

Abstract
Amyloid β peptides form fibrils that are commonly assumed to have a dry, homogeneous, and static internal structure. To examine these assumptions, fibrils under various conditions and different ages have been examined with multidimensional infrared spectroscopy. Each peptide in the fibril had a 13C=18O label in the backbone of one residue to disinguish the amide I′ absorption due to that residue from the amide I′ absorption of other residues. Fibrils examined soon after they formed, and reexamined after 1 year in the dry state, exhibited spectral changes confirming that structurally significant water molecules were present in the freshly formed fibrils. Results from fibrils incubated in solution for 4 years indicate that water molecules remained trapped within fibrils and mobile over the 4 year time span. These water molecules are structurally significant because they perturb the parallel β-sheet hydrogen bonding pattern at frequent intervals and at multiple points within individual fibrils, creating structural heterogeneity along the length of a fibril. These results show that the interface between β-sheets in an amyloid fibril is not a “dry zipper”, and that the internal structure of a fibril evolves while it remains in a fibrillar state. These features, water trapping, structural heterogeneity, and structural evolution within a fibril over time, must be accommodated in models of amyloid fibril structure and formation.
Keywords: Stable isotopes, 18O, 13C, spectral crosspeaks, linear excitons, ultrafast vibrational laser spectroscopy, 2D infrared photon echo experiment
The internal structure of amyloid beta (Aβ) fibrils (Figure 1) that accumulate in Alzheimer’s disease (AD) remains largely unknown despite extensive study. Early X-ray diffraction studies of fibrils isolated from diseased human brain tissue elicited reflections at 4.75 and 9.8–10.6 Å,1,2 which are characteristic of the cross-β structure that is found in other fibril-forming proteins.3 Solid-state NMR (ssNMR),4−6 hydrogen exchange,7−10 and EPR11 studies indicate that Aβ fibrils do indeed contain two in-register parallel β-sheets, most likely spanning residues 12–24 and 30–40. The distance between strands in a parallel β-sheet accounts for the 4.75 Å reflection from X-ray diffraction, while the distance between the two β-sheets is thought to account for the 9.8–10.6 Å reflection. In contrast to the full length peptides, X-ray crystallographic studies of short fibril-forming hexameric segments of Aβ12,13 reveal interstrand distances of 4.79–4.88 Å that are reasonably close to the diffraction result from naturally occurring fibrils, but these crystals are narrower by one-to-several angstroms than the 9.8–10.6 Å spacing observed in full-length Aβ peptides, due to close packing of side chains into a “dry steric zipper.”
Figure 1.
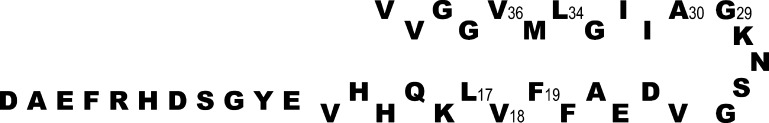
Amino acid sequence of Aβ40. The six residues examined in this work are denoted with their sequence numbers. Residues 1–11 are assumed to be random coil. Residues 12–24 and 30–40 are arranged to indicate that they are involved in β-sheets, and that the odd-numbered residue side chains in the two sheets are apposed to each other.
The net result of these data is that the secondary structure in an Aβ fibril is rather well-defined, but little is known about its tertiary and quaternary structure. Moreover, there is a significant degree of structural polymorphism among and within fibrils that is evident within a filament, in the number of filaments that comprise a fibril, in the morphology of fibrils by electron microscopy, and in the thermodynamic stability of fibrils (a filament consists of the Aβ peptide monomers that are hydrogen bonded to each other in the same β-sheet; there appear to be at least two filaments per fibril).14−25 This polymorphism makes biophysical characterization difficult, leaving broad questions about how the two β-sheets are packed against each other, the conformation of segments not involved in β-structure, the number and arrangement of individual filaments within a fibril, and the reason for their 10 nm width. Answers to these questions, and better understanding of polymorphism, are needed to develop therapeutic small molecules and antibodies that target amyloid fibrils for the treatment of AD.
Two-dimensional infrared (2D-IR) spectroscopy is a relatively new technology that is well poised to answer some of these questions. Compared to linear or Fourier transform infrared (FTIR) spectroscopy, 2D-IR has an inherently higher resolution and ability to discriminate between transitions with high and low extinction coefficients because signal strength in linear IR spectroscopy is proportional to the second power of the vibrational transition dipole, whereas signal strength in 2D-IR is proportional to the fourth power. Better resolution and sensitivity facilitates the identification of signals arising from isotope labels, so that 2D-IR can yield bond-specific information.26 In a manner that is analogous to 2D NMR spectroscopy, 2D-IR spectroscopy may be performed with a variety of pulse sequences to characterize events that evolve in time. Physical proximity may be revealed by off-diagonal peaks, and the technique may be expanded to three or more dimensions. However, 2D-IR also has several advantages over 2D NMR, such as much smaller sample requirements, much shorter data collection times, access to a second excited quantum state, variably polarized pulses, and a molecular instead of laboratory frame of reference. The short femtosecond pulses employed in 2D-IR permit time-resolved studies of ultrafast events such as electron transfer and solvent dynamics, or they may be applied in a “snapshot” mode over arbitrarily long time-scales. The studies reported herein represent the latter type of application.
