

Abstract
Modification of the epigenome may be a mechanism underlying toxicity and disease following chemical exposure. Animal and human data suggest that mercury (Hg) impacts DNA methylation. We hypothesize that methylmercury and inorganic Hg exposures from fish consumption and dental amalgams, respectively, may be associated with altered DNA methylation at global repetitive elements (long interspersed elements, LINE-1) and candidate genes related to epigenetic processes (DNMT1) and protection against Hg toxicity (SEPW1, SEPP1). Dental professionals were recruited at Michigan Dental Association (MDA) meetings in 2009 and 2010. Subjects (n=131) provided survey data (e.g. exposure sources, demographics) and biological samples for Hg measurement and epigenetic analysis. Total Hg was quantified via atomic absorption spectrophotometry in hair and urine, indicative of methylmercury and inorganic Hg exposures, respectively. Global repetitive and candidate gene methylation was quantified via pyrosequencing of bisulfite converted DNA isolated from buccal mucosa. Hair Hg (geometric mean (95% CI): 0.37 (0.31–0.44) μg/g) and urine Hg (0.70 (0.60–0.83) μg/L) were associated with sources of exposure (fish consumption and dental amalgams, respectively). Multivariable linear regression revealed a trend of SEPP1 hypomethylation with increasing hair Hg levels, and this was significant (p<0.05) among males. The trend remained when excluding non-dentists. No significant relationships between urine Hg and DNA methylation were observed. Thus, in a limited cohort, we identified an association between methylmercury exposure and hypomethylation of a potentially labile region of the genome (SEPP1 promoter), and this relationship was gender specific.
Keywords: epigenetics, environmental exposure, occupational exposure, selenoproteins
INTRODUCTION
Mercury (Hg) is a heavy metal pollutant of concern to public health (e.g., fish eaters, persons with mercury amalgam dental restorations) and to occupationally exposed workers (e.g., dentists, miners). Several regulatory bodies, including the U.S. Environmental Protection Agency, rank this potent toxic agent as a top three priority pollutant of concern [ATSDR, 2011]. Mercury remains a health risk as over 6,000 tons are estimated to be released into the environment annually from all sources, and concentrations continue to rise throughout the world [Swain et al., 2007].
The toxicity of Hg is complex and ultimately dictated by its chemical speciation. The public is primarily exposed to organic Hg (as methylmercury, MeHg) through fish consumption, and to inorganic Hg (I-Hg, as elemental Hg vapor) from dental amalgams [Clarkson and Magos, 2006; Mergler et al., 2007]. Dental professionals (dentists, dental hygienists, and dental assistants) are a potentially high risk group due to dual Hg exposures. In the workplace, dental professionals handle elemental Hg (present in amalgams), some have mercury amalgam dental restorations in their mouths, and at home they consume MeHg (present in fish). The toxic effects of high levels of MeHg and I-Hg exposure have been established, but underlying mechanisms and subtle health effects following lower level and chronic “real-world” exposures to Hg are not well characterized despite increasing relevance and concern to both public health and occupational health [Karagas et al., 2012].
Recent epidemiological and toxicological studies are beginning to show that epigenetic alterations may be key, subclinical mechanisms linking chemical exposure to toxicity and disease [Baccarelli and Bollati, 2009; Dolinoy et al., 2007a; Dolinoy et al., 2007b]. The epigenome consists of heritable alterations to the genome that influence gene expression and regulation without changing the underlying DNA sequence. Epigenetic changes have been linked to multiple diseases including hypertension, Alzheimer’s disease, and schizophrenia [Desplats et al., 2011; Millis, 2011; Nohesara et al., 2011]. DNA methylation is one mechanism of epigenetic regulation that involves the addition of a methyl group to cytosine residues upstream of guanine residues (CpG sites) in the DNA sequence. Exposure to many environmental chemicals (e.g., bisphenol A, arsenic, chromium, benzene, lead) is associated with alterations to DNA methylation in animal models, humans, and in vitro [Baccarelli and Bollati, 2009; Dolinoy et al., 2007a; Wright et al., 2010].
Health effects related to MeHg exposure often follow long-term chronic exposure or substantial latency periods (e.g., acute myocardial infarction risk [Guallar et al., 2002]; neurodevelopmental impacts associated with prenatal exposure [Grandjean et al., 2010]). As such, epigenetic alteration may underlie latent health effects from MeHg exposure. Recent animal studies support a connection between MeHg exposure and DNA methylation. Ecotoxicological studies of polar bears in the wild [Pilsner et al., 2010] and captive mink in the laboratory [Basu et al., under review] found associations between MeHg exposure and global CCGG hypomethylation in the brain. In rats exposed developmentally to MeHg, Desaulniers et al. observed decreased methylation in the promoter regions of specific genes as well as decreased expression of key enzymes in the DNA methylation pathway [Desaulniers et al., 2009], while in mice exposed prenatally, Onishchenko et al. found the opposite- hypermethylation of a candidate gene [Onishchenko et al., 2008]. Despite growing evidence of MeHg-associated epigenetic changes in a range of animal models, there is a paucity of data on the impact of MeHg on the human epigenome. Hypermethylation of a CpG site in women with high blood Hg levels compared with low blood Hg was noted in one study [Hanna et al., 2012]. In terms of I-Hg exposure, in vitro studies suggest I-Hg (as mercuric chloride) modifies the epigenome via changes to histone methylation [Gadhia et al., 2012] or inhibition of methionine synthase, a player in the methyl donor pathway [Waly et al., 2004]. Such epigenetic effects of I-Hg, to our knowledge, have yet to be established in a whole organism.
Risk assessment of Hg is largely based on health effects observed in populations exposed to high levels of MeHg (e.g., subsistence fish eaters) or I-Hg (e.g., chloralkali manufacturers). While several studies investigated chronic low dose Hg exposure and its association with subclinical effects on nervous, cardiovascular and immune systems, the true impact of chronic exposure to relevant and low doses of Hg remains uncertain and conflicted [Franzblau et al., 2012; Goodrich et al., 2012; Karagas et al., 2012; Roman et al., 2011]. While recent animal and in vitro studies suggest MeHg and I-Hg exposure may modify the epigenome, epidemiological evidence is limited to one cohort of women with blood Hg measurements [Hanna et al., 2012]. As such, the impact of MeHg and I-Hg on the epigenome (specifically DNA methylation), a subclinical measure that may give insight into mechanisms underlying toxic effects, remains uncertain in human populations.
Here we hypothesize that low dose exposures to MeHg and I-Hg via fish consumption and dental amalgams, respectively, will be associated with altered DNA methylation at multi-copy repeats (long interspersed element, LINE-1) and candidate genes (DNMT1, SEPW1, SEPP1) in a cohort of dental professionals. LINE-1 methylation is sensitive to environmental insults [Rusiecki et al., 2008; Wright et al., 2010], and is biologically important as LINE-1 hypomethylation is linked to genomic instability [Schulz et al., 2006]. We chose the candidate genes based on known Hg-induced expression changes [Chen et al., 2006; Desaulniers et al., 2009; Kim et al., 2005], and in the case of SEPW1, potential as an epigenetically labile region [Desplats et al., 2011]. Biological samples from members of the Michigan Dental Association (MDA) were used to test the hypothesis as this cohort is uniquely exposed both environmentally to MeHg and occupationally to I-Hg.
