

Abstract
Background
We recently reported a terminal deletion of approximately 2.4 Mb at chromosome 8p23.2-pter in a boy with autism. The deleted region contained the DLGAP2 gene that encodes the neuronal post-synaptic density protein, discs, large (Drosophila) homolog-associated protein 2. The study aimed to investigate whether DLGAP2 is genetically associated with autism spectrum disorders (ASD) in general.
Methods
We re-sequenced all the exons of DLGPA2 in 515 patients with ASD and 596 control subjects from Taiwan. We also conducted bioinformatic analysis and family study of variants identified in this study.
Results
We detected nine common single nucleotide polymorphisms (SNPs) and sixteen novel missense rare variants in this sample. We found that AA homozygotes of rs2906569 (minor allele G, alternate allele A) at intron 1 (P = 0.003) and CC homozygotes of rs2301963 (minor allele A, alternate allele C) at exon 3 (P = 0.0003) were significantly over-represented in the patient group compared to the controls. We also found no differences in the combined frequency of rare missense variants between the two groups. Some of these rare variants were predicted to have an impact on the function of DLGAP2 using informatics analysis, and the family study revealed most of the rare missense mutations in patients were inherited from their unaffected parents.
Conclusions
We detected some common and rare genetic variants of DLGAP2 that might have implication in the pathogenesis of ASD, but they alone may not be sufficient to lead to clinical phenotypes. We suggest that further genetic or environmental factors in affected patients may be present and determine the clinical manifestations.
Trial registration
ClinicalTrial.gov, NCT00494754
Keywords: Autism spectrum disorders, DLGAP2, Common variants, Rare variants
Background
Autism spectrum disorders (ASD) are a group of childhood-onset neurodevelopmental disorders characterized by impaired verbal/nonverbal communication, abnormal reciprocal social interaction, and the presence of stereotyped behaviors and restricted interests. Due to increased awareness and clinical sensitivity to ASD, broadening of the diagnostic criteria of ASD by including Asperger’s disorder and Pervasive Developmental Disorder, not otherwise specified in addition to autistic disorder, and other contributing factors, the prevalence of ASD has increased markedly in the past decade. Recent data showed that up to approximately 11 persons per thousand in the USA are affected with ASD, and males are more predominantly affected than females [1-3]. The literature documents strong evidence of a high degree of genetic influence in the etiology of autism with high heritability estimated to be more than 90% [4]. Genetic approaches such as cytogenetic analysis, genome-wide linkage and association scans, and candidate gene analysis, have been used to dissect the genetic complexity of ASD [5].
Traditional cytogenetic studies [6-9] and the recent array-based comparative genomic hybridization (array CGH) analysis have shown that chromosome abnormalities and rare copy number variants [10,11] are associated with ASD. Various chromosome abnormalities such as deletion, duplication, inversion, and translocation were identified in ASD patients. In particular, the advent of array CGH technology has greatly facilitated the detection of formerly undetectable submicroscopic copy number variants that are associated with ASD [10-14].
Our group previously identified two novel chromosome deletions in ASD using karyotyping analysis and array CGH [15]. One was a terminal deletion of approximately 2.4 Mb at 8p23.2-pter detected in a male patient with autistic disorder [15]. Several genes with neurobiological functions such as discs, large (Drosophila) homolog-associated protein 2 (DLGAP2), ceroid-lipofuscinosis, neuronal 8 (CLN8), the Rho guanine nucleotide exchange factor 10 (ARHGEF10) and F-box protein 25 (FBXO25), were mapped to this region. It is likely that haploinsufficiency of one or several of these genes might result in the clinical phenotypes of the affected patients. Hence, these genes might be considered as candidate genes of ASD patients in general.
DLGAP2 (GeneID 9298) encodes the discs, large (Drosophila) homolog-associated protein 2, which is also called PSD-95/SAP90-binding protein 2 and SAP90/PSD-95-associated protein 2 (SAPAP2). The protein is one of the membrane-associated guanylate kinases localized at the post-synaptic density that plays a role in the molecular organization of synapses and in neuronal cell signaling [16]. These kinases are a family of signaling molecules expressed at various submembrane areas, and contain the PDZ, SH3 and the guanylate kinase domains. Several studies have suggested that the synapse associated proteins (SAPs) localized at postsynaptic density are involved in the pathophysiology of psychiatric disorders [17-21]. Marshall et al. detected 13 loci with recurrent CNV in autism cases; they found that several postsynaptic density genes and synapse complex genes were mapped in these CNVs [22]. They also found that a genomic DNA duplication intersected the DLGAP2 gene in a patient with autism [22]. In addition, Ozgen et al. reported a classical inv dup del(8p) in a female patient with autism whose DLGAP2 gene was located within the 6.9 Mb terminal deletion [23]. Together with the finding in our previous study, these studies suggest that the DLGAP2 gene is an important candidate gene of ASD. To test this hypothesis, we conducted a deep resequencing of all the exons of the DLGAP2 in a sample of ASD from Taiwan. Herein, we present our findings of the genetic analysis of DLGAP2 in this report.
Methods
Subjects and procedures
All subjects were Han Chinese from Taiwan. Patients meeting the diagnostic criteria of either autistic disorder or Asperger’s disorder according to the DSM-IV and ICD-10 were enrolled in this study from the psychiatry department of a university hospital, a private medical center, and schools and early intervention centers in northern Taiwan. Subjects with ASD aged 3 to 25 years old, and with a clinical diagnosis of ASD confirmed by the structured interview using the Chinese version of the Autism Diagnostic Interview-Revised (ADI-R) [24,25]. They received clinical assessments and provided DNA data at the university hospital, no matter whether they were recruited from this university hospital or referred by other places. Patients with known chromosomal abnormalities and associated medical conditions were excluded from the study. Subjects receiving routine medical check-ups at the Department of Family Medicine of a medical center were recruited as controls. The mental status of the control subjects was screened by a senior psychiatrist. Individuals with major psychiatric disorders including ASD were excluded.
The Research Ethics Committee of the research sites approved this study. The patients and their parents were informed that participation in this study was completely voluntary and that nonparticipation would not influence their treatment. Written informed consent was obtained from the patients if they were able to understand the contents of the study, the parents of all the patients, and all the control subjects after the procedures were fully explained. A total of 515 patients with ASD were recruited into this study, including 449 male patients (mean age ± standard deviation (SD) = 8.9 ± 4.6 years) and 66 female patients (mean age ± SD = 8.5 ± 4.4 years). The control group comprised 596 individuals including 263 males (mean age ± SD = 42.5 ± 15.1 years) and 333 females (mean age ± SD = 45.2 ± 13.4 years).
The ADI-R data of the 515 patients revealed the scores as 21.19 ± 5.78 in the ‘qualitative abnormalities in reciprocal social interaction’ (cut-off = 10), 15.35 ± 4.19 in the ‘qualitative abnormalities in communication, verbal’ (cut-off = 8), 8.58 ± 3.28 in the ‘qualitative abnormalities in communication, nonverbal’ (cut-off = 7), and 7.14 ± 2.42 in the ‘restricted, repetitive and stereotyped patterns of behaviors’ (cut-off = 3). All patients with ASD were noted to have had abnormal development at or before 36 months.
