

Abstract
Exposure to ethanol in neonatal rats results in reduced neuronal numbers in the cerebellar cortex and deep nuclei of juvenile and adult animals. This reduction in cell numbers is correlated with impaired delay eyeblink conditioning (EBC), a simple motor learning task in which a neutral conditioned stimulus (CS; tone) is repeatedly paired with a co-terminating unconditioned stimulus (US; periorbital shock). Across training, cell populations in the interpositus (IP) nucleus model the temporal form of the eyeblink conditioned response (CR). The hippocampus, though not required for delay EBC, also shows learning-dependent increases in CA1 and CA3 unit activity. In the present study, rat pups were exposed to 0, 3, 4, or 5 mg/kg/day of ethanol during postnatal days (PD) 4–9. As adults, CR acquisition and timing were assessed during 6 training sessions of delay EBC with a short (280 msec) interstimulus interval (ISI; time from CS onset to US onset) followed by another 6 sessions with a long (880 msec) ISI. Neuronal activity was recorded in the IP and area CA1 during all 12 sessions. The high-dose rats learned the most slowly and, with the moderate-dose rats, produced the longest CR peak latencies over training to the short ISI. The low dose of alcohol impaired CR performance to the long ISI only. The 3E (3 mg/kg/day of ethanol) and 5E (5 mg/kg/day of ethanol) rats also showed slower-than-normal increases in learning-dependent excitatory unit activity in the IP and CA1. The 4E (4 mg/kg/day of ethanol) rats showed a higher rate of CR production to the long ISI and enhanced IP and CA1 activation when compared to the 3E and 5E rats. The results indicate that binge-like ethanol exposure in neonatal rats induces long-lasting, dose-dependent deficits in CR acquisition and timing and diminishes conditioning-related neuronal excitation in both the cerebellum and hippocampus.
Keywords: fetal alcohol syndrome disorders, eyeblink conditioning, postnatal ethanol, cerebellum, hippocampus
Introduction
Fetal alcohol spectrum disorders (FASD) represent a range of neurocognitive and neurobehavioral impairments caused by in utero alcohol exposure (Mattson et al., 2011; Schaefer and Deere, 2011). In the decades since fetal alcohol syndrome (FAS) was first formally described (Jones and Smith, 1975; Jones et al., 1973), a multitude of studies have documented the wide-ranging and deleterious effects of alcohol on the developing central nervous system (CNS) in humans (Niccols, 2007; Riley and McGee, 2005). FAS accounts for the children most severely afflicted by prenatal alcohol exposure, with a birth prevalence of at least 2 to 7 per 1000 births in the United States (May and Gossage, 2001; May et al., 2009). At the opposite end of the spectrum, alcohol-related neurodevelopmental disorder (ARND) refers to individuals that express the cognitive but not physical abnormalities associated with prenatal alcohol exposure (Chasnoff et al., 2010; Stratton et al., 1996). Due to difficulties in diagnosing ARND children in the absence of physical dysmorphology, however, the number of FASD children may actually be up to 10 times greater than reported (May and Gossage, 2001; Sampson et al., 1997). Individuals across the FASD spectrum exhibit pervasive deficits in higher order cognitive functions such as attention, learning, memory, and executive function (Kodituwakku, 2007; Mattson et al., 2011; Niccols, 2007; Streissguth, 2007).
FASD animal models, in which alcohol is administered to rodents during gestation or in the days immediately following birth, have been developed for investigating the teratogenic effects of alcohol on the developing brain (Stanton and Goodlett, 1998; West et al., 1989). The extent of alcohol’s neurotoxic effects is principally determined by the timing of exposure and the resulting blood alcohol concentration (BAC). The current study utilized ethanol exposure from PD 4 to PD 9, a period when the brain growth and development that occur in the rat are roughly equivalent to human brain growth and development that occur in the third trimester of gestation (Bayer et al., 1993; Goodlett and Johnson, 1999). Binge-like exposure of rats to alcohol (i.e., a large dose of alcohol administered over a short time period) during the “brain growth spurt” period of early postnatal life induces structural and functional CNS deficits across multiple brain areas, including the cerebellum and hippocampus (Hamilton et al., 2010; Hamre and West, 1993; Livy et al., 2003). An important goal of animal research is to better characterize these deficits across the full range of BACs associated with FASD in humans, particularly at low-to-moderate alcohol concentrations (Murawski and Stanton, 2011; Valenzuela et al., 2012).
Eyeblink conditioning (EBC) is a simple form of motor learning and arguably one of the best understood models of mammalian associative learning due to its well characterized behavior (Gormezano et al., 1983) and neural circuitry (Christian and Thompson, 2003; Freeman and Steinmetz, 2011; Steinmetz and Lindquist, 2009). In delay EBC, a conditioned stimulus (CS) of an innocuous light or tone overlaps and (typically) co-terminates with a mildly aversive unconditioned stimulus (US), such as a corneal air puff or periorbital electric shock. Repeated CS-US pairings result in a learned or conditioned eyeblink response (CR) to the CS. In well-trained subjects, the CR is timed such that the eyelid is maximally extended just before onset of the US. Sensory information related to the CS and US is relayed to the cerebellum via granule cells and inferior olive cells, respectively, converging in a cerebellar deep nucleus, the interpositus (IP), and 2 regions of the cortex, lobule HVI and the anterior lobe (Steinmetz and Lindquist, 2009). Recordings of neuronal activity from the anterior dorsolateral IP during delay EBC have revealed populations of cells that discharge just prior to execution of the eyeblink CR, modeling the temporal form of the behavioral response in both rabbits and rats (Berthier and Moore, 1990; Freeman and Nicholson, 2000; McCormick and Thompson, 1984; Rogers et al., 2001). The increase in CS-elicited spiking is proposed to activate downstream motor nuclei responsible for generation of the conditioned blink (McCormick and Thompson, 1984; Thompson, 2005).
The cerebellum of the neonatal rat is highly vulnerable to ethanol during early postnatal development (Bauer-Moffett and Altman, 1977), especially with binge-like doses that produce peak BACs in excess of 350 mg/dL. Ethanol-induced neuronal depletion is observed throughout the EBC brainstem-cerebellar neural circuit, including granule cells, inferior olive cells, Purkinje neurons, and IP neurons (Bonthius and West, 1991; Goodlett and Johnson, 1997; Green et al., 2002b; Pierce et al., 1993; Tran et al., 2005; Young et al., 2010). Indeed, even a single ethanol dose administered on PD 4 is sufficient to produce Purkinje cell death when these cells are counted in rats sacrificed just 10 hours after exposure (Idrus and Napper, 2012). Ethanol exposure in early development is proposed to induce rapid apoptosis in the cerebellum (and associated brainstem structures), leading to long-lasting impairments in the acquisition of delay EBC in both juvenile and adult rats (Brown et al., 2007; Green et al., 2000; Lindquist et al., 2007; Stanton and Goodlett, 1998; Tran et al., 2007), although some differences in CR timing have been seen in ethanol-exposed rats across the 2 age groups with more complex forms of EBC (Brown et al., 2009; Brown et al., 2008). IP damage may preferentially affect delay EBC performance, with acquisition deficits — defined by percentage of CRs or the blink’s peak amplitude — significantly correlated to ethanol-induced IP cell loss (Green et al., 2002b; Young et al., 2010). PD 4–9 ethanol exposure (5.25 g/kg/day) also produces slower-than-normal increases in learning-dependent IP unit activity to the CS (Green et al., 2002a), possibly due to a reduction in the number of plastic cells capable of encoding the CS-US association (Green, 2004).
An important factor related to the rate of acquisition and proper timing of the eyeblink CR is the interval between CS onset and US onset, known as the interstimulus interval (ISI). A series of studies in the rabbit have demonstrated that ISIs of 200–500 msec produce the most robust delay EBC and the best timed CRs relative to the expected US, compared to shorter or longer ISIs (Coleman and Gormezano, 1971; Frey and Ross, 1968; Schneiderman, 1966; Smith, 1968). More recent work suggests that similar CS-US intervals result in optimal CR acquisition and timing in rats as well as rabbits (e.g., Lindquist et al., 2009). During training with short or long ISIs, the temporal aspects of the conditioned blink must be adjusted to conform to the non-optimal stimulus configuration (Port et al., 1986). Numerous studies have reported maladaptively timed CRs in eyeblink-conditioned subjects with an inactivated or damaged cerebellar cortex (Garcia et al., 1999; Medina et al., 2000; Perrett et al., 1993), supporting a role for the cerebellar cortex, and the anterior lobe in particular, in regulating proper CR timing via precisely timed inhibition and disinhibition of the IP (Green and Steinmetz, 2005; Vogel et al., 2009). Ethanol-induced cerebellar damage is predicted to interfere with the cortical-interpositus interactions that control the adaptively timed behavioral response.
