

Abstract
Background
Genomic and transcriptomic sequence data are essential tools for tackling ecological problems. Using an approach that combines next-generation sequencing, de novo transcriptome assembly, gene annotation and synthetic gene construction, we identify and cluster the protein families from Favia corals from the northern Red Sea.
Results
We obtained 80 million 75 bp paired-end cDNA reads from two Favia adult samples collected at 65 m (Fav1, Fav2) on the Illumina GA platform, and generated two de novo assemblies using ABySS and CAP3. After removing redundancy and filtering out low quality reads, our transcriptome datasets contained 58,268 (Fav1) and 62,469 (Fav2) contigs longer than 100 bp, with N50 values of 1,665 bp and 1,439 bp, respectively. Using the proteome of the sea anemone Nematostella vectensis as a reference, we were able to annotate almost 20% of each dataset using reciprocal homology searches. Homologous clustering of these annotated transcripts allowed us to divide them into 7,186 (Fav1) and 6,862 (Fav2) homologous transcript clusters (E-value ≤ 2e-30). Functional annotation categories were assigned to homologous clusters using the functional annotation of Nematostella vectensis. General annotation of the assembled transcripts was improved 1-3% using the Acropora digitifera proteome. In addition, we screened these transcript isoform clusters for fluorescent proteins (FPs) homologs and identified seven potential FP homologs in Fav1, and four in Fav2. These transcripts were validated as bona fide FP transcripts via robust fluorescence heterologous expression. Annotation of the assembled contigs revealed that 1.34% and 1.61% (in Fav1 and Fav2, respectively) of the total assembled contigs likely originated from the corals’ algal symbiont, Symbiodinium spp.
Conclusions
Here we present a study to identify the homologous transcript isoform clusters from the transcriptome of Favia corals using a far-related reference proteome. Furthermore, the symbiont-derived transcripts were isolated from the datasets and their contribution quantified. This is the first annotated transcriptome of the genus Favia, a major increase in genomics resources available in this important family of corals.
Keywords: K-mer, Contig, Open reading frame, Fluorescent protein, Blast, Clustering, High-throughput sequencing, Illumina paired-end, Coral
Background
With the advent of Next-Generation Sequencing (NGS) technology, genomic data acquisition has become much easier, especially for non-model organisms [1]. The generation of transcriptomes from non-model organisms has also benefitted from NGS advances. Transcriptomic datasets can facilitate genome annotation, single-nucleotide polymorphism (SNP) analysis [2], marker development for population genetic and adaptive evolutionary studies [3], as well as functional classification [4] in non-model species. The application of transcriptome deep sequencing in metabolic pathway reconstruction and gene marker development has already shown great promise in Camellia sinesis[5], Cicer arietinum[6], Sphenodon punctatus[7], and Anopheles funestus[8].
This method is also valuable for relatively understudied species, such as Favia corals. Though corals are high in economic and ecological value, limited genomic resources are available, largely because samples are difficult to obtain. Because NGS requires only small amounts of animal tissue, it is possible get large amounts of information from very small samples (1–2 coral polyps). Recently, anthropogenic threats such as climate change, metal pollution and oceanic acidification [9] have led to rapid declines in worldwide coral populations, lending increased urgency to the need for genomic data. Detailed understanding at the genomic and transcriptomic level will allow for the development experimental studies to assess how the intensity and frequency of disturbances affects coral health and abundance.
Several studies have reported NGS long reads transcriptome sequencing of coral species such as Acropora millepora[10,11] and Pocillopora damicornis[12]. In addition, other recent studies have used the Short Sequence Reads (SSR) platform [13], or combined SSR and long reads approach to explore whole transcriptome modulation in response to low pH in adult Pocillopora damicornis[13], and in early life stages of Acropora millepora[14]. Yet, these coral clades are quite phylogenetically divergent from Favia[15].
This Favia genus is one of the most widely and uniformly distributed of all coral genera and is phenotypically presented as massive, dome-shaped and flat. In many cases Favia species exhibit cryptic species complexes and their phylogeny has been parodied as being a “Bigmessidae” [16]. Favia are in the Faviidae family that contains twenty-four genera, more than any other coral family [17]. Faviidae is one of the highly fragmented families and Indo-Pacific members appear to be distinct from Atlantic counterparts. Therefore, adding more molecular markers to resolve their phylogeny will add further resolution to coral systematics.
We sequenced and assembled 58 Mbp of Illumina cDNA reads from two coral Favia samples, termed “Fav1” and “Fav2,” that were collected at 65 m in the northern Red Sea (Figure 1). Reads were assembled into contigs and annotated to: 1) identify protein family clusters using the proteome of Nematostella vectensis as a reference; 2) assign functions to the protein family clusters using Nematostella vectensis GO, InterPro and KOG functional annotation; 3) identify homologous proteins in Acropora digitifera using sequence-based similarity searches; 4) identify symbiont-derived contigs in our assembly; and 5) conduct phylogenetic assessment using three molecular markers (Cytb, COI, 28S) and eleven full-length fluorescent proteins. The resulting data provide a valuable resource for future studies in Faviids and other corals.
Figure 1.
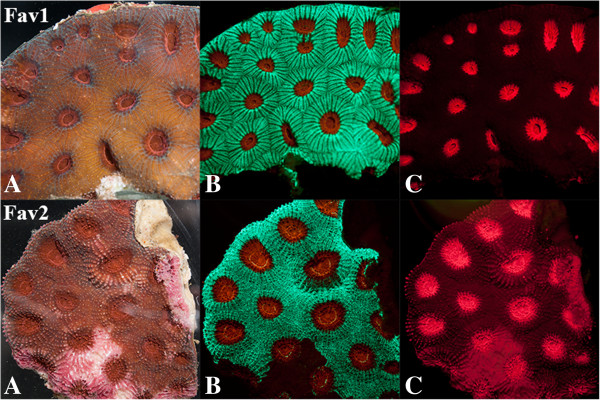
White light and fluorescent macrophotography of scleractinian coral samples. Samples of Favia sp. were placed in a narrow photography tank against a thin plate glass front. Fluorescent macro images (13.1 megapixel; Nikon D300S) were produced in a dark room by covering the flash (Vivitar 185) with interference bandpass excitation filters (Semrock, Rochester, NY). Longpass and bandpass emission filters (Semrock) were attached to the front of the camera. A) White light image; B) ex. 450–500 nm; em. 514LP; C) ex. 500–550 nm, em. 555 LP.
Results and discussion
De novo assembly
Holobiont cDNA libraries were synthesized from the RNA of two individual adult Favia sp. collected from the Gulf of Eilat in the Red Sea. Illumina runs performed on each separate, normalized, cDNA pool generated approximately 80 million reads per sample with average quality scores > Q20 at each base. The first step of assembly was carried out with ABySS [18,19], a de Brujin graph assembler. In order to recover transcripts across a range of expression levels, we carried out assembly across a range of k-mer values. Transcripts with low depth (i.e. weakly expressed) are best recovered with low k-mer values, while high depth (i.e. highly expressed) transcripts are best recovered with high k-mer values [20]. Using a range of k-mer values also allows for the identification of expressed splice variants arising from a single gene. As the Illumina read length was set to 75 bp, we chose initial k-mer values ranging from 29 to 45 bp for each sample run.
