

Abstract
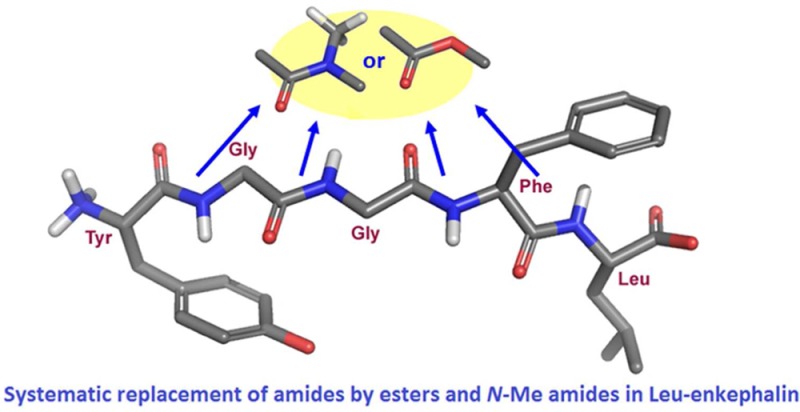
Leu-enkephalin analogues, in which the amide bonds were sequentially and systematically replaced either by ester or N-methyl amide bonds, were prepared using classical organic chemistry as well as solid phase peptide synthesis (SPPS). The peptidomimetics were characterized using competition binding, ERK1/2 phosphorylation, receptor internalization, and contractility assays to evaluate their pharmacological profile over the delta opioid receptor (DOPr). The lipophilicity (LogD7.4) and plasma stability of the active analogues were also measured. Our results revealed that the last amide bond can be successfully replaced by either an ester or an N-methyl amide bond without significantly decreasing the biological activity of the corresponding analogues when compared to Leu-enkephalin. The peptidomimetics with an N-methyl amide function between residues Phe and Leu were found to be more lipophilic and more stable than Leu-enkephalin. Findings from the present study further revealed that the hydrogen-bond donor properties of the fourth amide of Leu-enkephalin are not important for its biological activity on DOPr. Our results show that the systematic replacement of amide bonds by isosteric functions represents an efficient way to design and synthesize novel peptide analogues with enhanced stability. Our findings further suggest that such a strategy can also be useful to study the biological roles of amide bonds.
Keywords: Delta opioid receptor, enkephalin, amide bonds, degradation, lipophilicity, H-bonds
Our knowledge in the field of pain and analgesia has significantly improved over the past decades. The neuronal networks involved in pain processing are well-known and are among the best-characterized pathways. The investigation of these networks has led to the identification of new putative targets to alleviate pain. Despite the discovery of novel analgesic compounds for various targets, opioids are still among the most commonly used drugs for the treatment of moderate to severe pain. At least three major subtypes of opioid peptide receptors are defined, namely, mu (MOPr), delta (DOPr), and kappa (KOPr).1,2 In the clinic, commonly used opioids, such as morphine and its analogues, preferentially target and activate MOPr. The activation of this receptor is, however, also responsible for most of the side-effects of opioids. Several studies suggest that targeting DOPr may provide a strategy for developing new therapies to alleviate chronic pain without the usual adverse effects of narcotics.3−6 Based on these studies, the hypothesis that selective DOPr activation would lead to a better therapeutic profile is generally acknowledged.7,8 A number of useful DOPr agonists have been synthesized. Unfortunately, the progression of DOPr agonists toward human studies was prevented by the fact that the first generation of small molecules selectively activating this receptor produced nonlethal convulsions in rodents and nonhuman primates.9,10 Recent advance in the field however revealed that convulsions are not induced by all DOPr agonists.6,11,12 Indeed, as of to date, at least three nonpeptide delta compounds have reached clinical trials for the treatment of pain or depression. Due to their relative selectivity (1× to 5×) for DOPr over MOPr, their lack of MOPr-related unwanted effects, the simplicity of their structure, and their lack of toxicity, enkephalins (Tyr-Gly-Gly-Phe-Leu/Met) remain very attractive for the development of drugable compounds. However, because of their amide bonds, enkephalins are rapidly metabolized by peptidases (half-life ∼ 2 min)13 and are unable to cross the blood-brain barrier (BBB) to reach the opioid receptors located in the central nervous system, that is, the receptors playing a major role in analgesia.1
In endogenous bioactive peptides, the amide acts principally as a peptide chain holder. The amide bond is also frequently involved in important interactions as an H-bond donor or acceptor (or both). The nature of these interactions can be intramolecular (to stabilize an active conformation) or intermolecular (to promote binding with the receptor). In medicinal chemistry, amide bonds can be replaced with isosteric functions to (1) protect peptides from proteolysis, (2) increase peptide rigidity to favor the bioactive conformation, and (3) increase the lipophilicity of peptides. The exploitation of isosteric functions therefore represents an alternative to generating analogues featuring enhanced pharmacokinetic profiles.14
We previously reported the effects of the systematic replacement of amide bonds in Leu-enkephalin with E-alkenes.15 This strategy allowed us to demonstrate that the sole replacement of the first amide bond by an E-alkene did not affect the biological activity of Leu-enkephalin.15 Therefore, the first amide bond of enkephalins does not appear to be involved in either intramolecular or intermolecular interactions with DOPr, whereas the three subsequent amide bonds probably are. We now intend to extend our study to other amide surrogates, namely, ester and N-methyl amide functions that can be considered isosteric (i.e., having similar physical and/or chemical properties) to the amide bond. The existing synthetic analogues of Leu-enkephalins can be classified according to the way that they are obtained. These synthetic methods are mostly limited to the following four classes: (1) modification of the side chains in Tyr, Gly, Phe, and Leu,16,17 (2) inversion of the α chiral centers (e.g., DADLE),18 (3) rigidification of the enkephalin backbone through side chain-to-tail19 or side chain-to-side chain cyclization (e.g., DPDPE),20 and (4) introduction of constrained turn mimetics within the main chain.21 By comparison, few investigations have been devoted to the four peptide bonds. It is likely that most amides are involved either in binding to the receptor or in stabilizing the active conformer of Leu-enkephalin via hydrogen bonding. To our knowledge, systematic exploration of these important bonds has been achieved thrice, with thioesters,22 4-imidazolidinones,23 and alkenes.15 All of the other replacements of amides have only been partial. Some replacements involve N-methyl amides,24 triazoles and tetrazoles (via click chemistry),25 sulfonamides,26 phosphonamidates,27 ketomethylenes,28 and retro-inverso amides.29,30
Because the N-methyl amide and ester functions have the potential to act as hydrogen-bond acceptors and not as hydrogen-bond donors (Figure 1),14 they can be introduced in peptides to mimic some properties of the amide bond and to study the roles of amide bonds in receptor binding and activity. Nevertheless, no peptide bond surrogate is absolutely perfect;31N-methyl amides and esters therefore have their drawbacks. Indeed, both are devoid of hydrogen bond donor capabilities, a useful aspect to exploring the hydrogen bonding patterns of ligands of interest like Leu-enkephalin. It is however worth noting that additional properties are introduced when a normal secondary amide is turned into a tertiary amide or an ester. Indeed, the N-methyl group introduces steric bulk, distorts the planearity of the amide and restricts the conformational space around the modified amide.32 Occurrence of the cis isomer for tertiary amide bonds is also possible.33 The ester linkage appears to be a better mimic of a peptide bond as it does not suffer from that many geometric alterations.31,34 The Z conformer is the most stable as in amides, although the partial double bond character of the ester C(O)–O bond is not as pronounced as in the amide C(O)–NH bond.34 Consequently, esters are structurally more flexible. Despite these structural differences, we rely on induced fit phenomena or conformational selection35 to sample out minimally modified ligands with shapes approaching that of biologically active Leu-enkephalin. In this study, we sequentially and systematically replaced all of the amide bonds of Leu-enkephalin with either an ester or an N-methyl amide. The Leu-enkephalin analogues were then examined with respect to DOPr affinity, induction of extracellular signal-regulated kinase 1 and 2 (ERK1/2) phosphorylation, DOPr-internalization and inhibition of mouse vas deferens (MVD) contraction. The ability of the modified bonds to protect the synthesized analogues against degradation in plasma and to increase their lipophilicity was also assessed by measuring their half-life in diluted rat plasma and their LogD7.4 values.
Figure 1.
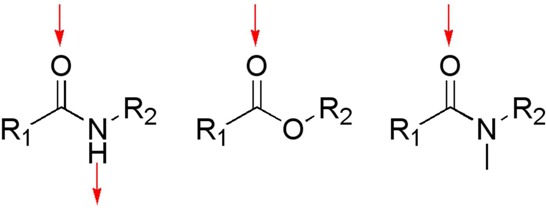
Illustration of the possible H-bonds. Possible hydrogen bonds in amide, ester and N-methyl amide functions are illustrated (red arrows). As opposed to the amide, the ester and the N-methyl amide are unable to interact as a hydrogen-bond donor, although they have the potential to accept a hydrogen bond.
Results and Discussion
Leu-enkephalin is considered an endogenous agonist for DOPr. However, like most peptides, Leu-enkephalin has a poor pharmacokinetic profile (i.e., short half-life and high hydrophilicity), which significantly hampers its usefulness as a drugable compound. A significant amount of work has been conducted to better understand the role of each amino acid in the biological activity of enkephalins. Because of their strong hydrophilic nature and their sensitivity to protease degradation, targeting amide bonds represents another strategy to improve the pharmacokinetic profile of Leu-enkephalin and to better understand the implications of these bonds in intra- and/or intermolecular interactions. As we have previously shown, the systematic and sequential replacement of amide bonds in Leu-enkephalin with isosteric functions can help in understanding their contributions to the biological activity of Leu-enkephalin at DOPr.15 In a continuous effort to assign the biological role of individual amide bonds in Leu-enkephalin and to increase the stability and the lipophilicity of this peptide, we have sequentially replaced all amide bonds first by an ester and then by an N-methyl amide function. These functions were selected because they are known to be isosteric to the amide bond and because they can participate in hydrogen bonds but only as hydrogen-bond acceptors,14 therefore offering a strategy to evaluate the role of an amide as a hydrogen-bond donnor.
Chemistry
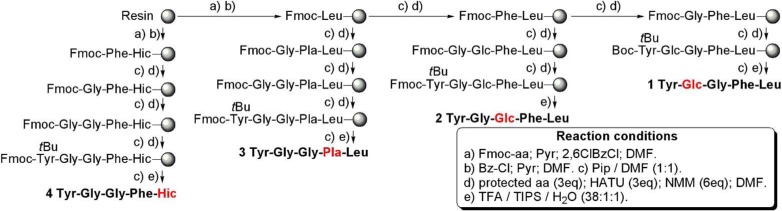
We have synthesized and thoroughly tested 8 Leu-enkephalin analogues (Figure 2). Four of these analogues (1–4) contain an ester bond in place of an amide bond. The abbreviations used to describe these analogues are based on the names of the corresponding hydroxyl acid replacing the amino acid: Glc, glycolic acid (glycine); Pla, phenyl lactic acid (phenylalanine); Hic, hydroxy isocaproic acid (leucine). The replacement of an amide bond with an ester (depsipeptides) has occasionally led to more stable compounds.36,37 Furthermore, few natural cyclic depsipeptides38,39 have anticancer (e.g., Rhomidepsin/FK-22840), antibacterial, anti-inflammatory, or antiviral (e.g., Valinomycin41) properties. Although less common, linear depsipeptides, such as Dolastatin 15, an anticancer drug,42 are also biologically active.
Figure 2.

Chemical structures of compounds 1–8.
The other four analogues (5–8) contain an N-methyl amide function in place of an amide, noted with −NMe– between the two amino acids or Sar, sarcosine (N-methyl glycine).43 The substitution of the normal secondary amides of Leu-enkephalin by N-methyl amide functions may reveal a number of positive effects. These more substituted amides can adopt cis conformations,32,44 which are extremely rare in secondary peptide bonds (Figure 1). For example, all known crystal structures of Leu-enkephalin have all of their amides in a trans conformation.45−50 Thus, these modified peptides could provide a way to explore additional modes of binding to DOPr. Among other benefits, increased selectivity toward receptor subtypes,51 enhanced potency,52 and increased permeability are also observed, the latter being a partial consequence of NH masking in the amide linkage. Importantly, N-methylated peptides have a tendency to cross the blood-brain barrier more easily than their nonmethylated parents.53 In the opioid field, the replacement of an amide with an N-methyl amide has already proved to be a valuable strategy to modify and improve peptide stability.54 The N-methyl amide bond-containing peptidomimetics DAMME (FK 33-824) and TAPS both have better resistance to peptidases when compared to endogenous opioid peptides.55,56 Among the opioid peptides containing an N-methyl amide, DAMGO, a highly selective MOPr agonist, is by far one of the most widely used pharmacological tools.
