

Abstract
Background
The dsRNA-activated protein kinase (PKR) phosphorylates the α subunit of eukaryotic translation initiation factor 2 (eIF2α), a global regulator of protein synthesis in mammals. In addition, PKR activates several signal transduction pathways including STAT3 and AKT. PKR is activated by a number of inflammatory stimuli that are induced in the inflamed intestine. In this study we intended to determine the role of PKR in colonic epithelial cells during experimental colitis in mice.
Methods
Age- and sex-matched PKR+/+,+/− and PKR−/− littermate mice were reconstituted with wildtype bone marrow cells and subjected to dextran sodium sulfate (DSS)-induced colitis.
Results
PKR−/− mice displayed more severe clinical and histological manifestations upon DSS colitis compared with their PKR+/+,+/− litter-mates. In response to DSS colitis, the colonic epithelial cells of PKR−/− mice exhibited impaired activation of the unfolded protein response (UPR) signaling, including eIF2α phosphorylation, endoplasmic reticulum (ER) chaperone response, and ER-associated degradation (ERAD) components, as well as antioxidative stress response. In addition, the phosphorylation of STAT3 and AKT, which are protective against epithelial cell death and colonic inflammation, was also impaired in the colonic epithelial cells of PKR−/− mice upon DSS colitis.
Conclusions
These data demonstrate that PKR is a physiologically relevant transducer of inflammatory response signaling in colonic epithelial cells. PKR may promote the homeostasis and survival of intestinal epithelial cells (IECs) through eIF2α-mediated UPR activation, as well as the activation of STAT3 and AKT pathways. In the absence of PKR, the survival and proliferation of IECs was impaired, thus exacerbating intestinal inflammation.
Keywords: PKR, DSS colitis, UPR, prosurvival signaling
The endoplasmic reticulum (ER) in eukaryotic cells is the site for the folding, assembly, modification, and maturation of polypeptides destined for secretion, membranes, and intracellular organelles. Protein folding in the ER is exquisitely sensitive to multiple environmental alterations and cellular disturbances including ER Ca2+ depletion, oxidative stress, and inflammatory stimuli. Disrupted ER protein folding homeostasis leads to accumulation of unfolded/misfolded proteins in the ER lumen, a condition termed ER stress. To cope with this cellular stress and restore ER homeostasis, eukaryotic cells have evolved the unfolded protein response (UPR). In mammalian cells, three ER-localized stress sensors signal the UPR: inositol-requiring kinase 1 (IRE1), activating transcription factor 6 (ATF6), and PKR-like ER kinase (PERK). The UPR protein sensors are activated in response to ER stress and signal downstream pathways that control transcriptional, translational, and posttranslational processes. During ER stress, the ER chaperone BiP releases from the luminal domain of PERK, which is activated through dimerization and trans-autophosphorylation. Activated PERK then phosphorylates the α subunit of eukaryotic translation initiation factor 2 (eIF2α) to reduce global protein synthesis. At the same time, phosphorylated eIF2α induces the translation of mRNA encoding activating transcription factor 4 (ATF4), which subsequently transactivates genes encoding other UPR-associated transcription factors, ER protein-folding machinery, and components of ER-associated protein degradation (ERAD), and promotes cellular homeostasis by inducing the biosynthesis and transport of amino acids, and spurring the antioxidative stress response.1
In addition to PERK, three cytosolic eIF2α kinases exist in mammals, including the double-stranded RNA-activated protein kinase (PKR).2 PKR was originally identified and characterized as a pathogen sensor and mediator of the interferon response to protect the host from viral infection. PKR-mediated eIF2α phosphorylation is a strategy to shut down viral protein synthesis in host cells.3 However, in contexts other than viral infection induction of downstream UPR signaling and inhibition of global translation through the same eIF2α phosphorylation by PKR activation may be crucial for ER homeostasis in response to various environmental alterations.2 Indeed, a number of reports have demonstrated that PKR does signal protection to ER stress. In addition to dsRNA, PKR can be activated by other inflammatory signals including lipopolysaccharide (LPS) and tumor necrosis factor alpha (TNF-α), as well as oxidative stress. When activated, PKR induces or acts in conjugation with a number of signal transduction pathways, including p38 mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinases (JNK), signal transducer and activator of transcription 3 (STAT3), and Akt/Protein kinase B.4 Therefore, PKR functions as a regulatory hub that coordinates inflammatory response signaling, pathogen sensing, and UPR signaling in mammalian cells.