2D-IR spectroscopy has been applied previously in two preliminary studies of Aβ fibrils with 13C=18O isotopes in the backbone carbonyl group of various residues. The first study showed that these labels were aligned in residues A21, G29, G33, and G38, forming a linear exciton as if in a one-dimensional crystal, while the label in residue G25 was disordered.27 The chief spectral characteristics of such an exciton are a red-shift of up to 30 cm–1, and a narrow well-defined absorption band. These results reported in this study confirmed the aforementioned secondary structure assignments, but they also revealed that fibrils have a strong internal electrostatic field gradient, arising from amino acid side chains of residues 30–40, and increasing in strength from residues G38 < G33 < G29.
The second 2D-IR study of Aβ fibrils involved the placement of 13C=18O labels in 18 out of 40 residues in Aβ, and revealed that several residues in ostensibly “dry” fibrils were perturbed by mobile water molecules trapped in cavities or channels in between the β-sheets.28 The perturbation pattern indicated that residue n in the N-terminal β-sheet (residues 12–24) was approximately apposed to residue 53-n in the slightly shorter C-terminal sheet (residues 30–40). The presence of trapped water in channels may also explain why the intersheet spacing of 9.8–10.6 Å in Aβ fibrils is larger than the intersheet spacing of dry steric zippers formed by hexameric Aβ segment crystals.
The present study brings 2D-IR spectroscopy to bear on questions about polymorphism and maturation in Aβ fibrils. One fibril sample was incubated for 4 weeks and prepared as a dry film. It was then examined shortly after being prepared and again after one year. The spectral changes observed over this period confirm the presence of structurally significant water molecules in the original fibrils. Other fibrils were examined after being allowed to form in solution for 10 weeks and again after being allowed to mature in solution for 4 years. The presence of structural heterogeneity in both young and old fibrils, and the evolution of this heterogeneity over time, has important implications for the design of therapeutic interventions aimed at preventing fibril formation.
Results
Fibrils after 1 Year in a Dry State
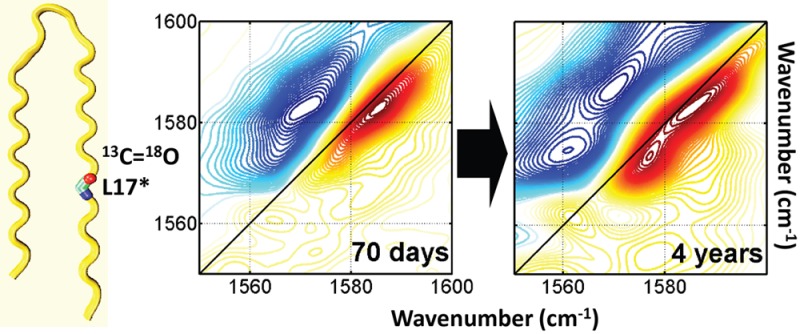
Aβ40 fibrils with 13C=18O labels in L34 were allowed to fibrillize in solution for 4 weeks, and then samples of this solution were evaporated onto a CaF2 window. Following the notation used in earlier papers, Aβ40 peptides containing such labels will be designated by the labeled residue identifier followed by an asterisk (i.e., L34*).27,28 The samples were examined shortly after evaporation, stored on the CaF2 window at room temperature (20 °C) in 2% relative humidity, and then examined after 1 year. 2D-IR spectra before and after this 1 year interval are shown in Figure 2.
Figure 2.

2D-IR spectra of 4-week-old L34* fibrils exposed to dry environment for different times. (a, b) 2D-IR spectra at T = 0 for the sample at (a) 0 day and (b) 1 year after solid fibril preparation. (c, d) 2D-IR spectra at T = 1 ps for the sample at (c) 0 day and (d) 1 year after solid fibril preparation. The pump frequencies (ωτ axis) span the range of the isotope labeled amide I′ transition (1560–1610 cm–1). The probe frequencies (ωt axis) span the range of 1550–1610 cm–1 to emphasize the weak isotope labeled amide I′ transition from L34 residue. The spectra are plotted with 60 evenly spaced contour lines. The intensities are shown with different colors from red (positive) to blue (negative). Note that the color bar scales are absolute values.
The spectral regions shown in all 2D-IR figures have been chosen to highlight the relatively weak amide I′ transitions (primes indicate that the sample was in D2O), and they exclude the much larger main amide I′ band from the unlabeled residues (e.g., Figure 3a). Amide I′ absorptions due to the labeled residues are shifted to 1550–1600 cm–1. The frequency shifts are due to the greater mass of the 13C=18O group, and their alignment in in-register parallel β-sheets. The alignment facilitates through-space coupling of the vibrational transition dipoles, and the formation of a linear exciton.27,28 The local electrostatic environment of 13C=18O and through-bond effects may also cause small shifts.
Figure 3.