Synthesis
For analogues 1–4, a protected depsipeptide was first synthesized and subsequently used in solid-phase peptide synthesis (SPPS). The NMe series of compounds 5–7 were synthesized from commercial substrates in SPPS. Compound 8 was synthesized in solution.
Starting from the amino acid, the corresponding alcool of phenylalanine and leucine, 9 and 10, were synthesized.57 The carboxylic acid was then protected in a benzyl ester,58 producing 11 and 12 in overall yields ranging from 39 to 66% (Scheme 1).
Scheme 1. Synthesis of Benzyl Esters 11 and 12.

For all four protected depsipeptides, the ester bond was created between a Boc protected amino acid 13–15 and an alcohol 11, 12, and 16, in yields ranging from 91% to quantitative.59 Hydrogenolysis of benzyl esters 17, 19, and 20 produced the related carboxylic acids 21–23 in yields ranging from 87 to 99% (Scheme 2).59 Without further modifications, protected depsipeptide 21 was used in the SPPS of analogue 1.
Scheme 2. Synthesis of Depsipeptides 18 and 21–23.

Cleavage of the Boc protecting group of 22 and 23 with TFA and subsequent introduction of an Fmoc protecting group produced 24 and 25 in yields ranging from 75% to quantitative (Scheme 3).15
Scheme 3. Synthesis of Depsipeptides 24 and 25.

The Boc protecting group of compound 18 was cleaved with TFA, and the amine obtained was coupled to a tyrosine Pfp ester60 to generate 26. Hydrogenolysis of the benzyl ester produced 27 in an overall yield of 77% (Scheme 4).
Scheme 4. Synthesis of Depsipeptide 27.

With all of the protected depsipeptides (21, 24, 25, and 27) in hand, the SPPS of the desired enkephalin analogues was conducted. Fmoc-Leu and 25 were coupled to the resin. The standard Fmoc methodology procedures were used with Fmoc-Phe, Fmoc-Gly, Fmoc-Tyr(tBu), 21, 24, and 27 to yield Leu-enkephalin analogues 1–7 (Scheme 5). After SPPS, all peptides were purified on preparative reverse phase-HPLC (RP-HPLC), and all fractions over 95% in purity were combined.
Scheme 5. Solid Phase Synthesis of Enkephalin Analogues 1–7.
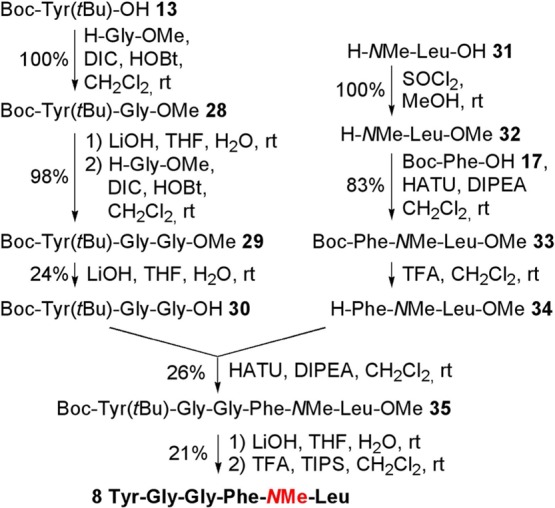
To avoid potential formation of diketopiperazine61 during the Fmoc deprotection of Phe, the analogue 8 was synthesized in solution. Starting from 13, the protected tripeptide 30(15) was obtained using two Gly-OMe couplings62 and two subsequent hydrolyses of the resulting methyl esters (28 and 29). Esterification of commercial amino acid 31 produced 32, which was coupled to Boc-Phe 15 using HATU to yield 33. Cleavage of the Boc protection of 33 gave dipeptide 34, which was then coupled with 30. Hydrolysis of the methyl ester and consecutive tBu cleavage with TFA produced the enkephalin analog 8 (Scheme 6). The crude peptide was purified on preparative RP-HPLC, and all fractions over 95% in purity were combined. HPLC purities of depsipeptides 1–8 are similar under two set of solvent systems, indicating that no epimerization occurred during synthesis of the Leu-enkephalin analogues (see the Supporting Information).
Scheme 6. Synthesis of Analogue 8.
Binding Properties and Activity at DOPr
In order to evaluate the ability of each compound to bind DOPr, we performed competitive binding assays using GH3/DOPr cell membrane preparations. Leu-enkephalin and compounds 1–8 inhibited the binding of 1 nM of [3H]-deltorphin II on DOPr in a concentration-dependent manner (Supporting Information Figure S1). Although Leu-enkephalin was found to have a better affinity than compounds 1, 2, and 5–7 for DOPr, replacing the fourth amide bond either by an ester bond (Figure S1A; compound 4) or an N-methyl amide bond (Figure S1B; compound 8) gave rise to peptidomimetics with similar affinities to Leu-enkephalin (Table 1). Replacing the third amide bond of Leu-enkephalin by an ester also retained most of the affinity for DOPr (Table 1 and Figure S1A; compound 3).
Table 1. Affinities of Leu-Enkephalin and Compounds 1–8 on DOPra.
The binding affinity (Ki) of each compound was determined by its ability to inhibit the binding of [3H]-deltorphin II (competitive binding), a selective DOPr agonist, to GH3/DOPr cell membrane extracts. Ki values are the means ± SEM of three separate experiments each performed in triplicate.
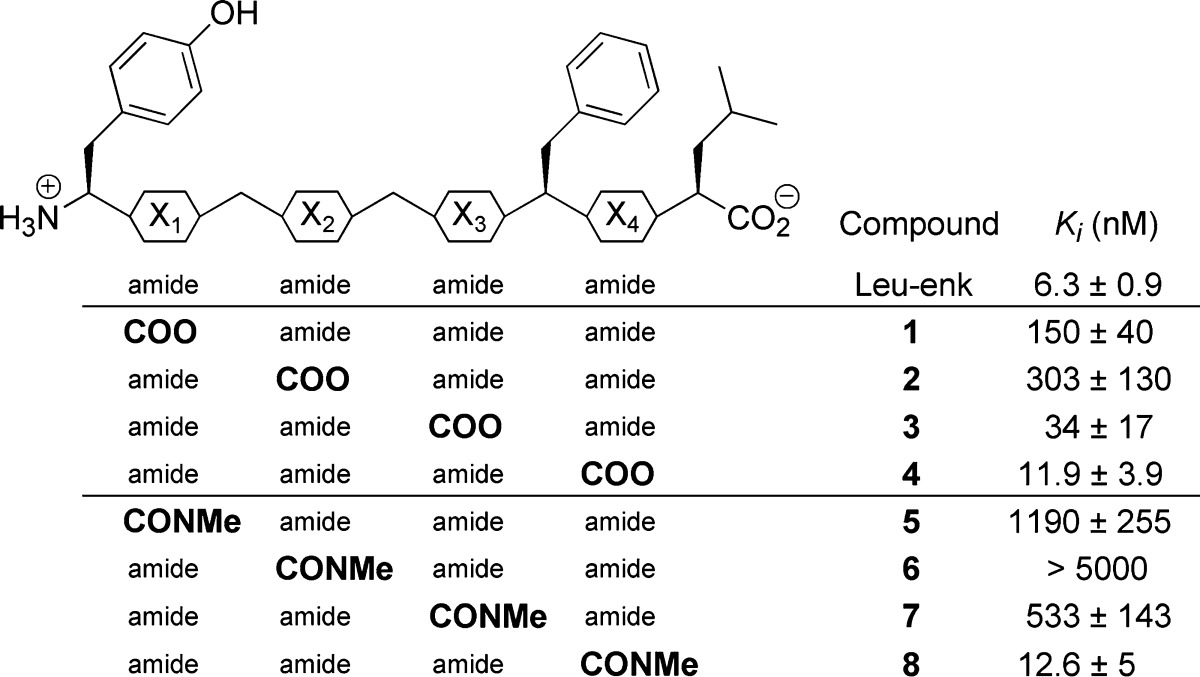
The activation of DOPr by an agonist induces rapid and transient phosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK1/2). As previously described, phosphorylation of ERK1/2 can be used as an indicator of the ability of a compound to behave as an agonist.15 Because the maximal effect of Leu-enkephalin appeared 5 min after stimulation (not shown), the effects of Leu-enkephalin (Figure 3A) and of compounds 1–8 were evaluated after 5 min of stimulation, with concentrations ranging from 10–9 to 10–5 M. We found that for three compounds (Figure 3B; compounds 1, 4, and 8) the lowest concentration that produced a significant phosphorylation of ERK1/2 was 10–7 M, which is similar to Leu-enkephalin. Compounds 3 and 7 required a concentration of 10–6 M, whereas for compound 2 the phosphorylation of ERK1/2 was only visible when the compound was used at 10–5 M. Finally, in accordance with observations made by others who showed that compounds 5 and 6 lack activity in the mouse vas deferens assay,63 a concentration of 10–5 M is not sufficient to induce a significant phosphorylation of ERK1/2.
Figure 3.
Leu-enkephalin and compounds 1–8 activate ERK1/2. Following agonist stimulation of DOPr, ERK1/2 proteins are rapidly and transiently phosphorylated (activated). (A) Western blot of ERK1/2 phosphorylation (pERK1 and pERK2) after 5 min of stimulation of DRGF11/DOPr-GFP cells with increasing concentrations (10–9–10–5 M) of Leu-enkephalin. (B) Densitometric analyses of Western blot results (pERK compared to control) obtained with increasing concentrations of Leu-enkephalin and compounds 1–8.
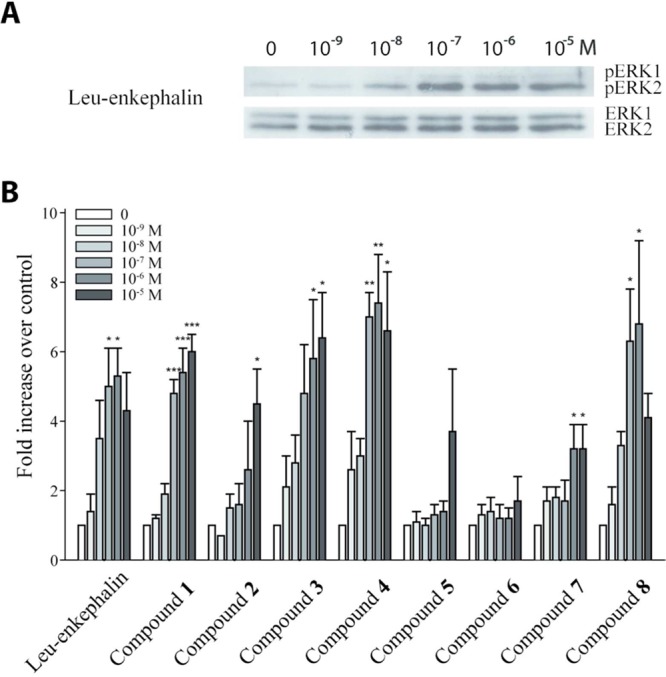
Most DOPr agonists induce a rapid internalization of the receptor. We have recently reported that the internalization of a fluorescent chimeric DOPr (DOPr-GFP) in transfected DRGF11 cells can be used to screen for active compounds.15 We therefore decided to measure the ability of compounds 4 and 8 to induce internalization of DOPr-GFP. At 1 μM, Leu-enkephalin produced a robust internalization of DOPr visible as soon as 5 min (not shown) following its application. As shown in Figure 4, the internalization is almost complete after 30 min, as revealed by the loss of fluorescence labeling at the membrane and by the accumulation of vesicle-like puncta in the cytoplasm of the cells. At 1 μM, compounds 4 and 8 also induced DOPr internalization (Figure 4). The level of internalization induced by these compounds was similar to the level observed with Leu-enkephalin at the same concentration. As indicated in Table 2, these analogues also potently inhibit electrically induced contractions of the mouse vas deferens, with compound 8 exhibiting the greatest activity. Furthermore, at the highest concentration tested (10–5 M), both analogues inhibited nerve-mediated contractions to a degree comparable to Leu-enkephalin (10–5 M), suggesting similar efficacy (not shown).
Figure 4.
Leu-enkephalin and compounds 4 and 8 induce DOPr internalization. Stimulation of DOPr with an agonist rapidly induces its internalization. Micrographs were taken before (control) and 30 min after treatment of DRGF11/DOPr-GFP cells with 1 μM of Leu-enkephalin or compound 4 or 8. All compounds induced robust internalization of DOPr. Indeed, while the membrane labeling is decreased, fluorescent vesicle-like structures are clearly visible inside the cells stimulated with the agonists.
