was assessed by determining the OD260/280 ratio with a Nanodrop 2000 (Thermo) and by visualization following agarose gel electrophoresis.
Genome sequencing and annotation
Whole-genome sequencing was performed with the Roche 454 genome sequencer FLX system and assembled with Newbler (version 2.0.01.14). The detailed sequencing and assembly methods have been described previously [52]. The complete genome sequence of S. zooepidemicus strain ATCC35246 has been deposited in the GenBank database with the accession number CP00290. The replication origin (oriC) was identified with Ori-Finder software [53]. Protein-coding genes were predicted with Glimmer 3.02 [54] using the default settings and a cutoff at 90 nt. Annotation of these genes was performed by homology searches in the NCBI nonredundant protein database with 80% overlap (E_value<1e-10), in the cluster of orthologous groups (COG) database [55], the InterPro member (InterProScan) databases [56] and the Kyoto encyclopedia of genes and genomes (KEGG) pathway database [57], respectively. The tRNA genes and rRNA genes were identified using the tRNAScan-SE tool [58], and RNAmmer 1.2 [59], respectively. Finally, genome annotation and the structure of the predicted genes were manually refined.
Comparative genomic analysis
Sequences and protein coding sequences for each strain (MGCS10565: CP001129.1; 4047:FM204883.1; H70:FM204884.1) were retrieved from NCBI (http://www.ncbi.nlm.nih.gov). The genomic co-linearity of four genome sequences was generated using the MUMmer 3 package [60]. Orthologous proteins were identified with Inparanoid and MultiParanoid [61]. The CLUSTAL W software [62] and MEGA4 software [63] were used to align the concatenated sequences from all orthologs and to construct phylogenetic trees. The Artemis Comparison Tool (ACT) [64] was used to view the overall comparison of S. zooepidemicus strain ATCC35246 and S. zoopidemicus MGCS10565, S. zooepidemicus H70 and S. equi 4047.
SOLiD RNA-seq library construction, sequencing and gene expression analysis
The standard protocol from SOLiDTM Small RNA Expression Kit (ABI) was used to construct the RNA-Seq library and sequencing was performed on an ABI SOLiD 4.0 sequencer. Reads with a quality value greater than 8 were mapped to the S. zooepidemicus strain ATCC35246 genome using the SOLiD™ System Analysis Pipeline Tool ,allowing mismatches up to five bases. The detailed mapping methods have been described previously [63] and rRNA reads were filtered before mapping. The expression level of genes was calculated by read counts normalized with the total mapped reads and the gene length was calculated using the RPKM method [65]. The differential expressions of genes between the in vitro and in vivo libraries were analyzed based on the DEGseq modeling methods [66].
Identification of pathogenicity islands (PAIs)
The PAIs of S. zooepidemicus strain ATCC35246 were identified according to the following criteria: First, GC content and GC skew were determined and regions showing differences from the average of the whole genome indicated potential PAI loci. Second, the PAI locus was present in ATCC35246, but was absent or scattered in the other three species. Third, mobility genes, such as integrases, transposases, IS elements were present at the boundaries of the locus. Four, virulence genes were located in the locus. Finally, these loci were confirmed using IslandViewer, an genomic island predictor that integrates three methods: IslandPick, IslandPath-DIMOB, and SIGI-HMM [67].
Construction of partial SeseCisland_3 knockout strain, ∆Island3-Sz35246
To construct ∆pSET4s-LR plasmid, the upstream (LA) and downstream (RA) fragments of the Sz35246 target region were amplified. These two fragments were linked by fusion PCR and inserted into the pSET4s plasmid. Competent Sz35246 cells were subjected to electrotransformation with ∆pSET4s-LR plasmid and positively transformed cells were selected at 28°C in the presence of spectinomycin. Bacteria at the mid logarithmic growth phase were diluted with THB containing spectinomycin and cultured at 28°C to the early logarithmic phase. The culture was then shifted to 37°C and incubated for 4 h. Subsequently, the cells were spread on THB and incubated at 28°C. Temperature resistant colonies were screened at 37°C for the loss of vector-mediated spectinomycin resistance. The putative double crossover homologous recombinant mutants and some of the deleted genes in SeseCisland_3 were detected by PCR.
