

Abstract
Background
The persistence of antibiotic resistance depends on the fitness effects of resistance elements in the absence of antibiotics. Recent work shows that the fitness effect of a given resistance mutation is influenced by other resistance mutations on the same genome. However, resistant bacteria acquire additional beneficial mutations during evolution in the absence of antibiotics that do not alter resistance directly but may modify the fitness effects of new resistance mutations.
Results
We experimentally evolved rifampicin-resistant and sensitive Escherichia coli in a drug-free environment, before measuring the effects of new resistance elements on fitness in antibiotic-free conditions. Streptomycin-resistance mutations had small fitness effects in rifampicin-resistant genotypes that had adapted to antibiotic-free growth medium, compared to the same genotypes without adaptation. We observed a similar effect when resistance was encoded by a different mechanism and carried on a plasmid. Antibiotic-sensitive bacteria that adapted to the same conditions showed the same pattern for some resistance elements but not others.
Conclusions
Epistatic variation of costs of resistance can result from evolution in the absence of antibiotics, as well as the presence of other resistance mutations.
Keywords: Antibiotic resistance, Epistasis, Experimental evolution, Escherichia coli
Background
The persistence of antibiotic-resistant bacteria depends on how resistance, in the form of chromosomal mutations or horizontally acquired elements such as plasmids, affects fitness relative to antibiotic-sensitive genotypes in the absence of antibiotics [1,2]. Recent work shows that the fitness effects of resistance mutations often vary depending on the presence of other resistance mutations on the same genome [3-7]. However, resistance evolution will often be accompanied by the fixation of additional mutations that do not confer resistance but increase fitness in the present environment, either because resistant bacteria evolve in heterogeneous hosts or natural environments where the optimal genotype changes over time, or because the cost of resistance causes selection for compensatory mutations (e.g. [8-13]). Any impact of adaptation in the absence of antibiotics on the subsequent cost of additional resistance mutations therefore potentially modulates costs of resistance in natural populations of pathogens.
Epistatic interactions have been observed among different resistance elements [3-6,14,15], and among different beneficial mutations during adaptation to novel environments [16-19]. However, interactions between the two types of mutations in terms of how they influence fitness in the absence of antibiotics are less clear. In this paper we ask whether adaptation of antibiotic-resistant and sensitive bacteria to the same antibiotic-free environment alters the fitness effects of additional resistance elements that confer resistance against other antibiotics. To test for this possibility, we experimentally evolved rifampicin-resistant (RifR) and rifampicin-sensitive (RifS) E. coli in liquid growth medium in the laboratory, before inserting mutations that conferred resistance to streptomycin (StrR), shown schematically in Figure 1.
Figure 1.

Construction of different genotypes and associated resistance phenotypes. (1) The wild type and three RifR genotypes evolved for approximately 200 generations in liquid LB. (2) Additional resistance elements were inserted in the form of StrR mutations on rpsL (K43N or K88R) or a SulR + StrR plasmid (RSF1010).
StrR mutations on rpsL change the structure of ribosomal protein S12 and interfere with target binding in the presence of streptomycin [20-22]. To determine whether observed effects were specific to rpsL mutations, we carried out parallel experiments with a plasmid that confers resistance to sulfonamides and streptomycin (SulR + StrR). The plasmid we used (RSF1010) has a broad host range and encodes resistance to streptomycin by enzymatic modification [23]. Therefore our experiment incorporated multiple distinct antibiotic-resistance mechanisms. We measured the fitness effects of resistance elements by pairwise competition assays, allowing us to quantify the effects of additional resistance elements for RifR and RifS bacteria both before and after adaptation. In several cases we found that the cost of a new resistance element was smaller in genotypes that had adapted to our experimental environment compared with the same genotypes without adaptation, although RifR and RifS bacteria showed different patterns for some resistance elements.
Methods
Bacterial strains and growth conditions
In all experiments we used E. coli MG1655 grown at 37°C. To isolate mutants resistant to either rifampicin or streptomycin (RifR or StrR), we plated independent cultures of the wild type onto LB agar supplemented with 50 mg/L rifampicin [24] or 25 mg/L streptomycin. After 24 h incubation at 37°C we picked individual colonies, restreaked to purify genotypes and confirm resistance, before growth for ~2 h in liquid LB and storage at −80°C in 25%v:v glycerol. Rifampicin-resistance mutations were identified by sequencing the central resistance-determining region of rpoB as described previously [24]; streptomycin-resistance mutations were identified by sequencing part of rpsL using primers fwd 5'-ATGATG GCGGGATCGTTG-3' and rev 5'-CTTCCAGTTCAGATTTACC-3' [5]. All three of the RifR mutations (D516G, S512F, I572S) and both StrR mutations (K43N and K88R) have been previously associated with resistance to these antibiotics in E. coli[5,12,25]. The SulR + StrR plasmid RSF1010 was obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany). We constructed double-resistant genotypes, with two resistance mutations or one resistance mutation plus the plasmid, by transduction or transformation as described below.