Table 2. Potency of Compounds 4 and 8 in the Mouse Vas Deferens Assaya.
| compd | EC50 (nM) |
|---|---|
| Leu-enkephalin | 74 ± 1 |
| 4 | 140 ± 5 |
| 8 | 35 ± 14 |
Potency (EC50) of each compound was determined by evaluating their ability to inhibit the electric-field induced contractions of the mouse vas deferens. EC50 values are the means ± SEM of three separate experiments.
Hydrophobicity and Plasma Stability
Because our underlying goal is to obtain analogues of Leu-enkephalin that are able to cross the BBB, we assessed the lipophilicity of the synthesized analogues by measuring some of their physicochemical properties (Table 3). The LogD7.4 values and retention times were measured for Leu-enkephalin, compounds 4, 8, and morphine. The cLogP and the topological polar surface area (tPSA) were also calculated for these compounds. For morphine and Leu-enkephalin, we obtained LogD7.4 values of −0.160 and −0.886, respectively. These values are similar to those previously reported by others for morphine (LogD7.4 = −0.0764) and Leu-enkephalin (LogD7.07 = −0.84565). The experimental LogD7.4 and calculated cLogP values show similarities. Our experimental data show an increased LogD7.4 for each isostere, indicating that our method is a valid way to obtain analogues with increased hydrophobicity. Of all of the modifications, the N-methyl amide led to compounds possessing the highest experimental lipophilicity. The HPLC retention time of a compound can be used to estimate its lipophilic nature; however, using our HPLC method, the retention times were not always in line with the experimental LogD7.4. Compounds 4 and 8 displayed high tPSA values, whereas CNS acting drugs usually exhibit smaller values.66,67 Nevertheless, there are some linear dermorphin analogues with tPSAs of ∼160 Å2 that produced antinociceptive effects after systemic administration.68
Table 3. Measured and Calculated Physicochemical Properties.
| compd | tR (min)a | LogD 7.4b | cLogPc | tPSA (Å2)c |
|---|---|---|---|---|
| Leu-enkephalin | 8.44 | –0.886 ± 0.030 | –0.851 | 199.95 |
| 4 | 9.93 | –0.400 ± 0.019 | –0.52 | 197.15 |
| 8 | 9.23 | –0.363 ± 0.004 | –0.230 | 191.16 |
| morphine | 1.22 | –0.160 ± 0.005 | 0.571 | 52.93 |
Retention time: RP-HPLC was performed using an Agilent Eclipse Plus C-18 column, 50 mm × 3.0 mm, 1.8 μm. Solvent A, 0.1% TFA in water; solvent B, 0.1% TFA in acetonitrile; 2–98% B in A over 20 min; flow rate, 0.4 mL/min.
Partition coefficient measured using the shake-flask method with PBS pH = 7.4.
Calculated using ChemBioDraw 12.0; tPSA, topological polar surface area.
As previously stated, replacing an amide bond with an ester or an N-methyl amide function has the potential to decrease peptide sensitivity toward protease-mediated degradation. We therefore assessed the stability of Leu-enkephalin and active compounds 4 and 8 in rat plasma diluted in an equal volume of saline. We used diluted plasma to decrease the rate of degradation of Leu-enkephalin. As expected, Leu-enkephalin was rapidly degraded (Figure 5). The LC-MS analysis revealed that the major degradation products of Leu-enkephalin, that is, Gly-Gly-Phe-Leu and Phe-Leu, appeared after 5 and 10 min of incubation in rat plasma, respectively (Table 4). These fragments are also generated in vivo following cleavage of the first and third amide bonds by aminopeptidase N and enkephalinase.13 Replacing the fourth amide bond with an N-methyl amide function (compound 8) significantly increased the half-life of this peptidomimetic, possibly by increasing the stability of the third amide bond (Figure 5 and Table 4). By contrast, introducing an ester in lieu of the fourth amide bond (compound 4) had no significant effect on the half-life of this analog in diluted rat plasma when compared to Leu-enkephalin (Figure 5). In fact, the ester replacement might even increase the sensitivity to proteolysis, because the Tyr-Gly-Gly-Phe fragment was also present in the LC-MS analysis soon after incubation in plasma (Table 4), suggesting that the fourth amide bond of Leu-enkephalin is less sensitive to degradation than its ester analog.
Figure 5.

Analogue containing N-methyl replacing an amide bond is more stable than Leu-enkephalin. Leu-enkephalin, compounds 4 and 8 were incubated in diluted rat plasma for 0–60 min. The amount of intact peptide/analogue was then determined by HPLC and expressed as the percentage of control. Experiments were performed in triplicate. Error bars represent the standard error of the mean (SEM).
Table 4. Plasma Stability of Enkephalin and Its Analogues.
| compd | half-life (min)a | 95% CI | degradation products (time for first appearance)b |
|---|---|---|---|
| Leu-enkephalin | 4.6 | [4.3–5.1] | Gly-Gly-Phe-Leu (5 min) |
| Phe-Leu (10 min) | |||
| 4 | 3.3 | [3.0–3.9] | Gly-Gly-Phe-OLeu (5 min) |
| Phe-OLeu (5 min) | |||
| Tyr-Gly-Gly-Phe (5 min) | |||
| Gly-Gly-Phe (20 min) | |||
| 8 | 10.7 | [8.7–12.7] | Gly-Gly-Phe-NMeLeu (5 min) |
| Phe-NMeLeu (20 min) |
Half-life: The stability of each compound was determined using an Agilent 1100 series Symmetry C-18 HPLC column, 150 mm × 4.6 mm, 5 μm, heated at 30 °C, flow, 1.2 mL/min, start with 0.1% TFA in water then 0 to 75% acetonitrile in 20 min, UV detection at 223 nm.
Degradation products: Degradation products in diluted rat plasma were identified by LC-MS analysis.
Conclusion
In the current study, we demonstrated that the fourth amide of Leu-enkephalin can be replaced by either an ester or an N-methyl amide bond, two functions with hydrogen-bond acceptor but no hydrogen-bond donor properties, without significantly affecting the biological activity of the analogues when compared to Leu-enkephalin. Taken together with our previous observations showing that the replacement of the fourth amide bond by an E-alkene impaired the binding and the biological activity of the analogue,15 these findings reveal that the hydrogen-bond acceptor properties of the fourth amide of Leu-enkephalin are important for its biological activity on DOPr. As expected, we also observed that the replacement of a single amide bond by an ester or an N-methyl amide function improves the lipophilicity of the analogues as compared to Leu-enkephalin. Finally, compound 8 has an improved stability in rat plasma. In conclusion, the replacement of the fourth amide bond of Leu-enkephalin by an N-methyl amide function appears as a promising strategy to increase the lipophilicity and the stability of enkephalin analogues. Our results also show that the systematic replacement of amide bonds by isosteric functions represents an efficient way to design and synthesize novel peptide analogues useful in studying the biological roles of amide bonds, a strategy that could be helpful for the design and the synthesis of novel Leu-enkephalin analogues.
Methods
Chemistry
For SPPS, commercial grade reagents were used without further purification. When necessary, all solvents were purified and dried prior to use. Optical rotation measurements were made on a Perkin-Elmer 241 polarimeter and are quoted in units of 10–1 deg cm2 g–1. Infrared spectra were recorded on a Perkin-Elmer Spectrum 1600 FTIR instrument. 1H and 13C NMR spectra were recorded in deuterated solvents on a Bruker AC 300 NMR instrument. The 1H and 13C NMR chemical shifts are reported in ppm (parts per million). The residual solvent peaks have been used as an internal reference. All coupling constants (J) are in Hertz. The abbreviations for the peak multiplicities are as follows: s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), qt (quintet), m (multiplet), and br (broad). Mass spectra were recorded on a VG Micromass ZAB-2F, on a MALDI-Tof, or on a ESI-Q-Tof (Maxis) instrument. HPLC preparative purification was done on a VYDAC 218TP C18 column.
L-(−)-3-Phenyllactic Acid (H-Pla-OH) (9)
l-Phenylalanine (15.0 g, 90.9 mmol) was dissolved in sulfuric acid (200 mL, 2 M), and sodium bromide (22.7 g, 220 mmol) was added. The mixture was stirred at −10 °C, and sodium nitrite (15.2 g, 220 mmol) was then added slowly. The reaction was allowed to reach rt and was stirred for 3 h. The crude mixture was extracted with ethyl ether (3 × 75 mL). The organic phases were pooled, concentrated under vacuum, and purified by flash chromatography on silica gel using EtOAc and hexanes (3:7). The bromide (17.0 g, 74.0 mmol) obtained was immediately dissolved in water (100 mL), and sodium carbonate (8.65 g, 82.0 mmol) was added slowly. The reaction was refluxed for 4 h. The resulting mixture was washed with ethyl ether. The aqueous phase was acidified using HCl 1 N until pH = 2 and was extracted with ethyl ether (3 × 50 mL). The combined organic phases were dried (MgSO4). The crude product was crystallized using ethyl ether and hexanes. A white crystalline solid was obtained (6.23 g, 41%). 1H NMR (300 MHz, CDCl3) δ (ppm) 7.36–7.24 (m, 5H), 4.53 (dd, 1H, J = 4.5 et 7.0 Hz), 3.21 (dd, 1H, J = 4.5 et 14.0 Hz), 3.00 (dd, 1H, J = 7.0 et 14.0 Hz). 13C NMR (75 MHz, CDCl3) δ (ppm) 177.2, 138.5, 129.5, 128.6, 127.2, 70.9, 40.2. IR (NaCl) ν (cm–1) 3450–2395, 3368, 2927, 1731, 1090. MS (m/e, rel intensity) 184 (MH4+, 23), 149 (9), 108 (26), 91 (100). Exact mass: calculated for C9H14N1O3, 184.0974; found, 184.0974. [α]D20 −33.8 (c = 0.98, CHCl3):69 [α]D21 −26.9 (c = 1.00, acetone).
L-(−)-2-Hydroxyisocaproic Acid (H-Hica-OH) (10)
l-Leucine (18.1 g, 138 mmol) was dissolved in a sulfuric acid solution (1 N, 324 mL, 324 mmol), and the temperature was lowered to −5 °C. Sodium nitrite (70.7 g, 1.02 mol) was dissolved in water (300 mL) and was added dropwise to the mixture. The reaction was stirred at rt for 65 h. Brine (300 mL) was added, and the crude mixture was extracted with ethyl acetate (3 × 250 mL). The organic phases were combined and dried (MgSO4). The title compound was obtained as a white solid (14.6 g, 80%). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.70–6.10 (br, 2H), 4.28 (t, 1H, J = 6.5 Hz), 2.00–1.70 (m, 1H), 1.62 (t, 2H, J = 7.0 Hz), 0.96 (d, 6H, J = 6.5 Hz). 13C NMR (75 MHz, CDCl3) δ (ppm): 180.4, 68.9, 43.1, 24.4, 23.2, 21.4. IR (NaCl) ν (cm–1): 3421, 2953, 2623, 1711, 1267. MS (m/e, rel intensity): 150 (MNH4+, 100), 99 (6), 85 (10). Exact mass: calculated for C6H16NO3, 150.1130; found, 150.1127. [α]D20 +6.0 (c = 0.15, CHCl3):70 [α]D20 +5.5 (c = 1.00, CHCl3).
H-Pla-OBn (General Protocol for Benzyl Protection) (11)
Phenyllactic acid (5.90 g, 35.5 mmol), benzyl bromide (4.64 mL, 39.1 mmol), and triethylamine (4.31 g, 42.6 mmol) were dissolved in acetone (50 mL). The reaction was refluxed for 16 h. The crude mixture was filtered and concentrated under vacuum. The crude solid was dissolved in ethyl acetate (100 mL) and washed with water (3 × 50 mL). The aqueous phases were combined and were extracted with ethyl acetate (3 × 50 mL). All the organic phases were combined and dried (MgSO4). The crude mixture was purified by flash chromatography on silica gel using EtOAc and hexanes (3:17). The title compound was obtained as colorless oil (8.60 g, 95%). 1H NMR (300 MHz, CDCl3) δ (ppm) 7.39–7.13 (m, 10H), 5.18 (s, 2H), 4.50 (dd, 1H, J = 4.5 et 6.5 Hz), 3.12 (dd, 1H, J = 4.5 et 14.0 Hz), 2.98 (dd, 1H, J = 6.5 et 14.0 Hz), 2.71 (d, 1H, J = 6.5 Hz). 13C NMR (75 MHz, CDCl3) δ (ppm) 174.1, 136.2, 135.1, 129.6, 129.5, 128.7, 128.7, 128.4, 126.9, 71.3, 67.4, 40.5. IR (NaCl) ν (cm–1) 3486, 3027, 2950, 1740, 1454. MS (m/e, rel intensity) 274 (MH4+, 100), 228 (8), 108 (17). Exact mass: calculated for C16H20N1O3, 274.1443; found, 274.1447. [α]D20 −45.0 (c = 3.55, CHCl3).