Genomic DNA was prepared from peripheral blood using the Puregene DNA purification system (Gentra Systems Inc. Minneapolis, MI, USA) according to the manufacturer’s instructions.
PCR amplification and sequencing
The genomic sequences of human DLGAP2 are available from the NCBI Reference Sequence: NM_004745.3. The human DLGAP2 comprises twelve exons that span approximately 207 kb on chromosome 8p23.2-23.3 [16]; the schematic genomic structure of the DLGAP2 is shown in Figure 1. Optimal PCR primer sequences were generated to amplify each exon of the DLGAP2 using Primer3 (http://bioinfo.ut.ee/primer3/). All the primer sequences, optimal annealing temperatures and size of each amplicon are listed in Additional file 1: Table S1. After PCR, aliquots of PCR products were processed using a PCR Pre-Sequencing Kit (USB, Cleveland, OH, USA) to remove residual primers and dNTPs following the manufacturer’s protocol. The purified PCR products were subjected to direct sequencing using the ABI Prism™ BigDye™ Terminator Cycle Sequencing Ready Reaction Kit Version 3.1, and the ABI autosequencer 3730 (Perkin Elmer Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s protocol. The quality of the sequencing results was visualized using Chromas 2.4.1 software (Technelysium Pty Ltd, South Brisbane, Australia). For variant identification, sequencing results of each subject were aligned and compared with the reference sequences using BioEdit software (http://www.mbio.ncsu.edu/bioedit/bioedit.html). To verify the authenticity of mutations identified in this study, repeated PCR from genomic DNA and re-sequencing of the amplicon in both directions were performed. The nomenclature of genetic variants follows the rules of the ‘Nomenclature for description of human sequence variations’ [26].
Figure 1.
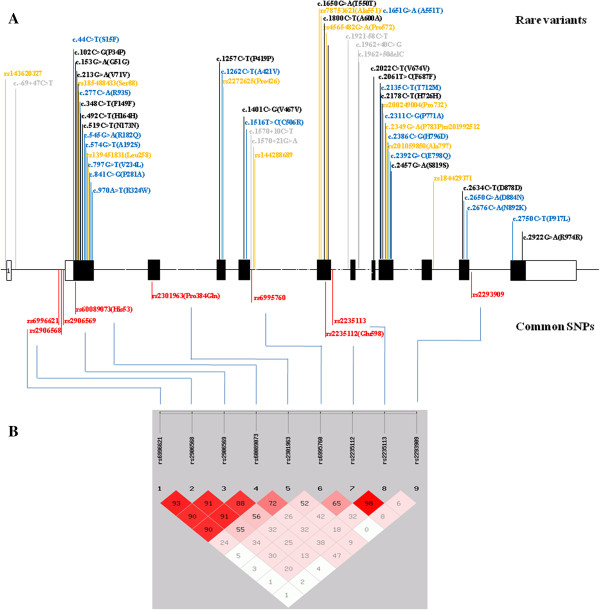
(A) Schematic genomic structure of the DLGAP2, and locations of the common single nucleotide polymorphisms (SNPs) and rare variants identified in this study. (B) Linkage disequlibrium (LD) analysis of nine common SNPs identified in this study.
Statistical analysis
For the common single nucleotide polymorphisms (SNPs), the differences in the allele and genotype frequencies between the patients and controls were assessed with the Chi-square test. Fisher's exact test was used to compare the combined frequency of rare variants between the patient and the control groups. It was also used to compare the frequency of damaging and functional rare missense variants between the ASD and control groups. Assessment of haplotype-based association analyses was performed using the SHEsis computer program [27]. A P value of less than 0.05 was considered statistically significant, and Bonferroni correction for multiple comparisons was performed when appropriate. We also calculated the Q value for each test with the false discovery test of 5% using QVALUE software (http://genomics.princeton.edu/storeylab/qvalue/) [28].
Results
After sequencing all the exons of DLGAP2 in 515 patients and 596 control subjects, we identified nine common SNPs (defined as with a minor allele frequency > 0.05), and a total of 49 rare variants (defined as with a frequency < 0.05) in this sample. The locations of these variants are listed in Figure 1.
Case-control association study of common SNPs
The genotype and allele frequencies of nine common variants in the patients and control subjects are listed in Table 1. Two SNPs (rs2906569 and rs2301963) were found to have significant differences in genotype frequency distribution between the patient and control groups, even after correction for multiple comparisons. The rs2906569 (A > G) was located at intron 1. The genotype AA homozygotes (G: minor allele, A: alternate allele) were significantly over-represented in the patient group compared to the control group (odds ratio: 1.46; 95% confidence interval, 1.13 to 1.87, P = 0.003) (Table 2). When the subjects were sub-grouped by gender, the over-representation of genotype AA homozygotes was still observed in male patients but not in female patients (Table 2). The rs2301963 (C > A) was a missense variant (P384Q) located at exon 3 (A: minor allele, C: alternate allele). The CC homozygotes were significantly over-represented in the patient group compared to the control group (odds ratio: 1.30; 95% confidence interval, 0.99 to 1.70; P = 0.0003) (Table 2). When the subjects were sub-grouped by gender, the over-representation of CC homozygotes was observed in male patients but not in female patients (Table 2).
Table 1.