MATERIALS AND METHODS
Study Population
During the 2009 and 2010 Michigan Dental Association (MDA) Annual Conventions, a convenience sample of 515 dentists, dental hygienists, dental assistants, and other related professionals was recruited as part of a study assessing the impact of genetic polymorphisms on Hg accumulation as previously described [Goodrich et al., 2011; Wang et al., 2012]. In brief, each subject completed a questionnaire and provided a urine sample, a hair sample, and four buccal swabs. A subset of participants with adequate total buccal mucosa DNA (>1 μg), DNA concentration (> 17.5 ng/μL) and quality (passed one or more epigenetic assays) were included in the present study. Further inclusion criteria consisted of available Hg biomarker data (hair and/or urine) and key demographic information (e.g., BMI, age). The final sample (n=131) was similar to the total MDA cohort in terms of Hg biomarker levels, exposure variables, and demographic variables (data not shown). Institutional Review Board approval from the University of Michigan was obtained for subject recruitment, sample collection, and all analyses performed (HUM00027621).
Survey Data
Participants completed a scantron survey providing detailed information on demographic variables (e.g., BMI, age), and sources of exposure to I-Hg (number of mercury amalgam dental restorations in their mouth, occupation, dental amalgams handled in the office, Hg spills in the office) and MeHg (fish consumption patterns: frequency, portion size, species). Estimated Hg intake from fish was calculated using fish consumption data as previously described [Goodrich et al., 2011; Wang et al., 2012].
Mercury Exposure Assessment
Urine and hair samples were collected as biomarkers of I-Hg and MeHg exposures, respectively [Berglund et al., 2005]. Spot urine samples were collected in Hg-free containers (Becton, Dickinson, and Company; NJ) and were stored frozen. Hair samples were cut from the occipital region of the scalp, and two cm closest to the scalp were used for analysis. Total Hg levels were quantified with a Direct Mercury Analyzer-80 according to U.S. Environmental Protection Agency Method 7473 as described by Nam and Basu [Nam and Basu, 2011]. All Hg concentrations exceeded the detection limit, and ample quality control checks were included in the analysis as previously detailed [Goodrich et al., 2011; Wang et al., 2012].
DNA Isolation and Bisulfite Treatment
Buccal mucosa samples were collected by swabbing the inside of both cheeks with four consecutive cotton swabs for 15 seconds each. Cotton swabs were immediately stored at room temperature in a preservative buffer [Goodrich et al., 2011; Min et al., 2006]. Buccal mucosa DNA was purified within one week of collection using the Promega SV Wizard Genomic DNA purification kit (Promega Cooperation, Madison, WI) and stored at −20°C. DNA concentration was quantified via absorbance at 260 nm with a NanoDrop 2000 (Thermo Fisher Scientific, Pittsburgh, PA). Genomic DNA was treated with sodium bisulfite, which converts unmethylated cytosines to uracils and leaves methylated cytosines unchanged [Grunau et al., 2001] with the EpiTect Bisulfite Kit (Qiagen, Valencia, CA).
Epigenetic Analysis
Global repetitive DNA methylation (LINE-1) and candidate gene methylation (DNMT1, SEPW1, SEPP1) were quantified via pyrosequencing based on approaches previously outlined [Anderson et al., 2012; Virani et al., 2012]. The pyrosequenced regions for LINE-1 and the candidate genes were less than 100 base pairs and each included four CpG sites. Pyrosequencing assays for SEPP1, SEPW1, and LINE-1 were designed with PyroMark Assay Design Software version 2.0. Pre-designed primers for DNMT1 were ordered from Qiagen (PyroMark CpG Assay # PM00075761). Approximately 50 ng bisulfite converted DNA was used in a PCR reaction for each assay along with HotStartTaq Master Mix (Qiagen), and primers (forward and biotinylated reverse) listed in Supporting Information Table S-I. The PyroMark MD Pyrosequencer platform (Qiagen) was utilized according to manufacturer’s suggested protocol to quantify DNA methylation at the selected CpG sites (computed by Pyro Q-CpG Software). The Pyro Q-CpG Software automatically incorporates internal quality controls by incorporation of C nucleotides into the dispensation order for non-CpG cytosines in the original sequence that should have been fully converted to Ts following modification by bisulfite to assess the efficiency of bisulfite conversion. Complete bisulfite conversion was required for samples to pass the pyrosequencing quality controls. Further quality control measures included quantifying methylation of commercially available 0% and 100% methylated human control DNA (Epitect Control DNA, Qiagen) on each 96-well plate, and measures of 100% control DNA were precise across plates (1.4 – 4.2% coefficient of variation, CV, for four pyrosequencing assays). At least one plate for each assay contained a standard curve of control DNA (0, 20, 40, 60, 80, 100% methylation) to ensure accuracy throughout, and control values fell in the expected ranges. MDA samples and controls were run in duplicate, and methylation values were averaged. Duplicate samples had low variability for LINE-1 (2.6% CV) and SEPP1 (4.9%) assays. Pyrosequencing is less precise at low methylation levels (<5%) and as such DNMT1 and SEPW1 had high repeated measures variability (29.8% and 41.7% CV, respectively), though precision improved greatly among samples with methylation values above 3% (DNMT1: 9.8% CV; SEPW1: 10.5%).
Statistical Analyses
Statistical analyses were performed with SPSS Statistics version 19 (IBM, Somers, NY) to assess relationships between biomarkers of MeHg (hair) and I-Hg (urine) and percent methylation of LINE-1 and the three candidate genes. All statistical analyses were run with both the mean methylation of four CpG sites and with methylation values of individual CpG sites. Hair and urine Hg levels were natural log-transformed to achieve normality.
Descriptive statistics were first calculated for variables of interest (e.g., percent methylation, Hg biomarker levels, Hg exposure variables, demographics). Key variables were stratified by gender. One-way analysis of variance (ANOVA) compared continuous variables across genders while Pearson’s chi-squared test compared categorical variables (e.g., occupation). Bivariate analyses (Pearson correlations) were run to assess relationships between DNA methylation, Hg biomarker levels, age, and BMI.
Multivariable linear regression was used to explore relationships between DNA methylation and Hg biomarker levels while adjusting for factors influencing methylation. Both mean percent methylation of four CpG sites of LINE-1, DNMT1, SEPW1, and SEPP1 and methylation of specific CpG sites were modeled. Manual backwards stepwise selection was used to form base models with available variables (e.g., BMI, age, gender) before testing the impact of Hg biomarker levels in the models. BMI and age were significant predictors (p<0.05) in some models and were ultimately retained in all final models for consistency. All models were performed using the total population and subgroups: males, females, dentists (dentist models not shown). Due to divergent findings for the relationship between hair Hg and SEPP1 methylation, when stratifying by gender, diagnostic parameters (deleted residuals, standardized dfbetas) were calculated to elucidate influential subjects in the SEPP1 models. Eight subjects strongly influenced the model (standardized dfbeta >0.25 for coefficients of hair Hg, urine Hg, or both). Statistical results were compared when including or excluding all eight influential subjects.