In vivo challenges of ICR mice
The Laboratory Animal Monitoring Committee of Jiangsu Province approved the experimental protocols. Two groups of eight-week-old ICR mice (10 animals per group) were used for in vivo infection studies. The wild-type Sz35246 and ∆Island3-Sz35246 were cultured with THB medium (Difco) at 37°C, with shaking at 180 rpm, separately. When the OD600 reached 0.6, bacteria were pelleted, resuspended in PBS and diluted appropriately to 1.25 × 106 CFU/ml (5×LD50 per 0.2 ml, LD50=5×104 CFU/ml) [68]. Mice were injected with 0.2 ml of liquid bacterial suspension. Survival was monitored for 5 days. Survival curves and statistical analysis were made by GraphPad Prism (Version 5.02).
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ZM and JG carried out the genome sequencing, participated in the virulence genes studies, sequence alignment, transcriptome analysis and drafted the manuscript. LY and YL prepared the bacterial DNA and RNA. ZM and BX carried out the gene knockout and animal experiments. RJ and QM participated in the analysis of PAIs. HF and SH conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Genome information for S. zoopedemicus ATCC35246.
Comparative gene expression analysis in vitro and in vivo for S. zoopedemicus ATCC35246.
S. zoopedemicus ATCC35246-specific genes.
Genes in SeseCisland_1 of the S. zoopedemicus ATCC35246 genome.
Genes in SeseCisland_2 of the S. zoopedemicus ATCC35246 genome.
Genes in SeseCisland_3 of S. zoopedemicus ATCC35246 genome.
Genes in SeseCisland_4 of the S. zoopedemicus ATCC35246 genome.
PCR detection of genes located in SeseCisland_3 deleted region. Red: Genes still existed in Sz35246 SeseCisland_3. Yellow: Knocked out genes of Sz35246 SeseCisland_3.
Contributor Information
Zhe Ma, Email: mazhe320@126.com.
Jianing Geng, Email: gengjianing@big.ac.cn.
Li Yi, Email: lilili123168@163.com.
Bin Xu, Email: 1091517919@qq.com.
Ruoyu Jia, Email: jiary@big.ac.cn.
Yue Li, Email: lye1987212@sina.com.
Qingshu Meng, Email: mengqingshu@big.ac.cn.
Hongjie Fan, Email: fhj@njau.edu.cn.
Songnian Hu, Email: husn@big.ac.cn.
Acknowledgments
This study was supported by Program from the National Basic Research Program (973) of China (2012CB518804), by grants from the National Natural Science Foundation of China (31172319, 31272581), the Research Fund for the Doctoral Program of Higher Education of China (2010097110005). The Jiangsu Agricultural Science and Technology Innovation Fund (CX(12)3078), and the project was funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
References
- Deng W, Puente JL, Gruenheid S, Li Y, Vallance BA, Vazquez A, Barba J, Ibarra JA, O'Donnell P, Metalnikov P. et al. Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc Natl Acad Sci USA. 2004;101(10):3597–3602. doi: 10.1073/pnas.0400326101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Shen X, Yan J, Han H, Zheng B, Liu D, Cheng H, Zhao Y, Rao X, Wang C. et al. GI-type T4SS-mediated horizontal transfer of the 89K pathogenicity island in epidemic Streptococcus suis serotype 2. Mol Microbiol. 2011;79(6):1670–1683. doi: 10.1111/j.1365-2958.2011.07553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutschler H, Meinhart A. epsilon/zeta systems: their role in resistance, virulence, and their potential for antibiotic development. J Mol Med (Berl) 2011;89(12):1183–1194. doi: 10.1007/s00109-011-0797-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Park JH, Inouye M. Toxin-antitoxin systems in bacteria and archaea. Annu Rev Genet. 2011;45:61–79. doi: 10.1146/annurev-genet-110410-132412. [DOI] [PubMed] [Google Scholar]