We evaluated various assembly parameters (e.g., total number of contigs, contigs longer than 100 bp, N50 length, and average contig length) as a function of k-mer length. The three k-mer values (35, 39, 45 for Fav1 and 31, 35, 39 for Fav2) with the highest N50 length [21] were selected as being most informative. In each sample, we eliminated contigs shorter than 150 bp [20] in the two k-mer assemblies with the shortest median contig length, but kept all the contigs in the assembly with the longest median contig length in order to retain any information useful for bridging in the subsequent assembly steps. Within each sample, the three k-mer assemblies were then combined, and the combined contigs were assembled with CAP3 (using default parameters), which computes overlaps to correct errors in constructing contigs and generates consensus sequences for contigs [22], thus eliminating redundant contigs. It has been suggested that assembly of ABySS followed CAP3 yield better contigs [19]. As a result, the N50 length distribution improved after using CAP3, and the best N50 values increased from 1027 to 1665 in Fav1, and 742 to 1439 in Fav2 (Figure 2). The final assembled datasets, which were used for all subsequent analyses, contained 58,848 sequences in Fav1 and 62,469 sequences in Fav2. The N50 values of these two datasets were higher than previous short-read publications [5,7,23] (675 bp, 1438 bp, 506 bp, respectively), suggesting that the quality of our data was comparable to results in other non-model species (For all commands and parameters, see Additional file 1: File S1).
Figure 2.
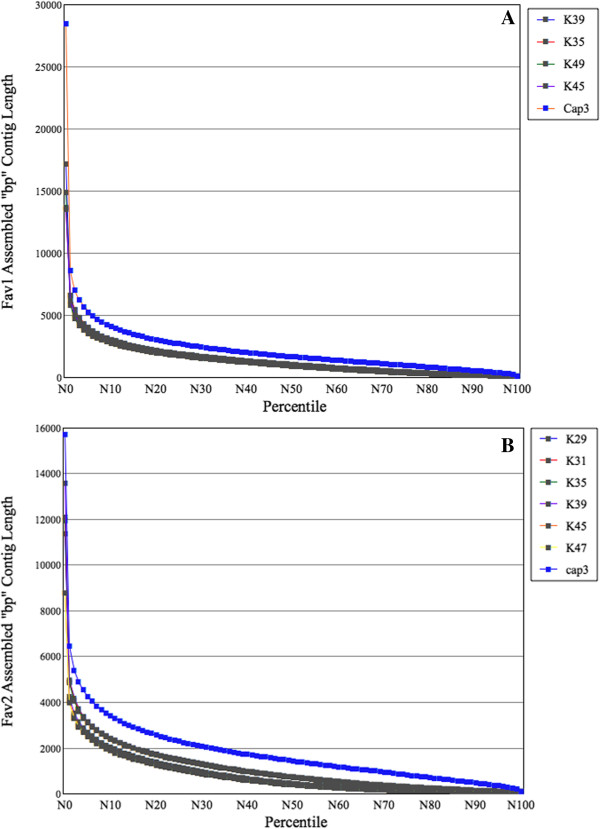
Contig length improvement after using CAP3. N50 (50% of the length of the assembled sequences) is a parameter to assess the contig length distribution (A)Fav1 contig length and N-values relationship. The thin lines represent the values for k-mer 35, 39, 45. The N50 length values were 1027, 1009, 949 bp, respectively. The line with cross represents the N-values after using CAP3, with N50 length of 1665. (B)Fav2 contig length and N-values relationship. The N50 length values for k-mer 39, 45, 49 were 453, 408, 391 bp, respectively. The N50 length values for k-mer 29, 31, 35 were 742, 734, 721 bp, respectively. The line with cross represents the N-values after using CAP3, with the N50 length of 1439 bp.
Homologous clustering of expressed coral transcripts
After using the EMBOSS package [24] to generate all possible open reading frames (ORFs) from stop to stop for each assembled contig, the resulting predicted ORFs were searched for sequence similarity against the N. vectensis proteome [25], using reciprocal BlastP (E-value ≤2e-30) [26] (Script 1). For the 519,766 predicted ORFs longer than 150 bp, 12,141 unique ORFs in Fav1 showed considerable sequence similarity to 7,186 existing protein sequences in N. vectensis. Similarly, 12,425 unique ORFs in Fav2 showed similarity to 6,862 N. vectensis protein sequences (Additional file 2: File S2, Additional file 3: File S3). The top Blast hits for each sample were saved in a pre-clustering list using a Perl script (Script 2; Output files reported in Additional file 4: File S4, Additional file 5: File S5). These lists were then used in TRIBE-MCL [27] to identify homologous protein family clusters in a comprehensive and uniform way (Additional file 6: File S6, Additional file 7: File S7). The main clustering parameter, inflation value (r), was selected as default (r = 2.5). Fav1 and Fav2 had similar numbers (7,186 and 6,862, respectively) of protein family clusters homologous to unique N. vectensis proteins. These clusters were subjected to further functional annotation.
In order to evaluate the completeness of our annotation using N. vectensis as the reference as opposed to using another available Cnidarian non-annotated proteome (A. digitifera), we applied a newly-developed completeness metric [28] (In prep.) to determine the proportion of the reference proteome covered by our sets of assembled transcripts. Only those ORFs with length coverage ≥80% of the matched protein from the N. vectensis or A. digitifera proteome were included. Completeness measurements in Fav1 and Fav2 compared to N. vectensis were 29.54% and 28.20%, respectively; when the same procedure was carried out using the unannotated proteome of A. digitifera as a reference (23,677 ORFs downloaded from http://marinegenomics.oist.jp/genomes/downloads?project_id=3. This showed an improvement of only 1-3%, thus validating our usage of N. vectensis as a reference proteome (Additional file 8: Table S1).
Functional annotation and characterization of the isoform clusters in Fav1
To identify the putative function of 7,187 isoform clusters, Gene Ontology (GO) and protein domain (KOG, InterPro) searches were performed using the functional annotation of the N. vectensis. (Data downloaded from the JGI genome project http://genome.jgi-psf.org/Nemve1/Nemve1.download.html). The clusters were assigned gene names based on the gene name annotation of the best Blast match for the sequences (Additional file 9: File S8). This process successfully assigned gene names for 6,632 (92.27%) clusters using GO term, KOG description, and InterPro description. Among 12,141 annotated best hits, 11,411 (93.98%) gene names were assigned to sequences. These provide a rough estimate of the number of different genes expressed in Fav1 libraries. Broadly, the putative homologs of genes involved in various cellular processes and pathways found to be functionally conserved.
Based on GO terms assignment to clusters, a total of 4,678 (65%) clusters were assigned at least one GO term, among which 11% were assigned at least one GO term in biological processes, 48% in molecular function and 6% in cellular component category (Additional file 10: Figure S1). Among the various biological processes, protein metabolism, and electron transport were mostly highly represented (Table 1). Protein metabolism is also highly represented in other transcriptome characterization studies [6,7,29].
Table 1.