RESULTS
Descriptive Statistics
Study population characteristics are summarized in Table I. The majority of participants were dentists (57%) while remaining participants had other related occupations (e.g., dental hygienist, dental assistant). Gender distribution was disparate across occupational groups as the majority of dentists were male (80%) and most non-dentists were female (93%; χ2 p-value <0.001). Nearly all MDA members self-identified as Caucasian (90%).
Table I.
Descriptive Statistics among All Subjects and Stratified by Gender. (Mean ± SD reported for BMI, age, dental amalgam, and fish Hg intake; geometric mean (95% CI) reported for urine Hg and hair Hg; percentage reported for dentists.)
| Total | Males | Females | |
|---|---|---|---|
| N | 131 | 64 | 67 |
| BMI (kg/m2) | 27.1 ± 4.7 | 27.8 ± 4.3 | 26.4 ± 5.1 |
| Age (yrs) | 55.8 ± 11.6 | 61.7 ± 10d | 50.1 ± 10.1d |
| Personal Dental Amalgam (#) | 4.4 ± 4.2 | 4.4 ± 4.3 | 4.4 ± 4.1 |
| Fish Hg Intake (μg/kg bw/ day)a | 0.08 ± 0.1 | 0.07 ± 0.09 | 0.09 ± 0.1 |
| Urine Hg (μg/L) | 0.70 (0.60–0.83) | 0.89 (0.71–1.12)c | 0.56 (0.45–0.70)c |
| Hair Hg (μg/g)b | 0.37 (0.31–0.44) | 0.46 (0.36–0.59)c | 0.30 (0.24–0.37)c |
| Dentist (%) | 57.3% | 93.8%e | 22.4%e |
Estimate based on self-reported fish consumption and average Hg values in commonly consumed fish species (see [Wang et al., 2012] for calculation details).
n=128 for hair Hg (62 males, 66 females).
p<0.05 ANOVA comparing males and females (log-transformed values).
p<0.001 ANOVA comparing males and females.
p<0.001 χ2 test comparing males and females.
Geometric means of urine Hg (0.70 μg/L) and hair Hg (0.37 μg/g) are similar to that of the general U.S. population according to the U.S. National Health and Nutrition Examination Survey (NHANES) [Centers for Disease Control and Prevention, 2009; McDowell et al., 2004] as discussed in our previous paper [Wang et al., 2012]. Personal dental amalgams and occupation were the biggest predictors of urine Hg levels while estimated Hg from fish consumption predicted hair Hg levels (see [Goodrich et al., 2011]). Males had significantly higher urine Hg and hair Hg levels compared with females (Table I), reflective of the difference observed between dentists and non-dentists (ANOVA p-value <0.001).
Global Repetitive Methylation (LINE-1)
Methylation at four CpG sites of the repetitive element, LINE-1, was quantified by pyrosequencing bisulfite converted DNA from buccal swab (n=131) samples. Table II lists mean methylation (± SD) of individual CpG sites along with the average of all sites. The Pyro Q-CpG Software has stringent internal quality control checks that indicate whether methylation readings pass or fail, and in the current study LINE-1 pyrosequencing had a low pass rate (38%). Of the 53 passing samples, mean LINE-1 methylation ranged from 61.4% to 76.9%, and CpG #4 had the biggest range among the specific sites (50.2% to 81%). Methylation of individual LINE-1 CpG sites was highly correlated (Pearson r=0.4 to 0.75). LINE-1 methylation was positively correlated with age (Pearson r=0.38, p=0.005) and did not differ significantly when stratified by gender, occupation, or race.
Table II.
Methylation (%) of LINE-1 and Candidate Genes (mean ± SD). The mean methylation of all CpG sites is reported along with % methylation at specific CpGs.
| N | Mean | CpG #1 | CpG #2 | CpG #3 | CpG #4 | |
|---|---|---|---|---|---|---|
| LINE-1 | 53 | 68.4 ± 3.7 | 66.1 ± 5.4 | 72.0 ± 3.0 | 67.6 ± 4.6 | 67.9 ± 4.9 |
| DNMT1 | 123 | 1.7 ± 1.4 | 0.6 ± 1.1 | 0.7 ± 0.9 | 0.7 ± 0.8 | 4.9 ± 4.4 |
| SEPW1 | 125 | 1.6 ± 0.9 | 0.7 ± 0.8 | 0.5 ± 0.7 | 4.1 ± 2.3 | 1.4 ± 1.0 |
| SEPP1 | 119 | 36.7 ± 18.2 | 30.3 ± 19.1 | 44.5 ± 18.5 | 33.9 ± 21.2 | 38.5 ± 15.0 |
Candidate Gene Methylation
Pyrosequencing of bisulfite converted DNA quantified methylation at four CpG sites in three candidate genes (DNMT1, SEPW1, SEPP1; see Table II). The candidate genes DNMT1 and SEPW1 exhibited low methylation levels (<10%), and the pyrosequencing assays were successful (88% and 89% passing rates, respectively). Average methylation of all CpG sites ranged from 0 to 7.4% for DNMT1 with CpG site #4 having the largest range among individual sites (0–29.6%). Mean methylation ranged from 0 to 4.7% for SEPW1 with CpG #3 displaying the most inter-individual variability (range 0–14.3%). Percent methylation of SEPP1 was quantified (85% passing rate), and results displayed high inter-individual variability (range 11.5 to 91.3%). Among individual sites, CpG #3 had the largest range (4–99.5%). Methylation values of individual CpG sites within each gene were correlated (Pearson r=0.17 to 0.98, p<0.05), though the majority of sites were significantly different compared to other sites in the same gene (Paired Samples T-Test, p-value <0.05). Stratification of mean DNMT1, SEPW1, and SEPP1 methylation revealed no significant differences by gender, occupation, or race (data not shown). Age and BMI were positively correlated with mean SEPP1 methylation (for both: Pearson r=0.26, p=0.004).
DNA Methylation and Hg: Bivariate Relationships
Bivariate analyses (Pearson correlations) revealed no significant (p<0.05) correlations between natural-log transformed hair or urine Hg levels and DNA methylation of LINE-1 or three candidate genes (Table III). Following stratification by gender, a significant negative correlation was observed between average SEPP1 methylation and hair Hg in males only (r=−0.28, p=0.04; depicted in Figure 1 A); this association persisted with individual SEPP1 CpG sites. The trend in males alone remained when excluding non-dentists (Figure 1 B) and after excluding eight influential subjects (Supporting Information Figure S1).