Experimental evolution in the absence of antibiotics
We initiated selection lines with each of the three RifR genotypes (D516G, I572S, S512F) and three replicate lines with the wild type (Figure 1). Each selection line was grown in 100 μl liquid LB medium and diluted 1000-fold into fresh medium approximately every 12 h, as described previously [24]. After 20 transfers, approximately 200 generations, we plated every population onto LB agar and isolated a single colony from each, which we then grew for 2 h in liquid LB and stored at −80°C. We checked for reversion to rifampicin-sensitivity (RifS) in the evolved clones derived from resistant genotypes by plating on LB agar supplemented with rifampicin, but observed none. This confirmed that the colony isolates used in the present study remained RifR, although it does not exclude the possibility of revertants at low frequency in evolved populations. This procedure yielded nine different genotypes in addition to the wild type (left-hand side of Figure 1): three RifR genotypes that had not adapted to LB, three RifR genotypes that had adapted to LB (RifR-evolved), and three RifS genotypes that had adapted to LB (RifS-evolved).
Addition of streptomycin resistance mutations and plasmid
We used P1 transduction to insert either K43N or K88R into each of the nine genotypes described above, plus three independent replicates of the wild type. We followed [26] with few modifications. Briefly, a lysate of each donor strain was prepared by growth with phage P1 at low multiplicity of infection until lysis was visible, then transduced to the relevant recipient genotype by growth in LB for 20 min, followed by addition of sodium citrate, further incubation for 1 h, plating on LB agar plus streptomycin and sodium citrate, and restreaking three times before isolating a single colony at random and storing at −80°C. All constructed genotypes were verified by resequencing rpsL. Transductions were done in a single temporal block; for one genotype (I572S + K88R) a different StrR mutation was acquired during the isolation procedure; for D516G + K43N, no colonies were obtained after repeated attempts; these are excluded from further analysis.
Plasmid RSF1010 is a natural, nontransmissible, broad-host range plasmid of the IncQ incompatibility group conferring resistance to sulfonamide and streptomycin [23]. We extracted the plasmid from cells grown in LB using the PureYield Plasmid Miniprep Kit (Promega). We then transformed the nine isolated genotypes and three replicates of the wild type using TSS Transformation [27]. Briefly, strains were grown in LB to OD600 0.3 and chilled on ice for 10 min before adding an equal volume of ice cold 2× TSS (TSS is LB with 10% w:v PEG8000, 5% v:v DMSO, 50 mM MgSO4 at pH 6.5) and incubation on ice for another 30 minutes. We then added 1 ml of competent cells to 1 μl (~100 ng) of the plasmid prep and incubated on ice for one hour. After incubation at 37°C for one hour to allow expression of the resistance genes, we plated cells on LB agar supplemented with 30 mg/L streptomycin. For each transformation a single colony was isolated at random, grown for 2 h in liquid LB and stored at −80°C.
The fitness effects of transduced mutations and the plasmid were reproducible in independently constructed replicates of the wild type: fitness did not differ significantly among different isolates for any of the resistance elements in our experiment (K43N: F2,4 = 3.00, P = 0.16; K88R: F1,4 = 0.05, P = 0.83; plasmid: F2,6 = 1.33, P = 0.33). To check that the plasmid was not lost during competition assays, we plated all competitions that included a plasmid-carrying genotype on tetrazolium arabinose (TA) plates, where streptomycin-resistant genotypes form white colonies as described below, and on LB agar supplemented with 30 mg/L streptomycin, on which plasmid-carrying genotypes can grow but the wild type cannot. We observed approximately the same number of colonies on streptomycin plates as white colonies on TA plates (paired t-test: t35 = 1.49, P = 0.14), indicating that the plasmid was maintained throughout the competition.
Fitness measurements
We measured the competitive fitness of each genotype against a marked strain: E. coli K12 MG1655 Δara, which is otherwise isogenic to the wild type and forms red colonies on TA agar (tryptone 1%, yeast extract 0.1%, NaCl 0.5%, L(+)arabinose 1%, TTC 0.005%). For each competition, we grew independent cultures of both competitors overnight in liquid LB, before mixing them 1:1 v:v and diluting 1000-fold into fresh LB media. We estimated the frequency of each competitor by plating the culture on TA agar before and after two growth cycles using the same protocol as during experimental evolution. We then calculated relative fitness, w, as 1 + s, where s is the selection coefficient s = ln(Rfinal / Rinitial) / t, where Rfinal and Rinitial are the ratios of the competing genotypes at the beginning and end of the assay and t is assay duration in generations [5,28,29]; in batch culture t can be approximated as log2(Nfinal/Ninitial), where N is total population size. We discounted each score by the cost of the Δara marker [5], which was not significantly different from zero (−0.025 on average; P = 0.06). To calculate the fitness effects of StrR mutations or the plasmid in a given genetic background, we took the difference in fitness (Δw) between the same strain with and without K43N, K88R or the plasmid.