Boc-Gly-Glc-OBn (General Protocol for Esterification) Glc: Glycolic Acid (18)
Boc-Gly-OH (4.60 g, 26.2 mmol), benzyl glycolate (3.69 g, 22.2 mmol), and DMAP (271 mg, 2.20 mmol) were dissolved in DCM (75 mL). DIC (4.12 mL, 26.2 mmol) was added dropwise at 0 °C. The reaction was allowed to warm up to rt and was stirred for 16 h. The reaction was filtered on Celite and adsorbed on silica gel. The mixture was purified by flash chromatography on silica gel using EtOAc and hexanes (3:7). The title compound was obtained as a white solid (6.58 g, 91%). 1H NMR (300 MHz, CDCl3) δ (ppm) 7.38–7.34 (m, 5H), 5.19 (s, 2H), 5.01 (br, 1H), 4.72 (s, 2H), 4.04 (d, 2H, J = 5.5 Hz), 1.45 (s, 9H). 13C NMR (75 MHz, CDCl3) δ (ppm) 170.0, 167.3, 155.7, 134.9, 128.6, 128.5, 128.3, 79.9, 67.2, 61.0, 42.1, 28.3. IR (NaCl) ν (cm–1) 3380, 2965, 1701, 1219. MS (m/e, rel intensity) 341 (MH4+, 22), 285 (100), 223 (72), 91 (15). Exact mass: calculated for C16H25N2O6, 341.1712; found, 341.1718.
Fmoc-Gly-Pla-OH (General Protocol for Fmoc Protection) (24)
Boc-Gly-Pla-OH (1.52 g, 3.80 mmol) was dissolved in DCM (8 mL) and TFA (2 mL). The solution was stirred for 1 h at rt. The resulting mixture was concentrated under vacuum and dried under vacuum using toluene (3 × 50 mL). The crude salt obtained was dissolved in water (8 mL), and sodium carbonate (1.30 g, 15.2 mmol) was added. A solution of Fmoc-Cl (1.03 g, 3.99 mmol) in THF (5 mL) was added slowly to the aqueous mixture at 0 °C. The reaction was allowed to warm at rt and stirred for 16 h. The THF in the mixture was evaporated under vacuum. The resulting mixture was diluted in water (150 mL), acidified with 1 N HCl to reach pH 2, and extracted using ethyl acetate (3 × 60 mL). The organic extract were combined, dried (MgSO4). The crude compound was purified by flash chromatography on silica gel eluting with ethyl acetate, hexanes, and acetic acid (40:59:1) to yield the title compound as a white solid (2.00 g, 100%). 1H NMR (300 MHz, CDCl3) δ (ppm) 7.75 (d, 2H, J = 7.5 Hz), 7.57 (d, 2H, J = 7.5 Hz), 7.39 (d, 2, J = 7.5 Hz), 7.32–7.19 (m, 7H), 5.33 (dd, 1H, J = 4.0 et 8.5 Hz), 5.24 (t, 1H, J = 5.0 Hz), 4.38 (t, 2H, J = 7.0 Hz), 4.20 (t, 1H, J = 7.0 Hz), 4.03 (d, 1H, J = 5.5 Hz), 3.96 (br, 1H), 3.26 (dd, 1H, J = 4.0 et 14.0 Hz), 3.13 (dd, 1H, J = 8.5 et 14.0 Hz). 13C NMR (75 MHz, CDCl3) δ (ppm) 177.7, 173.1, 169.8, 156.8, 143.8, 141.3, 135.6, 129.4, 128.7, 127.8, 127.3, 127.3, 125.2, 120.1, 73.5, 67.5, 47.1, 42.6, 37.1. IR (NaCl) ν (cm–1) 3577–2272, 3404, 3022, 2950, 1727, 1177. MS (m/e, rel intensity) 463 (MH4+, 38), 315 (28), 224 (93), 179 (100). Exact mass: calculated for C26H27N2O6, 463.1869; found, 463.1879. [α]D20 −6.99 (c = 2.75, CHCl3).
Boc-Tyr(tBu)-Gly-Glc-OBn (26)
Boc-Gly-Glc-OBn (500 mg, 1.32 mmol) was dissolved in DCM (7 mL) and TFA (3 mL). The reaction was stirred for 1 h at rt. The resulting mixture was concentrated under vacuum and dried under vacuum using toluene (3 × 20 mL). The crude product was dissolved in water (5 mL) and THF (10 mL). Potassium carbonate (365 mg, 2.64 mmol) was added, and the mixture was stirred at rt for 5 min. Boc-Tyr(tBu)-OPfp (664 mg, 1.32 mmol) was dissolved in THF (20 mL) and added to the reaction. The mixture was stirred for 2 h at rt. The crude mixture was diluted with ethyl acetate (125 mL) and then washed with saturated aq NaHCO3 (75 mL) and water (75 mL). The organic phase was dried (MgSO4). The crude mixture was purified by flash chromatography on silica gel eluting with EtOAc and hexanes (from 1:3 to 2:3). The title compound was obtained as a white solid (640 mg, 81%). 1H NMR (300 MHz, CDCl3) δ (ppm) 7.38–7.33 (m, 5H), 7.09 (d, 2H, J = 8.5 Hz), 6.91 (d, 2H, J = 8.5 Hz), 6.36 (br, 1H), 5.19 (s, 2H), 4.89 (br, 1H), 4.69 (s, 2H), 4.29 (br, 1H), 3.17 (qd, 2H, J = 19.0 et 5.0 Hz), 3.07–2.98 (m, 2H), 1.40 (s, 9H), 1.32 (s, 9H). 13C NMR (75 MHz, CDCl3) δ (ppm) 171.8, 169.0, 167.1, 154.2, 134.9, 131.4, 129.7, 128.6, 128.6, 128.4, 124.3, 80.2, 67.2, 55.6, 40.9, 37.7, 28.8, 28.2. IR (NaCl) ν (cm–1) 3340, 2989, 1752, 1690, 1160. MS (m/e, rel intensity) 543 (MH+, 23), 502 (19), 443 (100), 108 (18). Exact mass: calculated for C29H39N2O8, 543.02706; found, 543.02700. [α]D20 +2.1 (c = 1.69, CHCl3).
Boc-Tyr(tBu)-Gly-Glc-OH (General Protocol for Benzyl Ester Hydrogenolysis) (27)
Palladium 10% on activated charcoal (19 mg, 0.0180 mmol) was placed in a round-bottom flask, and ethyl acetate was added slowly under an argon atmosphere. Hydrogen was bubbled in the solution for 10 min. Boc-Tyr(tBu)-Gly-Glc-OBn (100 mg, 0.18 mmol) was dissolved in ethyl acetate (5 mL) and then added to the palladium suspension. The reaction was stirred under a hydrogen atmosphere for 1.5 h at rt. The reaction was filtered on Celite and concentrated under vacuum. The title compound was obtained as a white solid (90.5 mg, 95%). 1H NMR (300 MHz, CDCl3) δ (ppm) 7.08 (d, 2H, J = 8.5 Hz), 6.90 (d, 2H, J = 8.5 Hz), 6.76 (br, 1H), 5.16 (s, 2H), 4.67 (s, 2H), 4.52 (br, 1H), 4.17–4.06 (m, 2H), 3.01–2.87 (m, 2H), 1.36 (s, 9H), 1.32 (s, 9H). 13C NMR (75 MHz, CDCl3) δ (ppm) 172.1, 169.9, 169.0, 156.0, 154.1, 131.3, 129.7, 124.3, 80.9, 60.9, 40.9, 37.9, 28.8, 28.2. IR (NaCl) δ (cm–1) 3914–2731, 3402, 2977, 1661, 1507. MS (m/e, rel intensity) 453 (MH+, 8), 409 (12), 294 (63), 277 (100). Exact mass: calculated for C22H33N2O8, 453.2237; found, 453.2250. [α]D20 +2.9 (c = 0.80, CHCl3).
Boc-Tyr(tBu)-Gly-OMe (General Protocol for Glycine Coupling) (28)
H-Gly-OMe (1.11 g, 8.85 mmol) was dissolved in 1 M NaHCO3 (50 mL) and extracted with DCM (3 × 50 mL). The combined organic phases were dried (MgSO4) and concentrated under vacuum to a volume of 10 mL. Boc-Tyr(tBu)-OH (2.00 g, 5.90 mmol) and HOBt (160 mg, 1.18 mmol) were dissolved DCM (60 mL) and added to the solution. DIC (1.10 mL, 7.08 mmol) was added dropwise, and the reaction was stirred at rt for 16 h. The crude mixture was concentrated under vacuum and dissolved in ethyl ether (150 mL). The organic phase was washed with aqueous saturated NaHCO3 (2 × 100 mL), 0.5 N HCl (2 × 100 mL), and water (100 mL) and was dried (MgSO4). The crude compound was purified by flash chromatography on silica gel eluting with ethyl acetate and hexanes (1:1) to yield the title compound as a white solid (2.92 g, 100%). 1H NMR (300 MHz, CDCl3) δ (ppm) 7.13 (d, 2H, J = 8.5 Hz), 6.95 (d, 2H, J = 8.5 Hz), 5.06 (br, 1H), 4.39 (br, 1H), 4.75 (dd, 1H, J = 5.5, 18.5 Hz), 4.95 (dd, 1H, J = 5.0, 18.5 Hz), 3.76 (s, 3H), 3.15–2.90 (m, 2H), 1.44 (s, 9H), 1.36 (s, 9H). 13C NMR (75 MHz, CDCl3) δ (ppm) 172.7, 171.9, 155.2, 136.2, 129.6, 128.5, 128.4, 126.8, 79.7, 54.5, 52.1, 51.6, 38.9, 37.1, 30.9, 30.3, 28.2, 24.6, 23.76, 21.5. IR (NaCl) ν (cm–1) 3323, 2982, 1759, 1662, 1509, 1169. MS (m/e, rel intensity) 408 (M+, 2), 235 (100), 190 (48). Exact mass: calculated for C21H32N2O6, 408.2260; found, 408.2252. [α]20D +2.83 (c = 2.40, CHCl3).
Boc-Tyr(tBu)-Gly-OH (General Methyl Ester Hydrolysis Protocol)
Boc-Tyr(tBu)-Gly-OMe (2.32 g, 5.90 mmol) was dissolved in THF (90 mL). LiOH (990 mg, 23.6 mmol) was dissolved in water (10 mL) and was added to the reaction which was stirred for 16 h at rt. The resulting mixture was concentrated under vacuum, diluted in H2O (100 mL), and washed with ethyl ether (3 × 20 mL). The aqueous phase was acidified with 1 N HCl to reach pH 4 and extracted with ethyl acetate (3 × 50 mL). The organic extract were combined, dried (MgSO4), and concentrated under vacuum to yield the title compound as a white solid (2.20 g, 98%). 1H NMR (300 MHz, CDCl3) δ (ppm) 7.13 (d, 2H, J = 8.5 Hz), 6.95 (d, 2H, J = 8.5 Hz), 5.34 (br, 1H), 4.59 (br, 1H), 4.18–4.05 (m, 1H), 3.91 (dd, 1H, J = 4.0, 18.5 Hz), 3.02 (br, 2H), 1.40 (s, 9H), 1.35 (s, 9H). 13C NMR (75 MHz, CDCl3) δ (ppm) 172.0, 155.9, 154.0, 131.4, 129.7, 124.4, 80.7, 78.6, 55.5, 41.3, 38.2, 28.8, 28.2. IR (NaCl) ν (cm–1) 3375–2592, 3285, 2928, 2928, 1672, 1642, 1525, 1215, 1159. MS (m/e, rel intensity) 394 (M+, 1), 221 (100), 176 (95). Exact mass: calculated for C20H30N2O6, 394.2112; found, 394.2104. [α]20D +2.27 (c = 1.26, CHCl3).
