

Abstract
The next-generation antiandrogen MDV3100 prolongs overall survival of patients with metastatic castration-resistant prostate cancer (CRPC). However, patient responses are variable, and survival benefit remains relatively small. Developing effective modality to improve MDV3100 efficacy is urgently needed. Recent evidence suggests that constitutively active androgen receptor splice variants (AR-Vs) drive resistance to MDV3100. In our study, we show that methylselenol prodrug downregulates the expression and activity of both the full-length AR (AR-FL) and AR-Vs. The downregulation is independent of androgen and could be attributable to repressed transcription of the AR gene. Cotreatment with methylselenol prodrug and MDV3100 suppresses AR signaling more dramatically than either agent alone, and synergistically inhibits the growth of CRPC cells in vitro. The combinatorial efficacy is observed in not only AR-V-expressing cells but also cells expressing predominantly AR-FL, likely owing to the ability of the two drugs to block the AR signaling cascade at distinct steps. Ectopic expression of AR-FL or AR-V7 attenuates the combinatorial efficacy, indicating that downregulating AR-FL and AR-V7 is importantly involved in mediating the combinatorial efficacy. Significantly, methylselenol prodrug also downregulates AR-FL and AR-Vs in vivo and substantially improves the antitumor efficacy of MDV3100. These findings support a potential combination therapy for improving MDV3100 efficacy, and provide a rationale for evaluating the clinical application of combining methylselenol prodrug with MDV3100 for the treatment of CRPC.
Keywords: methylseleninic acid, methylselenocysteine, MDV3100, androgen receptor, castration-resistant prostate cancer
Relapse with castration-resistant prostate cancer (CRPC) after androgen deprivation therapy constitutes a major cause of prostate cancer mortality.1 There is now compelling evidence supporting resurgent androgen receptor (AR) signaling as a key driver of castration-resistant progression.1 This has led to the development of next-generation AR-targeting therapies that have recently received FDA approval for the treatment of metastatic CRPC, such as the potent AR antagonist MDV3100.
MDV3100 has fivefold to eightfold higher affinity to AR when compared to bicalutamide, the antiandrogen currently used in clinics.2 MDV3100 prolonged overall survival of metastatic CRPC patients who progressed after chemotherapy in a Phase III trial.3 However, many of the patients presented with therapy-resistant disease, and the vast majority who initially responded acquired resistance over time.3 The median survival was improved by only 4.8 months over placebo.3 Thus, developing more effective modality to improve the therapeutic outcome of MDV3100 is urgently needed.
A large number of androgen receptor splice variants (AR-Vs) that are devoid of the functional AR ligand-binding domain (LBD) as a result of alternative splicing have been recently identified.4–8 The majority of the AR-Vs identified to date display constitutive activity.4–8 Two major AR-Vs, AR-V7 (also named AR3) and ARv567es, are capable of regulating gene expression in the absence of AR-FL.4–7,9,10 AR-Vs are detected in most CRPC specimens,6,11 and the levels are significantly upregulated compared to hormone-naïve cancers.5–8,10,11 In fact, the AR-V proteins are expressed at a level comparable to that of AR-FL, or even represent the predominant form of AR, in a significant portion of metastatic CRPC tissues.8,11 Patients with high AR-V7 or detectable ARv567es expression have significantly shorter cancer-specific survival than other CRPC patients.11
Based on its AR-LBD-targeting nature, intuitively, MDV3100 may not be effective against AR-Vs. Although a study showed that ectopic expression of AR-V7 in LNCaP xenograft tumors does not alter the ability of MDV3100 to inhibit the growth of the tumors,12 this effect might be model specific. It was reported recently that, in CRPC cells expressing endogenous AR-FL and AR-Vs, AR-Vs drive resistance to MDV3100 by functioning as independent drivers of the AR transcriptional program.13 Thus, combining MDV3100 with an AR-V-targeting agent may represent a viable approach to overcome resistance to MDV3100.
Methylselenol is considered the active metabolite for the anticancer effect of methyl-selenium compounds.14 Previously, we showed that two methylselenol prodrugs, methylseleninic acid (MSA) and methylselenocysteine (MSC), are effective in downregulating AR-FL mRNA and protein.15–17 A marked downregulation of AR-FL mRNA could be detected as early as 3 hr post-treatment,15 while AR mRNA stability was not decreased within this duration,18 indicating that repressed transcription of the AR gene may contribute to the downregulation. As AR-Vs are generated by alternative splicing and utilize the same promoter as AR-FL, repressed transcription of the AR gene is also expected to lead to a reduction in AR-Vs. Accordingly, in our study, we tested the hypothesis that methylselenol prodrug downregulates AR-Vs and improves the therapeutic efficacy of MDV3100.
As the metabolism of MSC to methylselenol requires the activity of β-lyase, which is expressed in the liver and kidney, but not the prostate,14 MSC was used only in the animal experiments. In contrast, MSA, as an oxidized form of methylselenol, is readily reduced to methylselenol through a non-enzymatic reaction in cells.19 MSA can inhibit the growth of prostate cancer cells at in vivo-relevant concentrations (2–10 μM).19–23 Therefore, we used MSA in our in vitro experiments and compared the efficacy of MSA and MSC in improving MDV3100 efficacy in our in vivo experiments. It is important to point out that MSA and MSC have a very different biological and pharmacological activities than selenomethionine, the form of selenium used in the selenium and vitamin E chemoprevention trial,14,19,24–27 and that MSC has shown no evidence of toxicity in a single-dose Phase I trial.20
Material and Methods
Cell lines and reagents
The sources of the cell lines are described in Supporting Information. MDV3100 was from Selleck Chemicals (Houston, TX), and the 99% purity confirmed by NMR. MSA and MSC were from PharmaSe (Lubbock, TX). The following antibodies were used: anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Millipore, Billerica, MA), anti-AR (PG-21, Millipore, Billerica, MA) and anti-AR-V7 (Precision Antibody, Columbia, MD).
