

Abstract
Sampling alternative conformations is key to understanding how proteins work and engineering them for new functions. However, accurately characterizing and modeling protein conformational ensembles remains experimentally and computationally challenging. These challenges must be met before protein conformational heterogeneity can be exploited in protein engineering and design. Here, as a stepping stone, we describe methods to detect alternative conformations in proteins and strategies to model these near-native conformational changes based on backrub-type Monte Carlo moves in Rosetta. We illustrate how Rosetta simulations that apply backrub moves improve modeling of point mutant side chain conformations, native side chain conformational heterogeneity, functional conformational changes, tolerated sequence space, protein interaction specificity, and amino acid co-variation across protein-protein interfaces. We include relevant Rosetta command lines and RosettaScripts to encourage the application of these types of simulations to other systems. Our work highlights that critical scoring and sampling improvements will be necessary to approximate conformational landscapes. Challenges for the future development of these methods include modeling conformational changes that propagate away from designed mutation sites and modulating backbone flexibility to predictively design functionally important conformational heterogeneity.
Keywords: Protein design, protein dynamics, conformational heterogeneity, conformational sampling, alternative conformations, Rosetta, Ringer, Backrub
Introduction
Proteins are constantly fluctuating between alternative conformations (Frauenfelder et al., 1991). Processes including folding (Korzhnev et al., 2010), ligand binding (Boehr et al., 2009), and enzymatic catalytic cycles (Nagel and Klinman, 2009) depend on the movement across the energy landscape. While protein folding is generally driven by a large energy gap between the “native” state and the unfolded ensemble, functionally essential conformations within the “native” state are often separated by smaller energy differences (Fleishman and Baker, 2012).
Computational modeling of the “native” state can result in either a representative single structure or a limited ensemble of conformations. Several straightforward global and local metrics have been developed to compare computational predictions of a representative single structure to an experimentally derived X-ray structure (MacCallum et al., 2011). In contrast, modeling conformational heterogeneity within the “native” state presents significant complications. For example, conformational heterogeneity present in NMR structural ensembles can result from a lack of restraints, limitations in sampling methods, or genuine heterogeneity (Rieping et al., 2005; Schneider et al., 1999). Additionally, comparisons to simulations are often necessary to distinguish between multiple motional models suggested by NMR dynamics observables including Residual Dipolar Couplings (Meiler et al., 2001), CPMG relaxation dispersion (Bouvignies et al., 2011), and side chain order parameters (S2) (Li et al., 1996). X-ray crystallography, which is traditionally interpreted in terms of a single static structure, can also contain information about protein conformational heterogeneity (Best et al., 2006; Furnham et al., 2006; Lang et al., 2010; Levin et al., 2007). All of these experimental data types can be integrated to improve the computational modeling of protein conformational ensembles.
Ultimately, to connect protein conformational dynamics to function, simulations must be leveraged to provide structural mechanisms consistent with the experimental data. Molecular dynamics simulations present the most obvious solution to identify the structural mechanisms of conformational heterogeneity (Maragakis et al., 2008). However, other than in exceptional cases (Kelley et al., 2008; Shaw et al., 2010), the timescales accessible to molecular dynamics often preclude sampling functional motions. The computational requirements of molecular dynamics simulations also make it prohibitive to simultaneously sample sequence space for protein design.
Monte Carlo simulations, for example as used in Rosetta (Leaver-Fay et al., 2011), can also be used to sample protein conformations, but rely on having moves that result in energetically accessible conformations. Fixed backbone Monte Carlo simulations, where side chain conformations are sampled based on a rotamer library, can provide some indications of local flexibility (DuBay and Geissler, 2009). However, it is clear that both side chain and backbone flexibility are necessary to describe and design protein conformational heterogeneity (Friedland et al., 2008; Mandell and Kortemme, 2009). Backbone conformations are less easily discretized compared to side chain rotamers, leading to problems in both creating and validating Monte Carlo backbone moves. Many strategies to efficiently search through backbone space have been implemented in Rosetta including fragment insertion (Simons et al., 1997), loop closure with cyclic coordinate decent (CCD) (Canutescu and Dunbrack, 2003), local torsion sampling with kinematic loop closure (KIC) (Mandell et al., 2009), and backrub (Davis et al., 2006; Smith and Kortemme, 2008). Additionally, these moves can be combined and iterated with sequence design to enrich for proteins with desired conformational and functional properties that could not be explored without backbone flexibility.
Here, we describe the application and validation of the backrub sampling move, which was initially inspired by observations using high resolution X-ray crystallography (Davis et al., 2006), in Rosetta (Smith and Kortemme, 2008) (Figure 1 A-C). To test whether these moves accurately represent protein conformational heterogeneity, we provide example command lines and scripts that can be run using Rosetta version 3.5. These commands and scripts examine how backrub sampling affects predictions of mutant structures, alternative conformations observed by X-ray crystallography, peptide-ligand binding specificities, and evolutionary properties (Figure 1D). The continued development of flexible backbone sampling methods that agree with diverse experimental and evolutionary data will improve our ability to design and engineer new protein functions that depend on and exploit conformational heterogeneity.
Figure 1. The backrub move and its applications in Rosetta.

(A) The Richardson group originally described the “Backrub” move asa rotation around the Cαi-1 and Cαi +1axis by τdisp, along with simultaneous peptide plane rotations (τ1 and τ2), without disturbing other surrounding atom coordinates. (B) By changing the position of the Cαi-Cβi bond vector, this move can couple side chain rotameric changes with small local backbone adjustments.(C)In Rosetta, the generalized backrub move isa single rotation that can also include longer intervals and other backbone atom types as pivots for the rotations. (D) Implementing backrubs as a Monte Carlo move in Rosetta enables a variety of flexible backbone prediction and design applications that are described in this paper: predicting mutant conformations (Figure 2), modeling alternative conformations (Figures 3 and 4), coupling conformational and sequence plasticity (Figure 5), and designing amino-acid co-variation at protein interfaces (Figure 6).