Hippocampal damage does not affect the acquisition of delay EBC (Akase et al., 1989; Lee and Kim, 2004; Port et al., 1985; Schmaltz and Theios, 1972), though pyramidal neurons in subregions CA1 and CA3 do display coincident increases in excitatory activity, modeling the amplitude and time course of the eyeblink CR (Berger et al., 1980; Berger and Thompson, 1978). The hippocampus is also one of several forebrain areas sensitive to postnatal ethanol (reviewed in Gil-Mohapel et al., 2010). Binge-like exposure (peak BAC > 350 mg/dL) across PD 4–10 and PD 2–10 reduced CA1 cell numbers when counted, respectively, on PD 10 (Livy et al., 2003) and later in adults (Tran and Kelly, 2003). Interestingly, CA3 and dentate gyrus neuronal numbers were similarly reduced when counted on PD 10 but not in adult animals, suggesting compensatory mechanisms may selectively promote recovery in these 2 brain regions. Hippocampal-dependent learning is also impaired following early ethanol insult. For instance, trace EBC, in which the CS turns on and off prior to presentation of the US, relies on the hippocampus in addition to the basic brainstem-cerebellar circuit for successful learning (Woodruff-Pak and Disterhoft, 2008). Postnatal ethanol exposure is reported to disrupt trace EBC following exposure to high concentrations of ethanol in juvenile rats (Murawski et al., 2013; Thomas and Tran, 2012), but see (Lindquist, 2013)). Impaired hippocampal-dependent learning (Morris water maze), and a blockade of long-term potentiation (LTP) induction in area CA1, have also been reported following prenatal ethanol exposure in the guinea pig (Richardson et al., 2002). To our knowledge, hippocampal CA1 unit activity has not been previously recorded during delay EBC in adult rats administered ethanol during early postnatal life.
While PD 4–9 ethanol exposure is consistently reported to impair delay EBC acquisition across a range of ISIs, the effects on CR timing in adult rats have been more variable (Green et al., 2000; Lindquist, 2013; Tran et al., 2007). For instance, relative to the sham-intubation (SI) group — which is the critical comparison for all ethanol-induced effects involving intragastric intubation — Green et al. (2000) found significant lengthening of CR onset latencies in ethanol-exposed (5.25 g/kg/day) rats trained with a 350-msec ISI. Lindquist (2013) sequentially trained control and ethanol-exposed (5 g/kg/day) rats in trace and delay EBC. Significantly longer CR onset latencies were observed during delay EBC (580-msec ISI) in the ethanol-exposed rats compared to SI controls. Though no significant treatment group differences were seen in CR peak latency in either study, Green et al. (2000) saw a general reduction in peak latencies in ethanol-exposed rats whereas Lindquist (2013) saw an increase in peak latencies. Tran et al. (2007) also reported a general trend toward longer CR onset and peak latencies in adult rats trained with a 280- or 880-msec ISI, though no significant differences were found between ethanol-exposed (5.25 g/kg/day) and SI subjects.
The ISI can be manipulated within subjects to better accentuate putative timing abnormalities. Stanton and colleagues, for example, found dose-dependent impairments in both juvenile and adult rats concurrently trained with 2 distinct CSs (tone and light), each reinforced with a periocular shock US at a short, optimal (280 msec) ISI or a longer, non-optimal (880 msec) ISI (Brown et al., 2009; Brown et al., 2008). Only adult subjects displayed CR timing abnormalities and only to the long ISI, producing conditioned blinks with later onset and peak latencies than control subjects (Brown et al., 2008). The current experiment employed the ISI shift paradigm, in which subjects were trained at one ISI and then switched to a second, longer ISI. Plasticity in CR timing typically occurs with ISI shifts as the latency to CR peak amplitude systematically moves toward onset of the new US (Hoehler and Thompson, 1980).
In the present study, the training ISI was manipulated in order to assess the consequences of PD 4–9 ethanol exposure (3, 4, or 5 g/kg/day) on CR acquisition and timing in adult rats implanted with unilateral multiple-unit recording electrodes targeted at the cerebellar IP and area CA1 in the hippocampus. Control and ethanol-exposed rats were submitted to 6 training sessions of delay EBC with an optimal ISI (280 msec) followed by another 6 sessions of delay EBC with a non-optimal ISI (880 msec). The goals of the present study were to: 1) examine delay EBC in adult rats exposed to low, moderate, or high doses of ethanol as neonates, 2) examine predicted changes in CR timing following the ISI shift, and 3) contrast IP and CA1 neuronal activity across the 5 treatment groups as a function of the training ISI.
Materials and Methods
Subjects
Male and female Long Evans Blue Spruce rats (Harlan, Indianapolis, IN) were housed in breeding pairs in the animal colony room in the Department of Brain and Psychological Sciences at Indiana University. The morning after initial pairing (0900 hours), vaginal smears were obtained from each female with sterile saline. The smears were obtained daily until the determination of gestational day (GD) 0, as evidenced by the presence of sperm in the sample. After females were positive for insemination, breeding pairs were separated, and the females were individually housed and checked daily for births. Parturition typically occurred on GD 22, designated PD 0. Litters were culled by PD 3 to 8–10 pups, with equal numbers of males and females when possible. On PD 3 rats were randomly assigned a number and a small amount of non-toxic India black ink was injected subcutaneously (s.c.) into one or more paws for identification and group assignment. Pups were weaned on PD 25 and housed in same-sex groups of up to 4 rats in standard laboratory cages (48 cm × 20 cm × 26 cm) in the animal colony room. Food and water were available ad libitum. The animal colony was maintained on a 12:12 hour light/dark cycle (lights on at 0700 hours). Thirty-five rats (18 male and 17 female) from 18 litters were used in the current study. All experiments were performed in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals and approved by Indiana University’s Institutional Animal Care and Use Committee. All necessary measures were taken to minimize pain and discomfort.
Neonatal Treatment Groups
On PD 4, pups of a given litter were placed on clean bedding in a small plastic container atop a heating pad to minimize heat loss. Each pup was identified by paw tattoo and weighed prior to intubation. When possible, one male and one female pup from each litter were assigned to one of 3 ethanol treatment groups (3 g/kg/day [3E], 4 g/kg/day [4E], or 5 g/kg/day [5E]) or one control group, sham intubation (SI). Separate litters of pups comprised the Unintubated Control (UC) group. Pups in the ethanol-exposed and SI groups were intubated 3 times daily from PD 4 to PD 9. Intubations were separated by 2 hours and occurred at 0900, 1100, and 1300 hours. The first 2 intubations consisted of a solution of 6.80% (v/v), 9.06% (v/v), or 11.33% (v/v) ethanol in milk formula administered at a volume of 0.02778 mL/g body weight (West et al., 1984). The third intubation consisted of milk formula alone. Group SI was intubated 3 times daily but no formula was ever infused. UC pups were weighed once daily over the intubation period but not intubated.
Blood Alcohol Concentration (BAC)
Blood samples were collected from intubated rat pups exposed to 0, 3, 4, and 5 g/kg/day of ethanol 2 hours after the second ethanol/milk intubation on PD 4. At least 20 μL of blood was collected into heparinized capillary tubes following tail clip. Samples from the ethanol-exposed rats were centrifuged and plasma was separated and immediately frozen at −70°C until analysis. Thawed samples were subsequently analyzed using the Analox GL5 Analyzer (Analox Instruments, Lunenberg, MA). An oxygen-sensitive electrode measured the rate of oxygen consumption resulting from the oxidation of ethanol in each sample. BAC was calculated by comparing an experimental sample to a known alcohol standard used in the calibration procedure.
Surgical procedures
When rats were at least 70 days of age they were anesthetized with an intra-muscular injection of anesthetic cocktail consisting of 9 mg/kg sterile physiological saline, 74 mg/kg ketamine, 3.7 mg/kg xylazine, and 0.74 mg/kg acepromazine. Injection volume was 2.0 mL/kg body weight. Supplemental s.c. ketamine injections at a volume of 0.2 mL were administered every 90 minutes to maintain anesthesia throughout the surgery. When fully anesthetized, the rat was secured in a stereotaxic instrument and a midline scalp incision exposed the skull and the stereotaxic placements for implanting microelectrodes in the right CA1 (AP: −3.8 relative to Bregma; ML: +1.6; DV: −3.0) and the left interpositus (IP) nucleus (AP: −2.3 relative to the interaural line; ML: −2.7; DV: −5.7). CA1 microelectrodes were composed of a bundle of 4 microelectrodes (impedance ~0.5 – 1.0 MΩ) connected to gold pins housed in a 6-pin connector (NB Labs, Austin, TX). IP microelectrodes were constructed from stainless steel insect pins insulated with Epoxylite® and etched to a fine tip. A small current passing between the electrode tip and a counter electrode brought the tip impedance to between 1.5 – 4.0 MΩ. Two microelectrodes were fixed together with dental acrylic with the electrode tips separated by approximately 1 mm and connected to gold pins housed in a second 6-pin connector. After electrode implantation, a small amount of bone wax was used to secure the electrodes and seal the skull. Two Teflon®-coated stainless steel electromyographic (EMG) electrodes, each connected to one of the two 6-pin connecters, were implanted in the anterior portion of the orbicularis oculi muscle of the left eyelid. A ground wire from each connector was secured to 1 of 3 skull screws. A bipolar stimulating electrode (PlasticsOne, Roanoke, VA) was implanted subdermally, dorsocaudal to the left eye. Both connectors and the stimulating electrode were secured with dental acrylic. The surgical site was sutured around the headstage and the antibiotic Povidine was applied liberally to the scalp. Rats were housed individually after surgery and handled once daily across the 7-day recovery period.
