

Abstract
Background
Bilastine is a novel second-generation H1 antihistamine, which has not shown sedative or cardiotoxic effects in clinical trials and in post-marketing experience so far, developed for the symptomatic treatment of allergic rhinoconjunctivitis and urticaria. It has recently been granted marketing authorization for these therapeutic indications in adults and adolescents at a once-daily oral dose of 20 mg in several European countries.
Objective
This study was conducted to determine the pharmacokinetics of bilastine at a single oral dose of 20 mg in renally impaired subjects. The need for a dose adjustment in patients with renal insufficiency was assessed by comparing the exposure to bilastine in these subjects with the estimated exposure of a dose corresponding to the safety margin.
Methods
The study was an open-label, single-dose, parallel-group study of the pharmacokinetics and safety of a single dose of bilastine. The study was conducted as an in-patient setting at a clinical pharmacology facility. A total of 24 male or female subjects aged 18–80 years were to be enrolled in four groups of six subjects each. The groups were as follows: (1) healthy [glomerular filtration rate (GFR) >80 mL/min/1.73 m2]; (2) mild renal insufficiency (GFR 50–80 mL/min/1.73 m2); (3) moderate renal insufficiency (GFR 30–50 mL/min/1.73 m2); and (4) severe renal insufficiency (GFR ≤30 mL/min/1.73 m2). A single 20 mg bilastine tablet was administered in a fasted state. Blood and urine samples were collected from pre-dose up to 72 h post-dose for bilastine pharmacokinetic analysis. Pharmacokinetic results were summarized using appropriate descriptive statistics.
Results
There was a clear trend of increasing area under the plasma concentration–time curve (AUC) and maximum plasma concentration (Cmax) through the groups 1–4. The mean AUC from time zero to infinity (AUC∞) ranged from 737.4 to 1708.5 ng·h/mL in healthy subjects and severely impaired subjects, respectively. No significant differences among groups in median time to reach Cmax (tmax) or in the mean terminal disposition rate constants for bilastine were found. Renal and plasma clearance paralleled GFR. In all groups of renally impaired subjects the corresponding 90 % confidence interval of both AUC∞ and AUC from time zero to time of last measurable plasma concentration (AUClast) were not within the 0.8–1.25 interval, indicating that bioequivalence between groups could not be demonstrated. The majority of bilastine was excreted within the first 12 h, and elimination was essentially complete by 72 h.
Conclusion
An oral dose of bilastine (20 mg) was well-tolerated in renal insufficiency, despite the increase in exposure. The oral plasma clearance to renal clearance ratio [(CLP/F)/CLR] was approximately equal in the different groups, suggesting that renal excretion was the main elimination route for bilastine, and no alternative elimination routes were used even in severe renal insufficiency. Although exposure to bilastine was higher in renally impaired subjects, it remained well within the safety margins, thus allowing the conclusion that a 20-mg daily dose can be safely administered to subjects with different degrees of renal insufficiency without the need for dose adjustments.
Introduction
Bilastine, 2-[4-(2-(4-(1-(2-ethoxyethyl)-1H-benzimidazol-2-yl)piperidin-1-yl) ethyl) phenyl]-2-methylpropionic acid, is a novel second-generation H1 antihistamine, which has not shown sedative or cardiotoxic effects in clinical trials and in post-marketing experience so far, developed for the symptomatic treatment of allergic rhinoconjunctivitis and urticaria [1].
Bilastine has recently been granted marketing authorization for adults and adolescents (12 years and over) in most European countries. The approved dose is 20 mg once a day for symptomatic treatment of allergic rhinoconjunctivitis (seasonal and perennial) and urticaria.
Pharmacologic studies have shown bilastine to be highly selective for the histamine H1 receptor both in vitro and in vivo [2, 3]. Binding studies performed in animal tissues with a high density of H1 receptors showed that bilastine has a moderate to high affinity for H1 receptors, with values three times higher than those of cetirizine and five times higher than those of fexofenadine. These studies have also revealed that at a high concentration, bilastine does not show affinity for the 30 other receptors that have been assessed (including muscarinic receptors) nor for the other histamine receptor subtypes H2, H3, and H4. Bilastine binds specifically and selectively to the H1 receptor [2, 3].