Allele and genotype frequencies of common SNPs of the DLGAP2 gene in ASD patients and control subjects (MAF >5%)
| SNP | Location | Diagnosis | n | Genotype | P value | Q value | Allele | P value | Q value | Odds ratio (95 % CI) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs6996621 |
intron 1 |
|
|
|
C/C |
C/A |
A/A |
|
|
C |
A |
|
|
C versus A |
| c.-68-61C > A |
|
total |
autism |
454 |
387 (85.2%) |
62 (13.7%) |
5 (1.1%) |
0.619 |
0.312 |
836 (92.1%) |
72 (7.9%) |
0.401 |
0.204 |
1.11 (0.83 to 1.57) |
| |
|
|
control |
557 |
467 (83.8%) |
80 (14.4%) |
10 (1.8%) |
|
|
1014 (91.0%) |
100 (9.0%) |
|
|
|
| |
|
male |
autism |
394 |
336 (85.3%) |
55 (14.0%) |
3 (0.7%) |
0.593 |
0.203 |
727 (92.3%) |
61 (7.7%) |
0.611 |
0.611 |
|
| |
|
|
control |
246 |
208 (84.6%) |
34 (13.8%) |
4 (1.6%) |
|
|
450 (91.5%) |
42 (8.5%) |
|
|
|
| |
|
female |
autism |
60 |
51 (85%) |
7 (11.7%) |
2 (3.3%) |
0.663 |
0.266 |
109 (90.8%) |
11 (9.2%) |
0.956 |
0.956 |
|
| |
|
|
control |
311 |
259 (83.3%) |
46 (14.8%) |
6 (1.9%) |
|
|
564 (90.7%) |
58 (9.3%) |
|
|
|
| rs2906568 |
intron 1 |
|
|
|
C/C |
C/G |
G/G |
|
|
C |
G |
|
|
C versus G |
| c.-68-25C > G |
|
total |
autism |
456 |
188 (41.2%) |
169 (37.1%) |
99 (21.7%) |
0.142 |
0.112 |
545 (59.8%) |
367 (40.2%) |
0.125 |
0.105 |
1.15 (0.96 to 1.37) |
| |
|
|
control |
557 |
197 (35.4%) |
234 (42.0%) |
126 (22.6%) |
|
|
628 (56.4%) |
486 (43.6%) |
|
|
|
| |
|
male |
autism |
396 |
171 (43.2%) |
138 (34.8%) |
87 (22.0%) |
0.072 |
0.107 |
480 (60.6%) |
312 (39.4%) |
0.146 |
0.299 |
|
| |
|
|
control |
246 |
86 (35.0%) |
106 (43.0%) |
54(22.0%) |
|
|
278 (56.5%) |
214 (43.5%) |
|
|
|
| |
|
female |
autism |
60 |
17 (28.3%) |
31 (51.7%) |
12 (20%) |
0.316 |
0.266 |
65 (54.2%) |
55 (45.8%) |
0.671 |
0.897 |
|
| |
|
|
control |
311 |
111 (35.7%) |
128 (41.1%) |
72 (23.2%) |
|
|
350 (56.3%) |
272 (43.7%) |
|
|
|
| rs2906569 |
intron 1 |
|
|
|
A/A |
A/G |
G/G |
|
|
A |
G |
|
|
A versus G |
| c.-68-4A > G |
|
total |
autism |
458 |
219 (47.8%) |
138 (30.1%) |
101 (22.1%) |
0.0006 |
0.095 |
576 (62.9%) |
340 (37.1%) |
0.112 |
0.105 |
1.16 (0.97 to 1.38) |
| |
|
|
control |
557 |
215 (38.6%) |
232 (41.7%) |
110 (19.7%) |
|
|
662 (59.4%) |
452 (40.6%) |
|
|
|
| |
|
male |
autism |
398 |
199 (50.0%) |
109 (27.4%) |
90 (22.6%) |
0.0001 |
0.107 |
507 (63.7%) |
289 (36.3%) |
0.075 |
0.299 |
|
| |
|
|
control |
246 |
91 (37.0%) |
107 (43.5%) |
48 (19.5%) |
|
|
289 (58.7%) |
203 (41.3%) |
|
|
|
| |
|
female |
autism |
60 |
20 (33.3%) |
29 (48.3%) |
11 (18.4%) |
0.491 |
0.266 |
69 (57.5%) |
51 (42.5%) |
0.614 |
0.897 |
|
| |
|
|
control |
311 |
124 (39.9%) |
125 (40.2%) |
62 (19.9%) |
|
|
373 (60.0%) |
249 (40.0%) |
|
|
|
| rs60089073 |
exon 2 |
|
|
|
C/C |
C/T |
T/T |
|
|
C |
T |
|
|
C versus T |
| His73 |
|
total |
autism |
466 |
401 (86.1%) |
54 (11.6%) |
11 (2.3%) |
0.989 |
0.388 |
856 (91.8%) |
76 (8.2%) |
0.891 |
0.363 |
0.98 (0.71 to 1.35) |
| |
|
|
control |
557 |
481 (86.4%) |
63 (11.3%) |
13 (2.3%) |
|
|
1025 (92.0%) |
89 (8.0%) |
|
|
|
| |
|
male |
autism |
404 |
346 (85.6%) |
48 (11.9%) |
10 (2.5%) |
0.236 |
0.133 |
740 (91.6%) |
68 (8.4%) |
0.093 |
0.299 |
|
| |
|
|
control |
246 |
219 (89.0%) |
25 (10.2%) |
2 (0.8%) |
|
|
463 (94.1%) |
29 (5.9%) |
|
|
|
| |
|
female |
autism |
62 |
55 (88.7%) |
6 (9.7%) |
1 (1.6%) |
0.607 |
0.266 |
116 (93.5%) |
8 (6.5%) |
0.259 |
0.897 |
|
| |
|
|
control |
311 |
262 (84.3%) |
38 (12.2%) |
11 (3.5%) |
|
|
562 (90.4%) |
60 (9.6%) |
|
|
|
| rs2301963 |
exon 3 |
|
|
|
C/C |
C/A |
A/A |
|
|
C |
A |
|
|
C versus A |
| Pro384Gln |
|
total |
autism |
513 |
171 (33.3%) |
231 (45.1%) |
111 (21.6%) |
0.0005 |
0.095 |
573 (55.8%) |
453 (44.2%) |
0.026 |
0.105 |
1.21 (1.02 to 1.43) |
| |
|
|
control |
593 |
139 (23.4%) |
328 (55.%) |
126 (21.2%) |
|
|
606 (51.1%) |
580 (48.9%) |
|
|
|
| |
|
male |
autism |
448 |
150 (33.5%) |
202 (45.1%) |
96 (21.4%) |
0.068 |
0.107 |
502 (56.0%) |
394 (44.0%) |
0.102 |
0.299 |
|
| |
|
|
control |
260 |
66 (25.4%) |
136 (52.3%) |
58 (22.3%) |
|
|
268 (51.5%) |
252 (48.5%) |
|
|
|
| |
|
female |
autism |
65 |
21 (32.3%) |
29 (44.6%) |
15 (23.1%) |
0.124 |
0.266 |
71 (54.6%) |
59 (45.4%) |
0.420 |
0.897 |
|
| |
|
|
control |
333 |
73 (21.9%) |
192 (57.7%) |
68 (20.4%) |
|
|
338 (50.8%) |
328 (49.2%) |
|
|
|
| rs6995760 |
intron 5 |
|
|
|
A/A |
A/G |
G/G |
|
|
A |
G |
|
|
A versus G |
| c.1570 + 14A > G |
|
total |
autism |
500 |
346 (69.2%) |
141 (28.2%) |
13 (2.6%) |
0.913 |
0.388 |
833 (83.3%) |
167 (16.7%) |
0.925 |
0.363 |
1.01 (0.80 to 1.27) |
| |
|
|
control |
531 |
368 (69.3%) |
147 (27.7%) |
16 (3.0%) |
|
|
883 (83.1%) |
179 (16.9%) |
|
|
|
| |
|
male |
autism |
432 |
305 (70.6%) |
115 (26.6%) |
12 (2.8%) |
0.502 |
0.193 |
725 (83.9%) |
139 (16.1%) |
0.245 |
0.299 |
|
| |
|
|
control |
234 |
155 (66.3%) |
71 (30.3%) |
8 (3.4%) |
|
|
381 (81.4%) |
87 (18.6%) |
|
|
|
| |
|
female |
autism |
68 |
41 (60.3%) |
26 (38.2%) |
1 (1.5%) |
0.103 |
0.266 |
108 (79.4%) |
28 (20.6%) |
0.148 |
0.897 |
|
| |
|
|
control |
297 |
213 (71.7%) |
76 (25.6%) |
8 (2.7%) |
|
|
502 (84.5%) |
92 (15.5%) |
|
|
|
| rs2235112 |
exon 6 |
|
|
|
A/A |
A/G |
G/G |
|
|
A |
G |
|
|
A versus G |
| Glu598 |
|
total |
autism |
509 |
95 (18.8%) |
233 (45.7%) |
181 (35.5%) |
0.028 |
0.095 |
424 (41.6%) |
595 (58.4%) |
0.012 |
0.105 |