Table III.
Bivariate Analyses. Pearson correlations, r, are reported (with p-values in parentheses) for relationships observed in the total population. Urine Hg and hair Hg are natural log-transformed, and methylation values are averaged for all CpG sites in the amplicon.
| LINE-1 | DNMT1 | SEPW1 | SEPP1 | |
|---|---|---|---|---|
| BMI | 0.2 (0.14) | 0.09 (0.34) | −0.09 (0.34) | 0.26 (0.004) |
| Age | 0.38 (0.005) | 0.11 (0.23) | −0.05 (0.62) | 0.26 (0.004) |
| Urine Hg | 0.19 (0.18) | −0.01 (0.88) | 0.03 (0.73) | 0.11 (0.24) |
| Hair Hg | 0.04 (0.76) | −0.08 (0.38) | 0.01 (0.95) | −0.04 (0.67) |
Figure 1. Decreasing SEPP1 Methylation Levels with Hair Hg among Males.
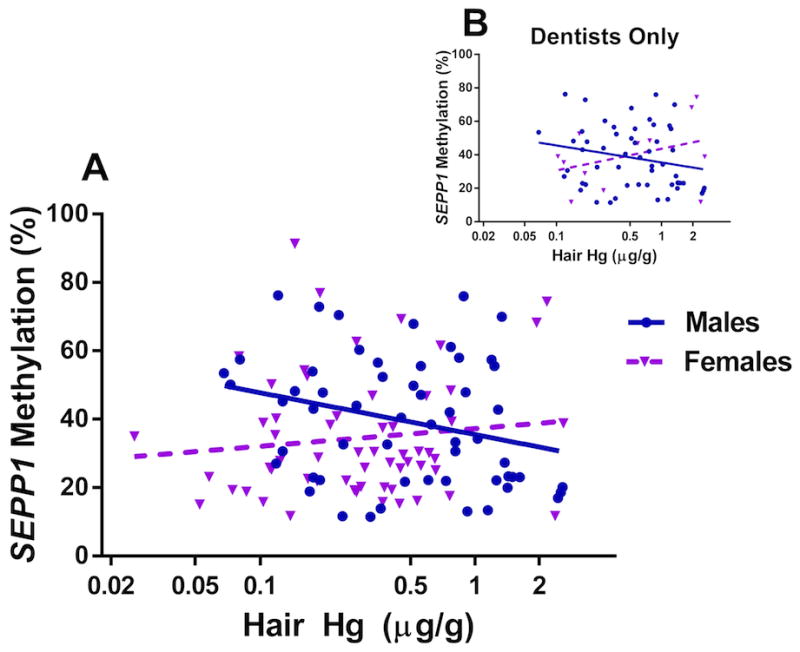
Mean % methylation of four CpG sites in SEPP1 is plotted against natural log-transformed hair Hg levels (μg/g) with gender stratification for A) all MDA subjects (n=57 males, 59 females) and B) dentists only (n=53 males, 12 females). SEPP1 is significantly hypomethylated with increasing hair Hg among males (Pearson r= −0.28, p=0.04), and the trend persists when excluding non-dentist males (r= −0.22, p=0.12). This trend is not apparent among all females (r= 0.12, p=0.37) or female dentists (r= 0.36, p=0.25).
DNA Methylation and Hg: Multivariable Linear Regression
Associations between global repetitive and gene specific DNA methylation and Hg biomarker levels were interrogated via multivariable linear regression adjusting for influential covariates (age, BMI). Table IV reports parameter estimates for hair Hg and urine Hg in models of average methylation at each region in the total population and stratified by gender. For consistency, age (significant predictor of LINE-1 and SEPP1 methylation) and BMI (significant predictor of SEPP1) were included in all final models, and results were similar to models including only significant predictors for each dependent variable (data not shown). Statistically significant relationships between hair Hg or urine Hg and methylation of LINE-1, DNMT1, or SEPW1 were not observed.
Table IV.
Linear Regression Models of Global Repetitive (LINE-1) and Candidate Gene Methylation. Mean methylation is modeled for each region in the total population and in gender groups. Beta coefficients, SE, and p-values are displayed for Hg biomarkers (natural log-transformed) after adjusting for age and BMI.
| Population | Hair Hg | Urine Hg | |||
|---|---|---|---|---|---|
| β (SE) | p-value | β (SE) | p-value | ||
| LINE-1 | Total | 0.12 (0.55) | 0.83 | 0.37 (0.57) | 0.52 |
| Males | −0.53 (0.72) | 0.47 | 0.04 (0.67) | 0.95 | |
| Females | 0.81 (0.84) | 0.35 | 1.36 (1.07) | 0.22 | |
|
| |||||
| DNMT1 | Total | −0.13 (0.14) | 0.36 | −0.03 (0.15) | 0.85 |
| Males | −0.15 (0.2) | 0.47 | 0.24 (0.2) | 0.24 | |
| Females | −0.02 (0.2) | 0.93 | −0.34 (0.23) | 0.15 | |
|
| |||||
| SEPW1 | Total | −0.01 (0.09) | 0.87 | 0.06 (0.09) | 0.55 |
| Males | 0.05 (0.14) | 0.73 | 0.13 (0.14) | 0.36 | |
| Females | −0.08 (0.12) | 0.49 | −0.05 (0.13) | 0.7 | |
|
| |||||
| SEPP1 | Total | −2.02 (1.8) | 0.26 | 2.38 (1.84) | 0.2 |
| Males | −5.14 (2.43) | 0.04 | 0.44 (2.46) | 0.86 | |
| Females | 0.86 (2.73) | 0.75 | 3.62 (2.83) | 0.21 | |
A significant association between hair Hg and SEPP1 hypomethylation was noted, though only among males (Figure 1). This trend persisted in SEPP1 site-specific models among males (Table V). In an effort to tease apart disparate results for males and females, standardized dfbetas were calculated and revealed eight subjects strongly influencing the parameter coefficients for hair Hg and/or urine Hg in the multivariable linear regression models of mean SEPP1 methylation. When the eight subjects (four male dentists, two females dentists, and two female non-dentists) were excluded, the link between average and site-specific SEPP1 hypomethylation and hair Hg concentrations was strengthened in the total population (hair Hg beta= −2.02, p=0.26 with all subjects; beta= −3.01, p=0.07 after exclusion) and males alone (hair Hg beta= −5.14, p=0.04 all males; beta= −6.21, p=0.007 after exclusion) while the association between hair Hg and SEPP1 methylation remained negligible in females (see Table S-II and Figure S1 in Supporting Information).
Table V.