Spectroscopic results from fibrillar Aβ peptides with 13C=18O labels in 6 different residues. (a) Amide I′ region of FTIR spectra from L17* (above) and V36* (below). The dominant feature at 1625 cm–1 in both spectra arises from peptide groups containing 12C=16O. Peptide groups containing 13C=18O labels yield subtle features in regions outlined by red rectangles. (b) 2D-IR spectra with zero waiting time (T = 0) from amyloid fibrils formed after 10 weeks. Aliquots were withdrawn from solutions of Aβ40 monomers with 13C=18O labels in the six different residues indicated, and dried on CaF2 windows. The vertical axis is the pump frequency (ωτ); the horizontal axis is the probe frequency (ωt). The pump frequencies span the range indicated by red rectangles in column (a). Red contours (positive) represent the 0 to 1 transition of the amide I′ vibration, while blue contours (negative) represent the 1 to 2 transition. The difference in ωt between the positive and negative peaks is due to anharmonicity of the vibrational transition energies. Diagonal lines are the locus of points where ωτ = ωt. The six spectra in this column have been previously published, and are reproduced here to facilitate comparisons across each row. The spectra in each row are from fibrils with labels in the same residue. (c) 2D-IR spectra with zero waiting time (T = 0) from amyloid fibrils formed after 4 years. The samples examined were prepared by withdrawing fresh aliquots from the same solutions that yielded the spectra in column (b) and drying them on CaF2 windows. The spectra shown are representative of those collected from two or more solutions. (d–f) 2D-IR spectra with selected waiting times between 250 and 3000 fs, collected from the same preparations yielding the spectra in column (c). Changes in band shape at various waiting times reflect fast dynamics near the 13C=18O labeled residue. (g) 2D-IR spectra with expanded scales, corresponding to the regions in column (f) outlined in black rectangles. The expanded scales aid in the identification of crosspeaks between absorption bands, which are relatively low in intensity; regions of high intensity absorption are off-scale and are rendered white. (h) Changes in the inverse slope of absorption band nodal line (IS) are plotted as a function of waiting time (T), and fitted to exponentially decaying frequency-frequency correlation functions. Decay constants are listed in Table 1. All 2D spectra are plotted with 60 evenly spaced contour lines with the maximum intensity of each spectrum normalized to 1. The relative intensities of different transitions are shown with different colors from red (positive) to blue (negative).
There are three bands arising from the labeled group centered at 1592, 1583, and 1574 cm–1, most clearly observed in spectra with a 1 ps waiting time (the waiting time, T, is the interval between the second and third pulses) and among the negative (blue) contours to the left of the diagonal (Figure 2c,d). The diagonal anharmonicity (i.e., roughly the horizontal separation between the red and the blue peaks) of the 1574 cm–1 band is conspicuously smaller than that of the other two bands. The smaller anharmonicity and relatively low frequency of this band both indicate that it arises from an exciton spanning more or better-aligned residues. After 1 year, the intensity of this band has increased in magnitude, whereas the other two bands have decreased.
Altogether, these changes indicate that L34 residues become better aligned after 1 year in the dry state. The simplest explanation for improved alignment is that trapped water molecules near residue L34 on day 0 escaped over the course of 1 year and ceased interfering with β-sheet structure. It has been suggested that amyloid formation may involve a structural annealing process,29 and it is possible that the trapped water molecules evinced by spectral diffusion facilitate this process, at least on a relatively long time scale.
Ten week versus 4 Year Comparison
A linear FTIR spectrum for L17* is shown in upper panel of Figure 3a. The corresponding spectrum for V36* is shown in the lower panel. The dominant feature between 1610 and 1640 cm–1 in both spectra is the amide I′ absorption band arising from the 39 unlabeled residues.
Of the 18 samples described in 2009,28 six of the solutions remained clear, retained their volume, and contained a normal number of Aβ fibrils with the same characteristic morphology by electron microscopy (EM; Supporting Information Figure S4). The 2D-IR spectra collected in 2009 for these six samples are reproduced in Figure 3b. Only the spectral region showing absorption due to 13C=18O labels is shown, and this region corresponds to the red boxes in Figure 3a. Spectra in Figure 3c were collected from fresh aliquots from the same samples that had been incubating at 20 °C in the interim. As is typical for 2D-IR spectra, absorption bands due to the isotope labels are well-resolved from the unlabeled bands (off-scale in these spectra), and they exhibit a signal/noise ratio that facilitates a detailed examination of band shapes.
A comparison of the spectra in Figure 3b and c indicates that L17* underwent a significant structural change between 10 weeks and 4 years, with a marked increase in the magnitude of the 1574 cm–1 band in the 4-year-old fibrils. Changes in V18* and F19* were relatively minor. G29* is not believed to take part in either of the parallel β-sheets of an Aβ fibril, and its absorption band is considerably broader after 4 years, suggesting greater heterogeneity at this location. V36* exhibits a similar absorption band shape after 4 years, but it has blue-shifted by 4 cm–1. This shift suggests that there has been a change in the alignment of the backbone of residue V36 in the older fibrils. Altogether, these changes indicate that portions of the fibril undergo slow structural change even when they are in the midst of the parallel β-sheets.