Absorption of bilastine is fast and proportional to dose, with its oral bioavailability being reduced by food and fruit juices [1, 4]. There is no evidence of sex or age effects on bilastine pharmacokinetics [5]. A pharmacokinetic/pharmacodynamic model based on data from 310 healthy volunteers suggested that bilastine pharmacokinetics follow a two-compartmental model with first-order absorption and elimination [6]. Safety margins were calculated from this model. No accumulation pattern was shown for bilastine after repeated dosing in a 14-day pharmacokinetic study of escalating daily doses from 10 to 100 mg [7]. Plasma protein binding has also been shown to be independent of plasma concentration (unpublished observation), and therefore to be a non-saturable process.
Exposure and accumulation of bilastine are modified when simultaneously administered with some membrane transport protein inhibitors. In particular, bioavailability of bilastine is significantly increased when co-administered with P-glycoprotein (P-gp) inhibitors and decreased when co-administered with organic anion transporting polypeptide (OATP) inhibitors [4, 8]. Bilastine is not a substrate of human breast cancer resistance protein (BCRP), organic anion transporter 1 and 3 (OAT1, OAT3), and organic cation transporter 2 (OCT2), and thus is not a potential “victim” of inhibitors of these transporters. Moreover, bilastine is not expected to cause drug–drug interactions because of inhibition (“perpetrator”) of the transport mediated by P-gp, multi-drug resistance protein 2 (MRP2), bile salt export pump (BSEP), OATP2B1, OCT1, Na-taurocholate cotransporting protein (NTCP), BCRP, OAT1-, OAT3-, OCT2-, OATP1B1-, or OATP1B3 [9]. Further, bilastine is not metabolized and does not interact significantly, either as an inhibitor or inducer, with the cytochrome P450 (CYP) enzyme system, suggesting a low propensity for drug–drug interactions involving this metabolic pathway [10].
The corrected QT (QTc) interval prolongations observed with combined treatments were equal to or lower than those with the CYP/P-gp inhibitors alone, suggesting bilastine may be safely co-administered with a CYP/P-gp inhibitor [11, 12].
No sedative or psychomotor effects were detected at doses up to 40 mg of bilastine. No significant anticholinergic effect was found at any bilastine dose tested [13, 14]. Clinical studies, including a 12-month long-term safety study with more than 500 (adult and adolescent) patients, have shown bilastine to be well-tolerated with no related serious adverse events (AEs) and no differences with respect to placebo up to doses twice the therapeutic dose [15–19].
According to the Note for Guidance on the evaluation of the pharmacokinetics of medicinal products in subjects with impaired renal function, the purpose of this study was to determine the pharmacokinetics of bilastine at a single oral dose of 20 mg in healthy and renally impaired subjects [20]. The need for dose adjustments in patients with renal impairment was assessed by comparing the exposure to bilastine in these subjects with the exposure of a dose corresponding to the estimated safety margin.
Methods
Study Design and Population
This study was conducted in accordance with the provisions of the Declaration of Helsinki and its amendments, US FDA and European Medicines Agency (EMA) guidelines, and the guidelines of the International Conference on Harmonization (ICH) and Good Clinical Practice (GCP) that were in place at the time of the study.
Male and female volunteers were enrolled in this open-label, single-dose, parallel-group study, undertaken at the facilities of Clinical Pharmacology of Miami, Inc., Miami, FL, USA, between September 2008 and April 2009.
The protocol and informed consent form were reviewed and approved by The Independent Investigational Review Board, Plantation, FL, USA, prior to the screening or enrolment of any study participants. All subjects gave their informed, written consent prior to undergoing any study-related procedures.