Inflammatory bowel diseases (IBDs) represent a set of inflammatory conditions in the gastrointestinal tract. Although IBDs were discovered dozens of years ago, the etiology of IBD is still unclear. Recently, some studies linked ER stress and UPR signaling to the pathogenesis of IBD by showing that patients with active Crohn's disease and ulcerative colitis exhibit signs of ER stress in their ileal and/or colonic epithelium.5–8 Genes that regulate ER homeostasis and the UPR including XBP1 have been identified to associate with Crohn's disease and/or ulcerative colitis. The murine and human intestinal epithelium contains Paneth and goblet cells, which have massive secretory functions and play essential roles in intestinal homeostasis and host defense. Intestinal epithelial cell (IEC)-specific deletion of XBP1 caused Paneth cell death and spontaneous inflammation in murine ileum.8 A previous study suggested that PKR may regulate the differentiation of a rat intestinal epithelial cell line by promoting the activity of alkaline phosphatase.9 However, the role of PKR in intestinal inflammation is unclear. Here we show that the deletion of PKR increases sensitivity to dextran sodium sulfate (DSS)-induced colitis in mice, due to reduced induction of adaptive UPR signaling and prosurvival signaling including the activation of STAT3 in colonic epithelial cells during inflammation.
MATERIALS AND METHODS
Mice
All mice were housed on 12/12 hour light/dark cycles at the Unit for Laboratory Animal Medicine (ULAM) at the University of Michigan Medical Center with free access to water and standard rodent chow. All animal care and procedures were conducted following the protocols and guidelines approved by the University of Michigan Committee on the Use and Care of Animals (UCUCA).
Generation of Bone Marrow Chimeras
Eight-week-old PKR−/− and PKR+/+,+/− littermate mice10 were lethally irradiated with 950 rad ionizing irradiation. After 2 hours, 5 × 106 bone marrow cells isolated from the tibias and femurs of wildtype mice were injected into the lethally irradiated recipient mice through the tail vein. After transplantation, mice were treated with a 6-week course of antibiotics in their drinking water and allowed to recover for another 2 weeks prior to treatment with DSS to induce colitis.
To generate chimeric mice with deletion of PKR in their hematopoietic compartment, 8-week-old wildtype mice on a C57BL/6 background (Jackson Laboratory, Bar Harbor, ME) were lethally irradiated with 950 rad ionizing irradiation. After 2 hours these mice received a tail vein injection of 5 × 106 bone marrow cells isolated from PKR−/− or PKR+/+ littermate mice. After transplantation mice were treated with a 6-week course of antibiotics in their drinking water and allowed to recover for another 2 weeks prior to experiment.
DSS-induced Colitis
For DSS-induced acute colitis, PKR−/− and PKR+/+,+/−mice reconstituted with wildtype bone marrow cells as well as wildtype mice reconstituted with PKR−/− or PKR+/+ bone marrow cells received 3% (w/v) DSS (MW 36,000–50,000; MP Biomedicals, Solon, OH) in drinking water for 7 days. Body weight change and rectal bleeding were monitored daily. After DSS administration the mice were euthanized, colon length was measured, and colon sections and colonic epithelial cells were isolated for histological and biochemical analyses.
Isolation of Colonic Epithelial Cells
The entire colons were cut open longitudinally, then feces was removed by washing with ice-cold phosphate-buffered saline (PBS). Colons were then cut into 2–3-mm pieces and incubated in Ca2+, Mg2+-free PBS buffer containing 10 mM EDTA in a 50 mL conical tube at 4°C for 1 hour with gentle rotation. Then the tubes were rigorously shaken to elute the epithelium from colon sections. The supernatant was removed and sieved through a cell strainer (500 μm; Fisher-brand, Pittsburgh, PA). The flow-through was centrifuged; the resulting cell pellet was washed twice in ice-cold PBS and snap-frozen in liquid nitrogen for protein and RNA extraction. The purity of isolated colonic epithelial cells was confirmed by FACS and quantitative reverse-transcription polymerase chain reaction (Q-RT-PCR). Trypan blue staining confirmed the presence of >85% viable epithelial cells after the 2-hour isolation procedure.