1.29 (1.05 to 1.50) |
| |
|
|
control |
513 |
66 (12.9%) |
239 (46.6%) |
208 (40.5%) |
|
|
371 (36.2%) |
655 (63.8%) |
|
|
|
| |
|
male |
autism |
444 |
85 (19.1%) |
205 (46.2%) |
154 (34.7%) |
0.049 |
0.107 |
375 (42.2%) |
513 (57.8%) |
0.014 |
0.299 |
|
| |
|
|
control |
224 |
28 (12.5%) |
102 (45.5%) |
94 (42.0%) |
|
|
158 (35.3%) |
290 (64.7%) |
|
|
|
| |
|
female |
autism |
66 |
11 (16.7%) |
28 (42.4%) |
27 (40.9%) |
0.671 |
0.266 |
50 (37.9%) |
82 (62.1%) |
0.825 |
0.928 |
|
| |
|
|
control |
289 |
38 (13.2%) |
137 (47.4%) |
114 (39.4%) |
|
|
213 (36.9%) |
365 (63.1%) |
|
|
|
| rs2235113 |
intron 6 |
|
|
|
C/C |
C/G |
G/G |
|
|
C |
G |
|
|
C versus G |
| c.1920 + 19C > G |
|
total |
autism |
510 |
156 (30.6%) |
233 (45.7%) |
121 (23.7%) |
0.050 |
0.095 |
545 (53.4%) |
475 (46.6%) |
0.030 |
0.105 |
1.21 (1.02 to 1.44) |
| |
|
|
control |
509 |
121 (23.8%) |
253 (49.7%) |
135 (26.5%) |
|
|
495 (48.6%) |
523 (51.4%) |
|
|
|
| |
|
male |
autism |
443 |
137 (30.9%) |
205 (46.3%) |
101 (22.8%) |
0.303 |
0.139 |
479 (54.1%) |
407 (45.9%) |
0.243 |
0.299 |
|
| |
|
|
control |
222 |
56 (25.2%) |
113 (50.9%) |
53 (23.9%) |
|
|
225 (50.7%) |
219 (49.3%) |
|
|
|
| |
|
female |
autism |
67 |
19 (28.3%) |
28 (41.8%) |
20 (29.9%) |
0.513 |
0.266 |
66 (49.3%) |
68 (50.7%) |
0.644 |
0.897 |
|
| |
|
|
control |
287 |
65 (22.6%) |
140 (48.8%) |
82 (28.6%) |
|
|
270 (47.0%) |
304 (53.0%) |
|
|
|
| rs2293909 |
intron 11 |
|
|
|
T/T |
T/C |
C/C |
|
|
T |
C |
|
|
T versus C |
| c.2709 + 71T > C |
|
total |
autism |
460 |
255 (55.4%) |
166 (36.1%) |
39 (8.5%) |
0.012 |
0.095 |
676 (73.5%) |
244 (26.5%) |
0.012 |
0.105 |
1.28 (1.06 to 1.55) |
| |
|
|
control |
573 |
265 (46.2%) |
254 (44.3%) |
54 (9.4%) |
|
|
784 (68.4%) |
362 (31.6%) |
|
|
|
| |
|
male |
autism |
403 |
230 (57.1%) |
138 (34.2%) |
35 (8.7%) |
0.117 |
0.107 |
598 (74.2%) |
208 (25.8%) |
0.165 |
0.299 |
|
| |
|
|
control |
256 |
127 (49.6%) |
108 (42.2%) |
21 (8.2%) |
|
|
362 (70.7%) |
150 (29.3%) |
|
|
|
| |
|
female |
autism |
57 |
25 (43.9%) |
28 (49.1%) |
4 (7.0%) |
0.719 |
0.266 |
78 (68.4%) |
36 (31.6%) |
0.698 |
0.897 |
|
| control | 317 | 138 (43.5%) | 146 (46.1%) | 33 (10.4%) | 422 (66.6%) | 212 (33.4%) | ||||||||
ASD, autistic spectrum disorders; MAF, minor allele frequency; SNPs, single nucleotide polymorphisms.
Table 2.
Odds ratio of rs2906569 and rs2301963 of the DLGAP2 gene in patients with autism and control subjects
| SNP | Location | Diagnosis | n | Odds ratio (95% CI) | P value | |
|---|---|---|---|---|---|---|
| rs2906569 |
intron 1 |
total |
|
|
A/A versus A/G + G/G |
|
| |
|
autism |
458 |
1.46 (1.13 to1.87) |
0.003a |
|
| |
|
control |
557 |
|
|
|
| |
|
male |
autism |
398 |
1.70 (1.23 to 2.36) |
0.001a |
| |
|
|
control |
246 |
|
|
| |
|
female |
autism |
60 |
0.75 (0.42 to 1.35) |
0.341 |
| |
|
|
control |
311 |
|
|
| rs2301963 |
exon 3 |
total |
|
|
C/C versus C/A + A/A |
P value |
| |
|
autism |
513 |
1.63 (1.25 to 2.13) |
0.0003a |
|
| |
|
control |
593 |
|
|
|
| |
|
male |
autism |
448 |
1.48 (1.05 to 2.08) |
0.024a |
| |
|
|
control |
260 |
|
|
| |
|
female |
autism |
65 |
1.70 (0.95 to 3.04) |
0.071 |
| control | 333 |
aP value < 0.05; SNP, single nucleotide polymorphism.
Further linkage disequilibrium (LD) analysis showed strong LD among rs6996621, rs2906568, rs2906569, and rs60089073. In addition, rs2235112 and rs2235113 also showed strong LD (Figure 1). In haplotype-based association analysis derived from nine SNPs, we found significant difference in the haplotype distribution of ACACAAGGT and CCACCAACT between the ASD patients and controls, but only haplotype CCACCAACT was sustained after correction for multiple comparisons (Table 3).
Table 3.
The distributions of haplotypes in ASD and controls
| Haplotype | Cases (frequency) | Controls (frequency) | Chi2 | Fisher's P | Pearson's P | Odds ratio (95%CI) | |
|---|---|---|---|---|---|---|---|
| 1 |
A C A C A A G G T |
8.84 (0.012) |
28.20 (0.036) |
7.081 |
0.007814a |
0.007806a |
0.367 (0.170 to 0.790) |
| 2 |
C C A C C A A C C |
41.17 (0.057) |
63.13 (0.080) |
1.548 |
0.213443 |
0.213365 |
0.768 (0.506 to 1.165) |
| 3 |
C C A C C A A C T |
84.15 (0.117) |
60.19 (0.076) |
11.981 |
0.000542a |
0.000541a |
1.880 (1.310 to 2.698) |
| 4 |
C C A C C A G G C |
12.99 (0.018) |
26.04 (0.033) |
2.331 |
0.126865 |
0.126793 |
0.592 (0.300 to 1.168) |
| 5 |
C C A C C A G G T |
86.66 (0.121) |
89.80 (0.114) |
1.380 |
0.240147 |
0.240071 |
1.218 (0.876 to 1.694) |
| 6 |
C C A T C A A C T |
27.74 (0.039) |
25.95 (0.033) |
0.973 |
0.324045 |
0.323985 |
1.319 (0.759 to 2.292) |
| 7 |
C G G C A A A C T |
31.96 (0.045) |
32.97 (0.042) |
0.465 |
0.495147 |
0.495131 |
1.192 (0.719 to 1.976) |
| 8 |
C G G C A A G G C |
30.14 (0.042) |
45.29 (0.057) |
0.902 |
0.342270 |
0.342214 |
0.792 (0.490 to 1.282) |
| 9 |
C G G C A A G G T |
60.91 (0.085) |
81.38 (0.103) |
0.381 |
0.537097 |
0.537090 |
0.892 (0.622 to 1.281) |
| 10 | C G G C A G G C T | 19.59 (0.027) | 37.66 (0.048) | 2.960 | 0.085418 | 0.085359 | 0.613 (0.349 to 1.076) |
ASD, autistic spectrum disorders; Global result: total control = 788.0, total case = 718.0; global chi2 is 26.804893 while df = 9 (frequency < 0.03 in both control and case has been dropped). Fisher's P value is 0.001537. Pearson's P value is 0.001507. Note: aP ≤ value 0.05.