RifR, RifR-evolved and RifS-evolved genotypes lacking either StrR mutations or the plasmid were each assayed in three different blocks: once after the evolution experiment to test for adaptation [24], once alongside genotypes with K43N or K88R, and once alongside genotypes carrying the plasmid. The correlation between fitness scores relative to the wild type measured for these genotypes in different blocks was high (r2 = 0.91, 0.94, 0.92), and there was no block × genotype interaction (F16,46 = 1.61, P = 0.10), indicating that fitness values of different genotypes relative to each other were repeatable across blocks of assays. In each block, competitions were replicated three times independently, and nine times for the wild type. Additional file 1: Table S1.
Testing for chaperone (DnaK and GroEL) overproduction
The best-described molecular mechanism for buffering against the fitness effects of deleterious mutations is the overproduction of molecular chaperones [30-32], enzymes that assist in correct protein folding [33]. To determine whether variation of costs of resistance among genotypes in our experiment could be explained by variation in chaperone production, we measured levels of the two key chaperones DnaK and GroEL [34] using western blots. Cells were harvested by centrifugation from mid-exponential cultures, resuspended in 1× SDS-PAGE sample buffer and boiled for 10 minutes. SDS-PAGE and immunoblotting with mouse-monoclonal anti-DnaK and anti-GroEL antibodies (Enzo Life Sciences) was carried out according to standard procedures. ImmunStar Western C Substrate Kit (BioRad) was used for detection in conjuction with horseradish peroxidase-conjugated anti-Mouse lgG secondary antibody (Amersham). Blots were imaged using the ChemiDoc XRS + CCD Sytem (BioRad) and analyzed using ImageJ 1.46 (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997–2012.).
Statistical analyses
We tested for adaptation to LB by a paired t-test using the average fitness of each genotype before and after experimental evolution. We tested for variation of the response to selection among genotypes by analysis of variance including genotype and assay block as factors. To test whether adaptation to LB altered the average fitness effects of additional resistance elements (StrR mutations or the plasmid) we conducted paired t-tests, taking the average fitness effect (Δw) of each resistance element on each genetic background before and after evolution, measured in the same block of competition assays. We analyzed variation of fitness effects among genotypes that had or had not adapted to LB by analyses of variance with genotype as a factor. Additionally, we tested pairwise epistatic interactions of RifR mutations with StrR mutations or the plasmid in genotypes that had not adapted to LB. We did this using the multiplicative model described by [5] and results are shown in Additional file 2: Table S2. Finally, to test for differences in levels of chaperones DnaK and GroEL we used pairwise t-tests with non-pooled standard deviation, correcting for multiple comparisons using the Bonferroni method.
Results
Adaptation in antibiotic-free conditions
Antibiotic-resistant (RifR) bacteria and the antibiotic-sensitive wild type increased in fitness over approximately 200 generations of evolution in LB growth medium (paired t-test: t5 = 6.20, P = 0.002; Figure 2). The change in fitness after experimental evolution varied among different evolved genotypes (F5,31 = 52.99, P < 0.0001). Among RifR genotypes, the response to selection was relatively large for genotypes derived from D516G and S512F, which had lower fitness than the wild type at the start of the experiment, compared with I572S that had a fitness advantage. After experimental evolution, RifR and RifS genotypes had similar levels of fitness on average (Welch’s t-test: t2.35 = 0.40, P = 0.72; Figure 2). Thus, despite initial variation, resistant and sensitive genotypes adapted to experimental conditions and converged upon similar levels of competitive fitness, as shown previously for a wider range of rpoB mutants including the three in this study [24].
Figure 2.

Adaptation of resistant and sensitive bacteria. Three RifR genotypes, denoted on the x-axis by their amino acid changes, and three replicates of the wild type (WT) were evolved for approximately 200 generations in antibiotic-free LB medium as described previously [24]. White and hatched bars show scores before and after evolution respectively. Each bar shows average ± s.e. competitive fitness relative to the wild type from nine independent assays, conducted in three temporal blocks; for the wild type before evolution standard error is shown for 27 assays.
Fitness effects of streptomycin resistance mutations
The fitness effects of StrR mutations were smaller on average in genotypes that had adapted to LB compared to the same genotypes without adaptation (paired t-test: t8 = 4.83, P = 0.001; Figure 3a,b). Across all genetic backgrounds, K43N was more costly on average than K88R (mean fitness effect ± s.d.-K43N:-0.11 ±0.07; K88R:-0.01 ±0.04; Figure 3a,b).