Rosetta Moves To Model Alternative Conformations In X-Ray Density
Modeling the Richardson Backrub in Rosetta
Unlike other methods for flexible backbone sampling implemented in Rosetta, which are based on fragment insertion or geometric constraints, the backrub move derives its motional model from conformational variation observed in high-resolution X-ray data (Davis et al., 2006). The Richardson group observed electron density consistent with a concerted backbone reorientation that moves a central side chain perpendicular to the main chain direction for 3% of total residues in a dataset of ultra-high resolution crystal structures. They noted that this move changed the accessible side chain conformations while leaving flanking structure undisturbed.
In Rosetta, the backrub consists of a rotation about an axis defined by the flanking backbone atoms that changes 6 internal backbone degrees of freedom in the protein, namely the Φ, ψ, and the N-Cα-C bond (α) angles at both pivots (Smith and Kortemme, 2008). In the Richardson formulation, the pivots were Cα atoms surrounding a single residue (Figure 1A), but the move can be performed over any backbone atom type over varying length scales (Figure 1C). Bond angle, rotational angle and Cβ/Hα placement constraints are included to eliminate the need for costly minimization steps after every move. In addition, the backrub move in Rosetta can be adapted so that it obeys detailed balance (Smith and Kortemme, 2008). One notable aspect of the backrub move is that it makes certain side chain conformations, which would not be accessible in the starting backbone conformation, accessible in the newly accepted backbone conformation (Figure 1B). Such moves alter the potential to accommodate new side chain conformations and mutations at the “backrubbed” position and its local neighbors.
Modeling the Response to Mutations
Subtle backbone adjustments are often necessary to accommodate differences between the wild type and mutant side chain. An initial test of the backrub move in Rosetta was to compare its performance to fixed backbone sampling in predicting the conformation of mutated side chains (Figure 2). Based on a template of the wild type structure, a successful prediction of a mutant structure would generate both the conformation of the mutant side chain and any changes that propagate away from the mutated residue. In general, backrub moves decrease the RMSD between the prediction and conformation observed in the mutant crystal structure (Figure 2). Particularly dramatic successes are achieved when there would be a clash to neighboring atoms that is relieved by a small backbone adjustment or when a local backbone move changes the probability of accessing a new conformation from a backbone dependent rotamer library.
Figure 2. Backrub sampling improves the prediction of mutant side chain conformation compared to fixed backbone simulations.

(A) Example predictions of mutant conformations given the wild type structure, with the indicated PDB codes and point mutations. The mutant crystal structure is shown in yellow compared to the wild type crystal structure (green, left), prediction based on fixed backbone simulations (magenta, center), or prediction based on backrub flexible backbone sampling (cyan, right). (B) The overall quantification of the results of fixed backbone and backrub predictions over a set of 136 buried (SASA <5%) side chains with conformations differing by more than 0.2 Å between mutant and wild type. The median RMSD decreases from 1.17 Å to 0.98 Å. Shown are box plots with the median as a black line and the 25-75th percentiles in the shaded box with outlier-corrected extreme values as dashed lines. (C) A scatter plot representation of the data in (B) shows that for many mutant structure predictions backrub leads to large improvements compared to fixed backbone simulations.
Due to the broad utility of this application for predicting the results of single or multiple point mutations, we have created a webserver that automates this task: https://kortemmelab.ucsf.edu/backrub/ (Lauck et al., 2010). On the server, the user must enter the desired PDB, the site of mutation and the new amino acid identity. By default, 10 independent simulations are performed, but this can be adjusted to 2-50 simulations. For each simulation, the server will attempt 10,000 moves that include backrub rotations of various lengths and angles centered on the mutated residue, and side chain moves in a 6Å shell around the mutated residue. The resulting conformations are scored with the Rosetta scoring function and accepted or rejected according to the Metropolis criterion using a kT of 0.6. The lowest scoring conformation out of all the simulations is returned to the user as the best prediction. Advanced users can exert greater control over these parameters by using the “backrub” Rosetta command line program or RosettaScripts (Fleishman et al., 2011). An example command line for point mutation prediction is as follows:
∼/rosetta/rosetta_source/bin/backrub.linuxgccrelease -database ∼/rosetta/rosetta_database/ -s 1CV1.pdb -ex1 -ex2 -extrachi_cutoff 0 -use_input_sc -backrub:ntrials 10000 -nstruct 10 -resfile 1CV1_M111I.resfile -pivot_residues 84 99 102 103 106 107 108 109 110 111 112 113 114 115 118
where the “resfile” sets positions to be mutated and repacked (allowing rotamer changes), and the pivot residues denote pivots allowed for backrub moves (necessary files to run the command line with Rosetta version 3.5 are included as example S1 at http://kortemmelab.ucsf.edu/resources/MIE_Supplement.tar.gzresults results shown in Figure 2 were obtained with Rosetta revision 18013). For details about command line flags and the resfile syntax, see the Rosetta manual at http://www.rosettacommons.org/.
Discovering and Modeling Alternative Conformations from X-ray data
The backrub move was inspired by manual examination of ultra-high (sub 1Å) resolution electron density maps (Davis et al., 2006) suggesting that conformational heterogeneity in X-ray data can be used to develop and validate new sampling methods. Subsequently, Alber and colleagues developed a method, Ringer (Lang et al., 2010) to automate the discovery of alternative side chain conformations in high (sub 2Å) resolution electron density maps by sampling around side chain dihedral angles (Figure 3A). Despite the limitation that Ringer uses a fixed backbone to define the sampling radius, they showed that 18% of side chains have evidence for unmodeled alternative conformations at electron density levels of 0.3-1σ. Concurrently, van den Bedem et al. (van den Bedem et al., 2009) developed a complementary method, qFit, which includes local backbone and side chain flexibility to compute an optimal fit to the electron density for each residue. The resulting 1-3 backbone and side chain conformations per residue are merged together in a multiconformer qFit model that improves R/Rfree and maintains excellent geometry statistics. Remarkably, despite an entirely different search procedure from the original observation by the Richardson group, many alternative conformations identified by qFit can be related by backrub-like moves (Figure 3B). Collectively, these studies suggest that electron density maps can provide a more informative representation of the “native” state than traditionally offered by static X-ray structures.
Figure 3. Backrub sampling improves the prediction of alternative side chain conformations observed in protein crystal structures.