Quantitative Real-time PCR
RNA from isolated colonic epithelial cells and IEC-6 cells were extracted by using RNeasy kit (Qiagen, Chatsworth, CA); RNA from 5 mm distal colons was isolated using TRIzol reagent (Invitrogen, La Jolla, CA). Q-RT-PCR was performed as previously described.11 Q-RT-PCR results were normalized to the levels of 18S rRNA or the mRNA encoding glyceraldehyde 3-phosphate dehydrogenase. Primer sequences are listed in Table 1.
TABLE 1.
Sequence of Primers
| Primer Name | Oligo Sequence (5′ to 3′) |
|---|---|
| mouse IL-1β (f) | CAACCAACAAGTGATATTCTCCATG |
| mouse IL-1β (r) | GATCCACACTCTCCAGCTGCA |
| mouse TNFα (f) | CCCTCACACTCAGATCATCTTCT |
| mouse TNFα (r) | GCTACGACGTGGGCTACAG |
| mouse iNOS (f) | CAGCTGGGCTGTACAAACCTT |
| mouse iNOS (r) | CATTGGAAGTGAAGCGTTTCG |
| mouse BiP (f) | TCATCGGACGCACTTGGA |
| mouse BiP (r) | CAACCACCTTGAATGGCAAGA |
| mouse GRP94 (f) | AATAGAAAGAATGCTTCGCC |
| mouse GRP94 (r) | TCTTCAGGCTCTTCTTCTGG |
| mouse ERp72 (f) | AGTCAAGGTGGTGGTGGGAAAG |
| mouse ERp72 (r) | TGGGAGCAAAATAGATGGTAGGG |
| mouse ERdj4 (f) | CCCCAGTGTCAAACTGTACCAG |
| mouse ERdj4 (r) | AGCGTTTCCAATTTTCCATAAATT |
| mouse P58IPK (f) | TCCTGGTGGACCTGCAGTACG |
| mouse P58IPK (r) | CTGCGAGTAATTTCTTCCCC |
| mouse calreticulum (f) | GAGTGGCTTGGACCAGAAGG |
| mouse calreticulum (r) | GGACCGCAGATGTCCGG |
| mouse Ero1α (f) | GCATTGAAGAAGGTGAGCAA |
| mouse Ero1α (r) | ATCATGCTTGGTCCACTGAA |
| mouse Ero1β (f) | GGGCCAAGTCATTAAAGGAA |
| mouse Ero1β (r) | TTTATCGCACCCAACACAGT |
| mouse PDI (f) | CAAGATCAAGCCCCACCTGAT |
| mouse PDI (r) | AGTTCGCCCCAACCAGTACTT |
| mouse XBP1 (f) | AAGAACACGCTTGGGAATGG |
| mouse XBP1 (r) | ACTCCCCTTGGCCTCCAC |
| mouse XBP1s (f) | GAGTCCGCAGCAGGTG |
| mouse XBP1s (r) | GTGTCAGAGTCCATGGGA |
| mouse ATF4 (f) | ATGGCCGGCTATGGATGAT |
| mouse ATF4 (r) | CGAAGTCAAACTCTTTCAGATCCATT |
| mouse Chop (f) | GTCCCTAGCTTGGCTGACAGA |
| mouse Chop (r) | TGGAGAGCGAGGGCTTTG |
| mouse Gadd34 (f) | CCCGAGATTCCTCTAAAAGC |
| mouse Gadd34 (r) | CCAGACAGCAAGGAAATGG |
| mouse ATF6α (f) | CTTCCTCCAGTTGCTCCATC |
| mouse ATF6α (r) | CAACTCCTCAGGAACGTGCT |
| mouse Herpud1 (f) | AGCAGCCGGACAACTCTAAT |
| mouse Herpud1 (r) | CTTGGAAAGTCTGCTGGACA |
| mouse Wfs1 (f) | CCATCAACATGCTCCCGTTC |
| mouse Wfs1 (r) | GGGTAGGCCTCGCCAT |
| mouse Nrf2 (f) | ACATCCTTTGGAGGCAAGAC |
| mouse Nrf2 (r) | GCCTTCTCCTGTTCCTTCTG |
| mouse GAPDH (f) | TTCAACGGCACAGTCAAGG |
| mouse GAPDH (r) | CATGGACTGTGGTCATGAG |
Western Blotting
The isolated colonic epithelial cells were lysed in RIPA buffer for protein extraction. Protein content was measured by Protein Assay (Bio-Rad, Hercules, CA), and protein samples were analyzed by 10%–20% reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS/PAGE) (Tris-HCl precast gels, Bio-Rad) and were detected with the following antibodies: anti-GRP94/BiP (StressGen, Vancouver, BC, Canada), anti-phosphor-PERK (Cell Signaling Technology, Beverly, MA), anti-ATF4 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-CHOP (Santa Cruz Biotechnology), anti-XBP1 (Santa Cruz Biotechnology), anti-LC3 (Novus Biologicals, Littleton, CO), and anti-glyceraldehyde 3-phosphate dehydrogenase (Millipore, Bedford, MA), and anti-phosphor-STAT3 (Cell Signaling Technology). After overnight incubation of primary antibodies at 4° C the membranes were washed and then incubated with secondary horseradish peroxidase (HRP)-coupled antibodies (GE Healthcare, Milwaukee, WI) and developed with a chemiluminescent detection system (GE Healthcare).