General Method for All Peptide Synthesis on Solid Support (Fmoc Methodology)15
The resins (Wang resin for the preparation of peptide 1 and Tenta Gel PHB resin for peptides 2–7) were washed using DMF (3×), iPrOH (3×), and DCM (3×), unless stated otherwise. These washing were made after every loading, benzyl protection, coupling, and Fmoc deprotection. Loading of the resin:71 The resin was placed in a sintered glass peptide synthesis vessel. The 2 equiv of the first amino acid, 2 equiv of 2,6-dichlorobenzoylchloride, and 4 equiv of pyridine were added to the resin. The suspension was agitated in a shaker for 16 h. The resin was then washed. All loadings were quantified by UV quantitation of Fmoc release: An aliquot (10 mg) of resin was dried under vacuum; a piperidine (1 mL) and DMF (1 mL) solution was added, and the suspension was agitated in a shaker for 30 min; a portion of the solution (0.5 mL) was diluted in DCM (4.5 mL) and was read with a UV spectrometer. Loading of the resin (mmol/g) = (absorbance 301 nm × 103 × 20)/(7800 × weight of the aliquot). The loadings obtained for each resin are mentioned. After the initial loading,72 the remaining free sites were protected using equal amounts of benzoyl chloride and pyridine (0.3 mL/g of resin) in DMF; the solution was agitated in a shaker for 3 h. For all Fmoc deprotections, the resin was treated with 50% piperidine in DMF and the suspension was agitated in a shaker for 20 min. All three couplings were performed using 3 equiv of protected amino acid and HBTU, with 6 equiv of NMM in a minimum volume of DMF, and the suspension was agitated in a shaker. All coupling procedures were stopped after 16 h or after the Kaiser’s test result was negative. The first coupling was done with Fmoc-Leu-OH, the second with Fmoc-Phe-OH, the third and fourth with Fmoc-Gly-OH, and the fifth with Fmoc-Tyr(tBu)-OH. The amount of modified amino acid (replacing the previously stated amino acids) used in coupling for each peptide is mentioned. All the final peptides were cleaved from their resin in a glass vial, and the suspension was stirred for 1 h 30 min with a magnetic stirrer. All cleavage solutions were done with 95% TFA, 2.5% H2O, and 2.5% TIPS. For each gram of resin, 3 mL of cleavage solution was used. After cleavage, the mixtures were filtered on cotton and dropped in a large amount of water (20 mL). The remaining solvents were concentrated under vacuum, and the aqueous solution was frozen and lyophilized. Purification and purity requirements: All crude peptides were purified using preparative reverse-phase HPLC, detecting at 280 nm, with a VYDAC 218TP C18 column and using ACN gradient in a 0.1% TFA aq solution (from 1:9 to 2:3), with a flow rate of 5 mL/min over 1 h. The purity of all fractions was analyzed using an Agilent 1100 series analytical HPLC, detecting at 214 nm, with a Phenomenex 5 μm 4.6.0 × 100 mm C18 column using method A, ACN gradient in a 0.1% TFA aq solution (from 0:1 to 1:0) over 60 min, with a flow rate of 1 mL/min; method B, ACN gradient in a 20 mM triethylammonium phosphate pH = 2.5 aq solution (from 0:1 to 4:6) over 60 min, with a flow rate of 1 mL/min. The fractions with purities of 95% or higher were combined, frozen, and lyophilized. Following these methods, compounds 1–7 were prepared.
H-Tyr-Gly-Glc-Phe-Leu-OH (1)
An amount of 320 mg of resin with a loading of 0.5 mmol/g was used. Boc-Tyr-Gly-Glc-OH (676 mg, 1.28 mmol) was used in the third coupling. The title peptide was obtained as a white solid (58.4 mg, 66%). 1H NMR (300 MHz, CD3OD) δ (ppm) 7.26–7.16 (m, 5H), 7.09 (d, 2H, J = 8.5 Hz), 6.76 (d, 2H, J = 8.5 Hz), 4.72 (dd, 1H, J = 5.0 et 9.5 Hz), 4.63 (d, 1H, J = 15.0 Hz), 4.42 (br, 2H), 3.20–3.11 (m, 2H), 2.98–2.90 (m, 2H), 1.65–1.59 (m, 3H), 0.90 (dd, 6H, J = 6.0 et 14.0 Hz). 13C NMR (75 MHz, CD3OD) δ (ppm) 174.3, 172.1, 169.5, 168.3, 167.8, 156.9, 136.7, 130.2, 129.0, 127.9, 126.4, 124.4, 115.4, 62.0, 54.5, 53.9, 50.8, 40.9, 40.1, 37.5, 36.4, 24.5, 21.9, 20.3. IR (NaCl) ν (cm–1) 3748–2741, 3423, 2965, 1653, 1457. MALDI-TOF (m/e, rel intensity) 556.7 (MH+, 100). [α]D20 +14.9 (c = 0.94, CH3OH).
H-Tyr-Glc-Gly-Phe-Leu-OH (2)
An amount of 750 mg of Tenta Gel S PHB resin with a loading of 0.250 mmol/g was used. Boc-Tyr-Glc-OH (296 mg, 0.750 mmol) was used in the fourth coupling. The title peptide was obtained as a white solid (46.3 mg, 44%). 1H NMR (300 MHz, CD3OD) δ (ppm) 8.40 (d, 1H, J = 8.0 Hz), 7.97 (d, 1H, J = 8.0 Hz), 7.24–7.14 (m, 5H), 7.08 (d, 2H, J = 8.5 Hz), 6.76 (d, 2H, J = 8.5 Hz), 4.76–4.64 (m, 2H), 4.41–4.31 (m, 2H), 3.97–3.67 (m, 3H), 3.10–3.02 (m, 2H), 2.90–2.82 (m, 1H), 1.67–1.59 (m, 3H), 0.91 (dd, 6H, J = 6.0 et 12.5 Hz). 13C NMR (75 MHz, CD3OD) δ (ppm) 174.3, 172.1, 169.3, 168.3, 167.8, 157.0, 136.8, 130.2, 128.9, 128.0, 126.3, 115.5, 115.4, 63.1, 61.2, 54.2, 53.9, 50.7, 41.4, 40.1, 37.5, 24.5, 24.5, 21.9, 20.4. IR (NaCl) ν (cm–1) 3600–2550, 3290, 2963, 1645, 1513. MALDI-TOF (m/e, rel intensity) 556.9 (MH+, 100). [α]D20 −1.88 (c = 0.55, CH3OH).
H-Tyr-Gly-Gly-Pla-Leu-OH (3)
An amount of 1.00 g of Tenta Gel S PHB resin with a loading of 0.250 mmol/g was used. Fmoc-Gly-Pla-OH (445 mg, 1.00 mmol) was used in the second coupling. A white solid was obtained (60.5 mg, 43%). 1H NMR (300 MHz, CD3OD) δ (ppm) 7.26–7.18 (m, 5H), 7.07 (d, 2H, J = 8.5 Hz), 6.74 (d, 2H, J = 8.5 Hz), 5.25 (dd, 1H, J = 4.0 et 8.0 Hz), 4.47–4.38 (m, 1H), 4.02–3.77 (m, 5H), 3.17–3.07 (m, 3H), 2.94 (dd, 1H, J = 8.0 et 13.0 Hz), 1.61–1.56 (m, 3H), 0.86 (dd, 6H, J = 6.0 et 11.5 Hz). 13C NMR (75 MHz, CD3OD) δ (ppm) 174.2, 170.3, 170.0, 168.9, 168.7, 156.8, 135.9, 130.1, 129.3, 128.0, 126.5, 124.6, 115.4, 74.6, 54.6, 50.3, 41.5, 40.4, 39.9, 37.2, 36.2, 24.3, 22.0, 20.3. IR (NaCl) ν (cm–1) 3695–2518, 3259, 2954, 1668, 1513. MALDI-TOF (m/e, rel intensity) 556.9 (MH+, 100). [α]D20 +9.7 (c = 0.99, CH3OH).
H-Tyr-Gly-Gly-Phe-Hica–OH (4)
An amount of 1.00 g of Tenta Gel R PHB resin with a loading of 0.100 mmol/g was used. Fmoc-Phe-Hica-OH (251 mg, 0.500 mmol) was used in the initial coupling. The title peptide was obtained as a white solid (51.0 mg, 91%). 1H NMR (300 MHz, CD3OD) δ (ppm) 7.29–7.16 (m, 5H), 7.07 (d, 2H, J = 8.5 Hz), 6.75 (d, 2H, J = 8.5 Hz), 5.03–4.97 (m, 1H), 4.74–4.69 (m, 1H), 4.02–3.73 (m, 5H), 3.11 (dd, 1H, J = 6.5 et 14.0 Hz), 3.00–2.88 (m, 2H), 1.82–1.63 (m, 3H), 0.92 (dd, 6H, J = 6.0 et 9.0 Hz). 13C NMR (75 MHz, CD3OD) δ (ppm) 171.2, 169.9, 169.1, 156.8, 140.0, 136.9, 130.1, 128.8, 128.1, 126.4, 124.6, 115.4, 71.8, 54.7, 53.6, 41.9, 41.5, 39.5, 36.6, 36.3, 24.4, 22.0, 20.4. IR (NaCl) ν (cm–1) 3713–2418, 3281, 2954, 1663, 1513. MALDI-TOF (m/e, rel intensity) 557.3 (MH+, 100). [α]D20 +12.6 (c = 1.03, CH3OH).
H-Tyr-Sar-Gly-Phe-Leu-OH (5)
An amount of 750 mg of Tenta Gel S PHB resin with a loading of 0.250 mmol/g was used. Sarcosine (233 mg, 0.750 mmol) was used in the fourth coupling. The title peptide was obtained as a white solid (47.8 mg, 45%). 1H NMR (300 MHz, D2O) δ (ppm) 7.31–7.20 (m, 5H), 7.13 (d, 2H, J = 8.5 Hz), 6.84 (d, 2H, J = 8.5 Hz), 4.71–4.62 (m, 1H), 4.61–4.51 (m, 1H), 4.31–4.21 (m, 1H), 3.91–3.82 (m, 4H), 3.09–3.07 (m, 4H), 2.83 (s, 3H), 1.56–1.54 (m, 3H), 0.84 (d, 3H, J = 6.0 Hz), 0.79 (d, 3H, J = 6.0 Hz). 13C NMR (75 MHz, D2O) δ (ppm) 176.3, 172.8, 170.7, 170.5, 155.3, 136.2, 131.1, 129.2, 128.8, 127.2, 125.1, 115.8, 54.7, 51.8, 51.6, 42.2, 41.8, 39.5, 37.2, 36.5, 35.3, 24.3, 22.2, 20.6. IR (NaCl) ν (cm–1) 3750–2850 (br), 1653, 1210, 1143. MS (m/e, rel intensity) 570 (MH+, 100), 304 (20), 282 (32), 214 (59). Exact mass: calculated for C29H39N5O7, 570.2928; found, 570.2928. [α]20D +16.4 (c = 0.97, MeOH).
H-Tyr-Gly-Sar-Phe-Leu-OH (6)
An amount of 750 mg of Tenta Gel S PHB resin with a loading of 0.250 mmol/g was used. Sarcosine (233 mg, 0.750 mmol) was used in the third coupling. The title peptide was obtained as a white solid (55.3 mg, 52%). 1H NMR (300 MHz, MeOH-d4) δ (ppm) 8.50–8.40 (m, 1H), 8.27–8.17 (m, 1H), 8.00–7.91 (m, 1H), 7.26–7.17 (m, 5H), 7.09 (d, 2H, J = 8.5 Hz), 6.75 (d, 2H, J = 8.5 Hz), 4.82–4.71 (m, 1H), 4.43–4.40 (m, 1H), 4.23–3.57 (m, 5H), 3.20–3.12 (m, 2H), 2.97–2.74 (m, 2H), 2.86 (s, 3H), 1.69–1.62 (m, 3H), 0.93 (d, 3H, J = 6.0 Hz), 0.89 (d, 3H, J = 6.0 Hz). 13C NMR (75 MHz, MeOH-d4) δ (ppm) 174.3, 172.1, 169.5, 168.9, 168.8, 156.9, 137.0, 130.1, 129.0, 128.0, 126.3, 124.6, 115.4, 54.3, 50.7, 40.1, 37.8, 37.5, 36.2, 34.5, 33.9, 24.5, 22.0, 20.4. IR (NaCl) ν (cm–1) 3575–2665 (br), 1660, 1461, 1203, 1140. MS (m/e, rel intensity) 570 (MH+, 100), 304 (43), 282 (45), 214 (83) Exact mass: calculated for C29H39N5O7, 570.2928; found, 570.2929. [α]20D +3.0 (c = 1.82, MeOH).