(A) Electron density sampling by Ringer around the χ1of R29 from PDB 1KWN reveals high electron density for the primary conformation 60° and a secondary peak (indicated by the black arrow), above the 0.3σ threshold that enriches for alternative conformations (shaded green area), near the 180° rotameric bin. (B)2mFo-DFc electron density surrounding R29 from PDB 1KWN contoured at 1σ (blue mesh) and 0.3σ (cyan mesh). The original PDB model is shown in yellow, with an alternative conformation identified by Ringer and modeled with qFit at 25% occupancy shown in green. (C) Example predictions with Rosetta, with the indicated PDB codes and residues. Sampling of side chain conformations (yellow) starting from alternative conformations (green, right) is improved by flexible backbone backrub moves (cyan, right) compared to fixed backbone side chain only sampling (magenta, center). (D) The overall quantification of the results, showing that backrub sampling increases identification of discrete side chain local minima modeled as alternative conformations by qFit compared to fixed backbone models over a set of 152 side chains with solvent accessibility less than 30%. The median RMSD decreases from 0.47 to 0.33. Box plots are shown as in Figure 2C. (E) A scatter plot representation of the data in (D) shows that backrub leads to large improvements compared to fixed backbone for many alternative conformation predictions.
By examining 30 pairs of matched room temperature and cryogenic X-ray datasets, Fraser et al. used Ringer and qFit to show that room temperature X-ray data increase the evidence for alternative conformations compared to data collected at conventional cryogenic temperatures (Fraser et al., 2011). We reasoned that sampling between these experimentally visualized alternative conformations would assess the ability of fixed backbone or backrub simulations to access a representative set of conformations that are significantly populated in the “native” state. To test this idea, we considered the A and B alternative conformations (Figure 3C) from 30 room temperature X-ray multiconformer models refined by qFit.
We focused our analysis on alternative conformations with Cβ deviations of 0.2 Å or greater, relative SASAs less than or equal to 30%, and different β1 rotameric bins (152 side chains). First, we split the multiconformer model into two separate PDB files, containing all residues without alternative conformations and either the “A” conformations or “B” conformations (for example, 1kwn_A.pdb and 1kwn_B.pdb. Next, we ran a RosettaScripts protocol that moves between the starting conformation specified by the flag –s (in this example, 1kwn_A.pdb) and the target alternative conformation specified by the flag –in:file:native (in this example, 1kwn_B.pdb). At the beginning of the protocol, the χ angles of a central side chain are switched from the starting conformation to the target conformation. This script tests whether changes in the surrounding side chains (fixed backbone) or both the backbone and surrounding side chains (backrub) better accommodate the new χ angles and find a side chain conformation close in RMSD to the target conformation. We tested this protocol in both directions between the A and B conformations. In the simple example below, the variables χ1 through χ4 provide the target χ angles (here the χ angles of conformation B), and piv1 through piv3 define the positions around the central residue allowed to be pivots for the backrub move.
The command to run the protocol is:
∼/rosetta/rosetta_source/bin/rosetta_scripts.linuxgccrelease –database ∼/rosetta/rosetta_database -s 1kwn_A.pdb -in:file:native 1kwn_B.pdb -parser:protocol model_alternate_conformation.xml -parser:script_vars pos=29 chi1=-145.522 chi2=-160.509 chi3=81.9108 chi4=175.816 piv1=28 piv2=29 piv3=30
The contents of model_alternate_conformation.xml are:
| <ROSETTASCRIPTS> |
| <SCOREFXNS> |
| Include the bond angle potential scoring term |
| <score12_backrub weights=score12_full> |
| <Reweight scoretype=mm_bend weight=1/> |
| </score12_backrub> |
| </SCOREFXNS> |
| <TASKOPERATIONS> |
| Define the restrictions on the sidechain moves that will occur in the simulation |
| <ExtraRotamersGeneric name=extra_rot ex1=1 ex2=2 extrachi cutoff=0/> |
| <IncludeCurrent name=input_sc/> |
| <DesignAround name=neighbors_only allow design=0 design_shell=0 repack_shell=6.0 resnums=%%pos%%/> |
| <RestrictToRepacking name=repack_only/> |
| <PreventRepacking name=fix_central_residue resnum=%%pos%%/> |
| </TASKOPERATIONS> |
| <FILTERS> |
| Calculates the side-chain RMSD before and after simulation |
| <SidechainRmsd name=rmsd threshold=10 include backbone=0 res1_res_num=%%pos%% res2_res_num=%%pos%%/> |
| </FILTERS> |
| <MOVERS> |
| Set the chi angles of the residue of interest |
| <SetChiMover name=setchi1 chinum=1 resnum=%%pos%% angle=%%chi1%%/> |
| <SetChiMover name=setchi2 chinum=2 resnum=%%pos%% angle=%%chi2%%/> |
| <SetChiMover name=setchi3 chinum=3 resnum=%%pos%% angle=%%chi3%%/> |
| <SetChiMover name=setchi4 chinum=4 resnum=%%pos%% angle=%%chi4%%/> |
| Set backrub moves to only occur near residue of interest |
| <Backrub name=backrub pivot_residues=%%piv1%%,%%piv2%%,%%piv3%% min_atoms=3 min_atoms=7/> |
| Set side-chain moves to only include residues within 6 angstrom shell |
| <Sidechain name=sidechain task_operations=extra_rot, input_sc, fix central residue, neighbors_only, repack_only/> |
| During Monte Carlo, alternate between backrub moves (75%) and side-chain moves (25%) |
| <ParsedProtocol name=backrub_protocol mode=single_random> |
| <Add mover_name=backrub apply_probability=0.75/> |
| <Add mover_name=sidechain apply_probability=0.25/> |
| </ParsedProtocol> |
| Set up Monte Carlo simulation with 10,000 steps and kT=0.6 |
| <GenericMonteCarlo name=backrub_mc mover_name=backrub_protocol scorefxn_name=score12_backrubtrials=10000 temperature=0.6 preapply=0/> |
| </MOVERS> |
| <PROTOCOLS> |
| Set the residue of interest to the desired chi angles |
| <Add mover_name=setchi1/> |
| <Add mover_name=setchi2/> |
| <Add mover_name=setchi3/> |
| <Add mover_name=setchi4/> |
| Calculate RMSD before simulation |
| <Add filter_name=rmsd/> |
| Run backrub simulation |
| <Add mover_name=backrub_mc/> |
| Calculate RMSD after simulation |
| <Add filter_name=rmsd/> |
| </PROTOCOLS> |
| </ROSETTASCRIPTS> |
Necessary files to run this script with Rosetta version 3.5 are included as example S2 at http://kortemmelab.ucsf.edu/resources/MIE_Supplement.tar.gzresults shown in Figure 3 were obtained with Rosetta revision 48648.