Figure 3.
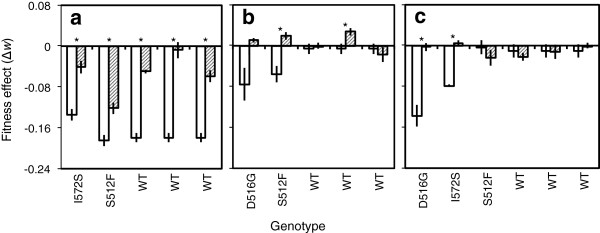
Effect of adaptation on the cost of additional resistance elements. Each bar shows the fitness effect of (a)rpsL K43N, (b)rpsL K88R or (c) plasmid RSF1010 either before (white bars) or after (hatched bars) adaptation to LB growth medium. Asterisks denote statistically significant effects of adaptation, as determined by pairwise t-tests assuming unequal variances. Each bar shows mean ± s.e. for three independent assays; for the wild type before evolution, bars show the average of three independently constructed genotypes, each assayed three times.
For K43N, all of the RifR-evolved and RifS-evolved genotypes we tested paid a smaller cost of resistance than the same genotypes without evolution in LB (Figure 3a). Among genotypes that had not adapted to LB, the cost was similar in S512F and the wild type, but comparatively small for I572S. This represents antagonistic pairwise epistasis between I572S and K43N: the cost of having both mutations was smaller than expected from their independent effects on the wild type (Additional file 2: Table S2). Among evolved genotypes, the cost of K43N also varied depending on genetic background (F4,10 = 12.84, P < 0.001), but in every case it was less costly than without evolution in LB.
When streptomycin resistance was due to K88R, the fitness cost was smaller after adaptation to LB for both of the RifR genotypes we tested, although this only represented statistically significant variation for S512F (Figure 3b). The fitness effect of K88R varied among genotypes that had not adapted to LB (F2,9 = 4.67, P = 0.04), being costly on average in RifR genotypes but not the wild type. K88R was also approximately neutral for two out of three evolved wild type (RifS) genotypes, and for the remaining one it had a small positive fitness effect. Consequently, adaptation to LB had no effect on average for the cost of K88R in RifS genotypes (F1,3 = 0.31, P = 0.62). By contrast, K88R was costly to RifR genotypes before, but not after experimental evolution in LB.
In summary, for one StrR mutation (K43N) the fitness cost was consistently lower for evolved genotypes than the same genotypes without adaptation to LB. A similar effect was observed for the other StrR mutation on RifR genetic backgrounds, but not for RifS genotypes, where K88R was approximately neutral both before and after evolution in LB on average. Thus, the fitness effects of StrR mutations varied epistatically depending on both adaptation in the absence of antibiotics and on the presence of other resistance mutations.
Fitness effects of an antibiotic-resistance plasmid
Insertion of the plasmid RSF1010, conferring resistance to sulfonamides and streptomycin, had a marginal cost on average (mean fitness effect ± s.d. = −0.03 ± 0.05), but this varied considerably among genotypes depending on adaptation to LB and RifR mutation (Figure 3c). There was no difference on average between the cost of RSF1010 in genotypes that had evolved in LB and those that had not (paired t-test: t5 = 1.29, P = 0.25), although in some cases the fitness effect of the plasmid was clearly lower in evolved genotypes. Specifically, RSF1010 was less costly for genotypes with RifR mutations D516G or I572S after they had evolved in LB (Figure 3c), but there was no difference for S512F or any of the RifS evolved genotypes. This variation was driven by a comparatively large fitness cost in two of the unevolved RifR genotypes, reflecting negative pairwise epistasis between the plasmid and D516G and I572S (Additional file 2: Table S2). Thus, the plasmid was costly to D516G and I572S before but not after evolution in antibiotic-free LB; other genotypes paid a comparatively small cost of carrying the plasmid.
Buffering of fitness effects is not due to overexpression of chaperones
The phenotypic effects of deleterious mutations can be buffered by overproduction of molecular chaperones, of which GroEL and DnaK are the two most important and best studied [32,34]. However, we found no upregulation of either chaperone in evolved genotypes (pairwise t-tests: all P values > 0.075; Additional file 3: Figure S1).
Discussion
We measured the fitness effects of three different antibiotic-resistance elements (two StrR mutations and a SulR + StrR plasmid) in RifR and RifS genotypes that had or had not adapted to antibiotic-free growth medium. In cases where additional resistance elements were costly to genotypes that had not evolved in LB, the cost was consistently smaller after adaptation to our experimental environment. For one of the StrR mutations this pattern was consistent across RifR and RifS genotypes, suggesting that the relatively small cost of new resistance mutations after adaptation in the absence of antibiotics is not specific to resistant bacteria. For the other StrR mutation and the plasmid, the cost was greatest in RifR genotypes that had not adapted to LB, and evolved genotypes showed a small cost or even a benefit of resistance by comparison. These results show that evolution in the absence of antibiotics can alter the fitness effects of new resistance elements, but this pattern depends on the identity of the new mutation and on the presence of other resistance mutations.