Behavioral training
Eyeblink conditioning took place in operant boxes (Coulburn Instruments, Allentown, PA) placed in sound attenuating chambers. Each operant box had 2 Plexiglas® and 2 stainless steel walls and a stainless steel floor grid. A speaker for the delivery of the tone CS was placed directly above the box. Electrodes and bipolar connecters on the rat’s head were attached to a tether composed of lightweight wire connected to a commutator (NB Labs Austin, TX), allowing the rat to move about freely. After approximately 7 days of recovery, all rats received one adaptation session (~1 hour) in which the tether was connected but no stimuli were presented. The next day, rats began the first of 12 training sessions, consisting of 100 trials delivered in 10 blocks of 10 trials. Trials were 80% reinforced (8 CS-US paired and 2 CS-alone trials per block), resulting in 80 paired trials and 20 CS-alone trials per session. The 2.8 kHz, 85 dB tone CS was 380 msec in duration (sessions 1–6) or 980 msec in duration (sessions 7–12) and the US for all paired trials was a co-terminating 1.5 mA, 100-msec periocular electric shock. Accordingly, the ISI for the initial 6 training sessions was 280 msec, and the ISI for the final 6 training sessions was 880 msec. The intertrial interval (ITI) was 30 ± 10 seconds.
Histology
After the last day of training, rats were overdosed with Euthasol® (Virbac Corp., Ft. Worth, TX). Direct current (100 μA) was passed through one electrode at each implantation site in the IP or area CA1 for approximately 10 seconds, producing a small electrolytic lesion to verify the neural recording location. Rats were subsequently perfused transcardially with physiologic saline, followed by a 10% formalin solution. Brains were extracted and placed in a sucrose formalin solution until fixed. Brains were sectioned (80 μm) with a cryostat throughout the deep nuclear region of the cerebellum and the entire hippocampal region and then mounted on gel-subbed slides. Sections were stained with cresyl violet followed by Prussian blue to visualize the location of the electrode placement and the presence of iron deposits following the lesion (Figure 1). A total of 35 rats was used in the current study, though data for two UC rats, two 3E rats, and one rat in each of the remaining groups (SI, 4E, and 5E) were excluded due to poor quality EMG records or improper electrode placement (i.e., missed both sites) for an N = 28.
Figure 1.

Photomicrographs of cresyl violet-stained sections at the level of the cerebellar interpositus nucleus (left) and area CA1 of the hippocampus (right). Electrode placement can be observed via the Prussian blue stain. Grey dashed ovals show the range of electrode placements for all subjects in both the IP and CA1 region. Cerebellar recordings were done ipsilateral to the trained eye and hippocampal recordings were done contralateral to the trained eye.
Behavioral Data
Trials were triggered for data acquisition and stimulus delivery was relayed to the chambers by a customized computer program. Raw EMG data were sampled for 1500 msec on each trial. The signal was amplified (10,000X), band-pass filtered (300–1000 Hz), digitized (500 Hz), rectified, smoothed (10-msec time constant), time-shifted (10 msec, to compensate for smoothing), and stored for offline analysis by a computer data acquisition system (Spike2, CED, London, UK). EMG data from each session were converted into comma-separated value files and then analyzed by a custom data analysis program (Datamunch program; King and Tracy, 1999) that computed the total number of trials in which a CR was detected. Thresholds for detecting CRs were set at 8 standard deviations above mean EMG activity during the 280-msec pre-CS period. Trials with elevated EMG activity during the 100-msec period immediately prior to CS onset were deemed bad trials and excluded from analysis. Any individual session that exceeded 25% bad trials was dropped. Elevated EMG activity during the 100-msec period immediately after CS onset was considered an alpha response (i.e., tone-evoked startle response) and not counted as a CR.
Neural Data
Neural activity was sampled for 1500 msec on each trial. The neural signal was passed through a JFET configured as a source follower, amplified (10,000X), band-pass filtered (500–5000 Hz), digitized (20 kHz), and stored for offline analysis. IP and CA1 neural activity (ipsilateral and contralateral to the recorded eye, respectively) were analyzed by isolating individual units using a moving time window with a threshold discrimination and template-matching algorithm (Spike2, CED, UK). For each brain region, up to 3 units with the best signal-to-noise ratio (~2:1) were separated for analysis on each session. Isolated spikes were considered independent events across sessions. Neural activity during the CS-US interval was converted into standard scores relative to the 280-msec pre-CS period. For the 280-msec ISI, one 280-msec bin was used. For the 880-msec ISI, three 293.3-msec bins were used to hold bin size across the 2 ISIs approximately constant. On each trial in which a behavioral CR was detected, difference scores were calculated by subtracting the mean neural activity in the pre-CS period from the mean neural activity for each bin of the ISI. Unit activity was averaged over 2 training sessions. Summary standard scores for each bin during the ISI were calculated by taking the mean of each difference score and dividing that mean by the corresponding standard error. The criterion for a significant change in neural activity for a given time bin was 2.02, the critical value for p ≤ .05 for a 2-tailed t test.
Statistical Analyses
Behavioral data were analyzed using repeated-measures analysis of variance (ANOVA) with a nested design to capture differences in CR production to the short and long ISI. Neural activity was also analyzed using repeated measures ANOVAs with a nested design. Simple repeated-measures ANOVAs, single factor ANOVAs, paired t tests, and Fisher’s PLSD were used as post hoc tests when significant main effects and interactions were obtained. A significant post hoc effect implies p ≤ .05. The alpha level was set at 0.05 for all tests. For missing session data, values were interpolated either from appropriate group means or from the individual values immediately prior to and after the missing value for a given subject. These instances of interpolation were rare (less than 5% of all training sessions) and were used only for repeated-measures analyses.
Results
Histology
Electrode tracks and marking lesions were used to confirm recording electrode placements. Figure 1 illustrates representative photomicrographs of recording electrode placements in the IP (left side) and area CA1 (right side). For each image, the dashed grey lines indicate the range of all hits. Only data from rats with placements in the IP and/or area CA1 are described (N = 28). Final distribution among groups was n = 5 for UC rats, n = 6 for SI rats, n = 5 for 3E rats, n = 6 for 4E rats, and n = 6 for 5E rats. Roughly equal numbers of males and females made up each treatment group. Sex was not examined in the current study, albeit sex differences are known to influence delay EBC, with females performing better than males (Dalla and Shors, 2009).
Blood Alcohol Concentrations
Mean BACs across the 3 ethanol groups (3E, 4E, and 5E) are shown in Figure 2A. A single-factor (treatment group) ANOVA yielded a significant main effect, F(2,14) = 24.0, p < .0001. Post hoc analyses revealed significant pair-wise contrasts across all comparisons, meaning the averaged BAC for each group was significantly different from all others. Peak BAC for each subject on PD 4 was correlated with the averaged CR percentage across the initial 6 training sessions with the 280-msec ISI (Figure 2B; see below for details), resulting in a significant negative correlation (R2 = 0.24, p < .05). As predicted, higher peak BACs impaired CR acquisition to a greater degree than lower peak BACs.
Figure 2.

(A) Average blood alcohol concentration (BAC; mean ± SE) for the 3E, 4E, and 5E treatment groups as measured on PD 4. (B) Scatterplot illustrating the significant negative correlation between PD 4 peak BAC and the averaged percentage of eyeblink CRs emitted across sessions 1 to 6, during training with the 280-msec ISI.
Eyeblink Conditioning
The percentage of emitted CRs across the 12 training sessions (Figure 3A: sessions 1 to 6; Figure 4A: sessions 7 to 12) was examined using a mixed-design 5 × 2 × 12 repeated measures ANOVA, with 1 between-subjects factor (treatment group) and 2 within-subject factors (ISI and session). Statistical analyses revealed a significant 3-way interaction, F(20,115) = 1.8, p < .05, indicating CR acquisition, and expression was differentially influenced across training to both ISIs by the various postnatal ethanol dosages. CR peak amplitude was measured across the same 12 sessions. A significant treatment-group main effect was observed [F(5,20) = 6.3, p < .0001], as well as a significant ISI × session interaction [F(5,20) = 7.9, p < .0001]. No other main effect or interaction reached significance. Follow-up post hoc analyses indicated no group differences in CR amplitude across any sessions except 3 and 4, with the UC rats exhibiting enhanced CR amplitudes relative to the remaining 4 groups (data not shown). Finally, to ensure no between-group differences exist in startle reactivity to the tone CS, alpha responses across the 12 training sessions were examined via single-factor (treatment) ANOVA. No effect of treatment group was observed for alpha peak (p = .64) or timing of the peak response (p = .81).
Figure 3.

(A) Percentage of emitted conditioned responses (%CR; mean ± SE) over training to the short 280-msec ISI. Groups UC and SI produced more CRs than the 4E and 5E rats over sessions 2–3, and more CRs than 5E rats over sessions 4–6. (B) Averaged CR peak latencies (mean ± SE) during CS-alone trials over training to the short 280-msec ISI. Postnatal ethanol exposure resulted in dose-dependent increases in CR peak latencies. ($ = significantly different from 4E; * = significantly different from 5E)
Figure 4.