Four-Year-Old Samples at Various Waiting Times
2D-IR spectra of 4-year-old fibrils at selected waiting times are shown in Figure 3c–f. In general, absorption bands tend to become more circular with longer waiting times due to spectral diffusion, that is, the averaging of transient effects that otherwise cause inhomogeneous broadening of the spectral bands (e.g., forming and breaking hydrogen bonds). To characterize the rate of spectral diffusion, values for the inverse slope (IS) were calculated as a function of waiting time T for various transitions in each sample (Table 1, Figure 3h, and Supporting Information Figure S2).30,31
Table 1. IS decay Constants for the High Frequency 13C=18O Absorption Bandsa.
| low frequency band (cm–1) | high frequency band (cm–1) | time constant (ps) | |
|---|---|---|---|
| L17* | 1570 | 1584 | 1.4 ± 0.3 |
| V18* | 1573 | 1581 | 1.0 ± 0.1 |
| F19* | 1573 | 1581 | 1.9 ± 0.5 |
| G29* | 1569 | – | – |
| A30* | 1570 | 1583 | 1.3 ± 0.3 |
| V36* | – | 1592 | 0.8 ± 0.4 |
Dashes indicate not present, not significant, or that only one frequency was observed for the residue.
Earlier studies have suggested that spectral diffusion on a picosecond time scale is due to mobile water molecules in the immediate environment of the isotope label.28,32,33 Spectral diffusion due to side chain conformational changes or backbone motions is markedly slower. Other mechanisms may contribute to waiting-time-dependent spectral shape changes. For example, different conformations may exhibit different excited vibrational state lifetimes due to hydration.34 A change in shape with waiting time may ensue, therefore, if the absorption bands from two conformations overlap, but decay at different rates. In the present study, all samples consisted of dry films on a CaF2 window. Therefore, differences in conformation and hydration of the films are unlikely to reflect differences in vibrational excited state lifetimes due to different hydration levels in the bulk solution.
For L17* at 10 weeks, we previously reported a spectral diffusion rate of ∼1 ps for the 1583 cm–1 band.28 After 4 years, the 1583 cm–1 band still exhibits a diffusion rate of ∼1.4 ps, while a new band at 1574 cm–1 exhibits little or no spectral diffusion on the picosecond time scale. At waiting times of 250–3000 fs, transitions are evident at 1592, 1583, and 1574 cm–1.35 Simple linear exciton theory suggests that these bands arise from isolated (i.e., infinitely diluted and unperturbed), dimeric, and multimeric units of labeled L17 residues, respectively.33,35,36 There is a distinct crosspeak between the 1574 and 1583 cm–1 bands at T = 3000 fs (Figure 3g), which indicates that these bands arise from within the same fibril (see Discussion and Supporting Information Figure S3).28
The various spectra of L17* present a self-consistent picture: mobile water molecules trapped near residue L17 occasionally perturb the alignment of 13C=18O groups in 10-week-old fibrils, giving rise to a 1583 cm–1 band and spectral diffusion. After 4 years, some of these water molecules are lost from near the residue, resulting in less perturbation in the excitonic structure and a red-shifted 1574 cm–1 band that exhibits no spectral diffusion.
V18* also exhibits at least three transitions at waiting times of 250–2500 fs, at similar frequencies. The lowest-frequency transition in V18* increased to a certain degree in the 4-year-old spectra, where the change pattern is similar to that of L17*.
For V18* at 10 weeks, we previously reported a spectral diffusion rate of 1.03 ps for the 1581 cm–1 band,28 indicating trapped water in close proximity to the residue. Four-year-old V18* fibrils have similar properties. A subpopulation with slightly lower frequency, evident at T = 250 and 1500 fs, shows by contrast that most of the 13C=18O groups in V18* are still perturbed by mobile water molecules. The subpopulation indicates that mobile-water-free excitonic structure of residue V18 start to form, but the relative population is smaller than that of residue L17 after 4 years.
The broad band arising from 10-week-old F19* fibrils at 1578 cm–1 persists after 4 years. However, this band evolves into a pair of distinct bands at 1573 and 1581 cm–1 at T = 2 ps with no evidence of a crosspeak. The proximity of these two bands makes it difficult to evaluate spectral diffusion, but the evolution of band shape with T may be due to spectral diffusion, and consistent with the presence of trapped mobile water molecules near residues V18 and L17. However, the absence of a crosspeak suggests that structural heterogeneity in F19* is not necessarily present within the same fibril. One possibility is that structural distortions are induced by the way that residue F19 side chains pack in between the β-sheets.
Four-year-old G29* fibrils at T = 0 exhibits a single broad transition at 1569 cm–1. Compared to the 10 week sample, the transition frequency remain unchanged, but the width of the transition increased. The nodal line slope for the 1569 cm–1 transition shows no evidence of spectral diffusion, indicating that this residue is not sensed by mobile water molecules. The broader width of the transition at this residue may be attributed to a more disordered structure near this residue after 4 years.
Ten-week-old A30* fibrils exhibited a dominant band at 1570 cm–1 and a weak band at 1583 cm–1. After 4 years, the bands and their shapes are essentially unchanged, although the grow-in crosspeak is clearly shown at T = 3000 fs. The 1583 cm–1 band exhibits a spectral diffusion rate of 1.29 ps, while the 1570 cm–1 band exhibits no discernible decay. These results indicate that most 13C=18O groups in A30* are well-aligned and form a low-energy exciton, while a relatively small number (relative population ∼5–15%) are perturbed by mobile water molecules.