Similarly to the mutation data set (Figure 2), backrub moves significantly improve the predictions (Figure 3D,E). Additionally, we tested the effect of including larger backrub moves or using C and N atoms as pivots in place of the normal Cα pivot. Applying these larger moves or using additional pivot atoms did not significantly affect the modeled alternate side-chain RMSD over the dataset. However, there may be certain residue types, secondary structures, or local environments that benefit from distinct move sets.
These results suggest that backrub moves help to model “native” state heterogeneity. While above we have described the validation of this procedure on high resolution room temperature X-ray data, similar strategies can be applied to cryogenic data, where models will likely contain fewer alternative conformations, or to low resolution data, where the electron density maps do not reveal discrete alternative conformations. Therefore, flexible backbone sampling strategies in Rosetta may help to improve the description of the “native” state offered by conventional or low-resolution X-ray crystallography experiments (Tyka et al., 2011). Such sampling methodologies will have many applications including flexible receptor docking in drug discovery (Sherman et al., 2006).
Sampling Functional Alternative Conformations in Cyclophilin A
While the preceding examples utilized backbone flexibility centered on a single residue, many protein motions require movement of a neighborhood of residues that may potentially spread across multiple elements of secondary structure. These movements can create loop, rigid body domain, or side chain rearrangements that are crucial for the biological mechanism.
The difficulty of discovering and simulating correlated motions is exemplified in the intrinsic conformational exchange of the proline isomerase cyclophilin A (CypA) (Fraser et al., 2009) (Figure 4A). Previous NMR studies by the Kern group identified a collective exchange process extending from the active site into the core of the protein and established a link between the rate of conformational exchange and the catalytic cycle of the enzyme (Eisenmesser et al., 2002; Eisenmesser et al., 2005). Room temperature X-ray crystallography and electron density interpretation using a combination of Ringer and manual inspection were used to reveal that the exchange was due to a coupled network of alternative side chain conformations (Fraser et al., 2009). The functional importance of the alternative conformation was tested by demonstrating a parallel reduction in dynamics and catalysis upon mutation of a residue outside the active site (Fraser et al., 2009). Intriguingly, the backbone movement of Phe113 renders its alternative conformation undetectable by Ringer, which is limited to fixed backbone sampling (Figure 4B).
Figure 4. Backrub sampling can be used to model functionally relevant alternative conformations.

(A) NMR relaxation experiments detect that residues in a dynamic network (cyan transparent surface) undergo a collective exchange between a major and minor conformation with and without substrate present (Eisenmesser et al., 2002; Eisenmesser et al., 2005; Fraser et al., 2009). Room temperature X-ray data collection and qFit multiconformer refinement identify a major (green) and minor (yellow) conformation providing a structural basis for the NMR observations. Additional alternative conformations are shown in orange. (B) Rosetta simulations can access the alternative conformation starting from either state (yellow/green) using backrub (right, cyan), but not fixed backbone (middle, magenta) sampling methods.
Due to the millisecond timescale of this correlated motion, the side chain conformational changes cannot be sampled in conventional molecular dynamics simulations. However, recent accelerated molecular dynamics simulations that reduce torsional barriers have recapitulated several key elements of the conformational dynamics during catalysis (Doshi et al., 2012). As an initial test of the ability of Rosetta to model correlated motions between neighboring side-chains in CypA, we modified the RosettaScripts protocol used to sample alternative conformations. We defined multiple positions that are allowed to be pivots for backrub and included a call to a “resfile” that specifies the positions whose side-chains can be repacked. This protocol improves sampling of the alternative conformation over fixed backbone approaches (Figure 4B).
The command to run the protocol is:
∼/rosetta/rosetta_source/bin/rosetta_scripts.linuxgccrelease -database ∼/rosetta/rosetta_database/ -s 3K0N_A.pdb -in:file:native 3K0N_B.pdb -parser:protocol model_alternate_conformation_F113.xml -resfile F113.resfile -parser:script_vars chi1=-53.763 chi2=-41.4727 chi3=0 chi4=0
The contents of model_alternate_conformation_F113.xml are:
| <ROSETTASCRIPTS> |
| <SCOREFXNS> |
| Include the bond angle potential scoring term |
| <score12_backrub weights=score12_full> |
| <Reweight scoretype=mm_bend weight=1/> |
| </score12_backrub> |
| </SCOREFXNS> |
| <TASKOPERATIONS> |
| <ExtraRotamersGeneric name=extra_rot ex1=1 ex2=2 extrachi_cutoff=0/> |
| <ReadResfile name=read_resfile filename=“F113.resfile”/> |
| </TASKOPERATIONS> |
| <FILTERS> |
| Calculates the side-chain RMSD before and after simulation |
| <SidechainRmsd name=rmsd threshold=10 include_backbone=0 res1_pdb_num=113A res2_pdb_num=113A/> |
| </FILTERS> |
| <MOVERS> |
| Set the chi angles of the residue of interest |
| <SetChiMover name=setchi1 chinum=1 resnum=113A angle=%%chi1%%/> |
| <SetChiMover name=setchi2 chinum=2 resnum=113A angle=%%chi2%%/> |
| <SetChiMover name=setchi3 chinum=3 resnum=113A angle=%%chi3%%/> |
| <SetChiMover name=setchi4 chinum=4 resnum=113A angle=%%chi4%%/> |
| Set backrub moves to only occur near residue of interest |
| <Backrub name=backrub pivot_residues=54A,55A,56A,59A,60A,61A,62A,63A,64A,65A,90A, 91A,92A,97A,98A,99A,100A,101A,102A,103A,110A,111A,112A,113A,114A,115A,116A,118A,119A,120A,121A,122A,123A,125A,126A,127 A,128A,129A,130A min_atoms=3 min_atoms=7/> |
| Set side-chain moves to only include residues within 6 angstrom shell |
| <Sidechain name=sidechain task_operations=read_resfile, extra_rot/> |
| During Monte Carlo, alternate between backrub moves (75%) and side-chain moves (25%) |
| <ParsedProtocol name=backrub_protocol mode=single_random> |
| <Add mover_name=backrub apply_probability=0.75/> |
| <Add mover_name=sidechain apply_probability=0.25/> |
| </ParsedProtocol> |
| Set up Monte Carlo simulation with 10,000 steps and kT=0.6 |
| <GenericMonteCarlo name=backrub_mc mover_name=backrub_protocol scorefxn name=score12 backrub trials=10000 temperature=0.6 preapply=0/> |
| </MOVERS> |
| <PROTOCOLS> |
| Set the residue of interest to the desired chi angles |
| <Add mover_name=setchi1/> |
| <Add mover_name=setchi2/> |
| <Add mover_name=setchi3/> |
| <Add mover_name=setchi4/> |
| Calculate RMSD before simulation |
| <Add filter_name=rmsd/> |
| Run backrub simulation |
| <Add mover_name=backrub_mc/> |
| Calculate RMSD after simulation |
| <Add filter_name=rmsd/> |
| </PROTOCOLS> |
| </ROSETTASCRIPTS> |
Necessary files to run this script with Rosetta version 3.5 are included in the as example S3 at http://kortemmelab.ucsf.edu/resources/MIE_Supplement.tar.gzresults shown in Figure 4 were obtained with Rosetta revision 48648.