(A) Percentage of emitted conditioned responses (%CR; mean ± SE) over training to the long 880-msec ISI. (B) The averaged percent CR for each treatment group across sessions 7 to 12. Groups UC and SI produced significantly more CRs than groups 3E and 5E and group 4E generated significantly more CRs than group 5E. (C) The averaged CR peak latencies for each treatment group across sessions 7 to 12. (D) The averaged change in CR peak latency from sessions 1–6 (280-msec ISI) to sessions 7–12 (880-msec ISI). (+ = significantly different from 3E; * = significantly different from 5E)
Figure 3A illustrates CR learning curves over training to the short (280 msec) ISI. The 5E rats were severely impaired in their acquisition of the behavioral response relative to the remaining 4 groups. The repeated measures ANOVA revealed a significant treatment × session interaction [F(20,115) = 1.8, p < .05], and post hoc testing indicated significant increases in CR frequency across sessions 1 to 6 for all treatment groups, except group 5E (p = .69). A session-by-session analysis revealed no significant treatment-group differences in CR production for the first training session. Diminished response was seen in groups 4E and 5E across sessions 2 and 3 relative to groups UC and SI. Beginning on session 4, the 4E rats rebounded (i.e., no significant differences from controls) whereas the 5E rats continued to emit significantly fewer CRs. CR peak latency, averaged across sessions 1 to 6, was analyzed next based on CS-alone trials (Figure 3B). Postnatal ethanol exposure led to dose-dependent increases in CR peak latency [F(4,23) = 5.2, p < .005], with significantly longer latencies in the 4E and 5E rats compared to the UC and SI rats. Peak latencies in the 5E rats were also significantly longer than latencies in the 3E rats.
Postnatal ethanol exposure continued to affect CR production after transition to the 880-msec ISI (Figure 4A), as evidenced by a significant treatment effect [F(4,23) = 5.5, p < .005]. There was no effect of training session, however, suggesting that asymptotic CR performance was established in all groups prior to the ISI switch. Figure 4B illustrates the averaged percent CR for each treatment group across sessions 7 to 12. Groups UC, SI, and 4E generated significantly more blinks than group 5E, whereas the 2 control groups generated more blinks than group 3E. The averaged CR peak latency for sessions 7 to 12 is shown in Figure 4C. No significant group differences were observed (p = .95). Figure 4D shows the averaged change in CR peak latency from sessions 1–6 to sessions 7–12. Groups UC and SI demonstrated greater shifts (i.e., lengthening) in CR peak latencies than groups 3E, 4E, or 5E, though the difference only approached statistical significance [F(4,21) = 1.9, p = .15].
Unit Activity
Across each of the 12 training sessions, 1 to 3 units were acquired per subject in the IP and/or CA1, resulting, on average, in 9–18 recorded units per treatment group, per session, in each brain region. IP and CA1 unit activity were examined in blocks of 2 training sessions. Similar to the behavioral results, postnatal ethanol exerted a significant effect on neuronal activity during training to the short ISI in both the cerebellar IP [F(4,66) = 5.0, p < .001] and hippocampal subregion CA1 [F(4,40) = 5.5, p < .0005] (Figure 5). There was also a significant main effect of session in both brain regions, IP [F(2,132) = 13.5, p < .0001] and CA1 [F(2,80) = 7.9, p < .0001], indicating significant enhancement in IP and CA1 neuronal activity over training to the short ISI (Figure 5A).
Figure 5.

(A) Average standard scores for IP unit activity during training to the 280-msec ISI. The UC rats demonstrated the greatest amount of conditioning-related changes in excitatory unit activity over training. The significance threshold (dashed line) was crossed by the UC, SI, and 4E rats, but not the 3E or 5E rats. (B) Average standard scores for CA1 unit activity over training to the 280-msec ISI. The 2 control groups exhibited the highest activity levels, whereas 3E and 5E rats showed attenuated activity. The significance threshold (dashed line) was crossed by the UC and SI rats, but not by any of the ethanol-exposed rats. (! = significantly different from UC; # = significantly different from SI; + = significantly different from 3E; $ = significantly different from 4E; * = significantly different from 5E)
Significant conditioning-related enhancements in unit activity, as evidenced by standard scores (dashed line in Figures 5 and 6), were seen in the UC and SI rats by the end of session 2 (Figure 5A). In contrast, significantly enhanced unit activity was not seen in the 3E or 5E rats across sessions 1–6 with the short ISI, suggesting a slower-than-normal engagement of these neurons by the EBC procedure. Comparing across treatment groups, IP spiking in the UC rats was significantly higher than in the 5E rats over sessions 1–6, the 3E rats over sessions 1–4, and the 4E rats over sessions 3–4. Recall that the 3E rats did not display acquisition deficits over training to the short ISI. Unlike the 3E or 5E rats, the 4E rats did show significant increases in IP activity across sessions 3–6. Their activity rate was significantly elevated relative to 5E rats on sessions 1–2 and (along with the SI rats) on sessions 5–6. Analysis of CA1 recordings demonstrated significantly enhanced unit activation across sessions 1–4 in the UC rats and sessions 3–6 in the SI rats. Moreover, all 3 ethanol groups were significantly impaired compared to groups UC and SI across sessions 1–2 (Figure 5B). The SI animals continued to exhibit more CA1 activation across sessions 3–6 compared to the 3E, 4E, and 5E rats, whereas unit activity in UC rats was significantly enhanced relative to group 5E only. Finally, as seen in the IP, group 4E exhibited increased CA1 activation across sessions 5–6 compared to group 5E.
Figure 6.
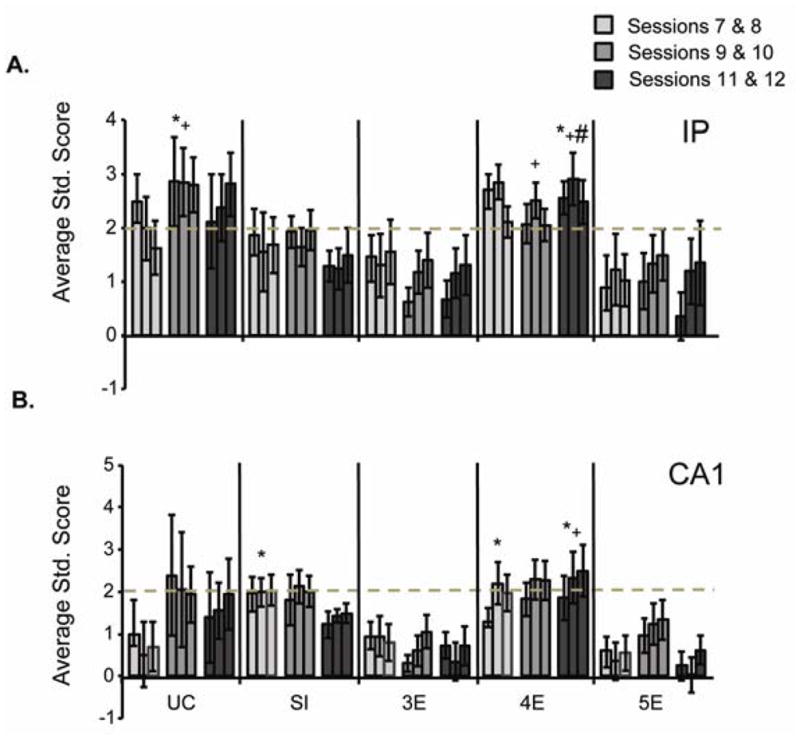
(A) Average standard scores for IP unit activity during training to the long 880-msec ISI. Three bins of ~293 msec are shown, with the first bin approximating the 280-msec ISI. IP activity was significantly enhanced (dashed line) in UC and 4E rats, and approached significance for the SI rats. IP unit activity was attenuated in groups 3E and 5E. (B) Average standard scores for CA1 unit activity over training to the long 880-msec ISI. CA1 activity was significantly enhanced (dashed line) in UC, SI, and 4E rats. CA1 unit activity was attenuated in groups 3E and 5E. (# = significantly different from SI; + = significantly different from 3E; * = significantly different from 5E)
Figure 6 illustrates IP and CA1 unit activity (3 bins, each ~293 msec) across sessions 7 to 12, when subjects were trained to the 880-msec ISI. Postnatal ethanol exerted a significant effect on both IP and CA1 neuronal responding [F(4,51) = 2.9, p < .05, and F(4,40) = 3.5, p < .05, respectively]. There was no significant effect of training session, however. As seen with the short ISI, the UC rats exhibited significant increases in conditioning-related unit activity across sessions 7–12 whereas unit activity in the SI rats exceeded significance across sessions 9–10 only. The 4E rats also showed significantly enhanced unit activity across sessions 7–12. Figure 6A shows enhanced IP unit activity in group UC relative to groups 3E and 5E over sessions 9–10. Group 4E had elevated IP activity relative to group 3E across sessions 9–10 and groups SI, 3E, and 5E across sessions 11–12. Thus, groups 3E and 5E generated significantly fewer CRs when trained to the 880-msec ISI and also displayed less excitatory IP unit activity. In Figure 6B, significant enhancement of the CA1 unit was seen in the UC, SI, and 4E rats. Comparing across groups, enhanced activity can be seen across sessions 7–8 in groups SI and 4E relative to group 5E. Over sessions 11–12, CA1 activity in the 4E rats remained significantly elevated compared to the 3E and 5E rats.