V36* fibrils at both 10 weeks and 4 years exhibited a broad inhomogeneous absorption band at ∼1590 cm–1. Two sharp transitions which are assumed to originate from liner-chain-exciton bands are observed at 1567 and 1574 cm–1, which indicates that there are two major allignment of V36 in a fibril. The broad band shown at T = 0 separates into two transitions located at 1592 and 1584 cm–1 at T = 2000 fs. The apparent blue shift of peak center by 4 cm–1 observed in the 4-year-old sample is caused by the decrease of the magnitude of 1584 cm–1 transition and increase in the magnitude of the a lower-frequency transition at 1567 cm–1, whereas the magnitude of the transitions at 1592 and 1574 cm–1 are unchanged. This observation suggests that the density of water molecules in the vicinity of V36 in a specific type of alignment decreased. At 10 weeks and after 4 years these fibrils both exhibited fast spectral diffusion (the 1592 cm–1 transition) caused by nearby mobile water.
It is unlikely that the changes observed after 4 years were a consequence of sample deterioration because the presence of the isotope peaks at these frequencies in each case indicates a high degree of sample order; the changes indicate only that the extent of this order has changed. Spectra in which labeled residues are diluted with unlabeled residues (i.e., nonadjacent) absorb in the vicinity of 1600 cm–1, with relatively small residue-dependent shifts.27 It is also unlikely that these fibrils disaggregated and then reaggregated into a different structure over the span of 4 years because the equilibrium monomer concentrations for these samples were measured and found to be undetectable.25 Chemical changes such as deamidation are possible, and may alter the pathways and kinetics of fibril formation.37 However, the excitonic structure and morphological appearance of the 4-year-old fibrils in this study are unchanged, and the changes observed are often associated with fast spectral diffusion suggesting that they are due to the presence of mobile water molecules.
Discussion
These results provide three key insights into Aβ40 fibril structure. First, they indicate that water molecules are a significant structural component in amyloid fibrils. Isolated residues containing 13C=18O labels exhibit absorption bands ∼1600 cm–1. When 13C=18O groups are part of an in-register parallel β-sheet within an amyloid fibril, vibrational transition dipole coupling results in the formation of narrow excitonic absorption bands with fundamental frequencies of 1575 cm–1 or lower. When water molecules are present, however, they can perturb dipolar coupling and cause the absorption band to occur at intermediate frequencies (i.e., between 1575 and 1600 cm–1). The inhomogeneities in Aβ fibrils due to such perturbations (measured as an IS) tend to decay on a time scale that can only be explained by water molecules that are mobile and that are perturbing the alignment of residues in the β-sheets of the fibril. The results from 1-year-old dry fibrils confirm that water molecules were trapped inside the fibril and that they slowly escaped over the span of a year. The pattern of perturbation and IS decays previously observed in 10-week-old fibrils suggested that the side chains of odd-numbered residues were trapped between the two β-sheets, and that residues n and 53-n were apposed to each other in different parallel β-sheets. These new results from 4-year-old fibrils indicate that these water molecules may be lost or may migrate to other locations in the fibril. The spectra documenting these changes yield a self-consistent picture that underscores the value of 2D-IR spectroscopy for characterizing these structures.
An important implication of these results is that efforts to model the structure or formation of Aβ40 fibrils must incorporate these water molecules into the interface between β-sheets, and not force the formation of a dry zipper interface as found in crystals of short Aβ peptide segments. The incorporation of water into the interface should facilitate the creation of models with the 9.8–10.6 Å intersheet spacing observed by X-ray diffraction.
The second key insight afforded by these spectra is that Aβ40 peptides undergo slow structural changes, spanning years, while remaining in a fibrillar state. This result was not obvious because a structure that appears so regular by EM might be expected to exhibit a high degree of structural cooperativity. These slow changes, coupled with picosecond-scale fast changes due to trapped mobile waters, challenges the notion that fibrils (even their core secondary structure) are static structures. Investigators using fluorescent dyes to report on the internal structure of amyloid fibrils over a ∼22 month span in animal models of Alzheimer’s disease have concluded that an unspecified type of structural maturation also occurs in vivo.38 Therefore, it is becoming clear that efforts to target theranostic agents that bind to fibrils or inhibit their formation must hit a moving target.
The third key insight is that structural heterogeneity exists within single fibrils. The presence of two or more distinct bands in the 1550–1600 cm–1 region is evidence of two or more exciton structures within a fibril. The grow-in crosspeak (Figure 3g and Supporting Information Figure S3) at nonzero waiting times between two transitions in the 2D-IR spectrum may arise from either fast chemical exchange between two conformations39,40 or energy transfer between coupled transitions.28,41 However, chemical exchange would imply changes in the chemical structure of the fibril, and would be too slow for a picosecond experiment to capture. Therefore, the grow-in crosspeak most likely arises from transition dipole coupling and the transfer of energy between two adjacent excitonic bands within the same fibril.
Even under ideal circumstances, this coupling is weak compared to the absorption bands involved. When present, however, it constitutes strong evidence for end-to-end relationships among the 13C=18O groups that participate in the exciton, that is, a linear chain. Coupling might also occur within the excitonic vibrational band structure. However, the complexity of the excitonic band in these fibrils will cause such cross-peaks to overlap and obscure each other. Therefore, the contribution of couplings within excitonic bands is likely to be negligible.