Here, we have specified neighboring residues that can undergo backrub moves. Similarly, the ability of backrub to sample functionally important loop conformations has been demonstrated for triosephosphate isomerase (TIM) (Smith and Kortemme, 2008). To efficiently sample these enzymatic motions, we used prior knowledge of residues that need conformational adjustments. Therefore, these strategies present an immediate challenge: sampling large correlated motions without prior knowledge of what residues are involved in the motion. One approach is to use unbiased simulations to identify flexible regions. In the case of TIM, unbiased simulations identify that the catalytically important loop region is highly flexible, but only simulations that focus on the loop have been shown to sample the entire range of motion. Another intermediate on the road to this goal is to include constraints from NMR relaxation dispersion experiments, which specify residues that are experiencing an exchange in chemical environment, but do not provide direct structural information about the exchange. Recent work using T4 Lysozyme (Bouvignies et al., 2011) suggests that Rosetta fragment insertion methods biased by experimental chemical shifts can generate structural descriptions of alternative conformations discovered by NMR. The success of backrub moves in sampling the enzyme motions of CypA and TIM indicate that “native” state sampling using backrub moves can likely be exploited in a similar fashion to link conformational dynamics discovered by NMR relaxation dispersion experiments with structural mechanisms.
Sequence Plasticity and Conformational Plasticity are Intertwined
The improvements offered by backrub moves in predicting point mutant structures (Figure 2) suggest that subtle backbone rearrangements can significantly alter the prediction of tolerated mutations. It follows that conformational ensembles created through backrub moves would also change the potential for sequences predicted to be consistent with a given protein fold. Indeed, incorporating backbone flexibility increased the overlap between sequences predicted to be consistent with the ubiquitin fold and the evolutionary record (Friedland et al., 2009). These results provided further evidence that the relationship between sequence and structural variability can be leveraged to develop and validate new conformational sampling methods. Both sequence alignments of orthologous proteins (natural selection) and sequences enriched in high-throughput binding experiments, such as phage display or peptide arrays (artificial selection), can been used to define the sequence variability that design methods can target.
Modeling Peptide Binding Specificity
In addition to predicting sequences tolerated by a single protein fold, flexible backbone methods can improve the prediction of binding specificity in peptide binding domains such as PDZ, SH3 and WW domains. Phage display coupled with next-generation sequencing techniques can generate experimental position weight matrices (PWMs) based on large numbers of potential sequences (Huang and Sidhu, 2011). A challenge for interpreting these datasets is to define the structural basis for specificity in binding pockets that are quite similar. To test how well Rosetta can recapitulate the binding specificities discovered by these experiments, we sampled conformations of both the peptide and receptor protein using backrub moves. As observed previously for sampling the ubiquitin fold family sequences, the temperature parameter is key for controlling the conformational diversity sampled by the ensemble (Figure 5A). For applications where there is a larger degree of backbone flexibility and corresponding sequence variability, higher temperatures can be explored. For PDZ domain-peptide interactions, a temperature of 0.6 kT was used (Smith and Kortemme, 2010, 2011). After generating an ensemble using backrub moves, design can be used to sample sequence changes of either the receptor or the peptide.
Figure 5. Rosetta generates near native ensembles using backrub sampling.
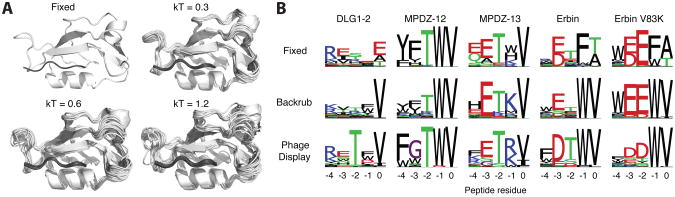
(A) Cα cartoons of Rosetta generated conformational ensembles using backrub sampling at different temperatures, compared to the fixed backbone (top left). Higher temperatures increase the conformational diversity and can increase agreement with experimental data. The PDZ domain structure is shown in white and peptide in grey. (B) Example results from the sequence tolerance protocol to predict peptide specificity for 4 PDZ domains (DLG1-2, MPDZ-12, MPDZ-13 and Erbin) and 1 PDZ domain point mutant (Erbin V83K); peptide positions are indicated using the standard nomenclature for PDZ domain motifs, with 0 denoting the C-terminal residue, followed by -1, -2, etc. Without backbone flexibility Rosetta fails to predict important residue preferences observed in experimental phage display selections, such as valine at the 0 position or tryptophan at the -1 position for DLG1-2 and Erbin.