Top 30 frequent annotated functions of homologous protein clusters in Fav1
|
Top frequent GO-annotated homologous protein clusters in Fav1 | ||
|---|---|---|
| Go categories/Description | Count | Percentage |
| Total clusters |
7,187 |
|
| Total (GO-annotated) |
4,677 |
65.1% |
|
Molecular function |
3,477 |
48.37% |
| 1-Nucleic acid binding |
241 |
5.15% |
| 2-Protein kinase activity |
218 |
4.66% |
| 3-DNA binding |
208 |
4.45% |
| 4-Catalytic activity |
173 |
3.70% |
| 5-Calcium ion binding |
158 |
3.38% |
| 6-ATP binding |
129 |
2.76% |
| 7-Protein binding |
119 |
2.54% |
| 8-GTP binding |
114 |
2.44% |
| 9-Transporter activity |
96 |
2.05% |
| 10-Structural constituent of ribosome |
82 |
1.75% |
|
Biological process |
776 |
16.59% |
| 1-Metabolism |
122 |
2.61% |
| 2-Electron transport |
88 |
1.88% |
| 3-Intracellular signaling cascade |
54 |
1.15% |
| 4-Proteolysis and peptidolysis |
48 |
1.03% |
| 5-Protein folding |
47 |
1.00% |
| 6-Protein modification |
31 |
0.66% |
| 7-Cell adhesion |
29 |
0.62% |
| 8-Intracellular protein transport |
26 |
0.56% |
| 9-Carbohydrate metabolism |
21 |
0.45% |
| 10-Regulation of cell cycle |
18 |
0.38% |
|
Cellular component |
424 |
6% |
| 1-Ubiquitin ligase complex |
68 |
1.45% |
| 2-Integral to membrane |
58 |
1.24% |
| 3-Membrane |
58 |
1.24% |
| 4-Nucleus |
46 |
0.98% |
| 5-Intracellular |
41 |
0.88% |
| 6-Cytoplasm |
26 |
0.56% |
| 7-Cytoskeleton |
24 |
0.51% |
| 8-Nucleosome |
16 |
0.34% |
| 9-Chromatin |
10 |
0.21% |
| 10-Extracellular region | 8 | 0.17% |
Top 30 high frequent annotated homologous protein clusters under cellular component, molecular function and biological processes. Full annotation included in Additional file 9: File S8.
According to assigned KOG descriptions to Fav1 clusters, a total of 6,326 (88%) clusters were assigned at least one KOG description. However, this was 4,489 (62.45%) with InerPro description assignment. This implies that the KOG description was most useful in assigning domain description to our dataset compared to InterPro. The top most frequently detected domain, associated with KOG and InterPro assignment, include conserved domain associated with predicted E3 ubiquitin ligase, fibrillins and related proteins containing Ca2 + −binding EGF-like domains, FOG: Zn-finger, GPCR Rhodopsin, and Ras GTPase superfamily. One of the utilities of domain annotation is that it provides quick access to homologs of genes with known roles in intercellular signaling pathway. The representation of genes involved in intracellular signaling pathway was very similar to that of A. millepora[10]. However, a few families showed the events of expansion (for example, Patched, Hepatocyte nuclear factor 4 and Activin-like kinase) and contraction (for example, Notch-delta, Frizzled, Wnt etc.) indicating their functional significance (Table 2).
Table 2.
Intracellular signaling pathway genes annotated in Fav1
|
Intracellular signaling pathway proteins annotated in Fav1 | ||
|---|---|---|
| Pathway | Protein name | Sequences (n) |
| Hedgehog |
Patched |
27 |
| |
Sonic |
2 |
| |
Fused |
1 |
| |
Receptor activity (IFRD-C) |
1 |
| |
DUF699 |
2 |
| |
Smoothened |
12 |
| JAK/STAT |
STAT protein |
1 |
| NFKB/Toll |
Nuclear factor NF-kappa-B |
1 |
| |
Intermediate in Toll-signaling |
1 |
| |
Toll-like receptor |
1 |
| NHR |
Hepatocyte nuclear factor 4 |
2 |
| Notch |
Notch |
4 |
| |
TACE |
3 |
| RTK |
RTK signaling protein |
1 |
| TGF-beta |
Activin-like kinase |
8 |
| |
SMAD |
9 |
| |
TGF-beta-receptor |
1 |
| WNT |
Frizzled |
9 |
| Wnt | 2 | |
Further, we identified major transcription factors encoding transcripts. In comparison to A. millerpora[10], the represented genes were somewhat similar. However, a few families were newly reported in our dataset (For example, HMG box, T-box, ETSDomain, MADS) (Table 3).
Table 3.
Major transcription factor families identified by conserved domain annotation
|
Transcription factors identified by KOG/InterPro/GO annotation in Fav1 | |
|---|---|
| Sequence description | Sequences(n) |
| CBF |
1 |
| Transcriptional Coactivator P50 |
1 |
| Transcriptional Coactivator P100 |
6 |
| Transcriptional Coactivator CAPER |
2 |
| Homeobox domain |
7 |
| HSF-type DNA-binding |
1 |
| P53 DNA-binding domain |
2 |
| NF-X1-type zinc finger protein |
3 |
| Dimerization partner (TDP) |
2 |
| Fork head |
15 |
| Basic region leucine zipper & bZIP |
6 |
| Helix-loop-helix DNA binding domain |
12 |
| Myb-like DNA-binding domain |
3 |
| Zinc finger C2H2 type |
3 |
| Zinc finger MIZ type |
1 |
| HMG box |
12 |
| TBOX |
5 |
| ETS domain |
12 |
| MADS domain | 4 |
Annotation of Symbiodinium-derived contigs
Holobiont coral tissues also contain eukaryotic dinoflagellate endosymbionts of the genus Symbiodinium[30,31]. We therefore determined the contribution of symbiont-derived transcripts in our analysis. First, we extracted the regions of cDNA contigs that corresponded to each individual annotated ORF in two datasets (For commands, see Additional file 1: File S1). Furthermore their similarity search against two Symbiodinium transcriptomes (http://medinalab.org/zoox/) was performed using BlastN. In order to define an E-value as a cutoff threshold, a reciprocal BlastN search between the N. vectensis genome and the two Symbiodinium transcriptomes showed an average E-value of e-80. Thus all contigs with similarity higher than this threshold to Symbiodinium were defined as likely to be symbiont-derived. Based on these results, 9% of the annotated ORFs (1.34% of the total assembled contigs) of Fav1 were labeled as symbiont sequences, and 8.7% (1.61% of total assembled contigs) of Fav2. FASTA files of these non-symbiont transcripts are reported (Additional file 11: File S9, Additional file 12: File S10). Finally, we performed BlastX (E-value equal to at least e-30) on the non-symbiont derived cDNA fragments against the N. vectensis proteome to confirm correct initial annotation by BlastP. All the cDNA sequences matched to the same N.vectensis IDs that were predicted using BlastP.