- Brzozowska I, Brzozowska K, Zielenkiewicz U. Functioning of the TA cassette of streptococcal plasmid pSM19035 in various Gram-positive bacteria. Plasmid. 2012;68(1):51–60. doi: 10.1016/j.plasmid.2012.01.010. [DOI] [PubMed] [Google Scholar]
- Timoney JF. The pathogenic equine streptococci. Vet Res. 2004;35(4):397–409. doi: 10.1051/vetres:2004025. [DOI] [PubMed] [Google Scholar]
- Abbott Y, Acke E, Khan S, Muldoon EG, Markey BK, Pinilla M, Leonard FC, Steward K, Waller A. Zoonotic transmission of Streptococcus equi subsp. zooepidemicus from a dog to a handler. J Med Microbiol. 2010;59(Pt 1):120–123. doi: 10.1099/jmm.0.012930-0. [DOI] [PubMed] [Google Scholar]
- Eyre DW, Kenkre JS, Bowler IC, McBride SJ. Streptococcus equi subspecies zooepidemicus meningitis--a case report and review of the literature. Eur J Clin Microbiol Infect Dis. 2010;29(12):1459–1463. doi: 10.1007/s10096-010-1037-5. [DOI] [PubMed] [Google Scholar]
- Feng ZG, Hu JS. Outbreak of swine streptococcosis in Sichan province and identification of pathogen. Anim Husbandry Vet Med Lett. 1977;2:7–12. [Google Scholar]
- Liu PH, Shen FS, Wang YK, Zhang SH. Identification of swine Streptococcus isolates in Shanghai. Chin J Vet Med. 2001;21:42–46. [Google Scholar]
- Ma Z, Geng J, Zhang H, Yu H, Yi L, Lei M, Lu CP, Fan HJ, Hu S. Complete genome sequence of Streptococcus equi subsp. zooepidemicus strain ATCC 35246. J Bacteriol. 2011;193(19):5583–5584. doi: 10.1128/JB.05700-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beres SB, Sesso R, Pinto SW, Hoe NP, Porcella SF, Deleo FR, Musser JM. Genome sequence of a Lancefield group C Streptococcus zooepidemicus strain causing epidemic nephritis: new information about an old disease. PLoS One. 2008;3(8):e3026. doi: 10.1371/journal.pone.0003026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden MT, Heather Z, Paillot R, Steward KF, Webb K, Ainslie F, Jourdan T, Bason NC, Holroyd NE, Mungall K. et al. Genomic evidence for the evolution of Streptococcus equi: host restriction, increased virulence, and genetic exchange with human pathogens. PLoS Pathog. 2009;5(3):e1000346. doi: 10.1371/journal.ppat.1000346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelburne SA 3rd, Sumby P, Sitkiewicz I, Okorafor N, Granville C, Patel P, Voyich J, Hull R, DeLeo FR, Musser JM. Maltodextrin utilization plays a key role in the ability of group A Streptococcus to colonize the oropharynx. Infect Immun. 2006;74(8):4605–4614. doi: 10.1128/IAI.00477-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giammarinaro P, Paton JC. Role of RegM, a homologue of the catabolite repressor protein CcpA, in the virulence of Streptococcus pneumoniae. Infect Immun. 2002;70(10):5454–5461. doi: 10.1128/IAI.70.10.5454-5461.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Xiao J, Pan L, Yang M, Zhang G, Jin S, Yu J. A systematic survey of mini-proteins in bacteria and archaea. PLoS One. 2008;3(12):e4027. doi: 10.1371/journal.pone.0004027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne LG. Dormancy of Mycobacterium tuberculosis and latency of disease. Eur J Clin Microbiol Infect Dis. 1994;13(11):908–914. doi: 10.1007/BF02111491. [DOI] [PubMed] [Google Scholar]
- Pandey DP, Gerdes K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005;33(3):966–976. doi: 10.1093/nar/gki201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RB, Gerdes K. Programmed cell death in bacteria: proteic plasmid stabilization systems. Mol Microbiol. 1995;17(2):205–210. doi: 10.1111/j.1365-2958.1995.mmi_17020205.x. [DOI] [PubMed] [Google Scholar]