Discussion
Utilizing an ISI shift paradigm, binge-like PD 4–9 ethanol administration resulted in delay EBC acquisition deficits in 5E rats trained to the 280-msec ISI and reduced responses in 3E and 5E rats following the ISI switch to 880 msec (Figures 3A and 4A). The 4E and 5E rats also produced mis-timed CRs, with longer peak amplitude latencies than control subjects during training to the 280-msec ISI (Figure 3B). The shift in CR timing following the ISI shift was reduced in all ethanol-exposed rats, though the effect only approached significance. Multiple unit recordings in the cerebellar IP showed lower levels and slower-than- normal learning-dependent excitatory activity in the 3E and 5E rats (Figures 5 and 6). Hippocampal CA1 unit activity showed a similar pattern across treatment groups. Importantly, no differences between ethanol-exposed and control rats in their responsiveness to the aversive US or in their performance during explicitly unpaired training have been reported (Green et al., 2000; Lindquist, 2013; Lindquist et al., 2007; Tran et al., 2007). Nor were significant differences in startle reactivity to the tone CS found in the current study between the various treatment groups, in agreement with other reports (e.g., Stanton and Goodlett, 1998). The data indicate the EBC deficit observed in ethanol-exposed rats is not due to impairments in processing the CS or US signal. Rather, deficiencies in CR acquisition and timing are proposed to be the result of ethanol-induced alterations in CS-US associative processes within the brainstem-cerebellar EBC neural circuit (Green, 2004).
Behaviorally, peak BAC on PD 4 in ethanol-exposed rats was negatively correlated with the averaged CR percentage across training to the short ISI (Figure 2B). CR acquisition was most severely impaired in the 5E rats, which produced significantly fewer conditioned blinks across sessions 2 to 6 compared to control animals (Figure 3A). The moderate ethanol dose retarded CR acquisition across sessions 2 and 3 in the 4E rats, whereas the low dose of alcohol had no significant impact on learning in the 3E rats. Following the switch to the 880-msec ISI the CR rate did not significantly change for any treatment group across the remaining sessions, suggesting the response rate was near or at asymptote at the completion of training to the 280-msec ISI. Averaged across sessions 7 to 12, the 3E and 5E rats produced significantly fewer CRs than the SI and UC rats. Interestingly, the 4E rats were not impaired relative to controls and emitted significantly more conditioned blinks than the 5E rats (Figure 4B). CR peak latencies were also selectively lengthened in the 4E and 5E rats during training to the 280-msec ISI (Figure 3B) but not to the 880-msec ISI (Figure 4C). The results support our hypothesis that the dynamic interplay between the cortex and IP may be altered by early ethanol exposure, resulting in impaired learning and maladaptively timed conditioned blinks.
Unlike CR frequency, there were clear changes in CR topography following the ISI shift for the 2 control groups, with the peak amplitude latency shifting forward in time by approximately 100–200 msec (Figures 4C and 4D). This result is consistent with that reported by Hoehler and Thompson (1980), who found that the peak amplitude of the nictitating membrane response in rabbits moved toward the new US onset time when switched from a 250-msec to a 500-msec ISI. Relative to the two control groups, the ethanol-exposed rats displayed changes that were more modest in CR timing following the ISI shift. The CR peak latency increased by about 50–100 msec over sessions 7 to 12 (Figures 4C and 4D), although peak latencies in the 4E and 5E rats were already significantly longer than controls prior to the switch (Figure 3B). This latter point may partially explain the somewhat exaggerated increase in CR percentage across session 6 (last 280-msec ISI session) to session 7 (first 880-msec ISI session) in the 4E (~12%) and 5E rats (~19%). The remaining groups showed changes that were more modest in CR frequency. With the longer CS-US interval, the 4E and 5E rats simply had more time to emit long-latency blinks which would be captured and counted as CRs. The longer CR peak latencies could also overshadow any deficits in the ethanol rats’ ability to move the blinks’ peak amplitude forward in time following the ISI shift (Figure 4D).
On average, control animals displayed higher rates of learning, better CR timing, and (UC rats only) more IP excitatory unit activity than the ethanol rats. It is important to note, however, that the unit data are presented as averaged standard scores, with dashed lines indicating significant increases in unit activity (Figures 5 and 6). Although the SI animals do not show significant differences in unit activity from ethanol animals, a higher proportion of neurons did show significant activation across sessions 3–6 during training to the short ISI (Figure 5A). Unit activity in the 3E and 5E rats, on the other hand, was below threshold throughout training to both the short and long ISI, although the 3E rats showed normal learning rates across sessions 1 to 6. As proposed by Green et al. (2002a), the data suggest that fewer conditioning-related units became plastic during delay EBC with the short ISI in rats administered the low or high ethanol doses over PD 4–9.
The current data reveal an interesting characteristic of the dose-response function — namely, the 4E rats perform better and show higher rates of IP and CA1 learning-related unit activity than the 3E rats during training to the long ISI (Figures 4, 5, and 6). The increase in CR frequency of the 4E rats relative to 3E rats may be partially explained by their long-latency conditioned blinks. And yet, using an ISI discrimination task — in which one CS is followed by the US after a short (280 msec) ISI and a second CS is followed by the US after a long (880 msec) ISI — Brown et al. (2008) reported a similar pattern for CR expression to the long ISI in adult rats. Specifically, rats dosed with 4 g/kg/day across PD 4–9 displayed impaired learning relative to controls across the early training sessions (to both the 280-msec and 880-msec ISI). This was not observed during the later sessions to the long ISI when they showed significantly enhanced performance relative to rats dosed with 3 g/kg/day on sessions 6, 10, 11, and 12 (Figure 2, in Brown et al., 2008). The 3E but not 4E rats were significantly impaired in the current study when averaged across sessions 7 to 12, relative to controls, though the 2 groups themselves did not significantly differ (Figure 4A). Nonetheless, it remains an interesting possibility that genuine differences in CR expression to the long ISI exist between the 3E and 4E rats and are perhaps mirrored in functional and/or structural differences.
As discussed above, postnatal ethanol exposure induces cell loss within the cerebellar deep nuclei, including the IP (Green et al., 2002b; Tran et al., 2005; Young et al., 2010), and the estimated number of remaining neurons is significantly correlated to the EBC learning rate (Green et al., 2002b; Young et al., 2010). Unbiased cell counts of the IP have been done with rats that had received ethanol concentrations of 1.0, 2.0, 3.0, and 5.25 g/kg/day (Green et al., 2006; Green et al., 2002a; Tran et al., 2005; Young et al., 2010), resulting in a general trend toward more IP neuronal depletion with higher peak BACs. Postnatal ethanol exposure in the 4E rats (peak BAC ~275 mg/dL) would therefore be predicted to induce greater IP cell loss than in the 3E rats (peak BAC ~210 mg/dL). As seen in Figure 6A, however, the 4E rats (but not the 3E or 5E rats) displayed significant enhancement in IP unit activity over sessions 9 to 12. Neurons recorded during each session were treated as independent populations, which yields information related to the number of neurons engaged by the behavioral task but does not address changes in firing rate (Maren, 2005). The IP recordings suggest, then, that a greater proportion of neurons within the IP of 4E rats were responsive to the CS than the 3E rats — a point that is hard to reconcile if 4 g/kg/day of ethanol induced more IP cellular damage than 3 g/kg/day. Alternatively, CR deficits may not be tied solely to absolute IP numbers, but may reflect an imbalance in function between the IP and cerebellar cortex. In other words, the 4 g/kg/day of ethanol may have produced more uniform cell loss in the IP and cortex than the low (3 g/kg/day) or high (5 g/kg/day) alcohol doses. Considering the importance of cortical-interpositus interactions in CR performance and timing, particularly with long ISIs, such an imbalance in cell numbers could prove more disruptive than uniform cell loss.
While the exact role of the cerebellar cortex in classical eyeblink conditioning remains an issue of debate (Attwell et al., 2002; Freeman and Steinmetz, 2011; Green and Steinmetz, 2005; Hong and Optican, 2008; Koekkoek et al., 2003; Kotani et al., 2006; Wang and Liu, 2011; Yamazaki and Nagao, 2012), one suggestion is that it aids in the establishment of IP plasticity and/or modulates topography of the behavioral response (Steinmetz and Lindquist, 2009; Villarreal and Steinmetz, 2005). Plasticity at the parallel fiber–Purkinje cell synapse occurs as a result of co-activation of the 2 primary sensory inputs to the cerebellum, mossy fibers from granule cells and climbing fibers from the dorsal accessory olive (Thompson and Steinmetz, 2009). Purkinje cells in the anterior lobe (which inhibit IP neurons) are proposed to fire vigorously in the early portion of the CS period followed by a decrease in firing, owing to long-term depression (LTD) during the later portion of the CS period — regulating the amplitude and temporal accuracy of the behavioral response (Green and Steinmetz, 2005; Hesslow and Ivarsson, 1994; Medina et al., 2000). Both in vivo and in vitro analyses indicate that acute ethanol exposure can disrupt LTD at the parallel fiber-Purkinje cell synapse, with observed reductions in LTD (Belmeguenai et al., 2008) as well as a reversal of synaptic plasticity, such that LTD is converted into LTP (Servais et al., 2007). Ethanol-induced disruptions in LTD at the parallel fiber-Purkinje cell synapse could contribute to the impaired EBC and timing deficits observed in the present experiment, provided such disruptions are demonstrated to occur following postnatal ethanol administration (e.g., see Backman et al., 1998).