Phylogenetic assessment
Molecular markers are essential tools for population genetic studies. Typically, combination of mitochondrial and nuclear markers are used to examine the species relationships. In order to generate a Favia molecular marker dataset, we downloaded Favia related sequences from NCBI. Similarity searches were carried out against this Favia dataset. Among various molecular markers, we chose COI, Cytb and 28S. Individual sequence regions were identified and extracted from the cDNA contig files in both samples. DNA alignments for each locus were generated using ClustalW2 with default parameters [32] (Additional file 13: File S11, Additional file 14: File S12, Additional file 15: File S13). Consequently, a matrix of these three loci was generated using FASconCAT [33]. A Maximum likelihood phylogenetic analysis (RaxML) was carried out [34]. Maximum likelihood phylogenetic analysis using three loci (COI, Cytb, 28S) suggests that these Favia samples belong to Faviids (Additional file 16: Figure S2). Morphological analysis places them as F. albidus[17], a species that is not yet represented in NCBI. For example, out of 18 Favia species that have been described morphologically, only 15 of them have molecular data in NCBI. F. albidus, F. helianthoides, and F. marshae lack molecular markers in NCBI. Based on geological distribution [17] and morphology, we suggest these two species belong to F. albidus. In fact, F. helianthoides has no morphological similarities with our samples, and F. marshae habitat has never been reported in Red Sea [17]. However, further skeletal samplings are required for final validation [35,36]. Regardless, this study increases the protein information of the Faviids from 496 proteins to over 12,000 proteins in NCBI.
Characterization of one exemplary homologous protein cluster
From the protein clustering results, we chose to characterize a protein family with a natural fluorescent property. One of the benefits of utilizing scleractinian corals as our model organism is that they posses genes for fluorescent proteins (FPs), a rare characteristic in most other Phyla’ besides Cnidaria [37-40]. In N. vectensis, six protein IDs encode for FPs [41]. A search among the homologous sequence clusters with E values of at least 2e-30 in each transcriptome led to the identification of one protein cluster group per sample representing potential fluorescent proteins (FPs). A total of 11 new potential FPs were identified, six belonging to the Fav1 sample and four belonging to the Fav2 sample. One additional sequence, s23Contig9635-2 was found by increasing the E-value to 2e-10 in Fav1. The alignment of these sequences with N. vectensis fluorescent protein sequences (JGI ID:205348, ID:206334), Branchiostoma GFPa1[42] and GFP of Aequorea victoria (GI:17943301) showed a considerable homology (Figure 3). The conserved chromophore region is located at the residues 303 to 305 based on the top sequence. Our data shows that one of the newly identified potential fluorescent protein sequences (Fav1 s23Contig16657-5) is 185 amino acids longer at the N-terminus (416 amino acids in total) and two of them were shown to be 49 (Fav2 s62Contig19888-6) and 41 amino acids (Fav2 s62Contig41210-3) longer than the consensus length of reported sequences in NCBI (wild-type GFP from Aequorea victoria is 236 amino acids) (Additional file 17: Figure S3). This extended region does not seem to interfere with the proper folding and expression of FP, however further studies are required to reveal the function of these upstream domains.
Figure 3.

Overlapping region of amino acid sequence alignment of one exemplary cluster of identified homologous protein clusters. This gene family belongs to naturally expressed fluorescent protein. Conserved chromophore region (XYG) is located at the position 303–305. The newly identified sequences with extended N-terminal are s23Contig16657-5, s23Contig40465-7 in Fav1, s62Contig19888-6, and s62Contig41210-3 in Fav2. The full-length alignment is reported in Additional file 17: Figure S3.
Furthermore, the maximum likelihood trees were generated from the alignment of 156 fluorescent sequences, including the 11 newly identified sequences (Additional file 18: Figure S4, Additional file 19: File S14 contains all the accession numbers). There was a strong bootstrap support for basal clade relationships within tree. This includes the order Ceriantipatharia, and Pennatulacea, although low bootstrap support for FPs within order Scleractinia. Ctenophore FP clustered with hydrozoan FP, therefore the cnidarian clade was not monophyletic. Others have shown that incongruence with taxonomy is not unusual in fluorescent proteins [43]. For better visualization, a smaller maximum likelihood sub-tree was generated from 46 scleractinian FP sequences (Figure 4). Although the bootstrap values improved compared to Additional file 18: Figure S4, some branches still exhibited low bootstrap values. Nonetheless, using RaXML [27], we categorized the newly identified sequences into four clades and using ProtTest [44] we identified “PROTGAMMAWAGF” as the best-fit model.
Figure 4.

Maximum likelihood tree of 46 known fluorescent proteins and 11 newly identified fluorescent protein sequences using RaxML. The alignment was 1,000 times bootstrapped and one FP sequence from N. vectensis was the out-group. The newly identified FP sequences are colored blue. Other colors represent different coral families; Faviidae, red; Acroporidae, orange; Oculinidae, brown; Pectiniidae, dark green; Meandrinidae, dark purple; Mussidae, pink; Poritidae, green; Node labels are bootstrap supports. See Additional file 19: File S14 for information on alignment.
In order to evaluate our assembly method and the possible impact of ABySS-specific errors on the annotation accuracy of the long candidate FP sequence, we performed both Trans-ABySS [20] and Trinity [45] on reads from Fav1. Both assembly programs led to the generation of sequences identical to Fav1 s23Contig16657-5 as predicted using ABySS and CAP3. (Additional file 20: File S15).
Validation of the identified protein clusters as fluorescent proteins
The intrinsic fluorescence of FPs includes a unique chromophore that is formed post-translationally within the protein upon autocatalytic cyclization and oxidation of residues X-Tyr-Gly [46]. The fluorophore is located almost at the center of the cylinder and is inaccessible to outside enzymes [46,47]. The GFP fluorophore is capable of forming under a wide range of conditions and once formed is highly stable. The entire structure is very resistant to denaturation by heat and denaturants. The three sequences with longer N-terminal domains (s23Contig16657-5, s62Contig19888-6 and s62Contig41210-3) were cloned into mammalian expression vectors. We used Kozak analysis [48] to pick the best potential start codon, and reading frames were generated using gene synthesis. The start codons are underlined in red in Additional file 21: Figure S5. The synthesized sequences were optimized for expression in mammalian cell lines. The synthesized sequences showed fluorescence when expressed in HEK-293 mammalian cells, thus validating them as genuine FPs (Figure 5).
Figure 5.
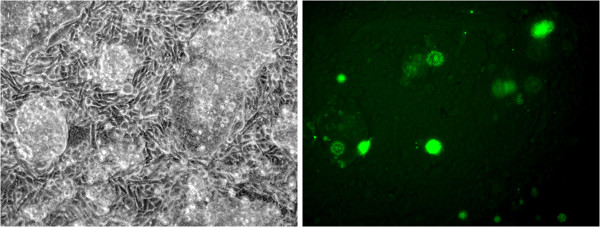
Expression of an assembled contig in HEK293 mammalian cells yields fluorescence. An open reading frame of contig 19888 from Fav2 was synthesized using mammalian preferred codon usage (887 bases of s62Contig19888) and subcloned into pcDNA 3.1, and transfected into HEK293 mammalian cells using Fugene (Boehringer-Mannheim). The left panel depicts a phase contrast image of transfected HEK293 cells, and the right panel depicts fluorescence (using FITC excitation and emission) from the same field. Scale bar = 100 microns.