Immunohistochemical Staining
Colonic tissues were fixed overnight in 10% formalin, embedded in paraffin, and cut into 5-lm sections. Paraffin sections were rehydrated and heat-induced antigen retrieval was then performed in 10 mM sodium citrate, 0.05% Tween-20, pH 6.0, for 10 minutes. Primary antibodies: anti-BiP antibody (Santa Cruz Biotechnology) at a dilution of 1:200; anti-ATF4 antibody (LifeSpan BioSciences, Seattle, WA) at a dilution of 1:200; anti-phosphor-STAT3 antibody (LifeSpan BioSciences) at a dilution of 1:200. Hematoxylin was performed for counterstaining.
Histological Scoring
Colonic tissues were fixed overnight in 10% formalin, embedded in paraffin, and cut into 5-μm sections for hematoxylin and eosin (H&E) staining using a standard protocol. H&E-stained sections were scored in a blinded manner by a licensed animal pathologist for the amount of tissue damage and inflammatory cell infiltration as previously described.12 The damaged area involved was scored on a scale of 0 to 4: 0 = no involvement, 1 ≥ 25%, 2 ≥ 50%, 3 ≥ 75%, 4 ≥ 100%. Inflammatory cell infiltration were scored on a scale of 0 to 3: 0 = occasional inflammatory cells in the lamina propria, 1 = increased numbers of inflammatory cells in the lamina propria, 2 = confluence inflammatory cells, extending into the submucosa, and 3 = transmural extension of the infiltrate.
Statistical Analysis
All data are presented as means ± SEM. Statistical significance of the difference between groups was analyzed using Student's t-test. P < 0.05 was considered significant.
RESULTS
Mice with PKR Deletion in Nonhematopoietic Cells Are More Sensitive to DSS-induced Colitis
In the absence of inflammatory insults, the colon of PKR−/− mice is morphologically indistinguishable from that of their wildtype littermates. To analyze a possible requirement for PKR in IECs to prevent IBD, lethally irradiated age- and sex-matched PKR+/+,+/− and PKR−/− littermate mice were reconstituted with wildtype bone marrow and then fed 3% DSS in the drinking water for 7 days to induce colitis. RT-PCR demonstrated that the level of Pkr mRNA in colonic epithelial cells of PKR−/− mice was less than 4% of that in PKR+/+ or PKR+/− littermate mice 8 weeks after bone marrow replacement (Fig. 1A), indicating that the intestinal epithelium was not repopulated by wildtype stem cells from the bone marrow transplantation. PKR−/− mice showed more severe clinical manifestations, as demonstrated by body weight loss, rectal bleeding, and colon shortening upon DSS challenge (Fig. 1B–D). In addition, PKR−/− mice displayed significantly higher levels of damaged area in the epithelium and inflammatory cell infiltration in both the proximal and distal colon (Fig. 2A,B). Consistent with the histological observations, the expression of proinflammatory genes Il-1β, Tnf-α, and Inos were dramatically induced in the colon of PKR−/− mice upon DSS colitis compared with their wildtype and heterozygous littermates (Fig. 3). In contrast, wildtype mice reconstituted with PKR−/− bone marrow cells showed similar body weight loss upon DSS colitis as compared with wildtype mice reconstituted with PKR+/+ bone marrow cells (Fig. 1E), indicating that the deletion of PKR in hematopoietic cells does not alter the sensitivity to DSS-induced colitis.