It has been proposed that the cerebellum and hippocampus might process different aspects of the conditioning task (Kim and Baxter, 2001). It has also been proposed that, early in training, the hippocampus associates the spatial and sensory features of the conditioning environment into a unitary representation (Rudy, 2009), which can then be associated with the US and used to gate or modulate the acquisition and expression of the conditioned blink (Lindquist, 2013; Penick and Solomon, 1991). Vogel et al. (2009) also hypothesized that delay EBC with long non-optimal ISIs (unlike optimal ISIs) may necessitate the convergence of stimulus information in the IP from other brain regions, including the cerebellar cortex or hippocampus, for robust learning-dependent synaptic plasticity to occur.
Consistent with previous studies (Berger et al., 1980; Berger and Thompson, 1978), CA1 recordings in UC and SI subjects did reveal, in response to the short ISI, significant learning-dependent increases in CA1 unit activity early in training (Figure 5B). That said, only group SI showed significant enhancement in unit activity compared to the 5E rats and it was limited to sessions 7–8. CS-mediated unit responses in the 3E and 5E rats did not cross the significance threshold over any of the 12 training sessions. CA1 unit activity in the 4E rats, in contrast, reached significance across sessions 5–6, and later, over sessions 9–12, mimicking the IP unit data discussed above. Thus, unit activity in the IP and CA1 was generally at levels consistent with diminished learning-dependent excitatory activity observed across all 12 training sessions in the 3E and 5E rats. Further research will be required to better understand how the diminished CA1 activity potentially contributes to impaired delay EBC.
In relation to our stated goals, we conclude that binge-like PD 4 to PD 9 ethanol exposure dose-dependently alters CR acquisition and timing in adult rats and that these effects are reflected in IP and CA1 neuronal activity. The 5E rats learned the most slowly and, with the 4E rats, produced the longest CR peak latencies over training to the short ISI. The low dose of alcohol selectively impaired CR performance to the long ISI only in 3E rats. Data from the 4E rats are more perplexing, considering their higher rate of CR production to the long ISI and enhanced IP and CA1 activation. This finding deserves more experimental attention to determine if the results are a sampling artifact or represent a true treatment group difference (cf., Brown et al., 2008). Following the ISI shift the CR peak latency in ethanol-exposed rats showed a trend toward less forward movement in time, relative to the 2 control groups. The lack of significant deficits might be partly explained, however, by the long-latency CRs generated by the 4E and 5E rats prior to the switch. The unit data generally support the behavioral results, indicating slower-than-normal increases in learning-dependent excitatory unit activity in both the IP and CA1 of 5E rats across all 12 sessions. The 3E rats, conversely, demonstrated normal CR acquisition to the 280-msec ISI but a lack of significant IP unit activity across the same period. Taken together, the results indicate that CR timing in eyeblink-conditioned subjects, particularly if different ISIs are used, may be a valuable measure for assessing ethanol-induced cerebellar dysfunction. This may aid in the development of clinical tools for the diagnosis of FASD individuals, including ARND children who exhibit a range of cognitive deficits but do not exhibit the gross physical dysmorphology typical of FAS (Jacobson et al., 2011; Jacobson et al., 2008).
Acknowledgments
This research was supported by National Institute on Alcohol Abuse and Alcoholism grant AA011945.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akase E, Alkon DL, Disterhoft JF. Hippocampal lesions impair memory of short-delay conditioned eye blink in rabbits. Behavioral Neuroscience. 1989;103:935–943. doi: 10.1037//0735-7044.103.5.935. [DOI] [PubMed] [Google Scholar]
- Attwell PJ, Ivarsson M, Millar L, Yeo CH. Cerebellar mechanisms in eyeblink conditioning. Annals of the New York Academy of Sciences. 2002;978:79–92. doi: 10.1111/j.1749-6632.2002.tb07557.x. [DOI] [PubMed] [Google Scholar]
- Backman C, West JR, Mahoney JC, Palmer MR. Electrophysiological characterization of cerebellar neurons from adult rats exposed to ethanol during development. Alcoholism, Clinical and Experimental Research. 1998;22:1137–1145. [PubMed] [Google Scholar]
- Bauer-Moffett C, Altman J. The effect of ethanol chronically administered to preweanling rats on cerebellar development: a morphological study. Brain Research. 1977;119:249–268. doi: 10.1016/0006-8993(77)90310-9. [DOI] [PubMed] [Google Scholar]
- Bayer SA, Altman J, Russo RJ, Zhang X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology. 1993;14:83–144. [PubMed] [Google Scholar]
- Belmeguenai A, Botta P, Weber JT, Carta M, De Ruiter M, De Zeeuw CI, Valenzuela CF, Hansel C. Alcohol impairs long-term depression at the cerebellar parallel fiber-Purkinje cell synapse. Journal of Neurophysiology. 2008;100:3167–3174. doi: 10.1152/jn.90384.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger TW, Laham RI, Thompson RF. Hippocampal unit-behavior correlations during classical conditioning. Brain Research. 1980;193:229–248. doi: 10.1016/0006-8993(80)90960-9. [DOI] [PubMed] [Google Scholar]
- Berger TW, Thompson RF. Neuronal plasticity in the limbic system during classical conditioning of the rabbit nictitating membrane response. I. The hippocampus. Brain Research. 1978;145:323–346. doi: 10.1016/0006-8993(78)90866-1. [DOI] [PubMed] [Google Scholar]
- Berthier NE, Moore JW. Activity of deep cerebellar nuclear cells during classical conditioning of nictitating membrane extension in rabbits. Experimental Brain Research. 1990;83:44–54. doi: 10.1007/BF00232192. [DOI] [PubMed] [Google Scholar]
- Bonthius DJ, West JR. Permanent neuronal deficits in rats exposed to alcohol during the brain growth spurt. Teratology. 1991;44:147–163. doi: 10.1002/tera.1420440203. [DOI] [PubMed] [Google Scholar]
- Brown KL, Burman MA, Duong HB, Stanton ME. Neonatal binge alcohol exposure produces dose dependent deficits in interstimulus interval discrimination eyeblink conditioning in juvenile rats. Brain Research. 2009;1248:162–175. doi: 10.1016/j.brainres.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KL, Calizo LH, Goodlett CR, Stanton ME. Neonatal alcohol exposure impairs acquisition of eyeblink conditioned responses during discrimination learning and reversal in weanling rats. Developmental Psychobiology. 2007;49:243–257. doi: 10.1002/dev.20178. [DOI] [PubMed] [Google Scholar]
- Brown KL, Calizo LH, Stanton ME. Dose-dependent deficits in dual interstimulus interval classical eyeblink conditioning tasks following neonatal binge alcohol exposure in rats. Alcoholism, Clinical and Experimental Research. 2008;32:277–293. doi: 10.1111/j.1530-0277.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- Chasnoff IJ, Wells AM, Telford E, Schmidt C, Messer G. Neurodevelopmental functioning in children with FAS, pFAS, and ARND. Journal of Developmental and Behavioral Pediatrics. 2010;31:192–201. doi: 10.1097/DBP.0b013e3181d5a4e2. [DOI] [PubMed] [Google Scholar]
- Christian KM, Thompson RF. Neural substrates of eyeblink conditioning: Acquisition and retention. Learning & Memory. 2003;11:427–455. doi: 10.1101/lm.59603. [DOI] [PubMed] [Google Scholar]
- Coleman SR, Gormezano I. Classical conditioning of the rabbit’s (Oryctolagus cuniculus) nictitating membrane response under symmetrical CS-US interval shifts. Journal of Comparative & Physiological Psychology. 1971;77:447–455. doi: 10.1037/h0031879. [DOI] [PubMed] [Google Scholar]
- Dalla C, Shors TJ. Sex differences in learning processes of classical and operant conditioning. Physiology & Behavior. 2009;97:229–238. doi: 10.1016/j.physbeh.2009.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman JH, Nicholson DA. Developmental changes in eye-blink conditioning and neuronal activity in the cerebellar interpositus nucleus. The Journal of Neuroscience. 2000;20:813–819. doi: 10.1523/JNEUROSCI.20-02-00813.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman JH, Steinmetz AB. Neural circuitry and plasticity mechanisms underlying delay eyeblink conditioning. Learning & Memory. 2011;18:666–677. doi: 10.1101/lm.2023011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey PW, Ross LE. Classical conditioning of the rabbit eyelid response as a function of interstimulus interval. Journal of Comparative Physiological Psychology. 1968;65:246–250. doi: 10.1037/h0025555. [DOI] [PubMed] [Google Scholar]