In Silico quantification of Faviids transcripts
In order to rule out the possibility of promiscuous domain assembly, we assessed the quality of the de novo assembly of FP sequences, as well as all other transcripts, by mapping reads on assembled contigs for each sample. Such read alignment to contigs is necessary to provide support for new transcript identification as well as for determining gene expression levels [49,50]. In order to measure the Reads Per Kilobase of exon model per Million mapped reads (RPKM) [50], a sub-fasta cDNA region, corresponding to each ORF, within each contig was generated. Reads were aligned to these annotated cDNA regions. Coverage (RPKM) measurements were determined using a Perl script (Script 3). The results are reported (Additional file 22: File S16, Additional file 23: File S17). The mapping of all the reads onto the annotated Faviids transcript showed that the number of reads corresponding to each transcript ranged from 10 to 47,189, with an average of 850 reads per transcript in Fav1, and 10 to 29,222, with an average of 766.37 reads per transcript in Fav2, indicating a wide range of expression level of Faviids transcripts. It also indicates that very low expressed annotated Faviids transcripts were also represented in our assembly. The minimum coverage (RPKM) of an annotated Fav1 transcript was 3.89 and maximum of 6,919.20 with an average of 68.61. The RPKM ranged from 3.60 to 8,576, with an average of 72.64 in Fav2. The average and the range of RPKM per transcript is similar and somewhat higher (25.7) than other whole transcriptome studies [26].
All the cDNA regions annotated for fluorescent property had reasonable coverage, including the long candidate cDNA sequence (Fav1 s23Contig16657-5) (Additional file 21: Figure S5). Based on the calculated RPKMs for each of the identified fluorescent protein in both samples, s23Contig19691-3 in Fav1, and s62Contig57475-7 in Fav2 had the highest coverage level (Figure 6).
Figure 6.
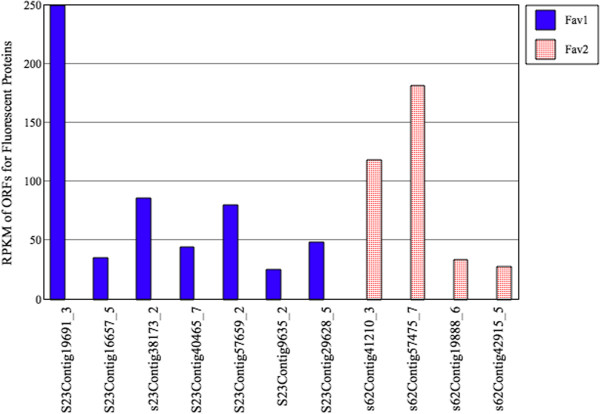
In silico coverage plot of the read-to-contig alignment measurements. The cDNA fragments with annotation for fluorescent protein coverage measurements.
Conclusions
In this study, we demonstrate a gene clustering strategy and utilize this in conjunction with NGS contig assembly, sequence conservation measurements, annotation and expression quantification for de novo assembled transcriptomic data. Working with two uncharacterized Faviid corals, we report 120,000 non-redundant transcripts to a genus whose sequence data was previously limited to 496 in public databases. These results provide greatly enhanced access to the expressed genes in Faviidae reef building corals, a potentially valuable resource of genetic/functional markers for population structure and functional genomic studies. We also took advantage of the optical properties of these corals expressed fluorescent proteins to validate our annotation methods to show that these sequences were indeed bonafide fluorescent protein genes. These methods reported in this study are available via Open Source software programs as well as our provided scripts.
Methods
Coral collection and total RNA isolation
This study was conducted during the period of May–June 2009 on a coral reef on the northern tip of the Gulf of Eilat, in the northern Red Sea (29º30′N, 34°55′E).
Samples were collected at 65 m, using closed-circuit trimix rebreather system (Megalodon™). The organisms were identified under water to the family level, Faviidae, and brought to the surface in a black mesh bag to avoid sun exposure. The organisms were immediately photographed and vouchered with white light and fluorescent photography as described in [51] and stored in a shaded running-seawater facility. Within 1–2 hours of collection, samples were rinsed in sterile-filtered artificial seawater and processed for RNA and DNA. The tissue of the coral was extracted from the skeleton using QiaShredder (Qiagen). For RNA, the TriZol method was used and stored as an ethanol precipitate for travel back to the US. DNA was extracted using Qiagen DNAeasy kit according to manufacturer’s protocol and stored in at 4°C. The specimens have been photo vouchered and their genomic and transcriptomic raw materials are stored in the American Museum of Natural History Ambrose Monell Cryo Collection.
Preparation and screening of cDNA library
Illumina sequencing using the GAII platform was performed at the Yale University W.M. Keck Biotechnology Resource Laboratory according to manufacturer’s instructions (Illumina, San Diego, CA) (Additional file 24: File S18) and using high quality RNA with a 28S rRNA band at 4.5 kb that is at least twice the intensity of the 18 s rRNA band at 1.9 kb. The cDNA library contained 77,804,306, 75-mer length reads. The sequencing data are deposited in NCBI Sequencing Read Archive [52]. (The BiosampleIDs = SAMN01761696, SAMN01761695).
De novo assembly
De novo assembly was carried out using ABySS with default settings across multiple k-mer values [18]. After assessing different k-mer values, the three best k-mer assemblies (35-mer, 39-mer, 45-mer for Fav1 and 31-mer, 35-mer, 39-mer for Fav2) were selected and concatenated for the second step of assembly. To evaluate the N50 length and the number of assembled contigs using different k-mer values, we used a Perl script. CAP3 [22] was used to remove redundancy across ABySS assemblies and to merge contigs into longer sequences. All assembled contigs were subjected to annotation and further protein homology searches. Trans-ABySS [20] and Trinity [45] were used to confirm the long ORFs, homologous to fluorescent proteins, which were identified with ABySS and CAP3.
Gene annotation and analysis
A set of possible Open Reading Frames (ORF), stop to stop from assembled sequences, was generated using EMBOSS [24]. To annotate the de novo assembled sequences, a similarity search against N. vectensis proteome was conducted using BLASTP with two E values of 2e-10 and 2e-30. The resulting data (E-value of 2e-30) was filtered and clustered using TRIBE-MCL [27]. Each homologous group was annotated using GO and KOG annotated N. vectensis data (http://genome.jgi-psf.org/Nemve1/Nemve1.download.html). For Symbiodinium peptide annotation, a homology search using BLASTN with E values of 2e-80 against the Symbiodinium transcriptome (http://medinalab.org/zoox/) was carried out. The final non-symbiont FASTA cDNA fragments were reported.
Completeness measurement
The BlastP (E-value of 2e-30) output list generated from homology search of both samples against N. vectensis[41] and A. digitifera[53] was organized for completeness measurements. The completeness formula according to [28] was implemented into a Perl script (In prep) to determine the percentage of the reference proteome that is covered by each of our sets of assembled transcripts. Length coverage of each of these reference ORFs by a hit from our data set had to be at least 80%.
Phylogenetic analysis of FPs
The maximum likelihood tree of identified fluorescent protein was generated using RaXML [34] under PROTGAMMAWAGF amino acid substitution model, selected based on the results from ProtTest [44]. The alignment was generated using MAFFT [54] and CLUSTALW2 [32] with minor adjustment at the N-terminus region, when long gaps were inconsistent with other isoforms. Bootstrap values were estimated based on 1,000 replicates and were given for all presented branches. The variant sites were visualized with geneious (http://www.geneious.com). Dendroscope was used for visualization [55].