Quantitative reverse transcription-PCR
The quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analysis was performed as described.15 The TaqMan® PCR primers and probes for β-actin, prostate-specific antigen (PSA) and kallikrein 2 (KLK2) were from Applied Biosystems, Grand Island, NY. The primer sequences for AR isoforms, ubiquitin-conjugating enzyme E2C (UBE2C) or cyclin A2 (CCNA2) were described in Refs. 5, 28, and 29.
Reporter gene assay
Three reporter plasmids were used. The androgen-responsive element (ARE)-luciferase reporter plasmid contains three ARE regions ligated in tandem to the luciferase reporter.30 The pGL4-ARpro8 and pGL4-ARpro1.7 AR-promoter-luciferase constructs contain an 8 or 1.7 kb fragment of the 5′-flanking region of the human AR gene, respectively.31 The backbone vector, pGL4.19[luc2CP/Neo], contains protein degradation sequences after luciferase to allow monitoring of rapid response. To ensure an even transfection efficiency, we conducted the transfection in bulk and then split the transfected cells for luciferase assay.16
22Rv1 tumor xenograft model
Male nude mice were obtained from NCI at 5–6 weeks of age. After 1 week of adaptation, mice were inoculated subcutaneously with 5 × 106 22Rv1 cells suspended in 50% Matrigel on the right dorsal flank. When tumor size reached ~100 mm3, mice were randomized to six groups, viz.r, vehicle control, MDV3100, MSA, MSC, MSA+MDV3100 or MSC+MDV3100. MDV3100 was prepared as in Ref. 2, and MSA and MSC as in Refs. 32 and 33. Drug administration was performed daily by an oral route.34 The dose was 10 mg/kg/day for MDV3100 and 3 mg selenium/kg/day for MSA and MSC. Tumor volume was calculated as 0.524 × width2 × length.35 At the termination of the experiment, mice were anesthetized, blood collected for serum PSA determination using quantitative ELISA (American Qualex, San Clemente, CA) and tumors removed for molecular analysis. All animal procedures were approved by Tulane University Institutional Animal Care and Use Committee.
Thin-layer chromatography
Solutions of MSA + MDV3100 or MSC + MDV3100 were incubated for 2 hr, and a 0.5-μl aliquot spotted on a Silica gel 60 F254 plate using glass capillary tubes. The plate was dried for 30 min, and chromatograms developed in solvent tank pre-equilibrated with running solvent mixer (methanol:-chloroform:NH4OH, 1:10:0.1, v/v/v) for 15 min. After development, the plate was dried for 10 min and visualized in an iodine/silicon bottle.
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry
Solutions of MSA + MDV3100 or MSC + MDV3100 were incubated for 2 hr. A 1-μl aliquot was then mixed with 1 μl of α-cyano-4-hydroxycinnamic acid matrix and spotted onto a stainless-steel plate for matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) analysis using Axima mass spectrometer (Shimadzu, Kyoto, Japan).
Statistical analysis
The Student’s two-tailed t-test was used to determine the mean differences between treatment and control. Data are presented as mean ± SEM. Drug interaction was characterized by using the Calcusyn software (Biosoft, Cambridge, UK), which calculates combination index values using the median-effect principle.36 A combination index value of <1, 1 or >1 denotes synergism, additivity or antagonism, respectively.
Results
MSA inhibits the transcription of the AR gene
To confirm that MSA downregulation of AR-FL mRNA is due to inhibited transcription of the AR gene, we performed a nuclear run-on assay in LNCaP cells. To differentiate the effect of MSA on transcriptional initiation vs. transcriptional elongation, we quantified the nascent transcripts by qRT-PCR using a primer-probe set corresponding to either the 5′-or the 3′-end of the AR-FL mRNA. As shown in Supporting Information Figure 1A, the levels of the nascent AR-FL mRNA at the 5′- and 3′-ends were reduced to a similar extent by MSA. The data indicate that MSA blocks AR transcriptional initiation, rather than slows down transcriptional elongation.
We further assessed the effect of MSA on the activity of the AR promoter using two reporter plasmids, pGL4-ARpro8 and pGL4-ARpro1.7.31 The plasmids were individually transfected into LNCaP or the castration-resistant 22Rv1 cells. Cells were then treated with MSA for 0.5, 1 or 2 hr, when the AR protein remained unchanged according to our published data.15 In both LNCaP (Supporting Information Fig. 1B) and 22Rv1 cells (Fig. 1a), MSA treatment significantly reduced the activities of both promoters. Together with the above data, these results demonstrate the ability of MSA to inhibit the transcription of the AR gene.
Figure 1.