Identification of rare genetic variants of DLGAP2
In this study, we found a total of 16 rare missense mutations in our ASD patients and control subjects. The locations of these variants are illustrated in Figure 1. These missense variants are novel and have not been reported before in the literature. Distributions of these rare missense variants are listed in Table 4. There was no significant difference in the combined frequency of rare missense mutations between the two groups (P = 1.00). Those individuals who carried these rare missense mutations had only one missense mutation; we found no individual who carried two rare missense variants simultaneously.
Table 4.
Unique nonsynonymous variants of the DLGAP2 gene identified in ASD patients and controls and their functional predictions
|
Location |
|
|
In silico analysis |
|
|
|
|
|
|---|---|---|---|---|---|---|---|---|
| Nucleotide position | Variants | PolyPhen-2 | SIFT | Autism | Control | rs2906569 | rs2301963 | |
| exon2 |
c.44 C > T |
S15F |
probably damaging |
affect protein function |
U1974 |
|
A/A |
C/A |
| c.277 C > A |
R93S |
probably damaging |
tolerated |
U1843 |
|
A/G |
C/A |
|
| c.545 G > A |
R182Q |
probably damaging |
tolerated |
U173 |
|
A/G |
C/A |
|
| c.574 G > T |
A192S |
benign |
tolerated |
U396 |
|
A/A |
C/C |
|
| c.797 G > T |
V234L |
benign |
tolerated |
|
ZN4215 |
G/G |
C/C |
|
| c.797 G > T |
V234L |
benign |
tolerated |
|
HN616 |
A/A |
C/A |
|
| c.841 C > G |
P281A |
probably damaging |
tolerated |
U323 |
|
A/G |
C/A |
|
| c.841 C > G |
P281A |
probably damaging |
tolerated |
U1519 |
|
G/G |
A/A |
|
| c.841 C > G |
P281A |
probably damaging |
tolerated |
U1988 |
|
G/G |
C/A |
|
| c.841 C > G |
P281A |
probably damaging |
tolerated |
|
ZN4014 |
A/G |
missing |
|
| c.841 C > G |
P281A |
probably damaging |
tolerated |
|
HN449 |
A/G |
C/A |
|
| c.841 C > G |
P281A |
probably damaging |
tolerated |
|
HN581 |
A/G |
C/A |
|
| c.970 A > T |
R324W |
probably damaging |
affect protein function |
U1803 |
|
A/A |
C/A |
|
| exon4 |
c.1262 C > T |
A421V |
benign |
tolerated |
|
ZN4205 |
G/G |
A/A |
| exon5 |
c.1516 T > C |
C506R |
probably damaging |
affect protein function |
U1247 |
|
missing |
C/A |
| exon9 |
c.2135 C > T |
T712M |
benign |
tolerated |
U2096 |
|
G/G |
C/A |
| |
c.2135 C > T |
T712M |
benign |
tolerated |
|
HN576 |
missing |
C/A |
| |
c.2135 C > T |
T712M |
benign |
tolerated |
|
HN278 |
A/A |
C/A |
| |
c.2311 C > G |
P771A |
benign |
tolerated |
|
ZN4182 |
A/A |
C/C |
| |
c.2386 C > G |
H796D |
probably damaging |
affect protein function |
|
ZN4053 |
A/G |
C/A |
| |
c.2392 G > C |
E798Q |
probably damaging |
affect protein function |
U1082 |
|
A/A |
C/C |
| exon11 |
c.2650 G > A |
D884N |
benign |
tolerated |
|
HN526 |
G/G |
C/A |
| |
c.2676 C > A |
N892K |
possibly damaging |
affect protein function |
|
ZN4107 |
missing |
missing |
| exon12 |
c.2750 C > T |
P917L |
benign |
tolerated |
U1000 |
|
A/G |
C/A |
| |
c.2750 C > T |
P917L |
benign (0.002) |
tolerated |
U2098 |
|
G/G |
C/A |
| |
c.2750 C > T |
P917L |
benign (0.002) |
tolerated |
|
HN444 |
A/A |
C/C |
| PolyPhen-2 |
autism |
control |
P (Fisher’s test) |
SIFT |
autism |
control |
P (Fisher’s test) |
|
| damaging |
9 |
5 |
0.12 |
functional |
4 |
2 |
0.32 |
|
| benign | 4 | 8 | tolerated | 9 | 11 | |||
Family study and functional prediction of rare variants
A total of 10 missense variants (S15F, R93S, R182Q, A192S, P281A, R324W, C506R, T712M, E798Q and P917L) were detected in 13 patients (Table 4). All the patients carrying these rare variants were heterozygotes. Four of the 13 patients (A, B, J, K) inherited the variant from their mothers and 6 (C, D, E, F, G, H) from their fathers (Figure 2). Three patients did not have enough genetic information from the parents. Among these ten missense variants, seven (S15F, R93S, R182Q, A192S, R324W, C506R and E798Q were found in the patient group only, while the other three (P281A, T712M and P917L) were detected also in the control group. Aside from these three variants that overlapped with the patient group, six missense variants (V234L, A421V, P771A, H796D, D884N, and N892K) were unique in the control group. The inheritance mode of missense variants found in the control group cannot be traced because we did not collect their parents’ DNA. In the analysis of functional prediction of these 16 rare missense variants, S15F, R93S, R182Q, P281A, R324W, C506R, H796D, E798Q, and N892K were predicted to have functional impact on the protein using the PolyPhen-2 or SIFT computer program (Table 4).
Figure 2.
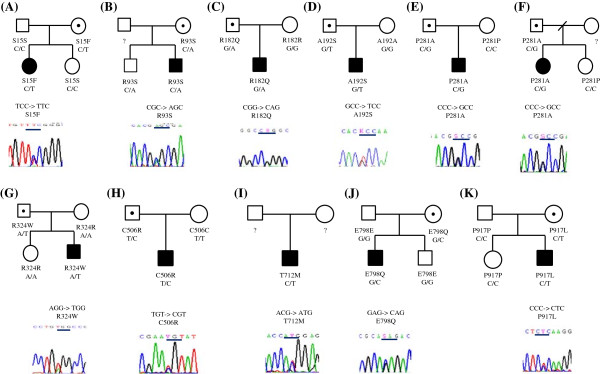
(A) Pedigree of S15F mutation, (B) Pedigree of R93S mutation, (C) Pedigree of R182Q mutation. (D) Pedigree of A192S mutation. (E,F) Pedigrees of P281A mutation. (G) Pedigree of R324W mutation. (H) Pedigree of C506R mutation. (I) Pedigree of T712M mutation. (J) Pedigree of E798Q mutation. (K) Pedigree of P917L mutation.