Ordinary electron microscopy with negative staining gives no indication of such heterogeneity (Supporting Information Figure S4), but measurements of mass-per-length from off-axis dark field electron microscopy suggest that morphological homogeneity is an illusion, at least with respect to the number of filaments per fibril.25 The 2D-IR measurements reported herein now indicate that there are heterogeneities and mobile water molecules within Aβ fibril filaments, and that they lead to an evolution of secondary internal fibril structure over long times. These conclusions are significant to molecular modeling efforts aimed at understanding the formation of amyloid fibrils, and at the design of diagnostic or therapeutic agents that target amyloid fibrils.
Methods
Sample Preparation
Synthetic 13C=18O labeled Aβ40 peptides (DAEFRHDSGY EVHHQKLVFF AEDVGSNKGA IIGLMVGGVV) were synthesized by the Small Scale Peptide Synthesis facility in the Keck Biotechnology Resource Laboratory at Yale University, and prepared as described previously. The presence of extra mass units due to the isotope labels, an overall isotopic purity of ≥95%, and in some cases the position of the isotope labels was confirmed for each peptide by mass spectrometry.28 The 4-year-old fibrils described in this paper were obtained from the same solutions described in 2009, which had been incubating at room temperature in the interim.27,28 Fresh aliquots of these solutions (∼5 μL) were evaporated on a CaF2 window, yielding dry films that required no further processing for spectroscopic study. The L34* sample was incubated for 4 weeks, evaporated on a CaF2 window, and stored for 1 year at room temperature with a desiccant.
2D-IR Spectroscopy
Spectra were collected in the same manner and on the same instrument as previously described.27,28 Three femtosecond IR pulses (k1, k2, and k3) were focused on the sample to induce a third order response (photon echo), which was heterodyned with a fourth IR pulse (the local oscillator), dispersed by a monochromator, and detected with a 64-pixel array detector. Rephasing and nonrephasing data were collected by scanning the time delay (τ) between k1 and k2 from −4 to +4 ps at 2 fs intervals. The waiting time (T) between k1/k2 and k3 pulses was zero except as indicated. Choppers were applied to the k3 and local oscillator pulses with a fixed phase difference to minimize scattering noise from the sample. The final spectrum, S(ωτ, T, ωt), was obtained from Fourier transforms of S(τ, T, λ), and only the real term from the transform result is shown. It has been reported that the arginine side chain has an absorption band that can overlap with that of 13C=18O, especially at longer waiting times.41 However, spectra collected from unlabeled fibrils do not reveal an absorption band with enough intensity to interfere with the analyses reported herein (Supporting Information Figure S1). These spectra also demonstrate the absence of interference due to natural abundance 13C=O groups.
Electron Microscopy
Aliquots of Aβ40-containing solutions were diluted by a factor of 10–20 and then adsorbed onto 300-mesh carbon-coated nickel grids, stained with 1% ammonium molybdate at pH 7.4, and visualized with a JEOL 1010 transmission electron microscope.
Acknowledgments
The authors are grateful to Martin Zanni for critical comments on a preliminary version of the manuscript.
Glossary
Abbreviations
- Aβ40
the 40-residue form of the amyloid β peptide
- 2D-IR
two-dimensional infrared spectroscopy
- FTIR
Fourier transform infrared spectroscopy
- AD
Alzheimer’s disease
- IS
inverse nodal line slope
Supporting Information Available
Figures S1–S4. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
L.L. synthesized the isotopically labeled residues; L.L. and H.K. prepared the fibrils; H.K. performed the electron microscopy; Y.S.K and J.M. collected and processed the 2D-IR spectra; J.M., Y.S.K, R.H, and P.H.A. designed the experiments, interpreted the data, and composed the paper.
The research was supported by grants to P.H.A. from the NIH (GM76201), the Alzheimer’s Association, and the Glenn Foundation, to Y.S.K. from NRF of Korea (2011-0015061), and to R.M.H. from the NIH (GM 12592) and NSF-CHE. The 2D-IR instrumentation was developed with a Research Resource grant from the NIH (P41 GM104605).
The authors declare no competing financial interest.