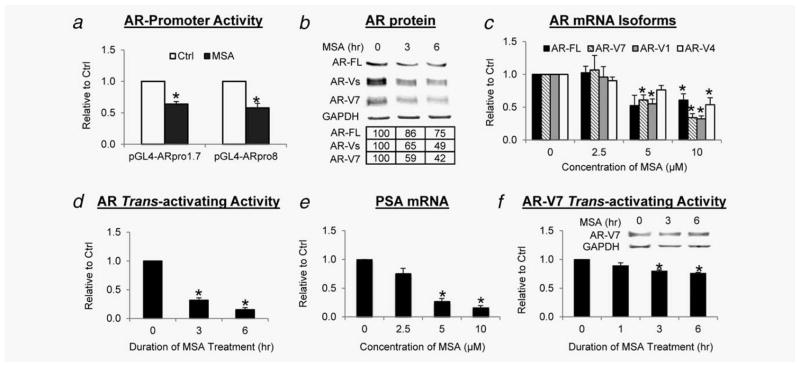
MSA downregulates AR-FL and AR-Vs and androgen-independent AR activity. Cells were cultured in androgen-deprived condition. (a) Inhibition of AR promoter activity. 22Rv1 cells transfected with AR-promoter-luciferase construct were treated with 10 μM MSA. (b) Down-regulation of AR-FL and AR-V proteins. 22Rv1 cells were treated with 10 μM MSA. The numbers in the tables denote relative normalized intensities compared to the control value of 100. (c) Downregulation of AR-FL and AR-V mRNAs. 22Rv1 cells were treated with MSA for 6 hr. (d) Suppression of androgen-independent AR activity. 22Rv1 cells transfected with the ARE-luciferase construct were treated with 10 μM MSA. (e) Downregulation of PSA mRNA. 22Rv1 cells were treated with MSA for 6 hr. (f) Repression of AR-V7 transactivation. PC-3 cells cotransfected with AR-V7-expressing and ARE-luciferase constructs were treated with 10 μM MSA. Inset, Western blot confirmation of AR-V7 level. *p < 0.05 from control.
MSA downregulates the expression of AR-Vs
As AR-Vs are generated by alternative splicing, the decrease in the transcription of the AR gene is expected to cause a reduction of not only AR-FL but also AR-Vs. Therefore, we investigated the effect of MSA on the expression of AR-Vs in 22Rv1 cells, which express AR-FL along with three ~80-KDa major AR-Vs, namely AR-V7, AR-V1 (also named AR4) and AR-V4 (also named AR5).4–6,37 Western blot analyses were conducted with an antibody recognizing all AR isoforms or specific for AR-V7. As the most abundant and active AR-V in the cells,5 AR-V7 is the only AR-V to which a specific antibody has been developed. As shown in Figure 1b, MSA downregulated both AR-FL and AR-Vs in a time-dependent manner, with the change in AR-Vs slightly more significant than that of AR-FL. Then, we examined the effect of MSA on the mRNA levels of different AR isoforms by qRT-PCR. As expected, MSA treatment significantly reduced AR-FL and AR-V transcripts in a dose-dependent fashion (Fig. 1c). AR-V7 and AR-V1 mRNA appeared to be more sensitive to MSA downregulation than AR-FL and AR-V4. These results indicate that MSA can indeed inhibit the expression of different AR-Vs at the transcriptional level.
MSA suppresses androgen-independent AR activity
As most of the AR-Vs identified to date display constitutive activity,4–8 we assessed the effect of MSA on androgen-independent AR activity by a reporter gene assay and the expression of an endogenous AR target, PSA, by qRT-PCR. As shown in Figures 1d and 1e, MSA depressed androgen-independent AR transactivation as a function of time and dose. We and others previously showed that although MSA suppression of the AR-FL signaling is attributable primarily to the reduction of AR-FL availability, additional mechanisms also contribute to the suppression.15,16,18 To determine whether this is also true for AR-V, we assessed the effect of MSA on the activity of AR-V7 that is exogenously expressed in PC-3 cells under the control of a CMV promoter. A modest, but statistically significant, decrease in AR-V7 trans-activating activity was evident after MSA treatment, whereas the protein level remained constant (Fig. 1f). The data implicate that MSA may affect AR-V signaling through a mechanism(s) beyond reducing the abundance of AR-Vs.
MSA potentiates MDV3100 efficacy in inhibiting AR signaling
As MSA inhibits the activity of AR-Vs, which lack the intended target of MDV3100—the AR LBD, it is reasonable to believe that MSA may synergize with MDV3100 to repress AR signaling in cells expressing both AR-FL and AR-Vs. This effect may not be limited to AR-V-expressing cells, because MSA also inhibits AR-FL and the mechanism is clearly distinct from that of MDV3100.15,16 To test this hypothesis, we quantified AR trans-activating activity by a reporter-gene assay and the mRNA levels of AR targets by qRT-PCR in 22Rv1 and the C4-2 castration-resistant derivative of LNCaP, which expresses predominantly AR-FL. As the ARE-luciferase activity was more sensitive to MDV3100 and MSA regulation than the endogenous targets, we reduced the doses of the drugs used for this assay. As shown in Figure 2a, the combination treatment produced a more pronounced inhibition of basal (upper panel) and dihydrotestosterone (DHT)-induced (lower panel) AR transactivation. MDV3100 was more effective in suppressing DHT-induced AR activity than basal AR activity, whereas MSA was equally effective in knocking down basal and DHT-induced AR transactivation. The effects on the expression of PSA and KLK2, two shared targets of AR-FL and AR-Vs, followed a similar trend (Fig. 2b and Supporting Information Fig. 2). However, only MSA, but not MDV3100, was able to decrease the expression of AR-V-specific targets, UBE2C and CCNA210 (Fig. 2c). This is consistent with their distinct mechanisms of action on AR signaling. Taken together, the data indicate that MSA can potentiate the ability of MDV3100 to shut down both ligand-dependent and ligand-independent AR signaling in CRPC cells.