FIGURE 1.

PKR−/− mice are more sensitive to DSS-induced colitis. PKR−/− and PKR+/+,+/− littermates were reconstituted with wildtype bone marrow cells. After 8 weeks the mice were fed 3% DSS in drinking water for 7 days; their body weight and rectal bleeding were monitored daily. (A) Colonic epithelial cells isolated from PKR−/− mice after bone marrow transplantation show very low levels of Pkr mRNA. (B) PKR−/− mice show more significant body weight loss during DSS colitis. (C) PKR−/− mice show more severe rectal bleeding during DSS colitis. (D) PKR−/− mice show more severe colon shortening after 7-day of DSS administration. PKR+/+,+/− : n = 13; PKR−/− : n = 11; *P < 0.05, **P < 0.01. (E) Two-month-old C57BL/6J wildtype mice were reconstituted with PKR+/+ or PKR−/− bone marrow cells. After 8 weeks the mice were fed 3% DSS in drinking water for 5 days followed by 2 days of free water; their body weight was monitored daily. PKR+/+ : n = 8; PKR−/−: n = 7.
FIGURE 2.

PKR−/− mice show more severe histological manifestation after 7 days of DSS administration. PKR−/− and PKR+/+,+/− littermates reconstituted with wildtype bone marrow cells were fed 3% DSS in drinking water for 7 days. (A) After DSS administration the colons were isolated and fixed for H&E staining. Representative images are shown (100×). (B) Histological scores including damage area involved and inflammatory cell infiltration taken from PKR−/− and PKR+/+,+/− littermates with DSS colitis. PKR+/+,+/− : n = 8; PKR−/− : n = 10; *P < 0.05, **P < 0.01.
FIGURE 3.
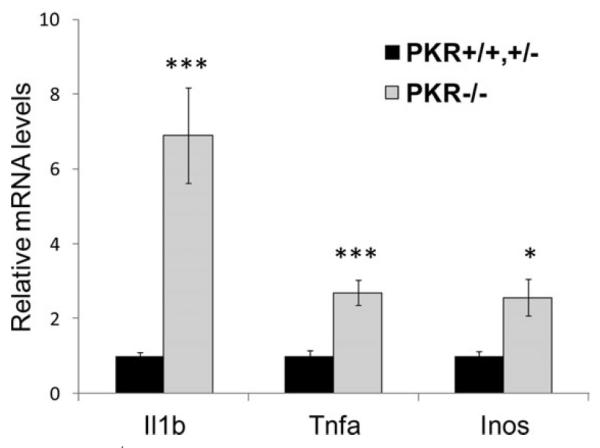
PKR−/− mice show hyperactivated inflammatory response in the colon upon DSS administration. PKR−/− and PKR+/+,+/− litter-mates with wildtype bone marrow cells were fed 3% DSS in drinking water for 7 days. After DSS administration the mice were euthanized and colonic epithelial cells were collected for RNA extraction, cDNA synthesis, and Q-RT-PCR. The mRNA levels were normalized to the expression of Gapdh. PKR+/+,+/− : n = 6; PKR−/− : n = 5; *P < 0.05, **P < 0.01, ***P < 0.001.