- Garcia KS, Steele PM, Mauk MD. Cerebellar cortex lesions prevent acquisition of conditioned eyelid responses. The Journal of Neuroscience. 1999;19:10940–10947. doi: 10.1523/JNEUROSCI.19-24-10940.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Mohapel J, Boehme F, Kainer L, Christie BR. Hippocampal cell loss and neurogenesis after fetal alcohol exposure: insights from different rodent models. Brain Research Reviews. 2010;64:283–303. doi: 10.1016/j.brainresrev.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Goodlett CR, Johnson TB. Neonatal binge ethanol exposure using intubation: timing and dose effects on place learning. Neurotoxicology & Teratology. 1997;19:435–446. doi: 10.1016/s0892-0362(97)00062-7. [DOI] [PubMed] [Google Scholar]
- Goodlett CR, Johnson TB. Temporal windows of vulnerability to alcohol during the third trimester equivalent: Why “knowing when” matters. In: Hanningan JH, Spear LP, Spear NE, Goodlett CR, editors. Alcohol and alcoholism: Effects on brain and development. Hillsdale, NJ: Lawrence Erlbaum; 1999. pp. 59–91. [Google Scholar]
- Gormezano I, Kehoe EJ, Marshall BS. Twenty years to classical conditioning with the rabbit. Progress in Psychobiology and Physiological Psychology. 1983;10:197–275. [Google Scholar]
- Green JT. The effects of ethanol on the developing cerebellum and eyeblink classical conditioning. Cerebellum. 2004;3:178–187. doi: 10.1080/14734220410017338. [DOI] [PubMed] [Google Scholar]
- Green JT, Arenos JD, Dillon CJ. The effects of moderate neonatal ethanol exposure on eyeblink conditioning and deep cerebellar nuclei neuron numbers in the rat. Alcohol. 2006;39:135–150. doi: 10.1016/j.alcohol.2006.09.002. [DOI] [PubMed] [Google Scholar]
- Green JT, Johnson TB, Goodlett CR, Steinmetz JE. Eyeblink classical conditioning and interpositus nucleus activity are disrupted in adult rats exposed to ethanol as neonates. Learning & Memory. 2002a;9:304–320. doi: 10.1101/lm.47602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JT, Rogers RF, Goodlett CR, Steinmetz JE. Impairment in eyeblink classical conditioning in adult rats exposed to ethanol as neonates. Alcoholism: Clinical and Experimental Research. 2000;24:438–447. [PubMed] [Google Scholar]
- Green JT, Steinmetz JE. Purkinje cell activity in the cerebellar anterior lobe after rabbit eyeblink conditioning. Learning & Memory. 2005;12:260–269. doi: 10.1101/lm.89505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JT, Tran T, Steinmetz JE, Goodlett CR. Neonatal ethanol produces cerebellar deep nuclear cell loss and correlated disruption of eyeblink conditioning in adult rats. Brain Research. 2002b;956:302–311. doi: 10.1016/s0006-8993(02)03561-8. [DOI] [PubMed] [Google Scholar]
- Hamilton GF, Whitcher LT, Klintsova AY. Postnatal binge-like alcohol exposure decreases dendritic complexity while increasing the density of mature spines in mPFC Layer II/III pyramidal neurons. Synapse. 2010;64:127–126. doi: 10.1002/syn.20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamre KM, West JR. The effects of the timing of ethanol exposure during the brain growth spurt on the number of cerebellar Purkinje and granule cell nuclear profiles. Alcoholism: Clinical and Experimental Research. 1993;17:610–622. doi: 10.1111/j.1530-0277.1993.tb00808.x. [DOI] [PubMed] [Google Scholar]
- Hesslow G, Ivarsson M. Suppression of cerebellar Purkinje cells during conditioned responses in ferrets. Neuroreport. 1994;5:649–652. doi: 10.1097/00001756-199401000-00030. [DOI] [PubMed] [Google Scholar]
- Hoehler FK, Thompson RF. Effect of the interstimulus (CS-UCS) interval on hippocampal unit activity during classical conditioning of the nictitating membrane response of the rabbit (Oryctolagus cuniculus) Journal of Comparative and Physiological Psychology. 1980;94:201–215. doi: 10.1037/h0077658. [DOI] [PubMed] [Google Scholar]
- Hong S, Optican LM. Interaction between Purkinje cells and inhibitory interneurons may create adjustable output waveforms to generate timed cerebellar output. PLoS One. 2008;3:e2770. doi: 10.1371/journal.pone.0002770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idrus NM, Napper RM. Acute and long-term Purkinje cell loss following a single ethanol binge during the early third trimester equivalent in the rat. Alcoholism, Clinical and Experimental Research. 2012;36:1365–1373. doi: 10.1111/j.1530-0277.2012.01743.x. [DOI] [PubMed] [Google Scholar]
- Jacobson SW, Stanton ME, Dodge NC, Pienaar M, Fuller DS, Molteno CD, Meintjes EM, Hoyme HE, Robinson LK, Khaole N, et al. Impaired delay and trace eyeblink conditioning in school-age children with fetal alcohol syndrome. Alcoholism, Clinical and Experimental Research. 2011;35:250–264. doi: 10.1111/j.1530-0277.2010.01341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SW, Stanton ME, Molteno CD, Burden MJ, Fuller DS, Hoyme HE, Robinson LK, Khaole N, Jacobson JL. Impaired eyeblink conditioning in children with fetal alcohol syndrome. Alcoholism, Clinical and Experimental Research. 2008;32:365–372. doi: 10.1111/j.1530-0277.2007.00585.x. [DOI] [PubMed] [Google Scholar]
- Jones KL, Smith DW. The fetal alcohol syndrome. Teratology. 1975;12:1–10. doi: 10.1002/tera.1420120102. [DOI] [PubMed] [Google Scholar]
- Jones KL, Smith DW, Ulleland CN, Streissguth AP. Pattern of malformation in offspring of chronic alcoholic mothers. Lancet. 1973;1:1267–1271. doi: 10.1016/s0140-6736(73)91291-9. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Baxter MG. Multiple brain-memory systems: the whole does not equal the sum of its parts. Trends in Neuroscience. 2001;24:324–330. doi: 10.1016/s0166-2236(00)01818-x. [DOI] [PubMed] [Google Scholar]
- King DA, Tracy J. DataMunch: A Matlab m-file collection for the analysis of trial-based spike and behavioral data. 1999. [Google Scholar]
- Kodituwakku PW. Defining the behavioral phenotype in children with fetal alcohol spectrum disorders: a review. Neuroscience Biobehavioral Reviews. 2007;31:192–201. doi: 10.1016/j.neubiorev.2006.06.020. [DOI] [PubMed] [Google Scholar]
- Koekkoek SK, Hulscher HC, Dortland BR, Hensbroek RA, Elgersma Y, Ruigrok TJ, De Zeeuw CI. Cerebellar LTD and learning-dependent timing of conditioned eyelid responses. Science. 2003;301:1736–1739. doi: 10.1126/science.1088383. [DOI] [PubMed] [Google Scholar]
- Kotani S, Kawahara S, Kirino Y. Purkinje cell activity during classical eyeblink conditioning in decerebrate guinea pigs. Brain Research. 2006;1068:70–81. doi: 10.1016/j.brainres.2005.10.090. [DOI] [PubMed] [Google Scholar]
- Lee T, Kim JJ. Differential effects of cerebellar, amygdalar, and hippocampal lesions on classical eyeblink conditioning in rats. The Journal of Neuroscience. 2004;24:3242–3250. doi: 10.1523/JNEUROSCI.5382-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist DH. Hippocampal-dependent Pavlovian conditioning in adult rats exposed to binge-like doses of ethanol as neonates. Behavioral Brain Research. 2013;242:191–199. doi: 10.1016/j.bbr.2012.12.030. [DOI] [PubMed] [Google Scholar]
- Lindquist DH, Sokoloff G, Steinmetz JE. Ethanol-exposed neonatal rats are impaired as adults in classical eyeblink conditioning at multiple unconditioned stimulus intensities. Brain Research. 2007;1150:155–166. doi: 10.1016/j.brainres.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist DH, Vogel RW, Steinmetz JE. Associative and non-associative blinking in classically conditioned adult rats. Physiology & Behavior. 2009;96:399–411. doi: 10.1016/j.physbeh.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livy DJ, Miller EK, Maier SE, West JR. Fetal alcohol exposure and temporal vulnerability: effects of binge-like alcohol exposure on the developing rat hippocampus. Neurotoxicology & Teratology. 2003;25:447–458. doi: 10.1016/s0892-0362(03)00030-8. [DOI] [PubMed] [Google Scholar]
- Maren S. Central and basolateral amygdala neurons crash the aversive conditioning party: Theoretical comment on Rorick-Kehn and Steinmetz (2005) Behavioral Neuroscience. 2005;119:1406–1410. doi: 10.1037/0735-7044.119.5.1406. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Crocker N, Nguyen TT. Fetal alcohol spectrum disorders: neuropsychological and behavioral features. Neuropsychological Review. 2011;21:81–101. doi: 10.1007/s11065-011-9167-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PA, Gossage JP. Estimating the prevalence of fetal alcohol syndrome: A summary. Alcohol Research & Health. 2001;25:159–167. [PMC free article] [PubMed] [Google Scholar]
- May PA, Gossage JP, Kalberg WO, Robinson LK, Buckley D, Manning M, Hoyme HE. Prevalence and epidemiologic characteristics of FASD from various research methods with an emphasis on recent in-school studies. Developmental Disabilities Research Reviews. 2009;15:176–192. doi: 10.1002/ddrr.68. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Thompson RF. Neuronal responses of the rabbit cerebellum during acquisition and performance of a classically conditioned nictitating membrane-eyelid response. The Journal of Neuroscience. 1984;4:2811–2822. doi: 10.1523/JNEUROSCI.04-11-02811.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina JF, Nores WL, Ohyama T, Mauk MD. Mechanisms of cerebellar learning suggested by eyelid conditioning. Current Opinion in Neurobiology. 2000;10:717–724. doi: 10.1016/s0959-4388(00)00154-9. [DOI] [PubMed] [Google Scholar]