Consistent with previous studies [3,5,6,15,35], we found pairwise epistatic interactions among resistance elements on the same genetic background: out of seven multiple-resistant genotypes with a RifR mutation and either an StrR mutation or the plasmid, three showed significant deviations from a multiplicative model of fitness (Additional file 2: Table S2). Moreover, our finding that evolution in the absence of antibiotics alters the fitness effects of additional resistance elements suggests that epistatic variation of costs of resistance can also be caused by beneficial mutations that fix in the absence of antibiotics.
To our knowledge, the best characterized mechanism that actively buffers against deleterious mutations is over-expression of molecular chaperones [34,36,37]. This did not explain our findings: the two main chaperones, DnaK and GroEL, were not overproduced in evolved genotypes. This is perhaps unsurprising, given that selection for chaperone-mediated buffering is expected to depend on intense genetic drift [38] or high mutation rates [39,40], which is supported by experiments with viruses [41]. In the absence of direct selection for a buffering mechanism, the impact of adaptation on the cost of StrR mutations and the plasmid is probably an indirect effect of mutations that were under positive selection for their effects on growth in LB. By analogy, rpoB mutations that fix under selection for rifampicin resistance have wide-ranging effects on the bacterial phenotype, owing to direct effects on the function of RNA polymerase and pleiotropic effects on the expression of other genes [42-44]. This is associated with altered growth phenotypes in unselected environments [44,45] and epistatic interactions with other resistance mutations [5,6]. Although we lack a physiological understanding of adaptation to LB, it is known to alter growth phenotypes in unselected environments to a similar degree as rpoB mutations in this experimental system [24]. This is consistent with beneficial mutations fixed in LB having indirect phenotypic effects that are unrelated to improved growth in LB but may generate epistatic interactions.
Our experiments included a limited set of resistance elements (three different types, including chromosomal mutations and a plasmid). Therefore we do not draw any general conclusions about how adaptation will influence the cost of new resistance elements. However, our results do show that adaptation in the absence of antibiotics can cause epistatic variation of fitness costs. In support, a recent study demonstrated variation of the fitness effects of rifampicin-resistance mutations among antibiotic-sensitive E. coli genotypes [46]. This is consistent with epistatic variation of fitness costs due to mutations at loci unrelated to drug resistance. We also stress that with only one selection line for each RifR genotype, variation in the outcomes of evolution among evolved RifR genotypes does not necessarily reflect an effect of starting genotype. Stochastic processes during adaptation, including the random appearance and loss of beneficial mutations [47], will also contribute to variation among individual selection lines. Therefore, while we found differences on average between RifR and RifS evolved genotypes, and between evolved and nonevolved genotypes, we did not test whether the effect of adaptation varies among genotypes with different rifampicin-resistance mutations.
Directions for further work include identifying the physiological mechanisms by which adaptation to antibiotic-free conditions can alter the costs of new resistance elements, and how general such effects are. For example, expression profiling of resistant and sensitive genotypes that have and have not been experimentally evolved could be used to find cellular functions that are altered by resistance mutations and adaptation, both independently and in combination. To determine whether adaptation has a general effect on the cost of additional resistance mutations would require experiments with other model organisms, antibiotics and growth media. In particular, testing whether compensatory mutations that ameliorate the deleterious effects of one resistance mutation [8,10-13] can buffer against the effects of resistance elements at other loci would be relevant to the broader question of how selection acts on antibiotic resistance across genetic backgrounds.
Conclusions
We observed epistatic variation of the fitness costs associated with antibiotic resistance, stemming from interactions between different resistance elements as observed previously [3,5,6] and from interactions between resistance elements and beneficial mutations that fixed during adaptation to drug-free conditions. This is potentially relevant for the cost of resistance in real populations of pathogenic bacteria. In chronic infections, such as those caused by Pseudomonas aeruginosa in cystic fibrosis patients, bacteria fix multiple mutations during a single infection, many of which are unrelated to resistance evolution [48-50]. Other pathogens such as Mycobacterium tuberculosis, which often carry antibiotic resistance elements, also fix mutations that are beneficial in the absence of antibiotics [51]. If such mutations influence the cost of subsequent resistance mutations, as they did in our experiment, then a better understanding of epistasis between resistance elements and other types of mutations will be important for predicting the likelihood that multi-drug resistant bacteria will persist following different types of treatment.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DCA and ARH designed and performed the experiments, analyzed data and wrote the paper. Both authors read and approved the final manuscript.