A bilastine 20 mg single oral dose was administered to four groups (n = 6 each) of subjects either healthy or with various degrees of renal insufficiency. Subjects were assigned to groups according to their glomerular filtration rate (GFR) during initial screening assessed by iothalamate clearance, as follows:
Group 1: healthy (GFR >80 mL/min/1.73 m2)
Group 2: mild renal insufficiency (GFR 50–80 mL/min/1.73 m2)
Group 3: moderate renal insufficiency (GFR 30–50 mL/min/1.73 m2)
Group 4: severe renal insufficiency (GFR ≤30 mL/min/1.73 m2)
It is recommended that renal function in pharmacokinetic studies is determined by measuring GFR using accurate and well established methods [20]. Therefore, the iothalamate method was used to obtain a highly accurate estimate of GFR because it is based on only passive glomerular filtration of an exogenous substance entirely excreted through the kidney [21]. Categories of renal insufficiency according to GFR values were based on the definition provided in the regulatory guidance in place at the time the study was conducted.
All participants and his/her partner were either infertile or willing to use an approved method of double-barrier contraception (hormonal plus barrier or barrier plus barrier) from the time of dose administration and for 1 month thereafter.
Healthy subjects with a similar distribution of age, bodyweight, and sex as the subjects with renal insufficiency (variability within ±10 %) were enrolled in the study after completion of the three groups of renal insufficiency. Healthy subjects were to be at least 18 years old with no evidence of significant organic or psychiatric illness. On clinical examination their systolic and diastolic blood pressure, heart rate, temperature, electrocardiogram (ECG), hematology, and biochemistry parameters were to be all within the normal range.
Eligible renally impaired subjects were to be between 45 and 110 kg, aged 18–80 years, and with a stable renal function for 3 months before inclusion in the study. Renally impaired subjects with end-stage renal disease on hemodialysis or peritoneal dialysis, with bilirubin levels >1.5 mg/dL and/or increment of aspartate aminotransferase (AST) or alanine aminotransferase (ALT) levels two times higher than the maximum limit of normality, with a platelet count lower than 75,000/mm3, or with serum potassium higher than 6 mEq/L were excluded.
Subjects treated with P-gp/CYP inhibitors or inducers within 30 days were excluded. Ingestion of grapefruit juice, cranberry juice, apple juice, Seville orange juice, any well-known P-gp inhibitor in the previous 7 days, or Saint John’s wort in the previous 15 days also excluded subjects from enrolment.
Screening evaluations were performed within 21 days prior to admission to the clinic. Blood and urine samples for the iothalamate GFR test were obtained at least 7 days prior to the anticipated clinic admission, and results were required to be obtained prior to the clinic visit. Subjects were accommodated in the clinic from the evening prior to dosing until released by the investigator subsequent to obtaining the 72-h blood sample and urine collection.
On the day of dosing, all subjects received a single oral 20-mg dose of bilastine with 200 mL of water by mouth following a fast of 8–10 h. Subjects continued fasting for 2 h after dosing and were provided with a standard diet at all other times.
Bilastine tablets were manufactured by FAES FARMA S.A., Leioa (Vizcaya), Spain, according to Good Manufacturing Practice guidelines and were supplied in blister strips containing 4 × 20 mg tablets of bilastine. The drug was securely stored during the study.
Pharmacokinetic Assessments
Blood samples were collected pre-dose and at 0.25, 0.50, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 8.0, 12, 24, 36, 48, and 72 h after bilastine administration. Approximately 7 mL of blood for each sample were collected into sodium heparin Vacutainer® tubes. Plasma was separated by centrifugation at 4–8 °C for 10 min at 1,500×g, and stored frozen within 1 h of collection.
Urine samples were collected at 0–12, 12–24, 24–48, and 48–72 h after bilastine administration. Urine was refrigerated during the collection intervals. Total urine volumes were measured for each time period and 25 mL aliquots were stored at −20 °C.