- Liu M, Zhang Y, Inouye M, Woychik NA. Bacterial addiction module toxin Doc inhibits translation elongation through its association with the 30S ribosomal subunit. Proc Natl Acad Sci USA. 2008;105(15):5885–5890. doi: 10.1073/pnas.0711949105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolhuis H, Palm P, Wende A, Falb M, Rampp M, Rodriguez-Valera F, Pfeiffer F, Oesterhelt D. The genome of the square archaeon Haloquadratum walsbyi : life at the limits of water activity. BMC Genomics. 2006;7:169. doi: 10.1186/1471-2164-7-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizenman E, Engelberg-Kulka H, Glaser G. An Escherichia coli chromosomal "addiction module" regulated by guanosine [corrected] 3',5'-bispyrophosphate: a model for programmed bacterial cell death. Proc Natl Acad Sci USA. 1996;93(12):6059–6063. doi: 10.1073/pnas.93.12.6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen SK, Maenhaut-Michel G, Mine N, Gottesman S, Gerdes K, Van Melderen L. Overproduction of the Lon protease triggers inhibition of translation in Escherichia coli: involvement of the yefM-yoeB toxin-antitoxin system. Mol Microbiol. 2004;51(6):1705–1717. doi: 10.1046/j.1365-2958.2003.03941.x. [DOI] [PubMed] [Google Scholar]
- Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL. et al. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–288. doi: 10.1093/nar/gkn226. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Pino A, Christensen-Dalsgaard M, Wyns L, Yarmolinsky M, Magnuson RD, Gerdes K, Loris R. Doc of prophage P1 is inhibited by its antitoxin partner Phd through fold complementation. J Biol Chem. 2008;283(45):30821–30827. doi: 10.1074/jbc.M805654200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komano T, Utsumi R, Kawamukai M. Functional analysis of the fic gene involved in regulation of cell division. Res Microbiol. 1991;142(2–3):269–277. doi: 10.1016/0923-2508(91)90040-h. [DOI] [PubMed] [Google Scholar]
- Lehnherr H, Maguin E, Jafri S, Yarmolinsky MB. Plasmid addiction genes of bacteriophage P1: doc, which causes cell death on curing of prophage, and phd, which prevents host death when prophage is retained. J Mol Biol. 1993;233(3):414–428. doi: 10.1006/jmbi.1993.1521. [DOI] [PubMed] [Google Scholar]
- Zekarias B, Mattoo S, Worby C, Lehmann J, Rosenbusch RF, Corbeil LB. Histophilus somni IbpA DR2/Fic in virulence and immunoprotection at the natural host alveolar epithelial barrier. Infect Immun. 2010;78(5):1850–1858. doi: 10.1128/IAI.01277-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielenkiewicz U, Ceglowski P. The toxin-antitoxin system of the streptococcal plasmid pSM19035. J Bacteriol. 2005;187(17):6094–6105. doi: 10.1128/JB.187.17.6094-6105.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WCN, McDougal LK, Hageman J, McDonald LC, Patel JB. Vancomycin-resistant Staphylococcus aureus isolates associated with Inc18-like vanA plasmids in Michigan. Antimicrob Agents Chemother. 2008;52:452–457. doi: 10.1128/AAC.00908-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JSGS, Spratt BG, Holden DW. A locus contained within a variable region of pneumococcal pathogenicity island 1 contributes to virulence in mice. Infect Immun. 2004;72:1587–1593. doi: 10.1128/IAI.72.3.1587-1593.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Tang J, Dong W, Wang C, Feng Y, Wang J, Zheng F, Pan X, Liu D, Li M. et al. A glimpse of streptococcal toxic shock syndrome from comparative genomics of S. suis 2 Chinese isolates. PLoS One. 2007;2(3):e315. doi: 10.1371/journal.pone.0000315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinhart A, Alonso JC, Strater N, Saenger W. Crystal structure of the plasmid maintenance system epsilon/zeta: functional mechanism of toxin zeta and inactivation by epsilon 2 zeta 2 complex formation. Proc Natl Acad Sci USA. 2003;100(4):1661–1666. doi: 10.1073/pnas.0434325100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioy VSMM, Camacho AG, Lurz R, Antelmann H. pSM19035-encoded zeta toxin induces stasis followed by death in a subpopulation of cells. Microbiology. 2006;152:2365–2379. doi: 10.1099/mic.0.28950-0. [DOI] [PubMed] [Google Scholar]