Supplementary Material
Relative Fitness of all Genotypes.
Epistasis between RifR mutations, StrR mutations and plasmid RSF1010.
Chaperone levels in different genotypes.
Contributor Information
Daniel C Angst, Email: daniel.angst@env.ethz.ch.
Alex R Hall, Email: alex.hall@env.ethz.ch.
Acknowledgements
We thank Martin Ackermann, Sebastian Bonhoeffer, Gabriel Perron and Olivier Tenaillon for helpful comments and discussion. Sequence data were generated in the Genetic Diversity Centre of ETH Zürich. The Δara marker was a gift from Arnaud Gutierrez and Ivan Matic. This work was funded by a Marie Curie Intra-European Fellowship within the 7th European Community Framework Programme.
References
- Andersson DI, Hughes D. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol. 2010;8(4):260–271. doi: 10.1038/nrmicro2319. [DOI] [PubMed] [Google Scholar]
- Andersson DI, Levin BR. The biological cost of antibiotic resistance. Curr Opin Microbiol. 1999;2(5):489–493. doi: 10.1016/S1369-5274(99)00005-3. [DOI] [PubMed] [Google Scholar]
- Rozen DE, McGee L, Levin BR, Klugman KP. Fitness costs of fluoroquinolone resistance in Streptococcus pneumoniae. Antimicrob Agents Chemother. 2007;51(2):412–416. doi: 10.1128/AAC.01161-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salverda ML, Dellus E, Gorter FA, Debets AJ, van der Oost J, Hoekstra RF, Tawfik DS, de Visser JA. Initial mutations direct alternative pathways of protein evolution. PLoS Genet. 2011;7(3):e1001321. doi: 10.1371/journal.pgen.1001321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trindade S, Sousa A, Xavier KB, Dionisio F, Ferreira MG, Gordo I. Positive epistasis drives the acquisition of multidrug resistance. PLoS Genet. 2009;5(7):e1000578. doi: 10.1371/journal.pgen.1000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward H, Perron GG, MacLean RC. The cost of multiple drug resistance in Pseudomonas aeruginosa. J Evol Biol. 2009;22(5):997–1003. doi: 10.1111/j.1420-9101.2009.01712.x. [DOI] [PubMed] [Google Scholar]
- Weinreich DM, Watson RA, Chao L. Perspective: sign epistasis and genetic constraint on evolutionary trajectories. Evolution. 2005;59(6):1165–1174. [PubMed] [Google Scholar]
- Cohan FM, King EC, Zawadzki P. Amelioration of the deleterious pleiotropic effects of an adaptive mutation in Bacillus subtilis. Evolution. 1994;48(1):81–95. doi: 10.2307/2410005. [DOI] [PubMed] [Google Scholar]
- Kugelberg E, Lofmark S, Wretlind B, Andersson DI. Reduction of the fitness burden of quinolone resistance in Pseudomonas aeruginosa. J Antimicrob Chemother. 2005;55(1):22–30. doi: 10.1093/jac/dkh505. [DOI] [PubMed] [Google Scholar]
- Levin BR, Perrot V, Walker N. Compensatory mutations, antibiotic resistance and the population genetics of adaptive evolution in bacteria. Genetics. 2000;154(3):985–997. doi: 10.1093/genetics/154.3.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulander W, Maisnier-Patin S, Andersson DI. Multiple mechanisms to ameliorate the fitness burden of mupirocin resistance in Salmonella typhimurium. Mol Microbiol. 2007;64(4):1038–1048. doi: 10.1111/j.1365-2958.2007.05713.x. [DOI] [PubMed] [Google Scholar]
- Reynolds MG. Compensatory evolution in rifampin-resistant Escherichia coli. Genetics. 2000;156(4):1471–1481. doi: 10.1093/genetics/156.4.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrag SJ, Perrot V, Levin BR. Adaptation to the fitness costs of antibiotic resistance in Escherichia coli. Proc R Soc Lond B Biol Sci. 1997;264(1386):1287–1291. doi: 10.1098/rspb.1997.0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva RF, Mendonça SC, Carvalho LM, Reis AM, Gordo I, Trindade S, Dionisio F. Pervasive sign epistasis between conjugative plasmids and drug-resistance chromosomal mutations. PLoS Genet. 2011;7(7):e1002181. doi: 10.1371/journal.pgen.1002181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich DM, Delaney NF, DePristo MA, Hartl DL. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science. 2006;312(5770):111–114. doi: 10.1126/science.1123539. [DOI] [PubMed] [Google Scholar]