Bilastine was extracted and quantified using a liquid chromatography with tandem mass spectrometer detection (LC/MS/MS) system of analysis. The lower and upper limits of quantification were 0.20 and 402.40 ng/mL, respectively, for bilastine in human plasma and 5.00 and 5,000 ng/mL, respectively, for bilastine in human urine [22]. Analysis was undertaken by Anapharm Inc., Québec City, QC, Canada.
The following pharmacokinetic parameters were derived for bilastine from individual plasma or urine concentrations: maximum plasma concentration (Cmax); time to reach Cmax (tmax); last measurable plasma concentration (Clast); area under the plasma concentration–time curve (AUC) from time zero to time of last measurable plasma concentration (AUClast), calculated according to the trapezoidal rule; AUC from time zero to infinity (AUC∞), calculated using the equation: AUC∞ = AUClast + Clast/ke, where ke is the elimination rate constant from the central compartment; terminal disposition rate constant (λz), calculated using linear regression on the terminal portion of the ln-concentration versus time curve; terminal elimination half-life (t½β), calculated as 0.693/λz; cumulative amount of unchanged drug excreted in urine (Ae), calculated as total amount of bilastine excreted unchanged in the urine over the entire period of sample collection; apparent total body clearance from plasma after oral administration (CLP/F, where F indicates bioavailability), calculated as dose/AUC∞; renal clearance (CLR), calculated as Ae/AUC over a defined period of time; apparent volume of distribution during terminal phase after non-intravenous administration (Vz/F), calculated as (CLp/F)/ke.
Safety Evaluations
Physical examinations were conducted on admission and at the end of the study. Vital signs (temperature, respiratory rate, supine/standing pulse rate, systolic and diastolic blood pressure, and bodyweight) were recorded daily. Twelve-lead ECG recordings were made pre-dose, and 1 and 24 h post-dose. Heart rate, PR interval, QRS interval, and QT interval were measured. QTc intervals were calculated using Fridericia’s (QTcF) and Bazett’s (QTcB) correction formulas [23].
Routine hematology, biochemistry, and urinalysis were conducted at screening, pre-dose, and 72 h post-dose.
AEs were collected for the duration of participation of the subject in the study and followed until satisfactory resolution. Information concerning AEs was solicited by the investigator by questioning subjects about any changes in their health condition, and as spontaneously reported by the subject. An AE was defined as any unfavorable and unintended sign (including an abnormal laboratory finding), symptom or disease, temporally associated with the use of the medicinal product, regardless of its nature, intensity, seriousness, or presumed relationship (causality) to the product or experimental procedure used. The term AE also applied to laboratory findings or results of other diagnostic procedures that were considered to be clinically relevant (e.g., required unscheduled diagnostic procedures or treatment measures or withdrawal from the study), unless directly related with the underlying disease of the participant (i.e., severe renal impairment).
Statistical Analyses
Pharmacokinetic and safety analyses were conducted by PharmaNet Specialized Pharmaceutical Services using SAS, release 8.2, Cary, NC, USA. Pharmacokinetic parameters were calculated using WinNonlin Enterprise, version 5.2 (Pharsight Corporation, Cary, NC, USA).
Pharmacokinetic results were summarized using appropriate descriptive statistics. Following ln-transformation, AUC and Cmax results were compared between groups using the two-one-sided t test procedure [24]. ANOVA was performed on the ln-transformations of AUC∞, AUClast, and Cmax. For each parameter, least-squares means for each group were derived from a linear fixed-effects ANOVA model (PROC MIXED) where cohort is a fixed effect. The 90 % confidence interval (CI) approach was used to compare AUCs and Cmax of groups 2–4 with those of group 1. The 90 % CIs were calculated from the ratio of test mean/control mean, where control was group 1 and tests were groups 2–4. A 90 % CI that fell entirely within the interval 0.8–1.25 led to the conclusion that the two cohorts were bioequivalent. An appropriate non-parametric test (Kruskal–Wallis) was applied to the comparison of tmax values.
AEs were coded in accordance with the Medical Dictionary for Regulatory Activities (MedDRA®), version 11.0, and were summarized by MedDRA® preferred term and system organ class.