Phylogenetic assessment
Molecular barcodes for all the Favia related sequences were downloaded from NCBI. A similarity search with sequences from our annotation was carried out against this Favia dataset. Cytb, COI and 28S sequences were identified and extracted from the cDNA contig files in both samples. DNA alignments for each locus were generated using ClustalW2 with default parameters [32]. Consequently, a matrix of these three loci was generated using FASconCAT [33]. A Maximum likelihood phylogenetic analysis (RaxML) was carried out [34]. Bootstrap values were estimated based on 10,000 replicates and were given for all presented branches. Dendroscope was used for visualization [55].
Cloning of fluorescent proteins
The three cDNA sequences (Fav1 s23Contig16657, Fav2 s62Contig19888-6 and Fav2 s62Contig41210-3) were synthesized and propagated in pUC57 (GenScript USA Inc.). Kozak [48] analysis was used to determine the location of the potential start codon. The genes were subcloned from pUC57 into the NotI-BamHI site of the mammalian expression vector pcDNA 3.1 (Invitrogen, Inc.) using standard recombinant techniques [56].
In Silico gene coverage measurements
Gene coverage levels were determined using a Perl script (Script 3). This script implements Bowtie [57] to map reads to an annotated reference cDNA, and calculates the RPKM according the formula used in [50]. For visualization, BWA [58] was used to generate the read-to-contig alignment. The annotated cDNA from individual samples were used as the reference contig, and SAMtools [59] was used to generate binary files to be visualized in the IGV [60] genome viewer (For commands, see Additional file 1: File S1).
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DFG, RD, VAP and SFPM designed the study. SFPM carried out the molecular genetic studies. SFPM and AN designed the bioinformatics pipeline and data handling scripts. RD coordinated the statistical analysis. H-T K, VAP and DFG designed synthetic genes for expression in mammalian cells. ZH, VAP, DT and DFG participated in sample collection and Illumina sequencing. DFG, RD, VAP and SFPM drafted the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Parameters and commands used in this manuscript.
BlastP parsed output files against N. vectensis proteome for sample Fav1 and Fav2 with 2e-30.
BlastP parsed output files against N. vectensis proteome for sample Fav1 and Fav2 with 2e-30.
TRIBE-MCL input files.
TRIBE-MCL input files.
Homologous protein clusters (TRIBE-MCL) output for sample Fav1 and Fav2.
Homologous protein clusters (TRIBE-MCL) output for sample Fav1 and Fav2.
Completeness metrics for two samples compared to N. ventensis and A. digitifera.
GO,KOG, InterPro annotation for homologous protein clusters in Fav1.
Distribution of Fav1 transcript clusters in different GO categories.
FASTA files for cDNA region encoding for non-symbiont annotated ORFs in Fav1 and Fav2.
FASTA files for cDNA region encoding for non-symbiont annotated ORFs in Fav1 and Fav2.
Alignment of Fav1 and Fav2 Cytb nucleotide sequences, including other Favia species.
Alignment of Fav1 and Fav2 COI nucleotide sequences, including other Favia species.
Alignment of Fav1 and Fav2 28S nucleotide sequences, including other Favia species.
Maximum likelihood tree of three loci (COI, Cytb, 28S). Data matrix was generated from 15 Favia species and Fav1 and Fav2. Nucleotide sequences were aligned using clustalw2 with default parameters, the 3 loci matrix was generated using FASconCAT, and the tree was constructed using RaxML (See methods). Montastrea cavernosa is selected as the out-group.
Amino acid sequence alignment of full-length fluorescent protein isoforms.
Maximum likelihood tree of 156 known fluorescent proteins, including our 11 newly identified sequences using RaxML. Shows the relationships of the major groups of known fluorescent proteins. Major lineages cluster together, although Ctenophore and Hydrozoa do not form a monophyletic group. Within Anthozoa class, order Ceriantharia (orange); Actinaria (red); Pennatulacea (dark green); and Scleractinia (black); Hydrozoa (purple); Copepoda (light green); Ctenophora (blue); Chordata (turquoise blue), most basal group; Newly identified sequences are colored blue within Scleractinia. The alignment was 1,000 times bootstrapped and B. floridae was the out-group.
Alignment of 156 known fluorescent proteins, including the 11 newly identified FP sequences.
Search result in Trans-ABySS and Trinity assembly output for homologous contig, similar to identified Fav1 s23Coting16657-5 produced by ABySS and CAP3.
Read-to-contig alignment. 75 bp read alignments to the coding region of s23Contig16657-5, 1,377 bp total length.
RPKM measurement for all annotated cDNA regions from Fav1 and Fav2.
RPKM measurement for all annotated cDNA regions from Fav1 and Fav2.
Protocol for preparing samples for sequencing of mRNA.
Scripts: Script 1: Perl script for performing blast search. Script 2: Perl script for pre-clustering the blast parsed file. Script 3: Perl script to calculate RPKM for the assembled file. Script S1: Perl script to shuffle short read sequences. Script S2: Perl script to measure the N50 statistics. Script S3: Unix shell script to remove Fasta files shorter than a threshold. Script S4: Generate the sub-Fasta file. Script S5: Extract the cDNA sequences corresponding to ORF files.
Contributor Information
Shaadi F Pooyaei Mehr, Email: fpooyaei-mehr@amnh.org.
Rob DeSalle, Email: desalle@amnh.org.
Hung-Teh Kao, Email: htkao2@gmail.com.
Apurva Narechania, Email: anarechania@amnh.org.
Zhou Han, Email: zhan@jbpierce.org.
Dan Tchernov, Email: dtchernov@univ.haifa.ac.il.
Vincent Pieribone, Email: vpieribo@jbpierce.org.
David F Gruber, Email: david.gruber@baruch.cuny.edu.
Acknowledgments
We thank Timor Katz and Tali Mass of the Interuniversity Institute of Eilat for their technical diving assistance in the collection of coral samples. Coral sample collections in this study have complied with the current laws of Israeli Natural Parks Authority; permit 2010/38008. Funding was provided by NSF grant # 0920572 and via a Baruch College Travel Grant to DFG.