- Chou HH, Chiu HC, Delaney NF, Segrè D, Marx CJ. Diminishing returns epistasis among beneficial mutations decelerates adaptation. Science. 2011;332(6034):1190–1192. doi: 10.1126/science.1203799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AI, Dinh DM, Schneider D, Lenski RE, Cooper TF. Negative epistasis between beneficial mutations in an evolving bacterial population. Science. 2011;332(6034):1193–1196. doi: 10.1126/science.1203801. [DOI] [PubMed] [Google Scholar]
- Kvitek DJ, Sherlock G. Reciprocal sign epistasis between frequently experimentally evolved adaptive mutations causes a rugged fitness landscape. PLoS Genet. 2011;7(4):e1002056. doi: 10.1371/journal.pgen.1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon O, Rodríguez-Verdugo A, Gaut RL, McDonald P, Bennett AF, Long AD, Gaut BS. The molecular diversity of adaptive convergence. Science. 2012;335(6067):457–461. doi: 10.1126/science.1212986. [DOI] [PubMed] [Google Scholar]
- Alekshun MN, Levy SB. Molecular mechanisms of antibacterial multidrug resistance. Cell. 2007;128(6):1037–1050. doi: 10.1016/j.cell.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Maisnier-Patin S, Paulander W, Pennhag A, Andersson DI. Compensatory evolution reveals functional interactions between ribosomal proteins S12 L14 and L19. J Mol Biol. 2007;366(1):207–215. doi: 10.1016/j.jmb.2006.11.047. [DOI] [PubMed] [Google Scholar]
- Ozaki M, Mizushima S, Nomura M. Identification and functional characterization of the protein controlled by the streptomycin-resistant locus in E. coli. Nature. 1969;222(5191):333–339. doi: 10.1038/222333a0. [DOI] [PubMed] [Google Scholar]
- Scholz P, Haring V, Wittmann-Liebold B, Ashman K, Bagdasarian M, Scherzinger E. Complete nucleotide sequence and gene organization of the broad-host-range plasmid RSF1010. Gene. 1989;75(2):271–288. doi: 10.1016/0378-1119(89)90273-4. [DOI] [PubMed] [Google Scholar]
- Hall AR. Genotype-by-environment interactions due to adaptation and antibiotic resistance in Escherichia coli. J Evol Biol. 2013. [DOI] [PubMed]
- Garibyan L, Huang T, Kim M, Wolff E, Nguyen A, Nguyen T, Diep A, Hu KB, Iverson A, Yang HJ. et al. Use of the rpoB gene to determine the specificity of base substitution mutations on the Escherichia coli chromosome. DNA Repair. 2003;2(5):593–608. doi: 10.1016/S1568-7864(03)00024-7. [DOI] [PubMed] [Google Scholar]
- Thomason LC, Costantino N, Court DL. E. coli genome manipulation by P1 transduction. Curr Protoc Mol Biol. 2007;1.17:1–8. doi: 10.1002/0471142727.mb0117s79. [DOI] [PubMed] [Google Scholar]
- Chung CT, Niemela SL, Miller RH. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc Natl Acad Sci U S A. 1989;86(7):2172–2175. doi: 10.1073/pnas.86.7.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullberg E, Cao S, Berg OG, Ilbäck C, Sandegren L, Hughes D, Andersson DI. Selection of resistant bacteria at very low antibiotic concentrations. PLoS Pathog. 2011;7(7):e1002158. doi: 10.1371/journal.ppat.1002158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenski RE, Rose MR, Simpson SC, Tadler SC. Long-term experimental evolution in Escherichia coli 1. Adaptation and divergence during 2,000 generations. Am Nat. 1991;138(6):1315–1341. doi: 10.1086/285289. [DOI] [Google Scholar]
- Fares MA, Ruiz-González MX, Moya A, Elena SF, Barrio E. Endosymbiotic bacteria-GroEL buffers against deleterious mutations. Nature. 2002;417(6887):398. doi: 10.1038/417398a. [DOI] [PubMed] [Google Scholar]
- Kerner MJ, Naylor DJ, Ishihama Y, Maier T, Chang HC, Stines AP, Georgopoulos C, Frishman D, Hayer-Hartl M, Mann M. et al. Proteome-wide analysis of chaperonin-dependent protein folding in Escherichia coli. Cell. 2005;122(2):209–220. doi: 10.1016/j.cell.2005.05.028. [DOI] [PubMed] [Google Scholar]
- Tokuriki N, Tawfik DS. Chaperonin overexpression promotes genetic variation and enzyme evolution. Nature. 2009;459:668–675. doi: 10.1038/nature08009. [DOI] [PubMed] [Google Scholar]
- Rutherford SL. Between genotype and phenotype: protein chaperones and evolvability. Nat Rev Genet. 2003;4(4):263–274. doi: 10.1038/nrg1041. [DOI] [PubMed] [Google Scholar]
- Maisnier-Patin S, Roth JR, Fredriksson A, Nyström T, Berg OG, Andersson DI. Genomic buffering mitigates the effects of deleterious mutations in bacteria. Nat Genet. 2005;37(12):1376–1379. doi: 10.1038/ng1676. [DOI] [PubMed] [Google Scholar]