H-Tyr-Gly-Gly-(NMe)Phe-Leu-OH (7)
An amount of 600 mg of Tenta Gel S PHB resin with a loading of 0.250 mmol/g was used. Fmoc-(NMe)Phe-OH (250 mg, 0.600 mmol) was used in the second coupling. The title peptide was obtained as a white solid (32.6 mg, 38%). 1H NMR (300 MHz, D2O) δ (ppm) 7.33–7.21 (m, 5H), 7.12 (d, 2H, J = 8.5 Hz), 6.83 (d, 2H, J = 8.5 Hz), 5.22–5.17 (m, 1H), 4.47–4.28 (m, 1H), 4.19–4.15 (m, 1H), 4.01–3.79 (m, 4H), 3.30–3.02 (m, 4H), 2.88 (s, 3H), 1.63–1.58 (m, 3H), 0.85 (d, 3H, J = 6.5 Hz) 0.81 (d, 3H, J = 6.5 Hz). 13C NMR (75 MHz, D2O) δ (ppm) 176.2, 172.1, 170.8, 170.5, 155.2, 136.8, 130.8, 129.2, 129.0, 128.7, 126.9, 125.4, 115.8, 59.6, 54.5, 51.4, 42.1, 41.1, 39.0, 35.9, 33.4, 24.4, 22.2, 20.3. IR (NaCl) ν (cm–1) 3610–2530 (br), 1669, 1203, 1139. MS (m/e, rel intensity) 570 (MH+, 34), 301 (39), 158 (61), 141 (100). Exact mass: calculated for C29H39N5O7, 570.2928; found, 570.2933. [α]20D −2.0 (c = 1.31, MeOH).
H-Tyr-Gly-Gly-Phe-(NMe)Leu-OH (8)
LiOH (26.0 mg, 0.612 mmol) dissolved in H2O (4 mL) was added to Boc-Tyr(tBu)-Gly-Gly-Phe-(NMe)Leu-OMe (113 mg, 0.153 mmol) dissolved in THF (4 mL). The mixture was stirred for 4 h at rt. The solvents were evaporated under reduced pressure. This crude solid was immediately dissolved in DCM (5 mL), TFA (2 mL), and TIPS (0.5 mL). The reaction was stirred at rt for 2 h and was concentrated under vacuum. The crude peptide was purified using preparative RP-HPLC with a C18 column and using ACN gradient in a 0.1% TFA aq solution (from 1:9 to 2:3). The purity of all fractions was analyzed using an analytical HPLC instrument, detecting at 214 nm, with a C18 column. All pure fractions were combined, frozen, and lyophilized. The title compound was obtained as a white solid (18.1 mg, 21%). 1H NMR (300 MHz, CD3OD) δ (ppm) 7.32–7.17 (m, 5H), 7.13 (d, 2H, J = 8.5 Hz), 6.80 (d, 2H, J = 8.5 Hz), 5.20–5.04 (m, 1H), 4.14–3.73 (m, 6H), 3.50–3.41 (m, 1H), 3.29–3.11 (m, 2H), 3.04–2.92 (m, 2H), 2.98 (s, 3H), 1.79–1.68 (m, 2H), 1.54–1.40 (m, 1H), 0.90 (dd, 1H, J = 6.5, 11.5 Hz). 13C NMR (75 MHz, CD3OD) δ (ppm) 172.9, 169.9, 169.6, 169.4, 156.9, 136.5, 130.1, 129.0, 128.2, 126.5, 124.5, 115.5, 55.1, 54.8, 51.1, 42.5, 41.6, 37.0, 36.8, 36.3, 30.7, 24.5, 22.2, 20.4. IR (NaCl) ν (cm–1) 3573–2502 (br), 3286, 3072, 2956, 1671, 1518, 1201, 1134. MS (m/e, rel intensity) 570 (M+, 100), 552 (14). Exact mass: calculated for C29H39N5O7, 570.2928; found, 570.2934. [α]D20 −5.92 (c = 0.49, CH3OH).
Cell Culture
GH3 cells (a somatomammotroph tumor cell line) stably expressing the mouse DOPr (GH3/DOPr) and DRGF11 cells (a fusion product of cells of mouse neuroblastoma cell line N18TG-2 with embryonic rat dorsal-root ganglion neurons) stably expressing green fluorescent protein (GFP)-tagged DOPr (DRGF11/DOPr-GFP) were grown at 37 °C in DMEM supplemented with 10% fetal bovine serum and 50 mg/L gentamicin in a humidified atmosphere of 95% air and 5% CO2.15
DOPr-Induced ERK1/2 Activation
The DRGF11/DOPr-GFP cells were stimulated for 5 min with increasing concentrations (10–9–10–5 M) of the different compounds. The stimulation was terminated by aspiration of the culture media and by the addition of ice-cold Hank’s buffered saline (HBS) (130 mM NaCl, 3.5 mM KCl, 1.8 mM CaCl2, 0.5 mM MgCl2, 2.5 mM NaHCO3, 5 mM HEPES, and 0.5 mM EGTA). The cells were then stabilized with ice-cold HBS containing 2 mM Na3VO4, 0.1 μM staurosporine, and complete protease inhibitors (Roche Diagnostics, Indianapolis, IN) for 10 min. Cell lysis was performed using a 50 mM HEPES pH 7.8 solution containing 1% Triton X-100, 2 mM Na3VO4, 0.1 μM staurosporine, and complete protease inhibitors (Roche Diagnostics). Cell lysates were centrifuged for 10 min at 13 500g and 4 °C. The supernatants were saved and stored at −20 °C until use. Protein extracts (15 μg per sample) were resolved with a 10% SDS-polyacrylamide gel and transferred on polyvinylidene fluoride membranes. The membranes were then blocked for 1 h using a Tris-buffered saline with 0.05% Tween 20 (TBS-T) containing 1% gelatin. After blocking, the membranes were incubated for 2 h at room temperature with anti-phosphoERK1/2 or anti-ERK1/2 rabbit antibodies (both 1:1000; Cell Signaling Technology, Danvers, MA). The membranes were washed three times with TBS-T and incubated for 1 h at room temperature with horseradish peroxidase conjugated anti-rabbit antibody (1:2000; GE Healthcare Life Sciences, Piscataway, NJ). Protein detection was performed using an enhanced chemiluminescence detection kit (Amersham ECL Western Blotting Detection Reagents from GE Healthcare Life Sciences). Densitometric analyses of pERK1/2 were performed using ImageJ, and the results were compared to the control.
Competitive Binding Assays
We evaluated the affinity of the different compounds for DOPr with GH3/DOPr membrane extracts. First harvested by a phosphate-buffered saline, cells were obtained by centrifugation. The resulting pellets were resuspended in a 10 mM potassium phosphate buffer pH 7.2 (buffer A) and centrifuged for 10 min at 40 000g. Cell pellets were resuspended in buffer A and left on ice for 20 min. Then, cells were centrifuged three times at 800g for 5 min, the supernatants were saved, and the pellets were resuspended in buffer A. Supernatants were pooled together and centrifuged for 10 min at 40 000g. The resulting pellet was finally resuspended in buffer A supplemented with 0.32 M sucrose and 5 mM EDTA and stored at −80 °C until use. Protein concentration was determined with Bio-Rad DC Protein Assay reagents (Bio-Rad Laboratories, Mississauga, ON, Canada).
[3H]-Deltorphin II (specific activity: 30–60 Ci/mmol; PerkinElmer, Waltham, MA) was used during competitive binding assays to selectively target DOPr highly expressed in membrane extracts. Experiments were performed using a membrane concentration of 100 μg of proteins/mL and a radiolabeled ligand concentration near the KD value previously obtained in saturation binding assays (∼1 nM). Nonspecific binding was determined using 10 μM Deltorphin II. Experiments were conducted with Tris buffer solution pH 7.4 in 5 mL polypropylene tubes for a final volume of 0.5 mL. Incubations were performed during 60 min at 37 °C, and the reaction was stopped by filtration using ice-cold assay buffer on a Whatman GF/C filter (GE Healthcare Life Sciences). Filters were placed in vials containing 8 mL of Ready Gel scintillation cocktail (Beckman Coulter Canada, Inc., Mississauga, ON, Canada), and the radioactivity was determined using a Beckman Coulter LS-6500 scintillation counter (Beckman Coulter Canada, Inc.). Data were analyzed using a nonlinear fitting analysis, and Ki values were calculated from IC50 determination using the Cheng-Prusoff equation.73
DOPr Internalization Assay
Using 35 mm glass bottom dishes (MatTek Corporation, Ashland, MA), DRGF11/DOPr-GFP cells were grown for 2–3 days in DMEM supplemented with 10% fetal bovine serum. Prior to the assay, the culture media was replaced by Earle’s buffer (140 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 0.9 mM MgCl2, 25 mM HEPES, 0.2% BSA, and 0.09% glucose). For each dish, three different cells were chosen and observed with an IX81 Olympus microscope (Olympus America, Center Valley, PA) equipped with a CSU-X1 confocal scanner unit (Yokogawa Electric Corporation, Newnan, GA) and a ProScan II motorized stage system (Prior scientific, Rockland, MA). Before cell stimulation with 1 μM of each compound, 3 min of baseline recording were performed. The images were obtained using a 60× objective and a QuantEM:512SC camera (Photometrics, Tucson, AZ) at room temperature at intervals of 25 s for 30 min.
Inhibition of the Mouse Vas Deferens Contraction
Mice were anesthetized with isoflurane and sacrificed by a cervical dislocation. Their vas deferens were dissected out, the semen was ejected, and the tissues were bathed in modified Krebs solution (mM): NaCl 119.3, KCl 4.7, CaCl2 2.5, KH2PO4 1.2, NaHCO3 25, and d-glucose 11.1, gassed with 95% O2 and 5% CO2 and kept at 34 °C. Longitudinal contractions were recorded isometrically at a constant reting tension of 0.5 gwt by a force displacement transducer FT03, amplified by a compact transducer amplifier P11T and displayed on a data acquisition system dash 4u (Grass Technologies, Astro-Med, Inc., Brossard, QC, Canada). Electric stimulation trains were generated by a Grass S88X dual output square pulse stimulator (Astro-Med, Inc.) coupled to universal coil platinum electrode (Radnoti, LLC, Monrovia, CA,) and repeated every 20 s. They consisted of six impulsions of 20 V during 1 ms with 9 ms intervals. A volume of 100 μL of the tested ligand was added in the 10 mL bath to obtain final concentrations ranging from 10–11 to 10–5 M. After 5 min of recording, the preparation was washed twice with 10 mL of fresh medium. As a control, Leu-enkephalin was added at a final concentration of 10–5 M. The data were analyzed with nonlinear fitting analysis, and the concentration necessary to produce a 50% inhibition (EC50) of the contraction was determined.
LogD7.4 Determination
The determination of the distribution coefficient (LogD) was performed using a modified version of the shake-flask method. Before the experiment, octanol and phosphate buffer (PBS pH 7.4) were mixed together for 24 h to allow saturation of each solution. The mixture was allowed to rest, and the phases were separated and used as solvents in the coefficient measurement. The experiment was performed at room temperature using triplicates for each measurement. Each peptide (0.1 mg) was placed in a vial to which saturated PBS (1 mL) and octanol (0.5 mL) were added. The vial was then shaken mechanically for 10 min. The mixture was allowed to rest for 30 min or until phase separation was completed. Aliquots of both phases were taken and injected in an HPLC instrument (10 μL of each aliquot was injected in an Agilent 1100 series HPLC, column: Agilent Eclipse Plus C-18 column, 50 mm × 3.0 mm, 1.8 μm; solvent A, 0.1% TFA in water; solvent B, 0.1% TFA in acetonitrile; 2–98% B in A over 20 min; flow rate, 0.4 mL/min; UV detection at 214 nm). The retention time of each peptide was already known from the HPLC purity analysis of each peptide. The octanol peak did not interfere with the experiment. The area under the curve (AUC) of the corresponding peak was integrated for each phase injected. The LogD for each peptide was calculated as follows: LogD7.4 = log10 (AUC octanol phase/AUC PBS phase).