Linear Regression Models of Individual SEPP1 CpG Sites. Beta coefficients, SE, and p-values are displayed for Hg biomarkers (natural log-transformed) after adjusting for age and BMI when modeling percent methylation of individual SEPP1 CpG site (in total population and stratified by gender).
| SEPP1 Site | Population | Hair Hg | Urine Hg | ||
|---|---|---|---|---|---|
| β (SE) | p-value | β (SE) | p-value | ||
| CpG #1 | Total | −2.57 (1.88) | 0.17 | 2.87 (1.92) | 0.14 |
| Males | −5.6 (2.46) | 0.03 | 0.51 (2.5) | 0.84 | |
| Females | 0.07 (2.91) | 0.98 | 4.48 (3.02) | 0.14 | |
|
| |||||
| CpG #2 | Total | −1.76 (1.83) | 0.34 | 2.27 (1.87) | 0.23 |
| Males | −5.11 (2.5) | 0.05 | 1.11 (2.54) | 0.66 | |
| Females | 1.61 (2.79) | 0.57 | 2.69 (2.89) | 0.36 | |
|
| |||||
| CpG #3 | Total | −2.61 (2.09) | 0.21 | 3.3 (2.13) | 0.13 |
| Males | −5.78 (2.81) | 0.04 | 0.89 (2.85) | 0.76 | |
| Females | 0.24 (3.17) | 0.94 | 5.03 (3.29) | 0.13 | |
|
| |||||
| CpG #4 | Total | −1.28 (1.49) | 0.39 | 0.97 (1.52) | 0.53 |
| Males | −4.55 (2.0) | 0.03 | −1.18 (2.03) | 0.57 | |
| Females | 1.53 (2.23) | 0.5 | 2.3 (2.31) | 0.32 | |
DISCUSSION
This work evaluates associations between MeHg and I-Hg exposures (estimated via hair and urine biomarkers, respectively) and buccal mucosa DNA methylation of LINE-1 and three genes (DNMT1, SEPW1, SEPP1) in a cohort of environmentally and occupationally exposed dental professionals. Though the cohort was limited by small sample size and differences between gender groups (primary occupation, age, Hg biomarker concentrations), a statistically significant relationship between SEPP1 hypomethylation and increasing hair Hg concentrations was apparent among males. Promoter methylation of SEPP1, which encodes a protein known to bind Hg, was highly variable and may be sensitive to modification by various environmental agents. In addition to assessing MeHg exposure, this research is the first epidemiological study to explore relationships between I-Hg and DNA methylation to our knowledge, though no significant findings surfaced.
The importance of SEPP1 in Hg toxicokinetics and protection against toxicity via direct binding and antioxidant properties is well established [Chen et al., 2006; Khan and Wang, 2009], though there is a scarcity of data on epigenetic regulation of SEPP1. Here, we quantified percent methylation within a CpG island in the promoter region of SEPP1 and found wide inter-individual variability. Metastable epialleles, which are stochastically methylated in underlying genetically identical regions, have high variability and are more vulnerable to environmental exposures. While several environmentally sensitive metastable epialleles have been discovered in mice [Dolinoy et al., 2007a; Dolinoy et al., 2007b; Weinhouse et al., 2011], few are known in humans [Waterland et al., 2010]. The range of SEPP1 methylation (11.5 – 91.3%) suggests it may be a metastable epiallele.
In the MDA cohort, age and BMI influenced SEPP1 methylation according to multivariable linear regression. Additionally, MeHg (via hair Hg levels) was associated with decreased SEPP1 methylation (mean and specific CpG site) – a relationship driven by the males. Previous studies identified selenium levels, gender, BMI, SEPP1 genotype, I-Hg exposure, and cancer status as predictors of SEPP plasma concentration, SEPP isoform prevalence, and/or Hg binding to the protein [Chen et al., 2006; Meplan et al., 2007; Meplan et al., 2009]. Genotype of SEPP1 was further associated with hair and urine Hg levels in the MDA cohort [Goodrich et al., 2011]. This is the first study, to our knowledge, to quantify SEPP1 methylation via pyrosequencing and report predictors that may underlie gene expression. SEPP1 may constitute an excellent target for future environmental epigenetic studies using DNA from various tissue types given the large inter-individual variability observed here, SEPP’s critical role in selenium homeostasis, and the complex interplay of selenium with many notable toxicants including Hg and arsenic. Future studies should examine MeHg and I-Hg exposure and SEPP1 methylation, mRNA levels, and protein levels in various tissues types while accounting for gender, BMI, age, SEPP1 genotype, and selenium status to assess the complex relationship between Hg and this vital antioxidant protein.
Methylation at LINE-1 and two candidate genes, selected for their known involvement with Hg and/or sensitivity to environmental stressors, was not significantly associated with Hg biomarker levels. Global repetitive DNA methylation was assessed via LINE-1, a transposable repetitive element that comprises 17% of the genome [Lander et al., 2001]. Repression of LINE-1 transposition via hypermethylation is crucial to protecting genomic stability [Schulz et al., 2006]. The LINE-1 promoter was heavily methylated in buccal mucosa DNA from dental professionals (mean ± SD: 68.4 ± 3.7%) as observed in previous studies with adult blood leukocyte DNA (78.9 ± 2.7% [Rusiecki et al., 2008]; 76.8 ± 1.9% [Wright et al., 2010]) and cord blood DNA (mean 81.6%) [Virani et al., 2012]. Previous studies found associations between hypomethylation of LINE-1 in blood leukocytes and exposures to lead or persistent organic pollutants implicating LINE-1 as a biomarker of environmental effects on the epigenome [Rusiecki et al., 2008; Wright et al., 2010]. In the MDA cohort, no trends were observed with LINE-1 methylation and Hg biomarkers, though the sample size was small (n=53) due to a low success rate of LINE-1 amplification and pyrosequencing. The MDA cohort buccal mucosa DNA samples were originally collected for genotyping purposes and underwent multiple freeze-thaw cycles before use in epigenetic analyses reported here. Thus, some DNA degradation occurred, and as previously observed in our laboratory, amplification of repetitive element sequences in compromised genomic DNA samples is less successful than amplification of unique genic regions.
The CpG island in SEPW1 may be an epigenetically important region as Desplats et al. [Desplats et al., 2011] noted methylation differences depending on neurological disease status in DNA from human brain tissue. Methylmercury treatment is linked to decreased expression of SEPW1 in human neuronal cells, suggesting possible epigenetic underpinnings [Kim et al., 2005]. However in this study, SEPW1 had low percent methylation in its CpG island that was not associated with urine Hg or hair Hg concentrations. The lack of association between Hg levels and methylation in peripheral buccal mucosa DNA does not discount SEPW1 methylation in target organs (e.g., brain) as a possible mechanism of Hg toxicity. Furthermore, the specific CpG sites interrogated here might not be the most sensitive loci to environmental agents.
The DNMT1 assay interrogated sites within a CpG island near the transcription start site, and found the region nearly unmethylated (<10%). Differential expression of DNA methyltransferases (e.g., DNMT1) may underlie aberrant methylation patterns such as the hypomethylation observed in MeHg exposed polar bears and mink [Pilsner et al., 2010]. Animal evidence suggests that MeHg impacts DNA methyltransferase expression [Desaulniers et al., 2009; Glover et al., 2009] and inhibits DNA methyltransferase activity [Basu et al., under review]. In the present study, no associations were observed between Hg biomarker levels and DNMT1 methylation in the assessed region of the gene among dental professionals with dual exposures to MeHg and I-Hg, though DNMT1 methylation at other CpG sites or in different tissue types may still be important.