Figure 2.
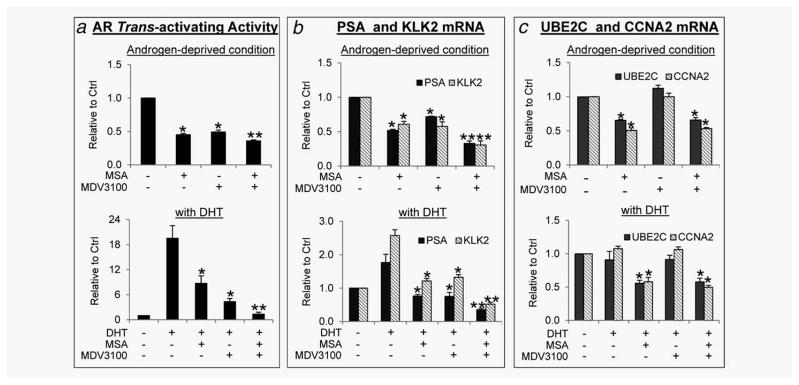
Combinatorial effect of MSA and MDV3100 on AR transactivation in 22Rv1 cells. (a) Cells transfected with ARE-luciferase construct were treated for 3 hr with 2 μM MSA and/or 5 μM MDV3100 in the absence of DHT, or with 2 μM MSA and/or 1 μM MDV3100 in the presence of 1 nM DHT. (b and c) Cells were treated for 6 hr with 10 μM MDV3100 and/or 4 μM MSA in the absence or presence of 1 nM DHT. *p < 0.05 from vehicle control or DHT control. **p < 0.05 from the MDV3100 group.
MSA synergistically enhances the growth-inhibitory efficacy of MDV3100
Given the critical role of AR signaling in sustaining the growth of CRPC cells, we assessed the combinatorial effect of MSA and MDV3100 on cell growth in vitro by an sulforhodamine B (SRB) assay.38 As shown in Table 1, in both 22Rv1 and C4-2 cells, MSA plus MDV3100 led to a more striking inhibition of cell growth than either agent alone. Furthermore, all the combinations produced combination index values of <1 (Table 2), suggesting a synergy between MSA and MDV3100 in inhibiting cell growth.
Table 2.
Combination index values for MSA and MDV3100
| MDV (μM) | MSA (μM)
|
|||||||
|---|---|---|---|---|---|---|---|---|
| 22Rv1 without DHT
|
22Rv1 with DHT
|
C4-2 without DHT
|
C4-2 with DHT
|
|||||
| 2 | 4 | 2 | 4 | 2 | 4 | 2 | 4 | |
| 5 | 0.57 | 0.199 | 0.549 | 0.304 | 0.631 | 0.665 | 0.463 | 0.727 |
|
| ||||||||
| 10 | 0.449 | 0.166 | 0.359 | 0.201 | 0.571 | 0.621 | 0.442 | 0.681 |
To delineate the functional role of AR downregulation in mediating the synergy between MSA and MDV3100, we transiently transfected 22Rv1 cells with an AR-FL or AR-V7 expression construct,5 and assessed the response of the transfected cells to MSA and MDV3100 growth inhibition. Of note, to ensure an even transfection efficiency, we performed the transfection in bulk and split the transfected cells for the SRB assay. To allow the cells to recover overnight before treatment and to avoid the possibility of losing the transiently transfected plasmids beyond 72 hr post-transfection, we conducted the SRB assay at 48 hr post-treatment, not at 72 hr as in Table 1. As shown in Figure 3, ectopically expressed AR-FL or AR-V7 not only weakened the growth-inhibitory ability of MSA but also significantly dampened the growth-inhibitory efficacy of the combination treatment. Therefore, the data indicate that the downregulation of AR-FL and AR-V is important to the combinatorial efficacy of MSA and MDV3100.
Table 1.
Effect of MSA and/or MDV3100 on the growth of 22Rv1 and C4-2 cells1
| MDV (μM) | MSA (μM)
|
MSA (μM)
|
C4-2 without DHT
|
C4-2 with DHT
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 22Rv1 without DHT
|
22Rv1 with DHT
|
|||||||||||
| 0 | 2 | 4 | 0 | 2 | 4 | 0 | 2 | 4 | 0 | 2 | 4 | |
| 0 | 100 | 69.2 ± 2.3* | 46.8 ± 5.9* | 100 | 67.0 ± 6.9* | 39.8 ± 5.2* | 100 | 76.4 ± 2.6* | 54.3 ± 1.9* | 100 | 48.9 ± 2.6* | 31.8 ± 1.0* |
|
| ||||||||||||
| 5 | 89.2 ± 1.5* | 64.9 ± 4.3* | 20.9 ± 5.3** | 78.1 ± 4.3* | 43.8 ± 3.1** | 28.5 ± 4.2* | 92.6 ± 0.6* | 62.7±2.3** | 47.3 ± 0.8** | 55.6 ± 2.9* | 36.5 ± 2.2** | 29.3 ± 2.1* |
|
| ||||||||||||
| 10 | 83.4 ± 1.6* | 54.9 ± 7.3* | 17.2 ± 4.0** | 71.9 ± 3.4* | 34.5 ± 3.2** | 20.4 ± 3.0** | 88.1 ± 1.0* | 58.2±2.3** | 43.7 ± 1.4** | 52.2 ± 2.3* | 33.7 ± 2.2** | 27.0 ± 2.1* |
SRB assay was performed at 72 hr after treatment, and data expressed as % of vehicle control (mean ± SEM).
p < 0.05 from vehicle group.
p < 0.05 from single-agent groups.