UPR Induction Is Impaired in Colonic Epithelial Cells of PKR−/− Mice
Our previous studies demonstrated that UPR molecules, e.g., BiP and ATF4, are induced in colonic epithelium in response to DSS colitis (unpublished data). Given the role of PKR as an inducer of UPR signaling, we then examined whether UPR activation is impaired in PKR−/− colonic epithelial cells during inflammation. Western blotting demonstrated that colonic epithelial cells of PKR−/− mice exhibited reduced levels of eIF2α phosphorylation and its downstream target ATF4 compared with their wildtype littermate mice. The induction of the ER chaperones BiP and GRP94 was also impaired at both the protein and RNA levels (Fig. 4A,C). In addition to ER chaperones, the induction of a number of UPR pathways were compromised in PKR−/− mice upon DSS colitis, including the UPR transcription factors XBP1s and ATF6, ER protein folding machinery ERp72, Ero1α, and PDI, ERAD components Herpud1 and Wfs1, and the antioxidative stress response nuclear factor-erythroid 2-related factor 2 (Nrf2).
FIGURE 4.

Activation of the UPR signaling and prosurvival pathways is impaired in PKR−/− colonic epithelial cells upon DSS colitis. PKR−/−and PKR+/+,+/− littermates with wildtype bone marrow cells were fed 3% DSS in drinking water for 6 days. (A) After DSS administration the mice were euthanized and the colonic epithelial cells were isolated for protein extraction and immunoblotting. (B) After DSS administration the mice were euthanized and the colonic epithelial cells were isolated for RNA extraction, cDNA synthesis, and Q-RT-PCR. The mRNA levels were normalized to the expression of Gapdh. (C) After DSS administration the mice were euthanized and the colons were isolated, fixed, and paraffin-embedded for immunohistochemical staining of BiP, ATF4, and phospho-STAT3 (200× or 400×). PKR+/+,+/− : n = 6; PKR−/− : n = 6; *P < 0.05, **P < 0.01.
Previous studies demonstrated that these adaptive UPR signaling pathways are essential during intestinal inflammation. The UPR-induced chaperone response in colonic epithelium has been shown to protect against DSS colitis in mice.13 Loss of Nrf2 increases susceptibility to DSS colitis, probably a consequence of enhanced oxidative damage and proinflammatory responses in the colonic mucosa.14 Therefore, PKR may protect colonic epithelial cells during inflammation through the induction of UPR signaling. In the absence of PKR, UPR signaling was impaired and may exacerbate ER stress and induce apoptosis. Consistently, the cleavage of caspase-3 was highly activated in colonic epithelial cells of PKR−/− mice after DSS challenge (Fig. 4A).
Prosurvival Signaling Is Compromised in the Absence of PKR During DSS Colitis
Given that PKR is an upstream inducer of STAT3 and Akt phosphorylation,4 we next examined whether the activation of these two signaling molecules was impaired in colonic epithelium of PKR−/− mice. Western blotting demonstrated that colonic epithelial cells isolated from PKR−/− mice challenged with DSS exhibited reduced phosphorylation of STAT3 and Akt compared with their wild-type littermate mice (Fig. 4A). Consistently, the nuclear staining of phosphor-STAT3 in the epithelial cells was diminished in the colon of PKR−/− mice (Fig. 4C).