Author Status
# R.M.H.: Deceased, February 27, 2013.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Eanes E. D.; Glenner G. G. (1968) X-Ray Diffraction Studies on Amyloid Filaments. J. Histochem. Cytochem. 16, 673–677. [DOI] [PubMed] [Google Scholar]
- Kirschner D. A.; Abraham C.; Selkoe D. J. (1986) X-Ray-Diffraction From Intraneuronal Paired Helical Filaments and Extraneuronal Amyloid Fibers in Alzheimer-Disease Indicates Cross-Beta Conformation. Proc. Natl. Acad. Sci. U.S.A. 83, 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunde M.; Serpell L. C.; Bartlam M.; Fraser P. E.; Pepys M. B.; Blake C. F. (1997) Common Core Structure of Amyloid Fibrils by Synchrotron X-Ray Diffraction. J. Mol. Biol. 273, 729–739. [DOI] [PubMed] [Google Scholar]
- Antzutkin O. N.; Balbach J. J.; Leapman R. D.; Rizzo N. W.; Reed J.; Tycko R. (2000) Multiple Quantum Solid-State NMR Indicates a Parallel, Not Antiparallel, Organization of Beta-Sheets in Alzheimer’s β-Amyloid Fibrils. Proc. Natl. Acad. Sci. U.S.A. 97, 13045–13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova A. T.; Ishii Y.; Balbach J. J.; Antzutkin O. N.; Leapman R. D.; Delaglio F.; Tycko R. (2002) A Structural Model for Alzheimer’s β-Amyloid Fibrils Based on Experimental Constraints From Solid State NMR. Proc. Natl. Acad. Sci. U.S.A. 99, 16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzutkin O. N.; Leapman R. D.; Balbach J. J.; Tycko R. (2002) Supramolecular Structural Constraints on Alzheimer’s β-Amyloid Fibrils From Electron Microscopy and Solid-State Nuclear Magnetic Resonance. Biochemistry 41, 15436–15450. [DOI] [PubMed] [Google Scholar]
- Kheterpal I.; Zhou S.; Cook K. D.; Wetzel R. (2000) Aβ Amyloid Fibrils Possess a Core Structure Highly Resistant to Hydrogen Exchange. Proc. Natl. Acad. Sci. U.S.A. 97, 13597–13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheterpal I.; Wetzel R.; Cook K. D. (2003) Enhanced Correction Methods for Hydrogen Exchange-Mass Spectrometric Studies of Amyloid Fibrils. Protein Sci. 12, 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheterpal I.; Lashuel H. A.; Hartley D. M.; Wlaz T.; Lansbury P. T.; Wetzel R. (2003) Aβ Protofibrils Possess a Stable Core Structure Resistant to Hydrogen Exchange. Biochemistry 42, 14092–14098. [DOI] [PubMed] [Google Scholar]
- Whittemore N. A.; Mishra R.; Kheterpal I.; Williams A. D.; Wetzel R.; Serpersu E. H. (2005) Hydrogen-Deuterium (H/D) Exchange Mapping of Aβ(1–40) Amyloid Fibril Secondary Structure Using Nuclear Magnetic Resonance Spectroscopy. Biochemistry 44, 4434–4441. [DOI] [PubMed] [Google Scholar]
- Torok M.; Milton S.; Kayed R.; Wu P.; McIntire T.; Glabe C. G.; Langen R. (2002) Structural and Dynamic Features of Alzheimer’s Aβ Peptide in Amyloid Fibrils Studied by Site-Directed Spin Labeling. J. Biol. Chem. 277, 40810–40815. [DOI] [PubMed] [Google Scholar]
- Sawaya M. R.; Sambashivan S.; Nelson R.; Ivanova M. I.; Sievers S. A.; Apostol M. I.; Thompson M. J.; Balbirnie M.; Wiltzius J. J. W.; McFarlane H. T.; Madsen A. O.; Riekel C.; Eisenberg D. (2007) Atomic Structures of Amyloid Cross-[Bgr] Spines Reveal Varied Steric Zippers. Nature 447, 453–457. [DOI] [PubMed] [Google Scholar]
- Colletier J. P.; Laganowsky A.; Landau M.; Zhao M. L.; Soriaga A. B.; Goldschmidt L.; Flot D.; Cascio D.; Sawaya M. R.; Eisenberg D. (2011) Molecular Basis for Amyloid-Beta Polymorphism. Proc. Natl. Acad. Sci. U.S.A. 108, 16938–16943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivaprasad S.; Wetzel R. (2004) An Intersheet Packing Interaction in Aβ Fibrils Mapped by Disulfide Cross Linking. Biochemistry 43, 15310–15317. [DOI] [PubMed] [Google Scholar]
- Wetzel R.; Shivaprasad S.; Williams A. D. (2007) Plasticity of Amyloid Fibrils. Biochemistry 46, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodali R.; Wetzel R. (2007) Polymorphism in the Intermediates and Products of Amyloid Assembly. Curr. Opin. Struct. Biol. 17, 48–57. [DOI] [PubMed] [Google Scholar]
- Petkova A. T.; Leapman R. D.; Guo Z.; Yau W.-M.; Mattson M. P.; Tycko R. (2005) Self-Propagating, Molecular-Level Polymorphism in Alzheimer’s β-Amyloid Fibrils. Science 307, 262–265. [DOI] [PubMed] [Google Scholar]
- Paravastu A. K.; Petkova A. T.; Tycko R. (2006) Polymorphic Fibril Formation by Residues 10–40 of the Alzheimer’s β-Amyloid Peptide. Biophys. J. 90, 4618–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachse C.; Xu C.; Wieligmann K.; Diekmann S.; Grigorieff N.; Fandrich M. (2006) Quaternary Structure of a Mature Amyloid Fibril From Alzheimer’s Aβ(1–40) Peptide. J. Mol. Biol. 362, 347–354. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M.; Coomaraswamy J.; Bolmont T.; Kaeser S.; Schaefer C.; Kilger E.; Neuenschwander A.; Abramowski D.; Frey P.; Jaton A. L.; Vigouret J. M.; Paganetti P.; Walsh D. M.; Mathews P. M.; Ghiso J.; Staufenbiel M.; Walker L. C.; Jucker M. (2006) Exogenous Induction of Cerebral β-Amyloidogenesis Is Governed by Agent and Host. Science 313, 1781–1784. [DOI] [PubMed] [Google Scholar]