- Hall AR, MacLean RC. Epistasis buffers the fitness effects of rifampicin-resistance mutations in Pseudomonas aeruginosa. Evolution. 2011;65:2370–2379. doi: 10.1111/j.1558-5646.2011.01302.x. [DOI] [PubMed] [Google Scholar]
- Fares MA, Barrio E, Sabater-Munoz B, Moya A. The evolution of the heat-shock protein GroEL from Buchnera, the primary endosymbiont of aphids, is governed by positive selection. Mol Biol Evol. 2002;19(7):1162–1170. doi: 10.1093/oxfordjournals.molbev.a004174. [DOI] [PubMed] [Google Scholar]
- Tokuriki N, Stricher F, Schymkowitz J, Serrano L, Tawfik DS. The stability effects of protein mutations appear to be universally distributed. J Mol Biol. 2007;369(5):1318–1332. doi: 10.1016/j.jmb.2007.03.069. [DOI] [PubMed] [Google Scholar]
- Gros PA, Tenaillon O. Selection for chaperone-like mediated genetic robustness at low mutation rate: impact of drift, epistasis and complexity. Genetics. 2009;182(2):555–564. doi: 10.1534/genetics.108.099366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser JA, Hermisson J, Wagner GP, Ancel Meyers L, Bagheri-Chaichian H, Blanchard JL, Chao L, Cheverud JM, Elena SF, Fontana W. et al. Perspective: evolution and detection of genetic robustness. Evolution. 2003;57(9):1959–1972. doi: 10.1111/j.0014-3820.2003.tb00377.x. [DOI] [PubMed] [Google Scholar]
- Wagner A. Robustness and evolvability in living systems. Princeton: Princeton University Press; 2005. [Google Scholar]
- Sanjuán R, Cuevas JM, Furió V, Holmes EC, Moya A. Selection for robustness in mutagenized RNA viruses. PLoS Genet. 2007;3(6):e93. doi: 10.1371/journal.pgen.0030093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst SA. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell. 2001;104(6):901–912. doi: 10.1016/S0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- Derewacz DK, Goodwin CR, McNees CR, McLean JA, Bachmann BO. Antimicrobial drug resistance affects broad changes in metabolomic phenotype in addition to secondary metabolism. Proc Natl Acad Sci U S A. 2013;110(6):2336–2341. doi: 10.1073/pnas.1218524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins AE, Nicholson WL. Uncovering new metabolic capabilities of Bacillus subtilis using phenotype profiling of rifampin-resistant rpoB mutants. J Bacteriol. 2008;190(3):807–814. doi: 10.1128/JB.00901-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trindade S, Sousa A, Gordo I. Antibiotic resistance and stress in the light of Fisher's model. Evolution. 2012;66:3815–3824. doi: 10.1111/j.1558-5646.2012.01722.x. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Verdugo A, Gaut BS, Tenaillon O. Evolution of Escherichia coli rifampicin resistance in an antibiotic-free environment during thermal stress. BMC Evol Biol. 2013;13:50. doi: 10.1186/1471-2148-13-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travisano M, Lenski RE. Long-term experimental evolution in Escherichia coli. III. Variation among replicate populations in correlated responses to novel environments. Evolution. 1995;49:189–200. doi: 10.2307/2410304. [DOI] [PubMed] [Google Scholar]
- Ciofu O, Mandsberg LF, Bjarnsholt T, Wassermann T, Høiby N. Genetic adaptation of Pseudomonas aeruginosa during chronic lung infection of patients with cystic fibrosis: strong and weak mutators with heterogeneous genetic backgrounds emerge in mucA and/or lasR mutants. Microbiology. 2010;156:1108–1119. doi: 10.1099/mic.0.033993-0. [DOI] [PubMed] [Google Scholar]
- Huse HK, Kwon T, Zlosnik JE, Speert DP, Marcotte EM, Whiteley M. Parallel evolution in Pseudomonas aeruginosa over 39,000 generations in vivo. MBio. 2010;1(4):e00199–10. doi: 10.1128/mBio.00199-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D'Argenio DA, Miller SI, Ramsey BW, Speert DP, Moskowitz SM. et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A. 2006;103(22):8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comas I, Borrell S, Roetzer A, Rose G, Malla B, Kato-Maeda M, Galagan J, Niemann S, Gagneux S. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat Genet. 2012;44(1):106–112. doi: 10.1038/ng.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Relative Fitness of all Genotypes.
Epistasis between RifR mutations, StrR mutations and plasmid RSF1010.
Chaperone levels in different genotypes.