DISCUSSION
In this study we demonstrate that PKR is a physiologically relevant activator of UPR signaling in colonic epithelium upon inflammation. In response to DSS colitis, PKR promoted homeostasis and survival of IECs through eIF2α-mediated UPR signaling. PKR−/− IECs showed reduced phosphorylation of eIF2α upon colitis, consistent with the diminished eIF2α phosphorylation observed in PKR−/− mouse embryo fibroblasts (MEFs) in response to inflammatory stimuli including IFNα/β.15 Reduced eIF2α phosphorylation in PKR−/− IECs upon colitis impaired the activation of UPR, e.g., transcription factors, chaperone response, ERAD components, and antioxidative stress response. This is consistent with our observations that mice expressing a nonphosphorylatable eIF2α in IECs are susceptible to DSS colitis and display diminished UPR induction in their colonic epithelial cells upon colitis (unpublished data). The UPR-activated transcription factors XBP1, ATF6α, and ATF4 are essential transactivators of UPR gene induction in response to ER stress. Mice deficient in XBP1 in the intestinal epithelium were sensitive to DSS colitis, although the underlying mechanism is not yet clear.8 ATF6α is activated upon ER stress by sequential cleavage by proteases S1P and S2P, yielding an active ATF6α p50 that functions as a potent activator of a number of ER chaperones including BiP, GRP94, and P58IPK. The hypomorphic mutation of S1P-encoding gene Mbtps1 impaired ATF6α-dependent induction of chaperone response, and increased susceptibility to experiment colitis in mice.13 ATF4 is induced at both the transcriptional and translational levels during ER stress and functions as an essential global UPR transactivator.2 In addition, ATF4 is activated by oxidative stress and plays an important role in the antioxidative stress response.16 Although there is little information about the role of ATF4 in IBD, it is possible that ATF4 is protective against ER stress and oxidative stress in IECs during intestinal inflammation. The importance of UPR-induced ER chaperones in DSS colitis is directly supported by our study using a murine model deficient in P58IPK, a heat-shock 40-kDa protein that normally resides in association with the ER chaperone BiP in the ER lumen and promotes proper protein folding.17P58IPK−/− mice are highly susceptible to DSS colitis, probably due to unresolved ER stress and selective activation of proapoptotic UPR signaling in colonic IECs during inflammation (unpublished data). There are few studies examining the role of ERAD pathways in IBD. However, given the function of ERAD in eliminating unfolded/misfolded protein and alleviating ER stress, it is possible that ERAD also impacts IEC function during intestinal inflammation. Therefore, the compromised induction of adaptive UPR pathways may exacerbate the dysfunction of colonic IECs in PKR−/− mice upon DSS challenge, and consequently worsen the symptoms of colitis.
PKR has been shown to physically interact with STAT3 and induce the phosphorylation at Tyr705 and Ser727, which is required for DNA binding and the transcriptional activation function of STAT3 upon PDGF stimulation.18 STAT3 is a pleiotropic transcription factor with crucial roles in a variety of cellular processes including proliferation and differentiation. Previous studies demonstrated that STAT3 activity is increased by phosphorylation in IECs during colonic inflammation, and transactivates the genes responsible for the stress response, apoptosis, and wound healing in IECs. IEC-specific deletion of STAT3 increased the susceptibility to DSS colitis.19 Similarly, the pleiotropic protein kinase AKT functions to inhibit IEC apoptosis and promote wound healing during intestinal inflammation.20 Therefore, compromised activation of STAT3 and AKT in PKR−/− mice may impair function and survival of IECs, thus promoting intestinal inflammation upon DSS challenge.
Several previous studies have linked PKR to apoptotic cell death. In NIH3T3 and COS-1 cells, TNF-α induces apoptosis through PKR-mediated eIF2α phosphorylation.21PKR−/− MEFs were resistant to apoptosis upon challenge with dsRNA, TNF-α, and LPS.22 Another study showed that dsRNA-activated PKR induced the expression of proapoptotic molecules including Fas and Bax in 3T3 L1 cells.23 In macrophages, cholesterol/7-ketocholesterol induced oxidative stress and activated the PKR-CHOP pathway that was required for the downstream apoptotic response.24 However, all of these studies were performed in vitro and the cells analyzed were very different from colonic epithelial cells at both the cell biological and physiological levels. Therefore, it seems the protective role of PKR in colonic epithelial cells may be both cell-type and/or disease-specific.
ACKNOWLEDGMENTS
The authors thank Dr. Bryan Williams of the Cleveland Clinic, Cleveland, OH, for providing PKR−/− mice, Dr. J. Erby Wilkinson of the Unit for Laboratory Animal Medicine, University of Michigan, Ann Arbor, MI, for histology scoring, Dr. Mary Davis at the University of Michigan Comprehensive Cancer Center, and Dr. Michael Shaw at the Department of Pathology, University of Michigan for help with murine bone marrow transplant, University of Michigan Comprehensive Cancer Center Tissue Core for histology, and Jenna Rousseau and Joseph Burzynski of the University of Michigan for technical support of biochemical analyses.
Funded by National Institutes of Health (NIH) grants P01 HL057346, R37 DK042394, R01 DK088227 and R01 HL052173 (to R.J.K.).