- Paravastu A. K.; Leapman R. D.; Yau W. M.; Tycko R. (2008) Molecular Structural Basis for Polymorphism in Alzheimer’s β-Amyloid Fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 18349–18354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravastu A. K.; Qahwash I.; Leapman R. D.; Meredith S. C.; Tycko R. (2009) Seeded Growth of Beta-Amyloid Fibrils From Alzheimer’s Brain-Derived Fibrils Produces a Distinct Fibril Structure. Proc. Natl. Acad. Sci. U.S.A. 106, 7443–7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinhardt J.; Sachse C.; Hortschansky P.; Grigorieff N.; Fandrich M. (2009) Aβ(1–40) Fibril Polymorphism Implies Diverse Interaction Patterns in Amyloid Fibrils. J. Mol. Biol. 386, 869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Hu X. Y.; Khant H.; Ludtke S. J.; Chiu W.; Schmid M. F.; Frieden C.; Lee J. M. (2009) Interprotofilament Interactions Between Alzheimer’s Aβ(1–42) Peptides in Amyloid Fibrils Revealed by CryoEM. Proc. Natl. Acad. Sci. U.S.A. 106, 4653–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu H.; Feingold-Link E.; Sharp K. A.; Rastogi T.; Axelsen P. H. (2010) Intrinsic Linear Heterogeneity of Amyloid Beta Protein Fibrils Revealed by Higher Resolution Mass-Per-Length Determinations. J. Biol. Chem. 285, 41843–41851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm P., and Zanni M. T. (2011) Concepts and Methods of 2D Infrared Spectroscopy, Cambridge University Press, Cambridge. [Google Scholar]
- Kim Y. S.; Liu L.; Axelsen P. H.; Hochstrasser R. M. (2008) Two-Dimensional Infrared Spectra of Isotopically Diluted Amyloid Fibrils From Aβ40. Proc. Natl. Acad. Sci. U.S.A. 105, 7720–7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. S.; Liu L.; Axelsen P. H.; Hochstrasser R. M. (2009) 2D IR Provides Evidence for Mobile Water Molecules in β-Amyloid Fibrils. Proc. Natl. Acad. Sci. U.S.A. 17751–17756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petty S. A.; Decatur S. M. (2005) Intersheet Rearrangement of Polypeptides During Nucleation of β-Sheet Aggregates. Proc. Natl. Acad. Sci. U.S.A. 102, 14272–14277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak K.; Park S.; Finkelstein I. J.; Fayer M. D. (2007) Frequency-Frequency Correlation Functions and Apodization in Two-Dimensional Infrared Vibrational Echo Spectroscopy: A New Approach. J. Chem. Phys. 127. [DOI] [PubMed] [Google Scholar]
- Fenn E. E.; Fayer M. D. (2011) Extracting 2D IR Frequency-Frequency Correlation Functions From Two Component Systems. J. Chem. Phys. 135. [DOI] [PubMed] [Google Scholar]
- Thirumalai D.; Reddy G.; Straub J. E. (2011) Role of Water in Protein Aggregation and Amyloid Polymorphism. Acc. Chem. Res. 12, 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A.; Hochstrasser R. M. (2011) A Peptide’s Perspective of Water Dynamics. Chem. Phys. 390, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton C. T.; Buchanan L. E.; Dunkelberger E. B.; Zanni M. T. (2011) Utilizing Lifetimes to Suppress Random Coil Features in 2D IR Spectra of Peptides. J. Phys. Chem. Lett. 2, 2357–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. S.; Hochstrasser R. M. (2009) Applications of 2D IR Spectroscopy to Peptides, Proteins, and Hydrogen-Bond Dynamics. J. Phys. Chem. B 113, 8231–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser R. M.; Whiteman J. D. (1972) Exciton Band Structure and Properties of a Real Linear Chain in a Molecular Crystal. J. Chem. Phys. 56, 5945–5958. [Google Scholar]
- Dunkelberger E. B.; Buchanan L. E.; Marek P.; Cao P.; Raleigh D. P.; Zanni M. T. (2012) Deamidation Accelerates Amyloid Formation and Alters Amylin Fiber Structure. J. Am. Chem. Soc. 134, 12658–12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyström S., Psonka-Antonczyk K. M., Ellingsen P. G., Johansson L. B. G., Reitan N., Handrick S., Prokop S., Heppner F. L., Wegenast-Braun B. M., Jucker M., Lindgren M., Stokke B. T., Hammarström P., and Nilsson K. P. R. (2013) Evidence for Age-Dependent in Vivo Conformational Rearrangement within Aβ Amyloid Deposits. ACS Chem. Biol. published online March, 25, 2013. DOI: 10.1021/cb4000376. [DOI] [PubMed] [Google Scholar]
- Kim Y. S.; Hochstrasser R. M. (2005) Chemical Exchange 2D IR of Hydrogen-Bond Making and Breaking. Proc. Natl. Acad. Sci. U.S.A. 102, 11185–11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J. R.; Kwak K. W.; Xie J.; Fayer M. D. (2006) Ultrafast Carbon-Carbon Single-Bond Rotational Isomerization in Room-Temperature Solution. Science 313, 1951–1955. [DOI] [PubMed] [Google Scholar]
- Ghosh A.; Tucker M. J.; Hochstrasser R. M. (2011) Identification of Arginine Residues in Peptides by 2D-IR Echo Spectroscopy. J. Phys. Chem. A 115, 9731–9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.