Plasma Stability
Plasma was prepared from two male Sprague–Dawley rats (300–350 g; Charles River Laboratories, St-Constant, Quebec, Canada). All animal procedures were approved by the Ethical Committee for Animal Care and Experimentation of the Université de Sherbrooke (protocol #234-10) and were performed according to the regulations of the Canadian Council on Animal Care (CCAC). Briefly, rats were anesthetized with isoflurane (3% isoflurane with 97% medical air) and exsanguinated using an 18G-11/2 needle connected to a 10 mL syringe. The blood was rapidly transferred to an EDTA-containing tube and centrifuged at 1600g for 15 min at 4 °C. The plasma was then stored at −80 °C in 750 μL aliquots until use. The stability of Leu-enkephalin and its analogues was determined using a modified procedure of a previously published protocol.74 In order to decrease the peptide degradation and to allow comparisons between Leu-enkephalin and its analogues, the experiments were performed in plasma diluted to 50% with saline. Nondiluted plasma (25 μL) was incubated at 37 °C for 15 min before the addition of the peptide solution (25 μL). All peptide solutions consisted of a 100 μM dilution of peptide in isotonic sodium chloride (0.9% w/v). After addition of the peptide, the solution was vortexed. The eight aliquots were incubated at 37 °C, and degradation was stopped at 0, 5, 10, 15, 20, 30, 40, and 60 min by the addition of methanol (100 μL). The resulting solutions were centrifuged at 13 000 rpm for 15 min at 4 °C. To 155 μL of the supernatant was added 5 μL of internal standard. The internal standard solution consisted of 500 μM Fmoc-leucine in methanol. Degradations were performed in triplicate, and the resulting solutions were analyzed by HPLC (40 μL of solution was injected in an Agilent 1100 series HPLC, column: Symmetry C18 5 μm 4.6 × 150 mm, heated at 30 °C, flow 1.2 mL/min, start with 0.1% TFA in water then 0 to 75% acetonitrile in 20 min, UV detection at 223 nm). A blank sample at the same dilution but containing no peptide was injected to identify background peaks due to plasma. A standard solution at the same dilution but containing no plasma was injected to ensure that no peptides were lost in the plasma precipitate. The percentage of nondegraded peptide was calculated by determining the ratio between the area under the curve (AUC) of Fmoc-leucine and the AUC of the tested peptide. For all points, the means of the ratios for the triplicates were calculated, and the mean of the 0 min triplicate was fixed at 100% peptide remaining. The AUC of the 0 min triplicate and the standard solution (no plasma) in all cases were not significantly different. For analysis of the appearance of peptide fragments, the same solutions were injected in an LC-MS instrument with the following method: 20 μL of solution was injected in an Waters Alliance/Waters micromass ZQ LC-MS; column, X terra MS C18 3.5 μM 2.1 × 50 mm, flow 1 mL/min, 0 to 50% acetonitrile in 8 min, mass detection between 150 and 700.
Acknowledgments
The authors are grateful to Robert Dumaine and Philippe Sarret for providing access to the Canadian Foundation for Innovation (CFI)-funded confocal microscope.
Supporting Information Available
All remaining experimental details, spectral characterization, HPLC analysis, and 1H NMR spectrum for the precursors and compounds 1–8. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ K.R. and A. P.-G. have contributed equally to this work. F.B., B.G., Y.L.D., and L.G. designed the study and wrote the manuscript. K.R., A.P.-G., P.B., J.-F.N., J.C., and V.B. performed the experiments and wrote the manuscript.
This work was supported by Grant #MOP-102612 from the Canadian Institute for Health Sciences (CIHR) awarded to B.G., Y.L.D. and L.G. A.P.-G. and K.R. were the recipients of the 2009 Dominico-Regoli/Institut de Pharmacologie de Sherbrooke fellowship and the 2010 Centre des Neurosciences/Institut de Pharmacologie de Sherbrooke fellowship, respectively. A.P.-G. and K.R. are, respectively, the recipients of a Ph.D. and a Master studentship from the Fonds de Recherche Québec – Santé (FRQS). L.G. is the recipient of a Junior 2-salary support from the FRQS. F.G., B.G., Y.L.D., and L.G. are members of the FRQS-funded Centre de Recherche Clinique Étienne Le Bel and of the Institut de Pharmacologie de Sherbrooke. L.G. is a member of the Centre des Neurosciences de Sherbrooke as well as of the FRQS-funded Quebec Pain Research Network (QPRN) and Réseau québécois de recherche sur le Médicament (RQRM).
The authors declare no competing financial interest.
Supplementary Material
References
- Bodnar R. J. (2012) Endogenous opiates and behavior: 2011. Peptides 38, 463–522. [DOI] [PubMed] [Google Scholar]
- Kieffer B. L. (1999) Opioids: first lessons from knockout mice. Trends Pharmacol. Sci. 20, 19–26. [DOI] [PubMed] [Google Scholar]
- Beaudry H.; Proteau-Gagne A.; Li S.; Dory Y.; Chavkin C.; Gendron L. (2009) Differential noxious and motor tolerance of chronic delta opioid receptor agonists in rodents. Neuroscience 161, 381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallantine E. L.; Meert T. F. (2005) A comparison of the antinociceptive and adverse effects of the mu-opioid agonist morphine and the delta-opioid agonist SNC80. Basic Clin. Pharmacol. Toxicol. 97, 39–51. [DOI] [PubMed] [Google Scholar]
- Mika J.; Przewlocki R.; Przewlocka B. (2001) The role of delta-opioid receptor subtypes in neuropathic pain. Eur. J. Pharmacol. 415, 31–37. [DOI] [PubMed] [Google Scholar]
- Petrillo P.; Angelici O.; Bingham S.; Ficalora G.; Garnier M.; Zaratin P. F.; Petrone G.; Pozzi O.; Sbacchi M.; Stean T. O.; Upton N.; Dondio G. M.; Scheideler M. A. (2003) Evidence for a selective role of the delta-opioid agonist [8R-(4bS*,8aα,8aβ, 12β)]7,10-Dimethyl-1-methoxy-11-(2-methylpropyl)oxycarbonyl 5,6,7,8,12,12b-hexahydro-(9H)-4,8-methanobenzofuro[3,2-e]pyrrolo[2,3-g]iso quinoline hydrochloride (SB-235863) in blocking hyperalgesia associated with inflammatory and neuropathic pain responses. J. Pharmacol. Exp. Ther. 307, 1079–1089. [DOI] [PubMed] [Google Scholar]
- Gaveriaux-Ruff C.; Kieffer B. L. (2011) Delta opioid receptor analgesia: recent contributions from pharmacology and molecular approaches. Behav. Pharmacol. 22, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderah T. W. (2010) Delta and kappa opioid receptors as suitable drug targets for pain. Clin. J. Pain 26(Suppl 10), S10–15. [DOI] [PubMed] [Google Scholar]
- Comer S. D.; Hoenicke E. M.; Sable A. I.; McNutt R. W.; Chang K. J.; De Costa B. R.; Mosberg H. I.; Woods J. H. (1993) Convulsive effects of systemic administration of the delta opioid agonist BW373U86 in mice. J. Pharmacol. Exp. Ther. 267, 888–895. [PubMed] [Google Scholar]
- Negus S. S.; Butelman E. R.; Chang K. J.; DeCosta B.; Winger G.; Woods J. H. (1994) Behavioral effects of the systemically active delta opioid agonist BW373U86 in rhesus monkeys. J. Pharmacol. Exp. Ther. 270, 1025–1034. [PubMed] [Google Scholar]
- Le Bourdonnec B.; Windh R. T.; Ajello C. W.; Leister L. K.; Gu M.; Chu G. H.; Tuthill P. A.; Barker W. M.; Koblish M.; Wiant D. D.; Graczyk T. M.; Belanger S.; Cassel J. A.; Feschenko M. S.; Brogdon B. L.; Smith S. A.; Christ D. D.; Derelanko M. J.; Kutz S.; Little P. J.; DeHaven R. N.; DeHaven-Hudkins D. L.; Dolle R. E. (2008) Potent, orally bioavailable delta opioid receptor agonists for the treatment of pain: discovery of N,N-diethyl-4-(5-hydroxyspiro[chromene-2,4′-piperidine]-4-yl)benzamide (ADL5859). J. Med. Chem. 51, 5893–5896. [DOI] [PubMed] [Google Scholar]
- Le Bourdonnec B.; Windh R. T.; Leister L. K.; Zhou Q. J.; Ajello C. W.; Gu M.; Chu G. H.; Tuthill P. A.; Barker W. M.; Koblish M.; Wiant D. D.; Graczyk T. M.; Belanger S.; Cassel J. A.; Feschenko M. S.; Brogdon B. L.; Smith S. A.; Derelanko M. J.; Kutz S.; Little P. J.; DeHaven R. N.; DeHaven-Hudkins D. L.; Dolle R. E. (2009) Spirocyclic delta opioid receptor agonists for the treatment of pain: discovery of N,N-diethyl-3-hydroxy-4-(spiro[chromene-2,4′-piperidine]-4-yl) benzamide (ADL5747). J. Med. Chem. 52, 5685–5702. [DOI] [PubMed] [Google Scholar]
- Roques B. P.; Noble F. (1995) Dual inhibitors of enkephalin-degrading enzymes (neutral endopeptidase 24.11 and aminopeptidase N) as potential new medications in the management of pain and opioid addiction. NIDA Res. Monogr. 147, 104–145. [PubMed] [Google Scholar]
- Venkatesan N.; Kim B. H. (2002) Synthesis and enzyme inhibitory activities of novel peptide isosteres. Curr. Med. Chem. 9, 2243–2270. [DOI] [PubMed] [Google Scholar]
- Proteau-Gagne A.; Bournival V.; Rochon K.; Dory Y.; Gendron L. (2010) Exploring the backbone of enkephalins to adjust their pharmacological profile for the delta opioid receptor. ACS Chem. Neurosci. 1, 757–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruby V. J.; Agnes R. S. (1999) Conformation-activity relationships of opioid peptides with selective activities at opioid receptors. Biopolymers 51, 391–410. [DOI] [PubMed] [Google Scholar]
- Morley J. S. (1980) Structure-activity relationships of enkephalin-like peptides. Annu. Rev. Pharmacol. Toxicol. 20, 81–110. [DOI] [PubMed] [Google Scholar]
- Janecka A.; Fichna J.; Janecki T. (2004) Opioid receptors and their ligands. Curr. Top. Med. Chem. 4, 1–17. [DOI] [PubMed] [Google Scholar]
- DiMaio J.; Nguyen T. M.; Lemieux C.; Schiller P. W. (1982) Synthesis and pharmacological characterization in vitro of cyclic enkephalin analogues: effect of conformational constraints on opiate receptor selectivity. J. Med. Chem. 25, 1432–1438. [DOI] [PubMed] [Google Scholar]
- Svensson C. I.; Rew Y.; Malkmus S.; Schiller P. W.; Taulane J. P.; Goodman M.; Yaksh T. L. (2003) Systemic and spinal analgesic activity of a delta-opioid-selective lanthionine enkephalin analog. J. Pharmacol. Exp. Ther. 304, 827–832. [DOI] [PubMed] [Google Scholar]
- Gosselin F.; Tourwe D.; Ceusters M.; Meert T.; Heylen L.; Jurzak M.; Lubell W. D. (2001) Probing opioid receptor-ligand interactions by employment of indolizidin-9-one amino acid as a constrained Gly(2)-Gly(3) surrogate in a leucine-enkephalin mimic. J. Pept. Res. 57, 337–344. [DOI] [PubMed] [Google Scholar]
- Lajoie G.; Lepine F.; Lemaire S.; Jolicoeur F.; Aube C.; Turcotte A.; Belleau B. (1984) Synthesis and biological activity of monothionated analogs of leucine-enkephalin. Int. J. Pept. Protein Res. 24, 316–327. [DOI] [PubMed] [Google Scholar]
- Rinnova M.; Nefzi A.; Houghten R. A. (2002) Opioid activity of 4-imidazolidinone positional analogues of Leu-Enkephalin. Bioorg. Med. Chem. Lett. 12, 3175–3178. [DOI] [PubMed] [Google Scholar]
- Schiller P. W.; St-Hilaire J. (1980) Synthesis, in vitro opiate activity, and intramolecular tyrosine–tryptophan distances of [4-tryptophan]enkephalin analogues. A reassessment of conformational models of enkephalin in solution. J. Med. Chem. 23, 290–294. [DOI] [PubMed] [Google Scholar]