- Kristoffersen P, Jensen GB, Gerdes K, Piskur J. Bacterial toxin–antitoxin gene system as a containment control in yeast cells. Application Environment Microbiology. 2000;66:5524–5526. doi: 10.1128/AEM.66.12.5524-5526.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower TR, Salmond GPC, Luisi B. Balancing at survival's edge: the structure and adaptive benefits of prokaryotic toxin-antitoxin partners. Curr Opin Struc Biol. 2011;21(1):109–118. doi: 10.1016/j.sbi.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Bolhuis H, Poele EM, Rodriguez-Valera F. Isolation and cultivation of Walsby's square archaeon. Environ Microbiol. 2004;6(12):1287–1291. doi: 10.1111/j.1462-2920.2004.00692.x. [DOI] [PubMed] [Google Scholar]
- Christensen SK, Pedersen K, Hansen FG, Gerdes K. Toxin-antitoxin loci as stress-response-elements: ChpAK/MazF and ChpBK cleave translated RNAs and are counteracted by tmRNA. J Mol Biol. 2003;332(4):809–819. doi: 10.1016/S0022-2836(03)00922-7. [DOI] [PubMed] [Google Scholar]
- Brocchi M, de Vasconcelos ATR, Zaha A. Restriction-modification systems in Mycoplasma spp. Genet Mol Biol. 2007;30(1):236–244. doi: 10.1590/S1415-47572007000200011. [DOI] [Google Scholar]
- Kobayashi I. Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 2001;29(18):3742–3756. doi: 10.1093/nar/29.18.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb K, Jolley KA, Mitchell Z, Robinson C, Newton JR, Maiden MC, Waller A. Development of an unambiguous and discriminatory multilocus sequence typing scheme for the Streptococcus zooepidemicus group. Microbiology. 2008;154(Pt 10):3016–3024. doi: 10.1099/mic.0.2008/018911-0. [DOI] [PubMed] [Google Scholar]
- Katz ME, Strugnell RA, Rood JI. Molecular characterization of a genomic region associated with virulence in Dichelobacter nodosus. Infect Immun. 1992;60(11):4586–4592. doi: 10.1128/iai.60.11.4586-4592.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomfield GA, Whittle G, McDonagh MB, Katz ME, Cheetham BF. Analysis of sequences flanking the vap regions of Dichelobacter nodosus: evidence for multiple integration events, a killer system, and a new genetic element. Microbiology. 1997;143(Pt 2):553–562. doi: 10.1099/00221287-143-2-553. [DOI] [PubMed] [Google Scholar]
- Romero P, Croucher NJ, Hiller NL, Hu FZ, Ehrlich GD, Bentley SD, Garcia E, Mitchell TJ. Comparative genomic analysis of ten Streptococcus pneumoniae temperate bacteriophages. J Bacteriol. 2009;191(15):4854–4862. doi: 10.1128/JB.01272-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay JA, Ruzin A, Ross HF, Kurepina N, Novick RP. The gene for toxic shock toxin is carried by a family of mobile pathogenicity islands in Staphylococcus aureus. Mol Microbiol. 1998;29(2):527–543. doi: 10.1046/j.1365-2958.1998.00947.x. [DOI] [PubMed] [Google Scholar]
- Ozeri V, Rosenshine I, Mosher DF, Fassler R, Hanski E. Roles of integrins and fibronectin in the entry of Streptococcus pyogenes into cells via protein F1. Mol Microbiol. 1998;30(3):625–637. doi: 10.1046/j.1365-2958.1998.01097.x. [DOI] [PubMed] [Google Scholar]
- Schwarz-Linek U, Hook M, Potts JR. The molecular basis of fibronectin-mediated bacterial adherence to host cells. Mol Microbiol. 2004;52(3):631–641. doi: 10.1111/j.1365-2958.2004.04027.x. [DOI] [PubMed] [Google Scholar]