We have automated the sequence tolerance protocol for using flexible backbone ensembles and sequence design for predicting peptide binding specificity on a webserver: https://kortemmelab.ucsf.edu/backrub/ (Lauck et al., 2010). Users can also download a “protocol capture” of the sequence tolerance method, complete with example input/output and scripts, in the Supplementary Materials accompanying (Smith and Kortemme, 2011). Given a peptide-bound structure, backrub sampling methods are used to generate an ensemble of conformations. For each conformation, a genetic algorithm is used to design sequences for high affinity binding. In this protocol, the interface energy is given greater weight (Smith and Kortemme, 2010, 2011). To compare these predictions to experimental data, we generate a sequence logo based on the positional frequencies in the resulting designed sequences (Figure 5B). Compared to fixed backbone methods, backrub sampling increases the agreement at several positions. Given the adaptable nature of many protein-protein interfaces, it is clear that flexible backbone methods will provide great insight into the structural and energetic basis for binding specificity. Additionally, as more datasets on mutant binding domains are collected, there is potential to look for co-variation between the sequences tolerated between receptor and peptide positions (Ernst et al., 2010).
Co-variation and Interface Design in Two-component Signaling
Testing computational protein design methods based on comparison with experimental PWMs is informative. However, it involves evaluating amino acid positions independently from each other and therefore may overlook some of the intricate details of pair-wise interactions between designed residues. In order to assess how well flexible backbone design protocols capture dependencies between designed residues, we directly compared designed residue co-variation to native residue co-variation. We chose to examine co-variation within the bacterial two-component signaling system, since it has previously been shown that sensor histidine kinases (HK) and their cognate response regulators (RR) exhibit significant intermolecular co-variation at their protein-protein interface (White et al., 2007).
Designed sequences were obtained by generating a backrub conformational ensemble of 500 structures starting from the co-crystal structure of HK853 and RR468 from Thermotoga maritima ((Casino et al., 2009) PDB 3DGE). Since bacterial HK and RR sequences are highly divergent, we used a temperature of 1.2 kT to produce a conformational ensemble that would yield sufficiently diverse designed sequences. We then performed sequence design using Monte Carlo simulated annealing on each structure, which resulted in 500 designed HK and RR sequences.
The command lines for this protocol are as follows:
Backrub ensemble generation: ∼/rosetta/rosetta_source/bin/backrub.linuxgccrelease -database ∼/rosetta/rosetta_database/ -s 3DGE.pdb -resfile NATAA.res -ex1 -ex2 -extrachi_cutoff 0 -backrub:mc_kt 1.2 -backrub:ntrials 10000 -nstruct 500 -backrub:initial_pack
Sequence design: ∼/rosetta/rosetta_source/bin/fixbb.linuxgccrelease -database ∼/rosetta/rosetta_database/ -s3DGE_0001_last.pdb -resfile ALLAA.res -ex1 -ex2 -extrachi_cutoff 0 -nstruct 1 -overwrite -linmem_ig 10 -no_his_his_pairE -minimize_sidechains
Necessary files to run these command lines with Rosetta version 3.5 are included as example S4 at http://kortemmelab.ucsf.edu/resources/MIE_Supplement.tar.gzresults shown in Figure 6 were obtained with Rosetta revision 39284.
Figure 6. Rosetta backrub design methods capture features of evolutionary amino-acid co-variation.

(A) Comparison between designed and natural intermolecular amino acid co-variation for histidine kinases (HK) and their cognate response regulators (RR). Each point represents a pair of amino acid positions. Natural co-variation was quantified using a mutual information based metric for all pairs of positions in a multiple sequence alignment of HKs concatenated to their cognate RRs. A backrub ensemble of 500 structures was generated for a HK/RR complex (PDB ID 3DGE) and RosettaDesign was used to predict one low energy sequence for each structure in the ensemble. Designed co-variation was quantified for all pairs of positions in the resulting multiple sequence alignment of 500 sequences. The red lines indicate the threshold cut-off for the top 30 designed co-varying intermolecular pairs (horizontal) and the top 30 natural co-varying pairs (vertical). The 12 intermolecular pairs of positions that are highly co-varying in both designed and natural sequences are highlighted in green. (B) The structure of a HK/RR complex with amino acids that are involved in highly co-varying intermolecular pairs in both natural and designed sequences are shown in green and stick representation. (C) Close-up of the 12 intermolecular co-varying pairs. Each of these 12 pairs of amino acids forms a physical interaction across the interface of the complex.
To compare the sequence features from interface design with those observed in naturally interacting proteins, we collected alignments of natural HK and RR sequences from Pfam (PF000512 for HK and PF00072 for RR) and concatenated all pairs of HKs and RRs that were adjacent in a particular genome (i.e., pairs with GI numbers differing by 1). To avoid bias from closely related sequences, we filtered the joint HK/RR alignment for redundancy using an 80% sequence identity cutoff. We quantified residue co-variation of all intermolecular pairs of amino acid positions in designed and natural sequences using a mutual information based statistic (Dickson et al., 2010).
We observed significant overlap between the designed and natural highly co-varying intermolecular pairs within the HK/RR complex (Figure 6A). Mapping the residue pairs that were highly co-varying in both designed and natural sequences onto the structure of the complex revealed that all of these pairs are localized to the HK/RR interface (Figure 6B). A closer examination of these pairs shows that each pair forms a physical interaction across the HK/RR interface (Figure 6C), suggesting that these pairs may be important for determining specificity in bacterial two-component signaling systems. Indeed, several of these positions have previously been mutated to alter the specificity of HK-RR interactions: HK-Thr in Pair 1, HK-Tyr in Pair 9 and HK-Val in Pair 11 (Skerker et al., 2008). The remaining pairs, including those that highly co-vary in designed sequences but not natural sequences, represent potential opportunities for rewiring two-component signaling specificity using computational protein design.
Future Challenges
The success of flexible backbone sampling methods in predicting mutant side chain (Figure 2) and alternative (Figure 3) conformations indicates the broad utility of these methods in designing sequences compatible with a target “native” structure. Previous studies have used Rosetta to provide structural mechanisms for NMR measures of protein dynamics (Friedland et al., 2009; Friedland et al., 2008) and to design mutations that stabilize specific conformations from a dynamic ensemble (Babor and Kortemme, 2009; Bouvignies et al., 2011). Additionally, the comparisons to naturally and artificially selected sequence data suggest that flexible backbone methods can be leveraged to design libraries for generating proteins with new or improved functions (Friedland and Kortemme, 2010).