Figure 3.
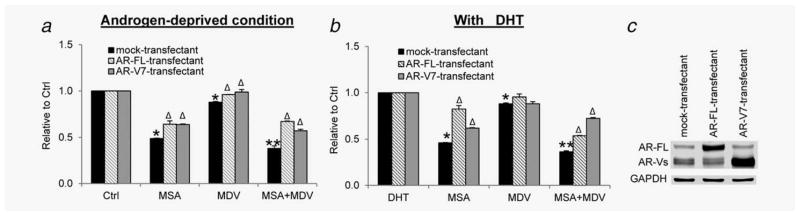
Attenuation of MSA and MDV3100 growth inhibition by ectopic expression of AR-FL or AR-V7. 22Rv1 cells were transfected with the expression construct of AR-FL or AR-V7 and treated with 4 μM MSA and/or 10 μM MDV3100 in the absence (a) or presence (b) of 1 nM DHT. *p < 0.05 from mock-transfectant. **p < 0.05 from single-agent groups. Δp < 0.05 from respective mock-transfectant in each treatment group. (c) Western blotting confirmation of transfection.
MSC, but not MSA, enhances MDV3100 efficacy in vivo
As described in Introduction, the requirement of β-lyase for the conversion of MSC to methylselenol limits its use in cell culture studies. Here, we assessed the ability of MSC, in comparison with MSA, to improve MDV3100 efficacy in the 22Rv1 xenograft model. As shown in Figure 4a, each of the three drugs individually inhibited the growth of 22Rv1 tumors, and statistical significance was achieved starting on Day 8 of the treatment. The average final tumor weight was 0.6–0.7 g in these groups, ~50% of that in the control group (Fig. 4b). Surprisingly, the tumor growth curve of the MSA + MDV3100 group was almost identical to that of the MDV3100 group, whereas the tumors in the MSC + MDV3100 group were significantly smaller than those in either of the single-agent-treated groups (Fig. 4a). The average final tumor weight in the MSC + MDV3100 group was 0.3 g, ~50% of that in the single-agent groups (Fig. 4b). All treatments appeared to be well tolerated, as there was no significant difference in body weight compared to the control group (Supporting Information Fig. 3).
Figure 4.

In vivo efficacy of MDV3100 and MSA/MSC in 22Rv1 xenograft model. (a) Mean tumor volumes (n = 6). (b) Mean tumor weights. (c) Mean AR-FL and AR-Vs protein levels. (d) Mean serum PSA levels. (e and f) Thin-layer chromatogram (e) or MALDI-TOF mass spectrum (f) of MSA and MDV3100 or MSC and MDV3100 mixtures. *p < 0.05 from control. **p < 0.05 from single-agent groups.
Then, we measured the levels of AR-FL and AR-Vs in the tumors by Western blotting and serum PSA by ELISA. We observed a statistically significant reduction in AR-FL protein level in response to MDV3100 treatment (Fig. 4c and Supporting Information Fig. 4). This effect, to our knowledge, has not been reported before. Consistent with the data on tumor growth (Figs. 4a and 4b), although MSA and MSC individually caused a significant decrease in the levels of AR-FL, AR-Vs (Fig. 4c and Supporting Information Fig. 4) and PSA (Fig. 4d), only MSC, when combined with MDV3100, further reduced their levels. Taken together, these results demonstrate that MSC, but not MSA, can improve the efficacy of MDV3100 in vivo.
Conjugation between MDV3100 and MSA
In an attempt to interpret why MSA is unable to potentiate MDV3100 efficacy in vivo, we examined the structures of MSA, MSC and MDV3100 (Supporting Information Fig. 5A), and found that MSA is much more likely than MSC to conjugate to MDV3100. This is because seleninic acid is a strong oxidizing agent and known to react with thiols under a mild condition.39,40 The selenium in MSA has an oxidation state of 4, and the hydroxyl group is a good leaving group. It is possible that MSA reacts with the thiourea group of MDV3100 to form the thioseleninate intermediate and further produces selenosulfide (Supporting Information Fig. 5B). In contrast, the selenium in MSC only displays an oxidation state of 2 without a good leaving group, and therefore less likely to react with MDV3100.
To test this possibility, we incubated MDV3100 with MSA or MSC in a test tube and performed thin-layer chromatography of the mixtures. Iodine-stained chromatogram is presented in Figure 4e. Evidently, new components shown as a streak above the MDV3100 band were present in the MSA + MDV3100 mixture, but not in the MSC + MDV3100 mixture (MSA band was invisible because iodine cannot stain MSA). We also analyzed the mixtures by MALDI-TOF MS. In addition to the MDV3100 peak at mass-to-charge ratio (m/z) of 465, two distinct peaks, at m/z 559 and 608, were present in the MSA + MDV3100 mixture (Fig. 4f). The m/z 559 peak, likely the selenosulfide product predicted above (Supporting Information Fig. 5B), was also detected in the MSC + MDV3100 mixture, but at a much lower intensity (Fig. 4f). Therefore, the data are consistent with the thin-layer chromatogram, supporting our predication that MSA is much more likely to conjugate to MDV3100 than MSC. Although further investigation is necessary to characterize the conjugates, it is possible that the conjugation may reduce the ability of MDV3100 or MSA to suppress AR signaling. The conjugation may be an issue particular to the treatment protocol used in our in vivo experiment, because the drugs were mixed together in high concentrations (mM range) before administered to mice.