References
- Metzker ML. Sequencing technologies—the next generation. Nat Rev Gen. 2009;11(1):31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Koepke T, Schaeffer S, Krishnan V, Jiwan D, Harper A, Whiting M, Oraguzie N, Dhingra A. Rapid gene-based SNP and haplotype marker development in non-model eukaryotes using 3'UTR sequencing. BMC Genomics. 2012;13:18. doi: 10.1186/1471-2164-13-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmer KR, Fan S, Gunter HM, Jones JC, Boekhoff S, Kuraku S, Meyer A. Rapid evolution and selection inferred from the transcriptomes of sympatric crater lake cichlid fishes. Mole Ecol. 2010;19(Suppl 1):197–211. doi: 10.1111/j.1365-294X.2009.04488.x. [DOI] [PubMed] [Google Scholar]
- Yang SS, Tu ZJ, Cheung F, Xu WW, Lamb JF, Jung HJ, Vance CP, Gronwald JW. Using RNA-Seq for gene identification, polymorphism detection and transcript profiling in two alfalfa genotypes with divergent cell wall composition in stems. BMC Genomics. 2011;12:199. doi: 10.1186/1471-2164-12-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi CY, Yang H, Wei CL, Yu O, Zhang ZZ, Jiang CJ, Sun J, Li YY, Chen Q, Xia T. et al. Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genomics. 2011;12:131. doi: 10.1186/1471-2164-12-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg R, Patel RK, Tyagi AK, Jain M. De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 2011;18(1):53–63. doi: 10.1093/dnares/dsq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller HC, Biggs PJ, Voelckel C, Nelson NJ. De novo sequence assembly and characterisation of a partial transcriptome for an evolutionarily distinct reptile, the tuatara (Sphenodon punctatus) BMC Genomics. 2012;13:439. doi: 10.1186/1471-2164-13-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford JE, Guelbeogo WM, Sanou A, Traore A, Vernick KD, Sagnon N, Lazzaro BP. De novo transcriptome sequencing in Anopheles funestus using Illumina RNA-seq technology. PloS One. 2010;5(12):e14202. doi: 10.1371/journal.pone.0014202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandolfi JM, Bradbury RH, Sala E, Hughes TP, Bjorndal KA, Cooke RG, McArdle D, McClenachan L, Newman MJ, Paredes G. et al. Global trajectories of the long-term decline of coral reef ecosystems. Science. 2003;301(5635):955–958. doi: 10.1126/science.1085706. [DOI] [PubMed] [Google Scholar]
- Meyer E, Aglyamova GV, Wang S, Buchanan-Carter J, Abrego D, Colbourne JK, Willis BL, Matz MV. Sequencing and de novo analysis of a coral larval transcriptome using 454 GSFlx. BMC Genomics. 2009;10:219. doi: 10.1186/1471-2164-10-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer E, Aglyamova GV, Matz MV. Profiling gene expression responses of coral larvae (Acropora millepora) to elevated temperature and settlement inducers using a novel RNA-Seq procedure. Mol Ecol. 2011;20(17):3599–3616. doi: 10.1111/j.1365-294X.2011.05205.x. [DOI] [PubMed] [Google Scholar]
- Traylor-Knowles N, Granger BR, Lubinski TJ, Parikh JR, Garamszegi S, Xia Y, Marto JA, Kaufman L, Finnerty JR. Production of a reference transcriptome and transcriptomic database (PocilloporaBase) for the cauliflower coral. Pocillopora damicornis. BMC Genomics. 2011;12:585. doi: 10.1186/1471-2164-12-585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal-Dupiol J, Zoccola D, Tambutte E, Grunau C, Cosseau C, Smith KM, Freitag M, Dheilly NM, Allemand D, Tambutte S. Genes related to ion-transport and energy production are upregulated in response to CO2-driven pH decrease in corals: new insights from transcriptome analysis. PloS One. 2013;8(3):e58652. doi: 10.1371/journal.pone.0058652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya A, Huisman L, Ball EE, Hayward DC, Grasso LC, Chua CM, Woo HN, Gattuso JP, Foret S, Miller DJ. Whole transcriptome analysis of the coral Acropora millepora reveals complex responses to CO(2)-driven acidification during the initiation of calcification. Mol Ecol. 2012;21(10):2440–2454. doi: 10.1111/j.1365-294X.2012.05554.x. [DOI] [PubMed] [Google Scholar]
- Kitahara MV, Cairns SD, Stolarski J, Blair D, Miller DJ. A comprehensive phylogenetic analysis of the Scleractinia (Cnidaria, Anthozoa) based on mitochondrial CO1 sequence data. PloS One. 2010;5(7):e11490. doi: 10.1371/journal.pone.0011490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Licuanan WY, Baird AH, Fukami H. Cleaning up the 'Bigmessidae': molecular phylogeny of scleractinian corals from Faviidae, Merulinidae, Pectiniidae and Trachyphylliidae. BMC Evol Biol. 2011;11:37. doi: 10.1186/1471-2148-11-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veron J. Corals World. 2000. pp. 100–131.
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19(6):1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahais V, Gayral P, Tsagkogeorga G, Melo-Ferreira J, Ballenghien M, Weinert L, Chiari Y, Belkhir K, Ranwez V, Galtier N. Reference-free transcriptome assembly in non-model animals from next-generation sequencing data. Mol Ecol Resour. 2012;12(5):834–845. doi: 10.1111/j.1755-0998.2012.03148.x. [DOI] [PubMed] [Google Scholar]
- Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, Mungall K, Lee S, Okada HM, Qian JQ. et al. De novo assembly and analysis of RNA-seq data. Nat Methods. 2010;7(11):909–912. doi: 10.1038/nmeth.1517. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Schwarz EM, Williams B, Schaeffer L, Antoshechkin I, Wold BJ, Sternberg PW. Scaffolding a Caenorhabditis nematode genome with RNA-seq. Genome Res. 2010;20(12):1740–1747. doi: 10.1101/gr.111021.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Res. 1999;9(9):868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadamoto H, Takahashi H, Okada T, Kenmoku H, Toyota M, Asakawa Y. De novo sequencing and transcriptome analysis of the central nervous system of mollusc Lymnaea stagnalis by deep RNA sequencing. PloS one. 2012;7(8):e42546. doi: 10.1371/journal.pone.0042546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16(6):276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, Salamov A, Terry A, Shapiro H, Lindquist E, Kapitonov VV. et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science. 2007;317(5834):86–94. doi: 10.1126/science.1139158. [DOI] [PubMed] [Google Scholar]
- Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36:W5–W9. doi: 10.1093/nar/gkn201. Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright A, Van Dongen S, Ouzounis C. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30(7):1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JA, Wang Z. Next-generation transcriptome assembly. Nat Rev Gen. 2011;12(10):671–682. doi: 10.1038/nrg3068. [DOI] [PubMed] [Google Scholar]
- Wang X-W, Luan J-B, Li J-M, Bao Y-Y, Zhang C-X, Liu S-S. De novo characterization of a whitefly transcriptome and analysis of its gene expression during development. BMC Genomics. 2010;11(1):400. doi: 10.1186/1471-2164-11-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney JC, Pettay DT, Sampayo EM, Warner ME, Oxenford HA, LaJeunesse TC. The relative significance of host-habitat, depth, and geography on the ecology, endemism, and speciation of coral endosymbionts in the genus Symbiodinium. Microbial Ecol. 2010;60(1):250–263. doi: 10.1007/s00248-010-9681-y. [DOI] [PubMed] [Google Scholar]
- Hagedorn M, Carter VL, Leong JC, Kleinhans FW. Physiology and cryosensitivity of coral endosymbiotic algae (Symbiodinium) Cryobiology. 2010;60(2):147–158. doi: 10.1016/j.cryobiol.2009.10.005. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R. et al. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Patrick Kück KM. FASconCAT: convenient handling of data matrices. Mole Phylogen Evol. 2010;56(2010):1115–1118. doi: 10.1016/j.ympev.2010.04.024. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, Ludwig T, Meier H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 2005;21(4):456–463. doi: 10.1093/bioinformatics/bti191. [DOI] [PubMed] [Google Scholar]