Despite these successes, exploiting backbone flexibility to design conformational heterogeneity, in contrast to design of a single target structure, remains largely unaddressed. A major challenge in the coming years will be to adapt these methods to design functionally important protein conformational dynamics. Examples of these design challenges include: designing loops to sample multiple conformations that exclude water and permit substrate flux during an enzymatic catalytic cycle, creating peptide binding domains where specificity is encoded by distinct binding modes, or generating coupled networks of side chain conformations that respond to an allosteric binding event.
To meet these lofty challenges, scoring functions must be sensitive to the small gaps that separate these conformations on the energy landscape (Fleishman et al., 2011). In addition, to avoid having populations biased by the sampling algorithm and provide better estimates of conformational entropy, the Monte Carlo move sets must obey detailed balance (Hastings, 1970). In addition to improvements in scoring and thermodynamics, more sophisticated sampling protocols will likely be needed. Here, we have primarily focused on backrub moves around Cα. However, sampling the “native” state of some protein environments may benefit from different strategies or iterations through a combination of sampling moves. Indeed, we have recently had success at modeling conformational changes that propagate away from a designed mutation by iteratively switching between different sampling and scoring strategies during the course of a single simulation (Kapp et al., 2012). Learning from the successes and failures of these new strategies will be essential to improve both protein design and our understanding of the relationship between protein conformational dynamics and function.
Acknowledgments
The Fraser lab is supported by the National Institutes of Health Early Independence Award (DP5 OD009180) and QB3. Rosetta development in the Kortemme lab is supported by awards from the National Science Foundation (NSF) to T.K. (NSF CAREER MCB-0744541; NSFEF-0849400) and the Synthetic Biology Engineering Research Center (NSF EEC-0540879; PI Keasling). Noah Ollikainen was additionally supported by an NSF graduate fellowship. We thank Russell Goodman and Henry van den Bedem for helpful discussions.
References
- Babor M, Kortemme T. Multi-constraint computational design suggests that native sequences of germline antibody H3 loops are nearly optimal for conformational flexibility. Proteins. 2009;75:846–858. doi: 10.1002/prot.22293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best RB, Lindorff-Larsen K, DePristo MA, Vendruscolo M. Relation between native ensembles and experimental structures of proteins. Proc Natl Acad Sci U S A. 2006;103:10901–10906. doi: 10.1073/pnas.0511156103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr DD, Nussinov R, Wright PE. The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvignies G, Vallurupalli P, Hansen DF, Correia BE, Lange O, Bah A, Vernon RM, Dahlquist FW, Baker D, Kay LE. Solution structure of a minor and transiently formed state of a T4 lysozyme mutant. Nature. 2011;477:111–114. doi: 10.1038/nature10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canutescu AA, Dunbrack RL., Jr Cyclic coordinate descent: A robotics algorithm for protein loop closure. Protein Sci. 2003;12:963–972. doi: 10.1110/ps.0242703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casino P, Rubio V, Marina A. Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell. 2009;139:325–336. doi: 10.1016/j.cell.2009.08.032. [DOI] [PubMed] [Google Scholar]
- Davis IW, Arendall WB, 3rd, Richardson DC, Richardson JS. The backrub motion: how protein backbone shrugs when a sidechain dances. Structure. 2006;14:265–274. doi: 10.1016/j.str.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Dickson RJ, Wahl LM, Fernandes AD, Gloor GB. Identifying and seeing beyond multiple sequence alignment errors using intra-molecular protein covariation. PLoS One. 2010;5:e11082. doi: 10.1371/journal.pone.0011082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi U, McGowan LC, Ladani ST, Hamelberg D. Resolving the complex role of enzyme conformational dynamics in catalytic function. Proc Natl Acad Sci U S A. 2012;109:5699–5704. doi: 10.1073/pnas.1117060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBay KH, Geissler PL. Calculation of proteins' total side-chain torsional entropy and its influence on protein-ligand interactions. J Mol Biol. 2009;391:484–497. doi: 10.1016/j.jmb.2009.05.068. [DOI] [PubMed] [Google Scholar]
- Eisenmesser EZ, Bosco DA, Akke M, Kern D. Enzyme dynamics during catalysis. Science. 2002;295:1520–1523. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- Ernst A, Gfeller D, Kan Z, Seshagiri S, Kim PM, Bader GD, Sidhu SS. Coevolution of PDZ domain-ligand interactions analyzed by high-throughput phage display and deep sequencing. Mol Biosyst. 2010;6:1782–1790. doi: 10.1039/c0mb00061b. [DOI] [PubMed] [Google Scholar]
- Fleishman SJ, Baker D. Role of the biomolecular energy gap in protein design, structure, and evolution. Cell. 2012;149:262–273. doi: 10.1016/j.cell.2012.03.016. [DOI] [PubMed] [Google Scholar]
- Fleishman SJ, Leaver-Fay A, Corn JE, Strauch EM, Khare SD, Koga N, Ashworth J, Murphy P, Richter F, Lemmon G, Meiler J, Baker D. RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS One. 2011;6:e20161. doi: 10.1371/journal.pone.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, Alber T. Hidden alternative structures of proline isomerase essential for catalysis. Nature. 2009;462:669–673. doi: 10.1038/nature08615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc Natl Acad Sci U S A. 2011;108:16247–16252. doi: 10.1073/pnas.1111325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- Friedland GD, Kortemme T. Designing ensembles in conformational and sequence space to characterize and engineer proteins. Curr Opin Struct Biol. 2010;20:377–384. doi: 10.1016/j.sbi.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Friedland GD, Lakomek NA, Griesinger C, Meiler J, Kortemme T. A correspondence between solution-state dynamics of an individual protein and the sequence and conformational diversity of its family. PLoS Comput Biol. 2009;5:e1000393. doi: 10.1371/journal.pcbi.1000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland GD, Linares AJ, Smith CA, Kortemme T. A simple model of backbone flexibility improves modeling of side-chain conformational variability. J Mol Biol. 2008;380:757–774. doi: 10.1016/j.jmb.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnham N, Blundell TL, DePristo MA, Terwilliger TC. Is one solution good enough? Nat Struct Mol Biol. 2006;13:184–185. doi: 10.1038/nsmb0306-184. discussion 185. [DOI] [PubMed] [Google Scholar]