REFERENCES
- 1.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 3.Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol. 2005;6:318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- 4.García MA, Meurs EF, Esteban M. The dsRNA protein kinase PKR: virus and cell control. Biochimie. 2007;89:799–811. doi: 10.1016/j.biochi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Shkoda A, Ruiz PA, Daniel H, et al. Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: impact on chronic inflammation. Gastroenterology. 2007;132:190–207. doi: 10.1053/j.gastro.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 6.Hu S, Ciancio MJ, Lahav M, et al. Translational inhibition of colonic epithelial heat shock proteins by IFN-gamma and TNF-alpha in intestinal inflammation. Gastroenterology. 2007;133:1893–1904. doi: 10.1053/j.gastro.2007.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heazlewood CK, Cook MC, Eri R, et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008;5:e54. doi: 10.1371/journal.pmed.0050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaser A, Lee AH, Franke A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sato N, Morimoto H, Baba R, et al. Functional expression of double-stranded RNA-dependent protein kinase in rat intestinal epithelial cells. J Cell Biochem. 2010;110:104–111. doi: 10.1002/jcb.22513. [DOI] [PubMed] [Google Scholar]
- 10.Yang YL, Reis LF, Pavlovic J, et al. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J. 1995;14:6095–6106. doi: 10.1002/j.1460-2075.1995.tb00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Back SH, Scheuner D, Han J, et al. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 2009;10:13–26. doi: 10.1016/j.cmet.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Otsuka M, Kang YJ, Ren J, et al. Distinct effects of p38alpha deletion in myeloid lineage and gut epithelia in mouse models of inflammatory bowel disease. Gastroenterology. 2010;138:1255–1265. 1265.e1–9. doi: 10.1053/j.gastro.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brandl K, Rutschmann S, Li X, et al. Enhanced sensitivity to DSS colitis caused by a hypomorphic Mbtps1 mutation disrupting the ATF6-driven unfolded protein response. Proc Natl Acad Sci U S A. 2009;106:3300–3305. doi: 10.1073/pnas.0813036106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khor TO, Huang MT, Kwon KH, et al. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res. 2006;66:11580–11584. doi: 10.1158/0008-5472.CAN-06-3562. [DOI] [PubMed] [Google Scholar]
- 15.Baltzis D, Li S, Koromilas AE. Functional characterization of pkr gene products expressed in cells from mice with a targeted deletion of the N terminus or C terminus domain of PKR. J Biol Chem. 2002;277:38364–38372. doi: 10.1074/jbc.M203564200. [DOI] [PubMed] [Google Scholar]
- 16.Harding HP, Zhang Y, Zeng H, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 17.Rutkowski DT, Kang SW, Goodman AG, et al. The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol Biol Cell. 2007;18:3681–3691. doi: 10.1091/mbc.E07-03-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deb A, Zamanian-Daryoush M, Xu Z, et al. Protein kinase PKR is required for platelet-derived growth factor signaling of c-fos gene expression via Erks and Stat3. EMBO J. 2001;20:2487–2496. doi: 10.1093/emboj/20.10.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pickert G, Neufert C, Leppkes M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koch S, Nava P, Addis C, et al. The wnt antagonist dkk1 regulates intestinal epithelial homeostasis and wound repair. Gastroenterology. 2011;141:259–268.e8. doi: 10.1053/j.gastro.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Srivastava SP, Kumar KU, Kaufman RJ. Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J Biol Chem. 1998;273:2416–2423. doi: 10.1074/jbc.273.4.2416. [DOI] [PubMed] [Google Scholar]
- 22.Der SD, Yang YL, Weissmann C, et al. A double-stranded RNA-activated protein kinase-dependent pathway mediating stress-induced apoptosis. Proc Natl Acad Sci U S A. 1997;94:3279–3283. doi: 10.1073/pnas.94.7.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balachandran S, Kim CN, Yeh WC, et al. Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signaling. EMBO J. 1998;17:6888–6902. doi: 10.1093/emboj/17.23.6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li G, Scull C, Ozcan L, et al. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol. 2010;191:1113–1125. doi: 10.1083/jcb.201006121. [DOI] [PMC free article] [PubMed] [Google Scholar]