- Murawski NJ, Jablonski SA, Brown KL, Stanton ME. Effects of neonatal alcohol dose and exposure window on long delay and trace eyeblink conditioning in juvenile rats. Behavioural Brain Research. 2013;236:307–318. doi: 10.1016/j.bbr.2012.08.025. [DOI] [PubMed] [Google Scholar]
- Murawski NJ, Stanton ME. Effects of dose and period of neonatal alcohol exposure on the context preexposure facilitation effect. Alcoholism, Clinical and Experimental Research. 2011;35:1160–1170. doi: 10.1111/j.1530-0277.2011.01449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niccols A. Fetal alcohol syndrome and the developing socio-emotional brain. Brain and Cognition. 2007;65:135–142. doi: 10.1016/j.bandc.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Penick S, Solomon PR. Hippocampus, context, and conditioning. Behavioral Neuroscience. 1991;105:611–617. doi: 10.1037//0735-7044.105.5.611. [DOI] [PubMed] [Google Scholar]
- Perrett SP, Ruiz BP, Mauk MD. Cerebellar cortex lesions disrupt learning-dependent timing of conditioned eyelid responses. The Journal of Neuroscience. 1993;13:1708–1718. doi: 10.1523/JNEUROSCI.13-04-01708.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce DR, Serbus DC, Light KE. Intragastric intubation of alcohol during postnatal development of rats results in selective cell loss in the cerebellum. Alcoholism: Clinical and Experimental Research. 1993;17:1275–1280. doi: 10.1111/j.1530-0277.1993.tb05241.x. [DOI] [PubMed] [Google Scholar]
- Port RL, Mikhail AA, Patterson MM. Differential effects of hippocampectomy on classically conditioned rabbit nictitating membrane response related to interstimulus interval. Behavioral Neuroscience. 1985;99:200–208. doi: 10.1037//0735-7044.99.2.200. [DOI] [PubMed] [Google Scholar]
- Port RL, Mikhail AA, Patterson MM. Failure of hippocampectomy to facilitate classical conditioning at an optimal interstimulus interval is not due to a “ceiling effect”. Behavioral Neuroscience. 1986;100:161–164. doi: 10.1037//0735-7044.100.2.161. [DOI] [PubMed] [Google Scholar]
- Richardson DP, Byrnes ML, Brien JF, Reynolds JN, Dringenberg HC. Impaired acquisition in the water maze and hippocampal long-term potentiation after chronic prenatal ethanol exposure in the guinea-pig. European Journal of Neuroscience. 2002;16:1593–1598. doi: 10.1046/j.1460-9568.2002.02214.x. [DOI] [PubMed] [Google Scholar]
- Riley EP, McGee CL. Fetal alcohol spectrum disorders: an overview with emphasis on changes in brain and behavior. Experimental Biology and Medicine. 2005;2306:357–365. doi: 10.1177/15353702-0323006-03. [DOI] [PubMed] [Google Scholar]
- Rogers RF, Britton GB, Steinmetz JE. Learning-related interpositus activity is conserved across species as studied during eyeblink conditioning in the rat. Brain Research. 2001;905:171–177. doi: 10.1016/s0006-8993(01)02532-x. [DOI] [PubMed] [Google Scholar]
- Rudy JW. Context representations, context functions, and the parahippocampal-hippocampal system. Learning & Memory. 2009;16:573–585. doi: 10.1101/lm.1494409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson PD, Streissguth AP, Bookstein FL, Little RE, Clarren SK, Dehaene P, Hanson JW, Graham JM., Jr Incidence of fetal alcohol syndrome and prevalence of alcohol-related neurodevelopmental disorder. Teratology. 1997;56:317–326. doi: 10.1002/(SICI)1096-9926(199711)56:5<317::AID-TERA5>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Schaefer GB, Deere D. Recognition, diagnosis and treatment of fetal alcohol syndrome. The Journal of the Arkansas Medical Society. 2011;108:38–40. [PubMed] [Google Scholar]
- Schmaltz LW, Theios J. Acquisition and extinction of a classically conditioned response in hippocampectomized rabbits (Oryctolagus cuniculus) Journal of Comparative & Physiological Psychology. 1972;79:328–333. doi: 10.1037/h0032531. [DOI] [PubMed] [Google Scholar]
- Schneiderman N. Interstimulus interval function of the nictitating membrane response of the rabbit under delay versus trace conditioning. Journal of Comparative & Physiological Psychology. 1966;62:397–402. [Google Scholar]
- Servais L, Hourez R, Bearzatto B, Gall D, Schiffmann SN, Cheron G. Purkinje cell dysfunction and alteration of long-term synaptic plasticity in fetal alcohol syndrome. Proceedings of the National Academy of Science, USA. 2007;104:9858–9863. doi: 10.1073/pnas.0607037104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MC. CS-US interval and US intensity in classical conditioning of the rabbit’s nictitating membrane response. Journal of Comparative & Physiological Psychology. 1968;66:679–687. doi: 10.1037/h0026550. [DOI] [PubMed] [Google Scholar]
- Stanton ME, Goodlett CR. Neonatal ethanol exposure impairs eyeblink conditioning in weanling rats. Alcoholism: Clinical and Experimental Research. 1998;22:270–275. [PubMed] [Google Scholar]
- Steinmetz JE, Lindquist DH. Neuronal basis of learning. In: Berntson GG, Cacioppo JT, editors. Handbook of Neuroscience for the Behavioral Sciences. Hoboken, NJ: John Wiley & Sons; 2009. pp. 507–527. [Google Scholar]
- Stratton K, Howe C, Battaglia F. Fetal alcohol syndrome: Diagnosis, epidemiology, prevention, and treatment. Washington, DC: National Academy Press; 1996. [Google Scholar]
- Streissguth AP. Offspring effects of prenatal alcohol exposure from birth to 25 years: The Seattle Prospective Longitudinal Study. Clincal Psychology in Medical Settings. 2007;14:81–101. [Google Scholar]
- Thomas JD, Tran TD. Choline supplementation mitigates trace, but not delay, eyeblink conditioning deficits in rats exposed to alcohol during development. Hippocampus. 2012;22:619–630. doi: 10.1002/hipo.20925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RF. In search of memory traces. Annual Review of Psychology. 2005;56:1–23. doi: 10.1146/annurev.psych.56.091103.070239. [DOI] [PubMed] [Google Scholar]
- Thompson RF, Steinmetz JE. The role of the cerebellum in classical conditioning of discrete behavioral responses. Neuroscience. 2009;162:732–755. doi: 10.1016/j.neuroscience.2009.01.041. [DOI] [PubMed] [Google Scholar]
- Tran TD, Jackson HD, Horn KH, Goodlett CR. Vitamin E does not protect against neonatal ethanol-induced cerebellar damage or deficits in eyeblink classical conditioning in rats. Alcoholism: Clinical and Experimental Research. 2005;29:117–129. doi: 10.1097/01.alc.0000150004.53870.e1. [DOI] [PubMed] [Google Scholar]
- Tran TD, Kelly SJ. Critical periods for ethanol-induced cell loss in the hippocampal formation. Neurotoxicology & Teratology. 2003;25:519–528. doi: 10.1016/s0892-0362(03)00074-6. [DOI] [PubMed] [Google Scholar]
- Tran TD, Stanton ME, Goodlett CR. Binge-like ethanol exposure during the early postnatal period impairs eyeblink conditioning at short and long CS-US intervals in rats. Developmental Psychobiology. 2007;49:589–605. doi: 10.1002/dev.20226. [DOI] [PubMed] [Google Scholar]
- Valenzuela CF, Morton RA, Diaz MR, Topper L. Does moderate drinking harm the fetal brain? Insights from animal models. Trends in Neuroscience. 2012;35:284–292. doi: 10.1016/j.tins.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal RP, Steinmetz JE. Neuroscience and learning: lessons from studying the involvement of a region of cerebellar cortex in eyeblink classical conditioning. Journal of the Experimental Analysis of Behavior. 2005;84:631–652. doi: 10.1901/jeab.2005.96-04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel RW, Amundson JC, Lindquist DH, Steinmetz JE. Eyeblink conditioning during an interstimulus interval switch in rabbits (Oryctolagus cuniculus) using picrotoxin to disrupt cerebellar cortical input to the interpositus nucleus. Behavioral Neuroscience. 2009;123:62–74. doi: 10.1037/a0014082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Liu SQ. Neural circuit and its functional roles in cerebellar cortex. Neurosci Bull. 2011;27:173–184. doi: 10.1007/s12264-011-1044-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JR, Goodlett CR, Bonthius DJ, Pierce DR. Manipulating peak blood alcohol concentrations in neonatal rats: review of an animal model for alcohol-related developmental effects. Neurotoxicology. 1989;10:347–366. [PubMed] [Google Scholar]
- West JR, Hamre KM, Pierce DR. Delay in brain growth induced by alcohol in artificially reared rat pups. Alcohol. 1984;1:83–95. doi: 10.1016/0741-8329(84)90101-0. [DOI] [PubMed] [Google Scholar]
- Woodruff-Pak DS, Disterhoft JF. Where is the trace in trace conditioning? Trends in Neuroscience. 2008;31:105–112. doi: 10.1016/j.tins.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Nagao S. A computational mechanism for unified gain and timing control in the cerebellum. PLoS One. 2012;7:e33319. doi: 10.1371/journal.pone.0033319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young BW, Sengelaub DR, Steinmetz JE. MK-801 administration during neonatal ethanol withdrawal attenuates interpositus cell loss and juvenile eyeblink conditioning deficits. Alcohol. 2010;44:359–369. doi: 10.1016/j.alcohol.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]