- Budd AF, Romano SL, Smith ND, Barbeitos MS. Rethinking the phylogeny of scleractinian corals: a review of morphological and molecular data. Integ Comp Biol. 2010;50(3):411–427. doi: 10.1093/icb/icq062. [DOI] [PubMed] [Google Scholar]
- Fukami H, Budd AF, Paulay G, Solé-Cava A, Allen Chen C, Iwao K, Knowlton N. Conventional taxonomy obscures deep divergence between Pacific and Atlantic corals. Nat Geosci. 2004;427(6977):832–835. doi: 10.1038/nature02339. [DOI] [PubMed] [Google Scholar]
- Ilagan RP, Rhoades E, Gruber DF, Kao HT, Pieribone VA, Regan L. A new bright green-emitting fluorescent protein–engineered monomeric and dimeric forms. FEBS J. 2010;277(8):1967–1978. doi: 10.1111/j.1742-4658.2010.07618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labas YA, Gurskaya NG, Yanushevich YG, Fradkov AF, Lukyanov KA, Lukyanov SA, Matz MV. Diversity and evolution of the green fluorescent protein family. Proc Natl Acad Sci USA. 2002;99(7):4256–4261. doi: 10.1073/pnas.062552299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matz MV, Fradkov AF, Labas YA, Savitsky AP, Zaraisky AG, Markelov ML, Lukyanov SA. Fluorescent proteins from nonbioluminescent Anthozoa species. Nat Biotech. 1999;17(10):969–973. doi: 10.1038/13657. [DOI] [PubMed] [Google Scholar]
- Bogdanov AM, Mishin AS, Yampolsky IV, Belousov VV, Chudakov DM, Subach FV, Verkhusha VV, Lukyanov S, Lukyanov KA. Green fluorescent proteins are light-induced electron donors. Nature chemical biology. 2009;5(7):459–461. doi: 10.1038/nchembio.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JC, Ryan JF, Watson JA, Webb J, Mullikin JC, Rokhsar D, Finnerty JR. StellaBase: the Nematostella vectensis genomics database. Nucleic acids research. 2006;34(Database issue):D495–D499. doi: 10.1093/nar/gkj020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomati EK, Manning G, Deheyn DD. Amphioxus encodes the largest known family of green fluorescent proteins, which have diversified into distinct functional classes. BMC evolutionary biology. 2009;9:77. doi: 10.1186/1471-2148-9-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddock SHD, Mastroianni N, Christianson LM. A photoactivatable green-fluorescent protein from the phylum Ctenophora. Proc Royal Soc B Biol Sci. 2010;277(1685):1155–1160. doi: 10.1098/rspb.2009.1774. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27(8):1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature biotechnology. 2011;29(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Moss LG, Phillips GN. , Jr. The molecular structure of green fluorescent protein. Dept of biochemistry and cell biology: Rice University; 1997. [DOI] [PubMed] [Google Scholar]
- Ormö M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 1996;273(5280):1392–1395. doi: 10.1126/science.273.5280.1392. [DOI] [PubMed] [Google Scholar]
- Kozak M. An analysis of 5'-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987;15(20):8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotech. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Gruber D, Kao H, Janoschka S, Tsai J, Pieribone V. Patterns of fluorescent protein expression in scleractinian corals. Biol Bull. 2008;215(2):143. doi: 10.2307/25470695. [DOI] [PubMed] [Google Scholar]
- Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Federhen S. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2010;38(Database issue):D5–D16. doi: 10.1093/nar/gkp967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinzato C, Shoguchi E, Kawashima T, Hamada M, Hisata K, Tanaka M, Fujie M, Fujiwara M, Koyanagi R, Ikuta T. et al. Using the Acropora digitifera genome to understand coral responses to environmental change. Nat Geosci. 2011;476(7360):320–323. doi: 10.1038/nature10249. [DOI] [PubMed] [Google Scholar]
- Katoh K, Asimenos G, Toh H. Multiple alignment of DNA sequences with MAFFT. Methods Mole Biol (Clifton, NJ) 2009;537:39–64. doi: 10.1007/978-1-59745-251-9_3. [DOI] [PubMed] [Google Scholar]
- Huson DH, Richter DC, Rausch C, Dezulian T, Franz M, Rupp R. Dendroscope: An interactive viewer for large phylogenetic trees. BMC Bioinforma. 2007;8(1):460. doi: 10.1186/1471-2105-8-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning, vol. 2. New York: Cold spring harbor laboratory press; 1989. [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotech. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Parameters and commands used in this manuscript.
BlastP parsed output files against N. vectensis proteome for sample Fav1 and Fav2 with 2e-30.
BlastP parsed output files against N. vectensis proteome for sample Fav1 and Fav2 with 2e-30.
TRIBE-MCL input files.
TRIBE-MCL input files.
Homologous protein clusters (TRIBE-MCL) output for sample Fav1 and Fav2.
Homologous protein clusters (TRIBE-MCL) output for sample Fav1 and Fav2.
Completeness metrics for two samples compared to N. ventensis and A. digitifera.
GO,KOG, InterPro annotation for homologous protein clusters in Fav1.
Distribution of Fav1 transcript clusters in different GO categories.
FASTA files for cDNA region encoding for non-symbiont annotated ORFs in Fav1 and Fav2.
FASTA files for cDNA region encoding for non-symbiont annotated ORFs in Fav1 and Fav2.
Alignment of Fav1 and Fav2 Cytb nucleotide sequences, including other Favia species.
Alignment of Fav1 and Fav2 COI nucleotide sequences, including other Favia species.
Alignment of Fav1 and Fav2 28S nucleotide sequences, including other Favia species.
Maximum likelihood tree of three loci (COI, Cytb, 28S). Data matrix was generated from 15 Favia species and Fav1 and Fav2. Nucleotide sequences were aligned using clustalw2 with default parameters, the 3 loci matrix was generated using FASconCAT, and the tree was constructed using RaxML (See methods). Montastrea cavernosa is selected as the out-group.
Amino acid sequence alignment of full-length fluorescent protein isoforms.
Maximum likelihood tree of 156 known fluorescent proteins, including our 11 newly identified sequences using RaxML. Shows the relationships of the major groups of known fluorescent proteins. Major lineages cluster together, although Ctenophore and Hydrozoa do not form a monophyletic group. Within Anthozoa class, order Ceriantharia (orange); Actinaria (red); Pennatulacea (dark green); and Scleractinia (black); Hydrozoa (purple); Copepoda (light green); Ctenophora (blue); Chordata (turquoise blue), most basal group; Newly identified sequences are colored blue within Scleractinia. The alignment was 1,000 times bootstrapped and B. floridae was the out-group.
Alignment of 156 known fluorescent proteins, including the 11 newly identified FP sequences.
Search result in Trans-ABySS and Trinity assembly output for homologous contig, similar to identified Fav1 s23Coting16657-5 produced by ABySS and CAP3.
Read-to-contig alignment. 75 bp read alignments to the coding region of s23Contig16657-5, 1,377 bp total length.
RPKM measurement for all annotated cDNA regions from Fav1 and Fav2.
RPKM measurement for all annotated cDNA regions from Fav1 and Fav2.
Protocol for preparing samples for sequencing of mRNA.
Scripts: Script 1: Perl script for performing blast search. Script 2: Perl script for pre-clustering the blast parsed file. Script 3: Perl script to calculate RPKM for the assembled file. Script S1: Perl script to shuffle short read sequences. Script S2: Perl script to measure the N50 statistics. Script S3: Unix shell script to remove Fasta files shorter than a threshold. Script S4: Generate the sub-Fasta file. Script S5: Extract the cDNA sequences corresponding to ORF files.