The ADI-R data of these 13 patients revealed scores of 22.82 ± 7.15 (range, 8 to 28) in the ‘qualitative abnormalities in reciprocal social interaction’ (cut-off = 10), 15.00 ± 4.84 (range, 5 to 22) in the ‘qualitative abnormalities in communication, verbal’ (cut-off = 8), 9.55 ± 4.39 (range, 4 to 14) in the ‘qualitative abnormalities in communication, nonverbal’ (cut-off = 7), and 6.00 ± 2.68 (range, 1 to 12) in the ‘restricted, repetitive and stereotyped patterns of behaviors’ (cut-off = 3). The average age at which the 13 patients said their first word was 37.1 ± 17.4 (range, 15 to 66) months old and at which they said their first phrase was 44.8 ± 18.5 (range, 19 to 78) months. Their current average intelligence quotients (IQ) were 82.8 ± 34.0 (range 40 to 126) for full-scale IQ, 84.9 ± 32.1 (range, 41 to 123) for performance IQ, and 82.3 ± 33.1 (range, 44 to 122 ) for verbal IQ, as assessed by the Wechsler Intelligence Scale for Children-third edition. Due to the lack of psychometric data for the control subjects who carried the rare missense mutations, we were not able to determine their clinical characteristics.
Clinical assessments of the parents of the 10 patients carrying rare missense mutations found that none of them achieved the clinical diagnosis of ASD. Their reports on the Chinese version of the Autism Spectrum Quotient [29] also revealed no evidence of an overt autistic trait.
Discussion
In this study, we found that rs2906569 at intron 1 and rs2301963 (P384Q) at exon 3 of DLGAP2 were associated with ASD. These two SNPs did not form significant LD in our genetic analysis. The functional significance of rs2906569 was difficult to infer, because it was located at intron 1. As to rs2301963 (C > A, P384Q), the A allele (Q allele) was the minor allele in our population, and was predicted to be probably damaging using the PolyPhen-2 computer program, but tolerated by SIFT. Based on the finding of significant over-representation of the CC homozygotes in the patient group, we suggest that the Q384 variant of the DLGAP2 might confer an increased risk of ASD, but the real mechanism and meaning of this association remain to be clarified. The small sample size of this study may have led to a false positive, which is also a limitation of this study. In a recent review of the role of common variants in autism, Devlin and colleagues reported their study on three large genome-wide association (GWA) studies of autism, each of which showed a single, non-overlapping risk locus. They found that there was no significant finding when all the data were combined. In their analysis, they found no definitive, replicated results, and they could not be certain that there was a role for common variants in autism risk [30]. Hence, an independent replication study is needed to verify our finding.
We also detected a total of 16 novel missense rare variants in the patient and control groups in this study. Given that some of these missense mutations were predicted to have functional impact on DLGAP2, we found no differences in their combined frequency between the two groups. In addition, most of these rare missense mutations found in the patients were inherited from their unaffected parents. Hence, the clinical relevance of these rare missense mutations to ASD is not straightforward, and needs further elucidation.
We attempted to assess the interactions between rs2906569 and rs2301963 and the missense rare variants in this study, but could not detect interactions between them. The small sample size of this study might be a major limiting factor. There were eight AA homozygotes of rs2906569 carrying rare missense variants, including four patients and four controls, and there were five CC homozygotes of rs2301963 carrying rare missense variants, including two patients and three controls. The genotype of rs2906569 and rs2301963 in subjects carrying the rare missense variants are listed in the Table 4.
To assess the phenotypic significance of two common SNPs associated with ASD, we compared the three core symptoms of ASD measured by the ADI-R and the Chinese version of Social Communication Questionnaire (SCQ) between patients with A/A and patients with A/G + G/G of rs2906569 (Additional file 2: Table S2), between patients with C/C and patients with C/A + A/A of rs2301963 (Additional file 3: Table S3). We also compared the three core symptoms of ASD measured by the ADI-R and SCQ between patients who carried rare missense mutations and those did not carry rare missense mutations (Additional file 4: Table S4). The Chinese SCQ is a screening tool based on the Autism Diagnostic Interview-Revised (ADI-R) algorithm which corresponds to DSM-IV diagnostic criteria. It examines the three functional domains of reciprocal social interaction, communication, and restricted, repetitive, and stereotyped patterns. The Chinese SCQ was translated under the approval of Western Psychological Services and was validated by the research group led by Gau and Wu [31]. The results showed that there were no statistical significant differences in all the comparisons (all P values > 0.1), regardless of gender with the following two exceptions. Patients with A/A of rs2906569 had less severe social interaction impairment than patients with A/G + G/G of rs2906569 (P = 0.0219) and such significant difference was only noted in male patients (P = 0.0285). But, the statistical significance did not sustain after correcting for multiple testing.
ASD is a complex disease with highly heterogeneous genetic underpinnings, and genotype-phenotype correlation remains a challenging task. According to the common variant hypothesis of complex psychiatric disorders, the effect size of common variants is usually considered small or modest. Moreover, it is not unusual to find inconsistent results among different studies, and the common variants can only explain a small proportion of the clinical variance. In a recent report on stage two of the autism genome project genome-wide association study, Anney and colleagues found that no single SNP showed a significant association with ASD or selected phenotypes at a genome-wide level after genotyping over a million SNPs, and the clinical variance explained by common variants en masse was small [32].
In contrast, the ‘rare variant hypothesis’ of complex psychiatric disorders suggests that rare variants are likely to have large effect sizes and to be de novo in their origin [33]. Given that several studies have provided evidence to support the large effect size of de novo rare mutations associated with ASD [34-36], emerging evidence suggests the multiple-hits model may be more appropriate to explain the incomplete penetrance and varied expressivity of genetic underpinnings of ASD [37,38]. Girirajan and colleagues proposed a ‘two-hit’ model in which a first hit may act in concert with some factors as a second hit, such as mutations in a single gene, micro-deletions/duplications, epigenetic factors, or environmental insults, and result in variable expressivity of phenotypes in complex neuropsychiatric disorders [38]. Furthermore, in a genetic study of high-functioning, idiopathic ASD, Schaaf and colleagues reported that in addition to de novo rare mutations, patients with ASD had a significantly higher proportion of multiple events of oligogenic heterozygosity than control subjects, suggesting oligogenic heterozygosity is a new potential mechanism in the pathogenesis of ASD [39]. In our previous study, we also reported a boy with ASD who carried two CNVs that were inherited respectively from his unaffected parents [40]. Our previous case report also lent support to the two-hit and compound heterozygosity models of ASD.
In a recent study examining the patterns and rates of exonic de novo mutations in ASD, Neale and colleagues found only a small increase in the rate of de novo events in ASD. They suggested an important but limited role for de novo point mutations in ASD, and supported polygenic models of ASD [41]. In the present study, we found that the putative deleterious missense variants occurred in both patient and control groups with equal chance, and that most of the rare missense mutations were inherited from their unaffected parents. We suggest that the missense mutations of DLGAP2 alone may not be sufficient for the clinical presentations of ASD, and additional hits such as environmental insults or further genetic mutations in the affected patients may be needed for the clinical presentations, which support ASD as likely a multifactorial disease. Hence, it is difficult to find a strong association with a single gene, like DLGAP2 in this study.