Discussion
Our study demonstrates a clear example of a rationally designed and potentially effective combination modality for improving MDV3100 efficacy against CRPC. We reported previously that MSA inhibits the expression and activity of AR-FL. Here, we showed that the inhibitory effect, which is independent of androgen, extends to AR-Vs, and could be attributable to MSA suppression of the transcription of the AR gene. Combined treatment of cultured CRPC cells with MSA and MDV3100 more significantly suppressed AR signaling and synergistically inhibited cell growth. The combinatorial efficacy was observed in not only AR-V-expressing cells but also cells expressing predominantly AR-FL, likely owing to the ability of the two drugs to block the AR signaling cascade at distinct steps. Ectopic expression of AR-FL or AR-V7 attenuated the combinatorial efficacy, indicating that downregulating AR-FL and AR-V7 is importantly involved in mediating the combinatorial efficacy.
Surprisingly, although MSA was able to downregulate AR-FL and AR-Vs in vivo and inhibited the growth of 22Rv1 tumors, it failed to enhance the antitumor effect when combined with MDV3100. In contrast, MSC, another methylselenol prodrug, significantly improved the antitumor efficacy of MDV3100. The lack of in vivo combinatorial efficacy between MSA and MDV3100 may be a result of conjugation between MSA with MDV3100, and the conjugation may be an issue particular to the treatment protocol used in our in vivo experiment because the drugs were mixed together at very high concentrations before administered to mice. To solve this problem, we will explore the possibility of sequential treatment with MSA and MDV3100 rather than using a cocktail. Nonetheless, the findings from this study support the potential of using methylselenol prodrug to improve MDV3100 efficacy.
In our study, we observed that MDV3100 decreases the level of AR-FL protein in the presence of androgen, an effect that, to our knowledge, has not been reported before. Similar effect was observed after bicalutamide treatment. In retrospect, this should have been expected of all antiandrogens on the basis of AR antagonism. Androgens are known to stabilize AR-FL protein by inducing AR N-C interaction.41 By blocking the binding of androgens to AR, antiandrogens would be expected to prevent androgen induction of AR N-C interaction and protein stabilization. We have found that both MDV3100 and bicalutamide can disrupt DHT-mediated AR N-C interaction, and MDV3100 is much more potent than bicalutamide in disrupting the interaction (data not shown). Thus, the decrease of AR-FL protein in response to MDV3100 and bicalutamide is likely owing to their AR antagonistic activity.
The prevalent upregulation of AR-Vs in CRPC tissues compared to hormone-naïve cancers has been unequivocally demonstrated by a number of groups.5–8,10,11 However, the clinical relevance of AR-Vs remains controversial. The skepticism comes mainly from the perception that AR-Vs represent a nominal percentage of the AR population in clinical specimens. Although this may be true at the mRNA level,11,12 significant discrepancy may exist between the abundance of AR-V mRNAs and proteins. Western blot analyses of 13 CRPC bone metastases demonstrated that the AR-V proteins are expressed at a level comparable to that of AR-FL in 38% of the samples.11 The relative high abundance of AR-V proteins was also shown by immunohistochemistry data. Using an AR-V7-specific antibody, three groups showed that AR-V7 is readily detectable in prostate cancer specimens.5,10,42 Another study of 50 primary prostate cancer tissues and 162 metastatic CRPC tissues with antibodies against either the N- or C-terminus of AR-FL demonstrated a significant decrease in nuclear C-terminal staining compared to the N-terminal intensity in CRPC tissues, with no apparent difference in primary prostate cancers.8 Thus, the clinical relevance of AR-Vs should not be dismissed simply because of their relative low mRNA abundance. Most importantly, AR-V7 or ARv567es expression level has been shown to be associated with adverse clinical outcomes.5,6,11
Treatment of VCaP cells or a castration-resistant subline of LNCaP with MDV3100 has been shown to increase the expression of AR-Vs.8,10 Although their lack of functional AR-LBD predicts resistance to MDV3100 inhibition, whether MDV3100 is indeed ineffective against AR-Vs has been controversial. Ectopic expression of AR-V7 in LNCaP xenograft tumors has been shown to produce minimal effect on the ability of MDV3100 to inhibit tumor growth.12 This has led the authors to conclude that the growth-promoting effects of AR-Vs are mediated through heterodimerization with AR-FL.12 However, studies by a number of other laboratories have shown that AR-Vs are capable of regulating gene expression in the absence of AR-FL.4–7,9,10 Additionally, in CRPC cells that express endogenous AR-FL and AR-Vs, AR-Vs have been reported to drive resistance to MDV3100.13 Our study demonstrates that methylselenol prodrug, which effectively downregulates AR-Vs, can significantly potentiate MDV3100 efficacy, and more importantly, overexpression of AR-V can attenuate the potentiating effect. Therefore, our finding further substantiates the contribution of AR-Vs to MDV3100 resistance and provides a foundation for the development of a more effective combinatorial modality for CRPC.
What’s new?