- Olczak J.; Kaczmarek K.; Maszczyńska I.; Lisowski M.; Stropova D.; Hruby V.; Yamamura H.; Lipkowski A.; Zabrocki J. (1998) Consequences of Cis-amide Bond Simulation in Opioid Peptides. Lett. Pept. Sci. 5, 437–440. [Google Scholar]
- de Bont D. B.; Sliedregt-Bol K. M.; Hofmeyer L. J.; Liskamp R. M. (1999) Increased stability of peptidesulfonamide peptidomimetics towards protease catalyzed degradation. Bioorg. Med. Chem. 7, 1043–1047. [DOI] [PubMed] [Google Scholar]
- Elliott R. L.; Marks N.; Berg M. J.; Portoghese P. S. (1985) Synthesis and biological evaluation of phosphonamidate peptide inhibitors of enkephalinase and angiotensin-converting enzyme. J. Med. Chem. 28, 1208–1216. [DOI] [PubMed] [Google Scholar]
- Almquist R. G.; Olsen C. M.; Uyeno E. T.; Toll L. (1984) Replacement of the peptide-backbone amides connecting Tyr-Gly and Gly-Gly in leucine-enkephalin with ketomethylene groups: synthesis and biological activity. J. Med. Chem. 27, 115–120. [DOI] [PubMed] [Google Scholar]
- Mammi N. J.; Goodman M. (1986) Conformational analysis of cyclic partially modified retro-inverso enkephalin analogues by proton NMR. Biochemistry 25, 7607–7614. [DOI] [PubMed] [Google Scholar]
- Richman S. J.; Goodman M.; Nguyen T. M.; Schiller P. W. (1985) Synthesis and biological activity of linear and cyclic enkephalins modified at the Gly3-Phe4 amide bond. Int. J. Pept. Protein Res. 25, 648–662. [DOI] [PubMed] [Google Scholar]
- Choudhary A.; Raines R. T. (2011) An evaluation of peptide-bond isosteres. ChemBioChem 12, 1801–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manavalan P.; Momany F. A. (1980) Conformational energy studies on N-methylated analogs of thyrotropin releasing hormone, enkephalin, and luteinizing hormone-releasing hormone. Biopolymers 19, 1943–1973. [DOI] [PubMed] [Google Scholar]
- Vitoux B.; Aubry A.; Cung M. T.; Marraud M. (1986) N-Methyl peptides. Int. J. Pept. Protein Res. 27, 617–632. [DOI] [PubMed] [Google Scholar]
- Stewart W. E.; Siddall T. H. III. (1970) Nuclear magnetic resonance studies of amides. Chem. Rev. 70, 517–551. [Google Scholar]
- Bosshard H. R. (2001) Molecular recognition by induced fit: how fit is the concept?. News Physiol. Sci. 16, 171–173. [DOI] [PubMed] [Google Scholar]
- Bramson H. N.; Thomas N. E.; Kaiser E. T. (1985) The use of N-methylated peptides and depsipeptides to probe the binding of heptapeptide substrates to cAMP-dependent protein kinase. J. Biol. Chem. 260, 15452–15457. [PubMed] [Google Scholar]
- Coombs G. S.; Rao M. S.; Olson A. J.; Dawson P. E.; Madison E. L. (1999) Revisiting catalysis by chymotrypsin family serine proteases using peptide substrates and inhibitors with unnatural main chains. J. Biol. Chem. 274, 24074–24079. [DOI] [PubMed] [Google Scholar]
- Davidson B. S. (1993) Ascidians: producers of amino acid-derived metabolites. Chem. Rev. 93, 1771–1791. [Google Scholar]
- Fusetani N.; Matsunaga S. (1993) Bioactive sponge peptides. Chem. Rev. 93, 1793–1806. [Google Scholar]
- Lemmens-Gruber R.; Kamyar M. R.; Dornetshuber R. (2009) Cyclodepsipeptides - potential drugs and lead compounds in the drug development process. Curr. Med. Chem. 16, 1122–1137. [DOI] [PubMed] [Google Scholar]
- Fenton D. E. (1977) Across the living barrier. Chem. Soc. Rev. 6, 325–343. [Google Scholar]
- Sarabia F.; Chammaa S.; Ruiz A. S.; Ortiz L. M.; Herrera F. J. (2004) Chemistry and biology of cyclic depsipeptides of medicinal and biological interest. Curr. Med. Chem. 11, 1309–1332. [DOI] [PubMed] [Google Scholar]
- Tulla-Puche J.; Marcucci E.; Prats-Alfonso E.; Bayo-Puxan N.; Albericio F. (2009) NMe amide as a synthetic surrogate for the thioester moiety in thiocoraline. J. Med. Chem. 52, 834–839. [DOI] [PubMed] [Google Scholar]
- Kessler H.; Anders U.; Schudok M. (1990) An unexpected cis peptide bond in the minor conformation of a cyclic hexapeptide containing only secondary amide bonds. J. Am. Chem. Soc. 112, 5908–5916. [Google Scholar]
- Aubry A.; Birlirakis N.; Sakarellos-Daitsiotis M.; Sakarellos C.; Marraud M. (1989) A crystal molecular conformation of leucine-enkephalin related to the morphine molecule. Biopolymers 28, 27–40. [DOI] [PubMed] [Google Scholar]
- Camerman A.; Mastropaolo D.; Karle I.; Karle J.; Camerman N. (1983) Crystal structure of leucine-enkephalin. Nature 306, 447–450. [DOI] [PubMed] [Google Scholar]
- Doi M.; Ishibe A.; Shinozaki H.; Urata H.; Inoue M.; Ishida T. (1994) Conserved and novel structural characteristics of enantiomorphic Leu-enkephalin. X-ray crystal analysis of Leu-enkephalin enantiomer, L-Tyr-Gly-Gly-L-Phe-L-Leu and D-Tyr-Gly-Gly-D-Phe-D-Leu. Int. J. Pept. Protein Res. 43, 325–331. [DOI] [PubMed] [Google Scholar]
- Griffin J. F.; Langs D. A.; Smith G. D.; Blundell T. L.; Tickle I. J.; Bedarkar S. (1986) The crystal structures of [Met5]enkephalin and a third form of [Leu5]enkephalin: observations of a novel pleated beta-sheet. Proc. Natl. Acad. Sci. U.S.A. 83, 3272–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastropaolo D.; Camerman A.; Ma L. Y.; Camerman N. (1987) Crystal structure of an extended-conformation leucine-enkephalin dimer monohydrate. Life Sci. 40, 1995–1999. [DOI] [PubMed] [Google Scholar]
- Smith D.; Griffin J. F. (1978) Conformation of [Leu5]enkephalin from X-ray diffraction: features important for recognition at opiate receptor. Science 199, 1214–1216. [DOI] [PubMed] [Google Scholar]
- Ron D.; Gilon C.; Hanani M.; Vromen A.; Selinger Z.; Chorev M. (1992) N-methylated analogs of Ac[Nle28,31]CCK(26–33): synthesis, activity, and receptor selectivity. J. Med. Chem. 35, 2806–2811. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Saperstein R.; Nutt R. F.; Freidinger R. M.; Brady S. F.; Curley P.; Perlow D. S.; Paleveda W. J.; Colton C. D.; Zacchei A. G.; et al. (1984) A super active cyclic hexapeptide analog of somatostatin. Life Sci. 34, 1371–1378. [DOI] [PubMed] [Google Scholar]
- Teixido M.; Zurita E.; Malakoutikhah M.; Tarrago T.; Giralt E. (2007) Diketopiperazines as a tool for the study of transport across the blood-brain barrier (BBB) and their potential use as BBB-shuttles. J. Am. Chem. Soc. 129, 11802–11813. [DOI] [PubMed] [Google Scholar]
- Gentilucci L. (2004) New trends in the development of opioid peptide analogues as advanced remedies for pain relief. Curr. Top. Med. Chem. 4, 19–38. [DOI] [PubMed] [Google Scholar]
- Huguenin R.; Maurer R. (1980) Resistance of FK 33–824 and other enkephalin analogues to peptidase degradation. Brain Res. Bull. 5, 47–50. [DOI] [PubMed] [Google Scholar]
- Sato T.; Sakurada S.; Sakurada T.; Furuta S.; Chaki K.; Kisara K.; Sasaki Y.; Suzuki K. (1987) Opioid activities of D-Arg2-substituted tetrapeptides. J. Pharmacol. Exp. Ther. 242, 654–659. [PubMed] [Google Scholar]
- Franz N.; Menin L.; Klok H.-A. (2009) A Post-Modification Strategy for the Synthesis of Uniform, Hydrophilic/Hydrophobic Patterned α-Hydroxy Acid Oligomers. Eur. J. Org. Chem. 2009, 5390–5405. [Google Scholar]
- Barrish J. C.; Lee H. L.; Mitt T.; Pizzolato G.; Baggiolini E. G.; Uskokovic M. R. (1988) Total synthesis of pseudomonic acid C. J. Org. Chem. 53, 4282–4295. [Google Scholar]
- van Dijk M.; Nollet M. L.; Weijers P.; Dechesne A. C.; van Nostrum C. F.; Hennink W. E.; Rijkers D. T. S.; Liskamp R. M. J. (2008) Synthesis and Characterization of Biodegradable Peptide-Based Polymers Prepared by Microwave-Assisted Click Chemistry. Biomacromolecules 9, 2834–2843. [DOI] [PubMed] [Google Scholar]
- Yamamoto N.; Takayanagi A.; Yoshino A.; Sakakibara T.; Kajihara Y. (2007) An approach for a synthesis of asparagine-linked sialylglycopeptides having intact and homogeneous complex-type undecadisialyloligosaccharides. Chemistry 13, 613–625. [DOI] [PubMed] [Google Scholar]
- Humphrey J. M.; Chamberlin A. R. (1997) Chemical Synthesis of Natural Product Peptides: Coupling Methods for the Incorporation of Noncoded Amino Acids into Peptides. Chem. Rev. 97, 2243–2266. [DOI] [PubMed] [Google Scholar]
- Creighton C. J.; Zapf C. W.; Bu J. H.; Goodman M. (1999) Solid-phase synthesis of pyridones and pyridopyrazines as peptidomimetic scaffolds. Org. Lett. 1, 1407–1409. [DOI] [PubMed] [Google Scholar]
- Beddell C. R.; Clark R. B.; Hardy G. W.; Lowe L. A.; Ubatuba F. B.; Vane J. R.; Wilkinson S. (1977) Structural requirements for opioid activity of analogues of the enkephalins. Proc. R. Soc. London, Ser. B 198, 249–265. [DOI] [PubMed] [Google Scholar]
- Avdeef A.; Barrett D. A.; Shaw P. N.; Knaggs R. D.; Davis S. S. (1996) Octanol-, chloroform-, and propylene glycol dipelargonat-water partitioning of morphine-6-glucuronide and other related opiates. J. Med. Chem. 39, 4377–4381. [DOI] [PubMed] [Google Scholar]
- DesMarteau D. D.; Lu C. (2007) Syntheses and lipophilicitymeasurement of Nα/N-terminus-1,1-dihydroperfluoroalkylated α-amino acids and small peptides. J. Fluorine Chem. 128, 1326–1334. [Google Scholar]
- Mahar Doan K. M.; Humphreys J. E.; Webster L. O.; Wring S. A.; Shampine L. J.; Serabjit-Singh C. J.; Adkison K. K.; Polli J. W. (2002) Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J. Pharmacol. Exp. Ther. 303, 1029–1037. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H. Y.; Smith B. R.; Ward K. W.; Kopple K. D. (2002) Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45, 2615–2623. [DOI] [PubMed] [Google Scholar]
- Ballet S.; Misicka A.; Kosson P.; Lemieux C.; Chung N. N.; Schiller P. W.; Lipkowski A. W.; Tourwe D. (2008) Blood-brain barrier penetration by two dermorphin tetrapeptide analogues: role of lipophilicity vs structural flexibility. J. Med. Chem. 51, 2571–2574. [DOI] [PubMed] [Google Scholar]
- Breuning M.; Hauser T.; Tanzer E. M. (2009) A novel one-pot procedure for the stereoselective synthesis of alpha-hydroxy esters from ortho esters. Org. Lett. 11, 4032–4035. [DOI] [PubMed] [Google Scholar]
- Koppenhoefer B.; Trettin U.; Figura R.; Lin B. (1989) Accurate determination of the intrinsic racemization in chiral synthesis via enantiomer resolution of underivatized vicinal diols. Tetrahedron Lett. 30, 5109–5110. [Google Scholar]
- Sieber P. (1987) An improved method for anchoring of 9-fluorenylmethoxycarbonyl-amino acids to 4-alkoxybenzyl alcohol resins. Tetrahedron Lett. 28, 6147–6150. [Google Scholar]
- Fields G. B.; Noble R. L. (1990) Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int. J. Pept. Protein Res. 35, 161–214. [DOI] [PubMed] [Google Scholar]
- Cheng Y.; Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Lalatsa A.; Lee V.; Malkinson J. P.; Zloh M.; Schatzlein A. G.; Uchegbu I. F. (2012) A prodrug nanoparticle approach for the oral delivery of a hydrophilic peptide, leucine(5)-enkephalin, to the brain. Mol. Pharmaceutics 9, 1665–1680. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.