Conclusions
We identified two common SNPs and some rare missense mutations of DLGAP2 that might be implicated in the pathogenesis of ASD. However, their clinical relevance is not straightforward. We suggest additional genetic or environmental factors in the affected patients might be present to determine the clinical presentations. The findings from this study can only be considered as preliminary as it has only a limited sample size. Further independent replication studies are needed to verify our findings in the present study.
Abbreviations
ADI-R: Autism diagnostic interview-revised; ARHGEF10: Rho guanine nucleotide exchange factor 10; Array CGH: Array-based comparative genomic hybridization; ASD: Autism spectrum disorders; CI: Confidence interval; CLN8: Ceroid-lipofuscinosis, neuronal 8; DSM-IV: Diagnostic and statistical manual of mental disorders, fourth edition; DLGAP2: Disc, large (Drosophila) homolog-associated protein 2; FBXO25: F-box protein 25; GWA: Genome-wide association; IQ: Intelligence quotient; LD: Linkage disequilibrium; MAF: Minor allele frequency; PCR: Polymerase chain reaction; SAPAP2: SAP90/PSD-95-associated protein 2; SAPs: Synapse associated proteins; SNP: Single nucleotide polymorphism; SD: Standard deviation.
Competing interests
The authors declare that they do not have competing interests.
Authors’ contributions
The contributions of the authors are as follows: SSG was the principle investigator in this project. SSG and CHC designed the study and wrote the protocol. SSG trained the clinical research team, supervised in research execution, and collected all the clinical data of the ASD cases. SSG and YYW were responsible for the ADI-R training and interviews. SSG, YYW, WCT, YNC, and YSH were responsible for clinical diagnoses and helped recruit the patients (in the order of number of subjects recruited) and CHC screened for mental disorders in the controls. WHC, HML and HMT performed the experimental works and analyzed the data. WHC drafted the manuscript. CHC and SSG critically revised the manuscript and all the authors approved the manuscript.
Web resources
Primer3: http://bioinfo.ut.ee/primer3/. SHEsis: http://analysis2.bio-x.cn/myAnalysis.php. PolyPhen-2: http://genetics.bwh.harvard.edu/pph2/. SIFT: http://sift.jcvi.org/.
Supplementary Material
Primer sequences, optimal annealing temperature (Ta) and size of PCR products of the DLGAP2 gene.
Comparison of the three core symptoms of autism spectrum disorder measured by the ADI-R and SCQ between patients with A/A versus patients with A/G + G/G of rs2906569 stratified by gender.
Comparison of the three core symptoms of autism spectrum disorder measured by the ADI-R and SCQ between patients with C/C versus patients with C/A + A/A of rs2301963 stratified by gender.
Comparison of the three core symptoms of autism spectrum disorder measured by the ADI-R and SCQ between 11 patients who were found to have rare missense mutations with the rest of the patients.
Contributor Information
Wei-Hsien Chien, Email: 082394@mail.fju.edu.tw.
Susan Shur-Fen Gau, Email: gaushufe@ntu.edu.tw.
Hsiao-Mei Liao, Email: hmllamda@gmail.com.
Yen-Nan Chiu, Email: philipch@ntch.gov.tw.
Yu-Yu Wu, Email: ruthyuyuwu@gmail.com.
Yu-Shu Huang, Email: hu1109s@yahoo.com.tw.
Wen-Che Tsai, Email: tsaiwenc@ntu.edu.tw.
Ho-Min Tsai, Email: homintsai@gmail.com.
Chia-Hsiang Chen, Email: cchen3801@gmail.com.
Acknowledgments
This work was supported by grants from National Science Council (NSC96-3112-B-002-033, NSC97-3112-B-002-009, NSC98-3112-B-002-004, and NSC 99-3112-B-002-036 to SS Gau), National Taiwan University (AIM for Top University Excellent Research Project:10R81918-03101R892103, 102R892103 to SS Gau), and an intramural grant from National Health Research Institutes (to CH Chen), Taiwan.
References
- Prevalence of autism spectrum disorders - Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. MMWR Surveill Summ. 2012;61(3):1–19. [PubMed] [Google Scholar]
- Fombonne E. Epidemiological surveys of autism and other pervasive developmental disorders: an update. J Autism Dev Disord. 2003;33(4):365–382. doi: 10.1023/A:1025054610557. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Fombonne E. Pervasive developmental disorders in preschool children: confirmation of high prevalence. Am J Psychiatry. 2005;162(6):1133–1141. doi: 10.1176/appi.ajp.162.6.1133. [DOI] [PubMed] [Google Scholar]
- Veenstra-VanderWeele J, Cook EH Jr. Molecular genetics of autism spectrum disorder. Mol Psychiatry. 2004;9(9):819–832. doi: 10.1038/sj.mp.4001505. [DOI] [PubMed] [Google Scholar]
- Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9(5):341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugge M, Bruun-Petersen G, Brondum-Nielsen K, Friedrich U, Hansen J, Jensen G, Jensen PK, Kristoffersson U, Lundsteen C, Niebuhr E. et al. Disease associated balanced chromosome rearrangements: a resource for large scale genotype-phenotype delineation in man. J Med Genet. 2000;37(11):858–865. doi: 10.1136/jmg.37.11.858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CL, Ledbetter DH. Autism and cytogenetic abnormalities: solving autism one chromosome at a time. Curr Psychiatry Rep. 2007;9(2):141–147. doi: 10.1007/s11920-007-0084-9. [DOI] [PubMed] [Google Scholar]
- Reddy KS. Cytogenetic abnormalities and fragile-X syndrome in autism spectrum disorder. BMC Med Genet. 2005;6:3. doi: 10.1186/1471-2350-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorstman JA, Staal WG, van Daalen E, van Engeland H, Hochstenbach PF, Franke L. Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol Psychiatry. 2006;11(1):1. doi: 10.1038/sj.mp.4001781. 18–28. [DOI] [PubMed] [Google Scholar]
- Christian SL, Brune CW, Sudi J, Kumar RA, Liu S, Karamohamed S, Badner JA, Matsui S, Conroy J, McQuaid D. et al. Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biol Psychiatry. 2008;63(12):1111–1117. doi: 10.1016/j.biopsych.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J. et al. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont ML, Sanlaville D, Redon R, Raoul O, Cormier-Daire V, Lyonnet S, Amiel J, Le Merrer M, Heron D, de Blois MC. et al. Array-based comparative genomic hybridization identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J Med Genet. 2006;43(11):843–849. doi: 10.1136/jmg.2006.043166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH Jr, Dobyns WB. et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17(4):628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T. et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Eng J Med. 2008;358(7):667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- Chien S-FG W-H, Wu Y-Y, Huang Y-S, Fang J-S, Chen Y-J, Soong W-T, Chiu Y-N, Chen C-H. Identification and molecular characterization of two novel chromosomal deletions associated with autism. Clin Genet. 2010;78(5):8. doi: 10.1111/j.1399-0004.2010.01395.x. [DOI] [PubMed] [Google Scholar]