Previous research on MeHg and DNA methylation has been limited with inconsistent results. Associations between MeHg exposure and DNA hypomethylation were observed in polar bear brains (at global CCGG sites) [Pilsner et al., 2010] and in rat liver following perinatal exposure (at Cdkn2a) [Desaulniers et al., 2009]. Decreased global DNA methylation (5-methylcytosine levels) occurred in rat embryonic neural stem cells treated with MeHg in vitro. Hypomethylation was accompanied by functional changes (decreased cell proliferation, increased senescence markers, and decreased expression of Dnmt3b - a DNA methyltransferase) in both parent and daughter cells (Bose et al., 2012). In contrast, perinatal MeHg treatment was associated with hypermethylation of Bdnf and subsequent decreased Bdnf expression in mice [Onishchenko et al., 2008]. Hypermethylation in the GSTM1 promoter among women with blood Hg >2.9 μg/L has been noted [Hanna et al., 2012]. The exact mechanism of MeHg interference with DNA methylation remains unknown, though inhibition of DNA methyltransferases such as DNMT1 is a possible mechanism that would be expected to cause hypomethylation, and inhibition of DNMTs by MeHg has recently been observed in mink [Basu et al., under review]. In addition Hg is known to inhibit many enzymes via interaction with sulfhydryl groups [Frasco et al., 2007; Omotayo et al., 2011], and inhibition of enzymes involved in the one-carbon metabolism pathway may also be associated with hypomethylation.
To our knowledge, this is the first study to assess the impact of I-Hg on DNA methylation in a whole organism, though unlike MeHg, no statistically significant relationships were found. Mouse embryonic stem cells treated in vitro with 1 μg/L I-Hg exhibited hypermethylation at a tissue-specific differentially methylated region (Rnd2) compared to controls [Arai et al., 2011]. Another in vitro study treated human neuroblastoma cells with 10 μM mercuric chloride, and the treatment eliminated methionine synthase activity, an enzyme involved in the single-carbon folate pathway that provides methyl groups for DNA methylation. If I-Hg impacts the DNA methylome, mechanisms may involve inhibition of enzymes such as methionine synthase or DNA methyltransferase (as observed with MeHg).
This study utilized a cohort originally designed to find genetic polymorphisms influencing Hg toxicokinetics and is thus limited by the source and quality of available DNA. The sample size for this work was further restricted by quality and quantity of remaining DNA available for epigenetic analyses. While DNA source is inconsequential in genetic research, epigenetic marks are tissue- and cell-type specific, though epigenetic epidemiology often relies on peripheral tissues. Methylation of peripheral sample DNA (e.g., blood leukocyte, saliva, buccal mucosa) has been successfully used as a biomarker associated with cancers, disease states, and environmental exposures [Cui et al., 2003; Nohesera et al., 2011; Ouellet-Morin et al., 2012; Rusiecki et al., 2008; Wright et al., 2010]. Recent studies observe epigenetic concordance between tissues for some, though not all, loci and specifically find high correlation of promoter region CpG islands (the genic feature interrogated in all candidate genes of this study) across tissue types [Davies et al., 2012; Thompson et al., 2010]. In the present study, methylation patterns in buccal mucosa could be indicative of alterations to the epigenome in targets of MeHg (cardiovascular and nervous systems) or I-Hg (kidney and nervous system), though further work is necessary to solidify this link. If epigenetic changes in peripheral tissues arise from similar mechanisms occurring in target tissues (e.g., inhibition of DNA methyltransferases or methionine synthase), relating DNA methylation patterns in buccal mucosa, saliva, or blood to toxicant exposure could serve as a first step in evaluating epigenetic change as a mechanism of toxicity.
The cross sectional design is a limitation as exposures throughout life can impact the epigenome. Specific life stages, such as gametogenesis, are known to be particularly vulnerable to epigenetic alterations by environmental agents, and the epigenome changes throughout the aging process [Dolinoy et al., 2007a; Dolinoy et al., 2007b; Gonzalo, 2010]. Although this study captured Hg biomarker levels at one time point, levels may also reflect previous exposures given the expected stability of exposure sources (e.g., personal amalgams, occupation, fish consumption) among most participants throughout adulthood. Due to multiple comparisons, it is possible that significant associations in this study occurred by chance alone, and relationships between MeHg and DNA methylation should be explored in future work.
This study was the first to relate both MeHg and I-Hg exposures to DNA methylation in an epidemiological cohort. Among males, higher hair Hg levels were significantly associated with decreased SEPP1 methylation (average and CpG site-specific) in a region discovered to have great inter-individual variability. Future studies should explore the relationships between Hg exposure and DNA methylation in a larger cohort with high quality DNA samples. Knowledge generated from this type of research has the potential to decipher the role of environmentally-induced epigenetic programming in the pathogenesis of toxicity and downstream disease.
Supplementary Material
Acknowledgments
We acknowledge the support of Dr. Kevin Sloan and Ms. Andrea Sunderman from the Michigan Dental Association. Dr. Yi Wang and the University of Michigan field staff were instrumental in sample collection.
GRANT SPONSORS
This research was funded by grants from the Center for Occupational Health and Safety Engineering at the University of Michigan (CDC/NIOSH Training Grant No. 2T42OH008455), the Michigan Institute for Clinical and Health Research (MICHR, UL1RR024986), the University of Michigan National Institutes of Environmental Health Sciences (NIEHS) Core Center P30 ES017885, and the University of Michigan School of Public Health. The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the funding agencies.
Footnotes
STATEMENT OF AUTHOR CONTRIBUTIONS
Drs. Basu and Franzblau applied for Research Ethics Board approval, and all co-authors formulated the study question and designed the study approach. Drs. Basu, Franzblau, and Goodrich recruited participants and collected data. Dr. Dolinoy oversaw epigenetic analyses. Dr. Goodrich conducted epigenetic analyses, analyzed the data, prepared draft figures and tables, and prepared the manuscript draft with important intellectual input from Drs. Basu, Franzblau and Dolinoy. All authors approved the final manuscript and had complete access to the study data.
No conflict of interest is declared.
References
- Anderson OS, Nahar MS, Faulk C, Jones TR, Liao C, Kannan K, et al. Epigenetic responses following maternal dietary exposure to physiologically relevant levels of bisphenol A. Environ Mol Mutagen. 2012;53:334–342. doi: 10.1002/em.21692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai Y, Ohgane J, Yagi S, Ito R, Iwasaki Y, Saito K, et al. Epigenetic assessment of environmental chemicals detected in maternal peripheral and cord blood samples. J Reprod Dev. 2011;57:507–517. doi: 10.1262/jrd.11-034a. [DOI] [PubMed] [Google Scholar]
- ATSDR. [Accessed September 28, 2012];Priority List of Hazardous Substances. 2011 Available at: http://www.atsdr.cdc.gov/SPL/index.html.