- Lannergard J, Flock M, Johansson S, Flock JI, Guss B. Studies of fibronectin-binding proteins of Streptococcus equi. Infect Immun. 2005;73(11):7243–7251. doi: 10.1128/IAI.73.11.7243-7251.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telford JL, Barocchi MA, Margarit I, Rappuoli R, Grandi G. Pili in gram-positive pathogens. Nat Rev Microbiol. 2006;4(7):509–519. doi: 10.1038/nrmicro1443. [DOI] [PubMed] [Google Scholar]
- Wilson RL, Elthon J, Clegg S, Jones BD. Salmonella enterica serovars gallinarum and pullorum expressing Salmonella enterica serovar typhimurium type 1 fimbriae exhibit increased invasiveness for mammalian cells. Infect Immun. 2000;68(8):4782–4785. doi: 10.1128/IAI.68.8.4782-4785.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Fan HJ, Lu CP. Molecular cloning and analysis of the UDP-Glucose Pyrophosphorylase in Streptococcus equi subsp. zooepidemicus. Mol Biol Rep. 2011;38(4):2751–2760. doi: 10.1007/s11033-010-0420-8. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Yoshida A, Nagata E, Hoshino T, Oho T, Awano S, Takehara T, Ansai T. Streptococcus anginosus l-cysteine desulfhydrase gene expression is associated with abscess formation in BALB/c mice. Mol Oral Microbiol. 2011;26(3):221–227. doi: 10.1111/j.2041-1014.2010.00599.x. [DOI] [PubMed] [Google Scholar]
- Gao F, Zhang CT. Ori-Finder: a web-based system for finding oriCs in unannotated bacterial genomes. BMC Bioinforma. 2008;9:79. doi: 10.1186/1471-2105-9-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27(23):4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28(1):33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder NJ, Apweiler R. The InterPro database and tools for protein domain analysis. Curr Protoc Bioinformatics. 2008;2:2–7. doi: 10.1002/0471250953.bi0207s21. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–357. doi: 10.1093/nar/gkj102. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexeyenko A, Tamas I, Liu G, Sonnhammer EL. Automatic clustering of orthologs and inparalogs shared by multiple proteomes. Bioinformatics. 2006;22(14):e9–15. doi: 10.1093/bioinformatics/btl213. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and Clustal X. Curr Protoc Bioinformatics. 2002;2:2–3. doi: 10.1002/0471250953.bi0203s00. [DOI] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24(8):1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21(16):3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Romualdi C, Bortoluzzi S, D'Alessi F, Danieli GA. IDEG6: a web tool for detection of differentially expressed genes in multiple tag sampling experiments. Physiol Genomics. 2003;12(2):159–162. doi: 10.1152/physiolgenomics.00096.2002. [DOI] [PubMed] [Google Scholar]
- Langille MG, Brinkman FS. IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics. 2009;25(5):664–665. doi: 10.1093/bioinformatics/btp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong-Jie F, Fu-yu T, Ying M, Cheng-ping L. Virulence and antigenicity of the szp-gene deleted Streptococcus equi ssp. zooepidemicus mutant in mice. Vaccine. 2009;27(1):56–61. doi: 10.1016/j.vaccine.2008.10.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genome information for S. zoopedemicus ATCC35246.
Comparative gene expression analysis in vitro and in vivo for S. zoopedemicus ATCC35246.
S. zoopedemicus ATCC35246-specific genes.
Genes in SeseCisland_1 of the S. zoopedemicus ATCC35246 genome.
Genes in SeseCisland_2 of the S. zoopedemicus ATCC35246 genome.
Genes in SeseCisland_3 of S. zoopedemicus ATCC35246 genome.
Genes in SeseCisland_4 of the S. zoopedemicus ATCC35246 genome.
PCR detection of genes located in SeseCisland_3 deleted region. Red: Genes still existed in Sz35246 SeseCisland_3. Yellow: Knocked out genes of Sz35246 SeseCisland_3.