The next-generation anti-androgen MDV3100 prolongs overall survival of patients with metastatic castration-resistant prostate cancer (CRPC). However, patient responses are variable and survival benefit remains small, making the improvement of MDV3100 efficacy urgently needed. This study provides the first example of a rationally-designed, potentially effective combination modality for improving MDV3100 efficacy against CRPC. Methylselenol prodrug potentiates MDV3100 efficacy, an effect that is associated with the ability of methylselenol prodrug to downregulate androgen receptor and its constitutively-active splice variants. The findings provide a rationale for evaluating the clinical application of combining methylselenol prodrug with MDV3100 for the treatment of castration-resistant prostate cancer.
Acknowledgments
The authors thank Drs. Yun Qiu and Zhiyong Guo (University of Maryland) for AR-FL and AR-V7 expression constructs, Dr. Shahriar Koochekpour (Roswell Park Cancer Institute) for C4-2 cells and Dr. Clement Ip (Roswell Park Cancer Institute), Dr. Guangdi Wang (Xavier University) and Miss Huifang Li (Vancouver Prostate Centre) for very helpful discussion.
Grant sponsor: NCI; Grant number: K01CA114252; Grant sponsor: ACS; Grant number: RSG-07-218-01-TBE; Grant sponsor: DOD; Grant numbers: W81XWH-12-1-0112, W81XWH-12-1-0275; Grant sponsor: Mary Kay Foundation; Grant number: 019-11; Grant sponsor: National Natural Science Foundation of China; Grant number: 81272851; Grant sponsor: NIH-NCI; Grant numbers: CA095441, CA 079721, CA129828, CA 172468; Grant sponsors: Louisiana Cancer Research Consortium Start-up Fund, Tulane Cancer Center Developmental Fund, Tulane University School of Medicine Pilot Fund and Bridge Fund
Abbreviations
- AR
androgen receptor
- AR-FL
full-length androgen receptor
- AR-Vs
androgen receptor splice variants
- ARE
androgen-responsive element
- CRPC
castration-resistant prostate cancer
- DHT
dihydrotestosterone
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- LBD
ligand-binding domain
- MSA
methylseleninic acid
- MSC
methylselenocysteine
- PSA
prostate-specific antigen
- qRT-PCR
quantitative reverse transcription-PCR
- SRB
sulforhodamine B
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Lamont KR, Tindall DJ. Minireview: alternative activation pathways for the androgen receptor in prostate cancer. Mol Endocrinol. 2011;25:897–907. doi: 10.1210/me.2010-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–90. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–97. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 4.Dehm SM, Schmidt LJ, Heemers HV, et al. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–77. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo Z, Yang X, Sun F, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–13. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu R, Dunn TA, Wei S, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun S, Sprenger CC, Vessella RL, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–30. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang X, Morrissey C, Sun S, et al. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS One. 2011;6:e27970. doi: 10.1371/journal.pone.0027970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu R, Isaacs WB, Luo J. A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. Prostate. 2011;71:1656–67. doi: 10.1002/pros.21382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu R, Lu C, Mostaghel EA, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457–62. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hornberg E, Ylitalo EB, Crnalic S, et al. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One. 2011;6:e19059. doi: 10.1371/journal.pone.0019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watson PA, Chen YF, Balbas MD, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci USA. 2010;107:16759–65. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Chan SC, Brand LJ, et al. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2012;73:483–9. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ip C. Lessons from basic research in selenium and cancer prevention. J Nutr. 1998;128:1845–54. doi: 10.1093/jn/128.11.1845. [DOI] [PubMed] [Google Scholar]
- 15.Dong Y, Lee SO, Zhang H, et al. Prostate specific antigen expression is down-regulated by selenium through disruption of androgen receptor signaling. Cancer Res. 2004;64:19–22. doi: 10.1158/0008-5472.can-03-2789. [DOI] [PubMed] [Google Scholar]
- 16.Dong Y, Zhang H, Gao AC, et al. Androgen receptor signaling intensity is a key factor in determining the sensitivity of prostate cancer cells to selenium inhibition of growth and cancer-specific biomarkers. Mol Cancer Ther. 2005;4:1047–55. doi: 10.1158/1535-7163.MCT-05-0124. [DOI] [PubMed] [Google Scholar]
- 17.Lee SO, Yeon CJ, Nadiminty N, et al. Monomethylated selenium inhibits growth of LNCaP human prostate cancer xenograft accompanied by a decrease in the expression of androgen receptor and prostate-specific antigen (PSA) Prostate. 2006;66:1070–5. doi: 10.1002/pros.20329. [DOI] [PubMed] [Google Scholar]
- 18.Chun JY, Nadiminty N, Lee SO, et al. Mechanisms of selenium down-regulation of androgen receptor signaling in prostate cancer. Mol Cancer Ther. 2006;5:913–18. doi: 10.1158/1535-7163.MCT-05-0389. [DOI] [PubMed] [Google Scholar]