- Baccarelli A, Bollati V. Epigenetics and environmental chemicals. Curr Opin Pediatr. 2009;21:243–251. doi: 10.1097/mop.0b013e32832925cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu N, Head JA, Nam DH, Pilsner JR, Carvan MJ, Chan HM, et al. Effects of methylmercury of epigenetic markers in a mammal, bird, and fish. [Under review, submitted October 2012] [Google Scholar]
- Berglund M, Lind B, Bjornberg KA, Palm B, Einarsson O, Vahter M. Inter-individual variations of human mercury exposure biomarkers: a cross-sectional assessment. Environ Health. 2005;4:20. doi: 10.1186/1476-069X-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. [Accessed November 1, 2010];Fourth National Report on Human Exposure to Environmental Chemicals. 2009 Available at: http://www.cdc.gov/exposurereport/pdf/FourthReport.pdf2009.
- Chen C, Yu H, Zhao J, Li B, Qu L, Liu S, et al. The roles of serum selenium and selenoproteins on mercury toxicity in environmental and occupational exposure. Environmental Health Perspectives. 2006;114:297–301. doi: 10.1289/ehp.7861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson TW, Magos L. The toxicology of mercury and its chemical compounds. Critical Reviews in Toxicology. 2006;36:609–662. doi: 10.1080/10408440600845619. [DOI] [PubMed] [Google Scholar]
- Cui H, Cruz-Correa M, Giardiello FM, Hutcheon DF, Kafonek DR, Brandenberg S, et al. Loss if IGF2 imprinting: a potential marker for colorectal cancer risk. Science. 2003;299:1753–1755. doi: 10.1126/science.1080902. [DOI] [PubMed] [Google Scholar]
- Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S, et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012;13:R43. doi: 10.1186/gb-2012-13-6-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desaulniers D, Xiao GH, Lian H, Feng YL, Zhu J, Nakai J, et al. Effects of mixtures of polychlorinated biphenyls, methylmercury, and organochlorine pesticides on hepatic DNA methylation in prepubertal female Sprague-Dawley rats. Int J Toxicol. 2009;28:294–307. doi: 10.1177/1091581809337918. [DOI] [PubMed] [Google Scholar]
- Desplats P, Spencer B, Coffee E, Patel P, Michael S, Patrick C, et al. Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J Biol Chem. 2011;286:9031–9037. doi: 10.1074/jbc.C110.212589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci. 2007a;104:13056–13061. doi: 10.1073/pnas.0703739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinoy DC, Weidman JR, Jirtle RL. Epigenetic gene regulation: linking early developmental environment to adult disease. Reprod Toxicol. 2007b;23:297–330. doi: 10.1016/j.reprotox.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Franzblau A, d’Arcy H, Ishak MB, Werner RA, Gillespie BW, Albers JW, et al. Low-level mercury exposure and peripheral nerve function. Neurotoxicology. 2012;33:299–306. doi: 10.1016/j.neuro.2012.02.009. [DOI] [PubMed] [Google Scholar]
- Frasco MF, Colletier JP, Weik M, Carvalho F, Guilhermino L, Stojan J, et al. Mechanisms of cholinesterase inhibition by inorganic mercury. FEBS J. 2007;274:1849–1861. doi: 10.1111/j.1742-4658.2007.05732.x. [DOI] [PubMed] [Google Scholar]
- Gadhia SR, Calabro AR, Barile FA. Trace metals alter DNA repair and histone modification pathways concurrently in mouse embryonic stem cells. Toxicol Lett. 2012;212:169–177. doi: 10.1016/j.toxlet.2012.05.013. [DOI] [PubMed] [Google Scholar]
- Glover CN, Zheng D, Jayashankar S, Sales GD, Hogstrand C, Lundebye AK. Methylmercury speciation influences brain gene expression and behavior in gestationally-exposed mice pups. Toxicol Sci. 2009;110:389–400. doi: 10.1093/toxsci/kfp105. [DOI] [PubMed] [Google Scholar]
- Gonzalo S. Epigenetic alterations in aging. J Appl Physiol. 2010;109:586–597. doi: 10.1152/japplphysiol.00238.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JM, Wang Y, Gillespie B, Werner R, Franzblau A, Basu N. Methylmercury and elemental mercury differentially associate with blood pressure among dental professionals. Int J Hyg Environ Health. 2012 doi: 10.1016/j.ijheh.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JM, Wang Y, Gillespie B, Werner R, Franzblau A, Basu N. Glutathione enzyme and selenoprotein polymorphisms associate with mercury biomarker levels in Michigan dental professionals. Toxicol Appl Pharmacol. 2011;257:301–308. doi: 10.1016/j.taap.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandjean P, Satoh H, Murata K, Eto K. Adverse effects of methylmercury: environmental health research implications. Environ Health Perspect. 2010;118:1137–45. doi: 10.1289/ehp.0901757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunau C, Clark SJ, Rosenthal A. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001;29:E65. doi: 10.1093/nar/29.13.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guallar E, Sanz-Gallardo MI, van’t Veer P, Bode P, Aro A, Gomez-Aracena J, et al. Mercury, fish oils, and the risk of myocardial infarction. N Engl J Med. 2002;347:1747–1754. doi: 10.1056/NEJMoa020157. [DOI] [PubMed] [Google Scholar]
- Hanna CW, Bloom MS, Robinson WP, Kim D, Parsons PJ, Vom Saal FS, et al. DNA methylation changes in whole blood is associated with exposure to the environmental contaminants, mercury, lead, cadmium and bisphenol A, in women undergoing ovarian stimulation for IVF. Hum Reprod. 2012 doi: 10.1093/humrep/des038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagas MR, Choi AL, Oken E, Horvat M, Schoeny R, Kamai E, et al. Evidence on the human health effects of low-level methylmercury exposure. Environ Health Perspect. 2012;120:799–806. doi: 10.1289/ehp.1104494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Wang F. Mercury-selenium compounds and their toxicological significance: toward a molecular understanding of the mercury-selenium antagonism. Environ Toxicol Chem. 2009;28:1567–1577. doi: 10.1897/08-375.1. [DOI] [PubMed] [Google Scholar]
- Kim YJ, Chai YG, Ryu JC. Selenoprotein W as molecular target of methylmercury in human neuronal cells is down-regulated by GSH depletion. Biochem Biophys Res Commun. 2005;330:1095–102. doi: 10.1016/j.bbrc.2005.03.080. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- McDowell MA, Dillon CF, Osterloh J, Bolger PM, Pellizzari E, Fernando R, et al. Hair mercury levels in U.S. children and women of childbearing age: reference range data from NHANES 1999–2000. Environ Health Perspect. 2004;112:1165–71. doi: 10.1289/ehp.7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meplan C, Crosley LK, Nicol F, Beckett GJ, Howie AF, Hill KE, et al. Genetic polymorphisms in the human selenoprotein P gene determine the response of selenoprotein markers to selenium supplementation in a gender-specific manner (the SELGEN study) The FASEB Journal. 2007;21:3063–74. doi: 10.1096/fj.07-8166com. [DOI] [PubMed] [Google Scholar]