- Ranta S, Zhang Y, Ross B, Takkunen E, Hirvasniemi A, de la Chapelle A, Gilliam TC, Lehesjoki AE. Positional cloning and characterization of the human DLGAP2 gene and its exclusion in progressive epilepsy with mental retardation. European journal of human genetics: EJHG. 2000;8(5):381–384. doi: 10.1038/sj.ejhg.5200440. [DOI] [PubMed] [Google Scholar]
- Kristiansen LV, Meador-Woodruff JH. Abnormal striatal expression of transcripts encoding NMDA interacting PSD proteins in schizophrenia, bipolar disorder and major depression. Schizophr Res. 2005;78(1):87–93. doi: 10.1016/j.schres.2005.06.012. [DOI] [PubMed] [Google Scholar]
- de Bartolomeis A, Fiore G. Postsynaptic density scaffolding proteins at excitatory synapse and disorders of synaptic plasticity: implications for human behavior pathologies. Int Rev Neurobiol. 2004;59:221–254. doi: 10.1016/S0074-7742(04)59009-8. [DOI] [PubMed] [Google Scholar]
- Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of AMPA receptor trafficking and signaling molecule transcripts in the prefrontal cortex in schizophrenia. Synapse. 2006;60(8):585–598. doi: 10.1002/syn.20329. [DOI] [PubMed] [Google Scholar]
- Boeckers TM. The postsynaptic density. Cell Tissue Res. 2006;326(2):409–422. doi: 10.1007/s00441-006-0274-5. [DOI] [PubMed] [Google Scholar]
- Wu K, Hanna GL, Easter P, Kennedy JL, Rosenberg DR, Arnold PD. Glutamate system genes and brain volume alterations in pediatric obsessive-compulsive disorder: a preliminary study. Psychiatry Res. 2013;211(3):214–220. doi: 10.1016/j.pscychresns.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y. et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82(2):477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgen HM, van Daalen E, Bolton PF, Maloney VK, Huang S, Cresswell L, van den Boogaard MJ, Eleveld MJ, Van't Slot R, Hochstenbach R. et al. Copy number changes of the microcephalin 1 gene (MCPH1) in patients with autism spectrum disorders. Clin Genet. 2009;76(4):348–356. doi: 10.1111/j.1399-0004.2009.01254.x. [DOI] [PubMed] [Google Scholar]
- Gau SS, Liu LT, Wu YY, Chiu YN, Tsai WC. Psychometric properties of the Chinese version of the social responsiveness scale. Res Autism Spectr Disord. 2013;7(2):349–360. doi: 10.1016/j.rasd.2012.10.004. [DOI] [Google Scholar]
- Gau SS-F, Lee C-M, Lai M-C, Chiu Y-N, Huang Y-F, Kao J-D, Wu Y-Y. Psychometric properties of the Chinese version of the social communication questionnaire. Res Autism Spectr Disord. 2011;5(2):809–818. doi: 10.1016/j.rasd.2010.09.010. [DOI] [Google Scholar]
- den Dunnen JT, Antonarakis SE. Nomenclature for the description of human sequence variations. Hum Genet. 2001;109(1):121–124. doi: 10.1007/s004390100505. [DOI] [PubMed] [Google Scholar]
- Shi YY, He L. SHEsis, a powerful software platform for analyses of linkage disequilibrium, haplotype construction, and genetic association at polymorphism loci. Cell Res. 2005;15(2):97–98. doi: 10.1038/sj.cr.7290272. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genome-wide studies. Proc Natl Acad Sci USA. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau WY, Gau SS, Chiu YN, Wu YY, Chou WJ, Liu SK, Chou MC. Psychometric properties of the Chinese version of the Autism Spectrum Quotient (AQ) Res Dev Disabil. 2013;34(1):294–305. doi: 10.1016/j.ridd.2012.08.005. [DOI] [PubMed] [Google Scholar]
- Devlin B, Melhem N, Roeder K. Do common variants play a role in risk for autism? Evidence and theoretical musings. Brain Res. 2011;1380:78–84. doi: 10.1016/j.brainres.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gau SS, Lee CM, Lai MC, Chiu YN, Huang YF, Kao JD, Wu YY. Psychometric properties of the Chinese version of the Social Communication Questionnaire. Research in Autism Spectrum Disorders. 2011;5(2):809–818. doi: 10.1016/j.rasd.2010.09.010. [DOI] [Google Scholar]
- Anney R, Klei L, Pinto D, Almeida J, Bacchelli E, Baird G, Bolshakova N, Bolte S, Bolton PF, Bourgeron T. et al. Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum Mol Genet. 2012;21(21):4781–4792. doi: 10.1093/hmg/dds301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DB, Cavalleri GL, Ahmadi KR. The genetics of common diseases: 10 million times as hard. Cold Spring Harb Symp Quant Biol. 2003;68:395–401. doi: 10.1101/sqb.2003.68.395. [DOI] [PubMed] [Google Scholar]
- Vaags AK, Lionel AC, Sato D, Goodenberger M, Stein QP, Curran S, Ogilvie C, Ahn JW, Drmic I, Senman L. et al. Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am J Hum Genet. 2012;90(1):133–141. doi: 10.1016/j.ajhg.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C. et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43(6):585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremer A, Giacobini M, Eriksson M, Gustavsson P, Nordin V, Fernell E, Gillberg C, Nordgren A, Uppstromer A, Anderlid BM. et al. Copy number variation characteristics in subpopulations of patients with autism spectrum disorders. Am J Med Genet B. 2011;156(2):115–124. doi: 10.1002/ajmg.b.31142. [DOI] [PubMed] [Google Scholar]
- Kumar RA. Two-hit wonder: a novel genetic model to explain variable expressivity in severe pediatric phenotypes. Clin Genet. 2010;78(6):517–519. doi: 10.1111/j.1399-0004.2010.01530_1.x. [DOI] [PubMed] [Google Scholar]
- Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, Vives L, Walsh T, McCarthy SE, Baker C. et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010;42(3):203–209. doi: 10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CP, Sabo A, Sakai Y, Crosby J, Muzny D, Hawes A, Lewis L, Akbar H, Varghese R, Boerwinkle E. et al. Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum Mol Genet. 2011;20(17):3366–3375. doi: 10.1093/hmg/ddr243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien WH, Gau SS, Wu YY, Huang YS, Fang JS, Chen YJ, Soong WT, Chiu YN, Chen CH. Identification and molecular characterization of two novel chromosomal deletions associated with autism. Clin Genet. 2010;78(5):449–456. doi: 10.1111/j.1399-0004.2010.01395.x. [DOI] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485(7397):242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer sequences, optimal annealing temperature (Ta) and size of PCR products of the DLGAP2 gene.
Comparison of the three core symptoms of autism spectrum disorder measured by the ADI-R and SCQ between patients with A/A versus patients with A/G + G/G of rs2906569 stratified by gender.
Comparison of the three core symptoms of autism spectrum disorder measured by the ADI-R and SCQ between patients with C/C versus patients with C/A + A/A of rs2301963 stratified by gender.
Comparison of the three core symptoms of autism spectrum disorder measured by the ADI-R and SCQ between 11 patients who were found to have rare missense mutations with the rest of the patients.