- 19.Ip C, Thompson HJ, Zhu Z, et al. In vitro and in vivo studies of methylseleninic acid: evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res. 2000;60:2882–6. [PubMed] [Google Scholar]
- 20.Marshall JR, Ip C, Romano K, et al. Methyl selenocysteine: single-dose pharmacokinetics in men. Cancer Prev Res (Phila) 2011;4:1938–44. doi: 10.1158/1940-6207.CAPR-10-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu H, Li GX, Wang L, et al. Methylseleninic acid enhances taxane drug efficacy against human prostate cancer and down-regulates antiapoptotic proteins Bcl-XL and survivin. Clin Cancer Res. 2008;14:1150–8. doi: 10.1158/1078-0432.CCR-07-4037. [DOI] [PubMed] [Google Scholar]
- 22.Dong Y, Zhang H, Hawthorn L, et al. Delineation of the molecular basis for selenium-induced growth arrest in human prostate cancer cells by oligonucleotide array. Cancer Res. 2003;63:52–9. [PubMed] [Google Scholar]
- 23.Jiang C, Wang Z, Ganther H, et al. Caspases as key executors of methyl selenium-induced apoptosis (anoikis) of DU-145 prostate cancer cells. Cancer Res. 2001;61:3062–70. [PubMed] [Google Scholar]
- 24.Li GX, Lee HJ, Wang Z, et al. Superior in vivo inhibitory efficacy of methylseleninic acid against human prostate cancer over selenomethionine or selenite. Carcinogenesis. 2008;29:1005–12. doi: 10.1093/carcin/bgn007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA. 2009;301:39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Bonorden MJ, Li GX, et al. Methyl-selenium compounds inhibit prostate carcinogenesis in the transgenic adenocarcinoma of mouse prostate model with survival benefit. Cancer Prev Res (Phila) 2009;2:484–95. doi: 10.1158/1940-6207.CAPR-08-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohta Y, Kobayashi Y, Konishi S, et al. Speciation analysis of selenium metabolites in urine and breath by HPLC- and GC-inductively coupled plasma-MS after administration of selenomethionine and methylselenocysteine to rats. Chem Res Toxicol. 2009;22:1795–801. doi: 10.1021/tx900202m. [DOI] [PubMed] [Google Scholar]
- 28.Chen Z, Zhang C, Wu D, et al. Phospho-MED1-enhanced UBE2C locus looping drives castration-resistant prostate cancer growth. EMBO J. 2011;30:2405–19. doi: 10.1038/emboj.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang C, Wang L, Wu D, et al. Definition of a FoxA1 Cistrome that is crucial for G1 to S-phase cell-cycle transit in castration-resistant prostate cancer. Cancer Res. 2011;71:6738–48. doi: 10.1158/0008-5472.CAN-11-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yeh S, Chang C. Cloning and characterization of a specific coactivator, ARA70, for the androgen receptor in human prostate cells. Proc Natl Acad Sci USA. 1996;93:5517–21. doi: 10.1073/pnas.93.11.5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao B, Liu X, Li J, et al. 20(S)-protopanaxadiolaglycone downregulation of the full-length and splice variants of androgen receptor. Int J Cancer. 2013;132:1277–87. doi: 10.1002/ijc.27754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao S, Durrani FA, Rustum YM. Selective modulation of the therapeutic efficacy of anticancer drugs by selenium containing compounds against human tumor xenografts. Clin Cancer Res. 2004;10:2561–9. doi: 10.1158/1078-0432.ccr-03-0268. [DOI] [PubMed] [Google Scholar]
- 33.Qi Y, Fu X, Xiong Z, et al. Methylseleninic acid enhances paclitaxel efficacy for the treatment of triple-negative breast cancer. PLoS One. 2012;7:e31539. doi: 10.1371/journal.pone.0031539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang W, Jiang C, Pei H, et al. In vivo molecular mediators of cancer growth suppression and apoptosis by selenium in mammary and prostate models: lack of involvement of gadd genes. Mol Cancer Ther. 2009;8:682–91. doi: 10.1158/1535-7163.MCT-08-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gleave ME, Hsieh JT, Wu HC, et al. Serum prostate specific antigen levels in mice bearing human prostate LNCaP tumors are determined by tumor volume and endocrine and growth factors. Cancer Res. 1992;52:1598–605. [PubMed] [Google Scholar]
- 36.Chou TC, Talaly P. A simple generalized equation for the analysis of multiple inhibitions of Michaelis-Menten kinetic systems. J Biol Chem. 1977;252:6438–42. [PubMed] [Google Scholar]
- 37.Tepper CG, Boucher DL, Ryan PE, et al. Characterization of a novel androgen receptor mutation in a relapsed CWR22 prostate cancer xenograft and cell line. Cancer Res. 2002;62:6606–14. [PubMed] [Google Scholar]
- 38.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–16. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 39.Abdo M, Knapp S. Mechanism of a redox coupling of seleninic acid with thiol. J Org Chem. 2012;77:3433–8. doi: 10.1021/jo300156x. [DOI] [PubMed] [Google Scholar]
- 40.Faehl LG, Kice JL. Oxidation of sulfides and phosphines by aromatic selenonic and seleninic acids. J Org Chem. 1979;44:2357–61. [Google Scholar]
- 41.Zhou ZX, Lane MV, Kemppainen JA, et al. Specificity of ligand-dependent androgen receptor stabilization: receptor domain interactions influence ligand dissociation and receptor stability. Mol Endocrinol. 1995;9:208–18. doi: 10.1210/mend.9.2.7776971. [DOI] [PubMed] [Google Scholar]
- 42.Yamashita S, Lai KP, Chuang KL, et al. ASC-J9 suppresses castration-resistant prostate cancer growth through degradation of full-length and splice variant androgen receptors. Neoplasia. 2012;14:74–83. doi: 10.1593/neo.111436. [DOI] [PMC free article] [PubMed] [Google Scholar]