- Meplan C, Nicol F, Burtle BT, Crosley LK, Arthur JR, Mathers JC, et al. Relative abundance of selenoprotein P isoforms in human plasma depends on genotype, Se intake, and cancer status. Antioxid Redox Signal. 2009;11:2631–40. doi: 10.1089/ARS.2009.2533. [DOI] [PubMed] [Google Scholar]
- Mergler D, Anderson HA, Chan LHM, Mahaffey KR, Murray M, Sakamoto M, et al. Methylmercury exposure and health effects in humans: a worldwide concern. Ambio. 2007;36:3–11. doi: 10.1579/0044-7447(2007)36[3:meahei]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Millis RM. Epigenetics and hypertension. Curr Hypertens Rep. 2011;13:21–28. doi: 10.1007/s11906-010-0173-8. [DOI] [PubMed] [Google Scholar]
- Min JL, Lakenberg N, Bakker-Verweij M, Suchiman E, Boomsma DI, Slagboom PE, et al. High microsatellite and SNP genotyping success rates established in a large number of genomic DNA samples extracted from mouth swabs and genotypes. Twin Research and Human Genetics. 2006;9:501–506. doi: 10.1375/183242706778024973. [DOI] [PubMed] [Google Scholar]
- Nam DH, Basu N. Rapid methods to detect organic mercury and total selenium in biological samples. Chem Cent J. 2011;5:3. doi: 10.1186/1752-153X-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohesara S, Ghadirivasfi M, Mostafavi S, Eskandari MR, Ahmadkhaniha H, Thiagalingam S, et al. DNA hypomethylation of MB-COMT promoter in the DNA derived from saliva in schizophrenia and bipolar disorder. J Psychiatr Res. 2011;45:1432–1438. doi: 10.1016/j.jpsychires.2011.06.013. [DOI] [PubMed] [Google Scholar]
- Omotayo TI, Rocha JB, Ibukun EO, Kade IJ. Inorganic mercury interacts with thiols at the nucleotide and cationic binding sites of the ouabain-sensitive cerebral electrogenic sodium pump. Neurochem Int. 2011;58:776–784. doi: 10.1016/j.neuint.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Onishchenko N, Karpova N, Sabri F, Castren E, Ceccatelli S. Long-lasting depression-like behavior and epigenetic changes of BDNF gene expression induced by perinatal exposure to methylmercury. J Neurochem. 2008;106:1378–1387. doi: 10.1111/j.1471-4159.2008.05484.x. [DOI] [PubMed] [Google Scholar]
- Ouellet-Morin I, Wong CC, Danese A, Pariante CM, Papadopoulos AS, Mill J, et al. Increase serotonin transporter gene (SERT) DNA methylation is associated with bullying victimization and blunted cortisol response in stress in childhood: a longitudinal study of discordant monozygotic twins. Psychol Med. 2012 doi: 10.1017/S0033291712002784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilsner JR, Lazarus AL, Nam DH, Letcher RJ, Sonne C, Dietz R, et al. Mercury-associated DNA hypomethylation in polar bear brains via the LUminometric Methylation Assay: a sensitive method to study epigenetics in wildlife. Mol Ecol. 2010;19:307–314. doi: 10.1111/j.1365-294X.2009.04452.x. [DOI] [PubMed] [Google Scholar]
- Roman HA, Walsh TL, Coull BA, Dewailly E, Guallar E, Hattis D, et al. Evaluation of the cardiovascular effects of methylmercury exposures: current evidence supports development of a dose-response function for regulatory benefits analysis. Environ Health Perspect. 2011;119:607–614. doi: 10.1289/ehp.1003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusiecki JA, Baccarelli A, Bollati V, Tarantini L, Moore LE, Bonefeld-Jorgensen EC. Global DNA hypomethylation is associated with high serum-persistent organic pollutants in Greenlandic Inuit. Environ Health Perspect. 2008;116:1547–1552. doi: 10.1289/ehp.11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz WA, Steinhoff C, Florl AR. Methylation of endogenous human retroelements in health and disease. Curr Top Microbiol Immunol. 2006;310:211–250. doi: 10.1007/3-540-31181-5_11. [DOI] [PubMed] [Google Scholar]
- Swain EB, Jakus PM, Rice G, Lupi F, Maxson PA, Pacyna JM, et al. Socioeconomic consequences of mercury use and pollution. Ambio. 2007;36:45–61. doi: 10.1579/0044-7447(2007)36[45:scomua]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Terry MB, Delgado-Cruzata L, Vin-Raviv N, Wu HC, Santella RM. DNA methylation in white blood cells: association with risk factors in epidemiologic studies. Epigenetics. 2011;6:828–837. doi: 10.4161/epi.6.7.16500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RF, Atzmon G, Gheorghe C, Liang HQ, Lowes C, Greally JM, et al. Tissue-specific dysregulation of DNA methylation in aging. Aging Cell. 2010;9:506–518. doi: 10.1111/j.1474-9726.2010.00577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virani S, Dolinoy DC, Halubai S, Jones TR, Domino SE, Rozek LS, et al. Delivery type not associated with global methylation at birth. Clin Epigenetics. 2012;4:8. doi: 10.1186/1868-7083-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waly M, Olteanu H, Banerjee R, Choi SW, Mason JB, Parker BS, et al. Activation of methionine synthase by insulin-like growth factor-1 and dopamine: a target for neurodevelopmental toxins and thimerosal. Mol Psychiatry. 2004;9:358–370. doi: 10.1038/sj.mp.4001476. [DOI] [PubMed] [Google Scholar]
- Wang Y, Goodrich JM, Gillespie B, Werner R, Basu N, Franzblau A. An Investigation of Modifying Effects of Metallothionein Single Nucleotide Polymorphisms on the Association between Mercury Exposure and Biomarker Levels. Environ Health Perspect. 2012;120:530–534. doi: 10.1289/ehp.1104079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterland RA, Kellermayer R, Laritsky E, Rayco-Solon P, Harris RA, Travisano M, et al. Season of conception in rural gambia affects DNA methylation at putative human metastable epialleles. PLoS Genet. 2010;6:e1001252. doi: 10.1371/journal.pgen.1001252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhouse C, Anderson OS, Jones TR, Kim J, Liberman SA, Nahar MS, et al. An expression microarray approach for the identification of metastable epialleles in the mouse genome. Epigenetics. 2011;6:1105–1113. doi: 10.4161/epi.6.9.17103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright RO, Schwartz J, Wright RJ, Bollati V, Tarantini L, Park SK, et al. Biomarkers of lead exposure and DNA methylation within retrotransposons. Environ Health Perspect. 2010;118:790–795. doi: 10.1289/ehp.0901429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.