- Hastings WK. Monte-Carlo Sampling Methods Using Markov Chains and Their Applications. Biometrika. 1970;57:97–&. [Google Scholar]
- Huang H, Sidhu SS. Studying binding specificities of peptide recognition modules by high-throughput phage display selections. Methods Mol Biol. 2011;781:87–97. doi: 10.1007/978-1-61779-276-2_6. [DOI] [PubMed] [Google Scholar]
- Kapp GT, Liu S, Stein A, Wong DT, Remenyi A, Yeh BJ, Fraser JS, Taunton J, Lim WA, Kortemme T. Control of protein signaling using a computationally designed GTPase/GEF orthogonal pair. Proc Natl Acad Sci U S A. 2012;109:5277–5282. doi: 10.1073/pnas.1114487109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley NW, Vishal V, Krafft GA, Pande VS. Simulating oligomerization at experimental concentrations and long timescales: A Markov state model approach. J Chem Phys. 2008;129:214707. doi: 10.1063/1.3010881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzhnev DM, Religa TL, Banachewicz W, Fersht AR, Kay LE. A transient and low-populated protein-folding intermediate at atomic resolution. Science. 2010;329:1312–1316. doi: 10.1126/science.1191723. [DOI] [PubMed] [Google Scholar]
- Lang PT, Ng HL, Fraser JS, Corn JE, Echols N, Sales M, Holton JM, Alber T. Automated electron-density sampling reveals widespread conformational polymorphism in proteins. Protein Sci. 2010;19:1420–1431. doi: 10.1002/pro.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauck F, Smith CA, Friedland GF, Humphris EL, Kortemme T. RosettaBackrub--a web server for flexible backbone protein structure modeling and design. Nucleic Acids Res. 2010;38:W569–575. doi: 10.1093/nar/gkq369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban YE, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popovic Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin EJ, Kondrashov DA, Wesenberg GE, Phillips GN., Jr Ensemble refinement of protein crystal structures: validation and application. Structure. 2007;15:1040–1052. doi: 10.1016/j.str.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Raychaudhuri S, Wand AJ. Insights into the local residual entropy of proteins provided by NMR relaxation. Protein Sci. 1996;5:2647–2650. doi: 10.1002/pro.5560051228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCallum JL, Perez A, Schnieders MJ, Hua L, Jacobson MP, Dill KA. Assessment of protein structure refinement in CASP9. Proteins. 2011;79(10):74–90. doi: 10.1002/prot.23131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell DJ, Coutsias EA, Kortemme T. Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat Methods. 2009;6:551–552. doi: 10.1038/nmeth0809-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell DJ, Kortemme T. Backbone flexibility in computational protein design. Curr Opin Biotechnol. 2009;20:420–428. doi: 10.1016/j.copbio.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Maragakis P, Lindorff-Larsen K, Eastwood MP, Dror RO, Klepeis JL, Arkin IT, Jensen MO, Xu H, Trbovic N, Friesner RA, Iii AG, Shaw DE. Microsecond molecular dynamics simulation shows effect of slow loop dynamics on backbone amide order parameters of proteins. J Phys Chem B. 2008;112:6155–6158. doi: 10.1021/jp077018h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiler J, Prompers JJ, Peti W, Griesinger C, Bruschweiler R. Model-free approach to the dynamic interpretation of residual dipolar couplings in globular proteins. J Am Chem Soc. 2001;123:6098–6107. doi: 10.1021/ja010002z. [DOI] [PubMed] [Google Scholar]
- Nagel ZD, Klinman JP. A 21st century revisionist's view at a turning point in enzymology. Nat Chem Biol. 2009;5:543–550. doi: 10.1038/nchembio.204. [DOI] [PubMed] [Google Scholar]
- Rieping W, Habeck M, Nilges M. Inferential structure determination. Science. 2005;309:303–306. doi: 10.1126/science.1110428. [DOI] [PubMed] [Google Scholar]
- Schneider TR, Brunger AT, Nilges M. Influence of internal dynamics on accuracy of protein NMR structures: derivation of realistic model distance data from a long molecular dynamics trajectory. J Mol Biol. 1999;285:727–740. doi: 10.1006/jmbi.1998.2323. [DOI] [PubMed] [Google Scholar]
- Shaw DE, Maragakis P, Lindorff-Larsen K, Piana S, Dror RO, Eastwood MP, Bank JA, Jumper JM, Salmon JK, Shan Y, Wriggers W. Atomic-level characterization of the structural dynamics of proteins. Science. 2010;330:341–346. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Simons KT, Kooperberg C, Huang E, Baker D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J Mol Biol. 1997;268:209–225. doi: 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- Skerker JM, Perchuk BS, Siryaporn A, Lubin EA, Ashenberg O, Goulian M, Laub MT. Rewiring the specificity of two-component signal transduction systems. Cell. 2008;133:1043–1054. doi: 10.1016/j.cell.2008.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CA, Kortemme T. Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant side-chain prediction. J Mol Biol. 2008;380:742–756. doi: 10.1016/j.jmb.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CA, Kortemme T. Structure-based prediction of the peptide sequence space recognized by natural and synthetic PDZ domains. J Mol Biol. 2010;402:460–474. doi: 10.1016/j.jmb.2010.07.032. [DOI] [PubMed] [Google Scholar]
- Smith CA, Kortemme T. Predicting the tolerated sequences for proteins and protein interfaces using RosettaBackrub flexible backbone design. PLoS One. 2011;6:e20451. doi: 10.1371/journal.pone.0020451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyka MD, Keedy DA, Andre I, Dimaio F, Song Y, Richardson DC, Richardson JS, Baker D. Alternate states of proteins revealed by detailed energy landscape mapping. J Mol Biol. 2011;405:607–618. doi: 10.1016/j.jmb.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bedem H, Dhanik A, Latombe JC, Deacon AM. Modeling discrete heterogeneity in X-ray diffraction data by fitting multi-conformers. Acta Crystallogr D Biol Crystallogr. 2009;65:1107–1117. doi: 10.1107/S0907444909030613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RA, Szurmant H, Hoch JA, Hwa T. Features of protein-protein interactions in two-component signaling deduced from genomic libraries. Methods Enzymol. 2007;422:75–101. doi: 10.1016/S0076-6879(06)22004-4. [DOI] [PubMed] [Google Scholar]