Vital signs, ECG, and laboratory results were summarized using descriptive statistics.
Outliers of ECG analysis were summarized and identified as follows:
Change in QT from baseline >30 ms or QT >450 ms for male, 430 ms for female;
Change in QT from baseline >60 ms;
Change in QTcF from baseline >30 ms or QTcF >450 ms for male, 430 ms for female;
Change in QTcF from baseline >60 ms.
Results
Demographics
Twenty-four subjects (six per group) were enrolled and completed the study as per protocol. A summary of the demographic characteristics of the subjects in this study are presented in Table 1. All groups were composed predominantly of male subjects. The mean (± standard deviation) age of the subjects was 68.3 ± 8.2 years, with a mean age across groups between 65 and 72 years. Height, bodyweight, and sex data were generally comparable across treatment groups. The mean height across groups was between 163 and 171 cm while mean bodyweight across groups was between 68 and 80 kg.
Table 1.
Demographic and other baseline characteristicsa
| Parameter | Group 1: healthy (n = 6) | Group 2: mild (n = 6) | Group 3: moderate (n = 6) | Group 4: severe (n = 6) | All (n = 24) |
|---|---|---|---|---|---|
| Age (years) [mean ± SD] | 65.7 ± 1.9 | 71.2 ± 5.4 | 71.3 ± 4.6 | 65.0 ± 14.6 | 68.3 ± 8.2 |
| Height (cm) [mean ± SD] | 165.6 ± 3.7 | 166.0 ± 8.7 | 170.3 ± 9.1 | 163.5 ± 10.4 | 166.4 ± 8.2 |
| Bodyweight (kg) [mean ± SD] | 74.2 ± 3.5 | 69.8 ± 9.8 | 79.3 ± 12.9 | 68.6 ± 12.4 | 72.9 ± 10.6 |
| Sex (n) | |||||
| Male | 5 | 5 | 5 | 4 | 19 |
| Female | 1 | 1 | 1 | 2 | 5 |
| GFR (mL/min/1.73 m2) [mean ± SD] | 110.0 ± 13.1 | 63.3 ± 7.8 | 38.5 ± 6.8 | 20.0 ± 6.7 | |
| Concomitant medications | |||||
| Number of subjects | 0 | 2 | 3 | 4 | 9 |
| Angiotensin II antagonists | 0 | 0 | 0 | 3 | 3 |
| Angiotensin II antagonists + diuretics | 0 | 0 | 1 | 0 | 1 |
| ACE inhibitors | 0 | 1 | 0 | 0 | 1 |
| β-Blocking agents | 0 | 1 | 0 | 0 | 1 |
| Amlodipine | 0 | 1 | 0 | 0 | 1 |
| Furosemide | 0 | 0 | 1 | 0 | 1 |
| Tamsulosin | 0 | 1 | 0 | 0 | 1 |
| Clopidogrel | 0 | 0 | 1 | 0 | 1 |
| Allopurinol | 0 | 0 | 0 | 1 | 1 |
| Metformin | 0 | 1 | 2 | 0 | 3 |
| Insulin | 0 | 0 | 1 | 1 | 2 |
| Alprazolam | 0 | 0 | 0 | 1 | 1 |
| Chlordiazepoxide/clidinium | 0 | 0 | 0 | 1 | 1 |
aGroups categorized according to degree of renal impairment
GFR glomerular filtration rate, SD standard deviation
At the time of enrolment, subjects in groups 2–4 were receiving concomitant medications for the treatment of renal impairment and its related symptoms. At least one subject in groups 2–4 received drugs for diabetes mellitus and half of the subjects in groups 3 and 4 received antihypertensive drugs. The incidence of these other pathologies increased in incidence from groups 2 to 4. These subjects continued to receive concomitant medication during the study.
Pharmacokinetics
Mean plasma concentrations of bilastine by group are shown graphically in Fig. 1.
Fig. 1.
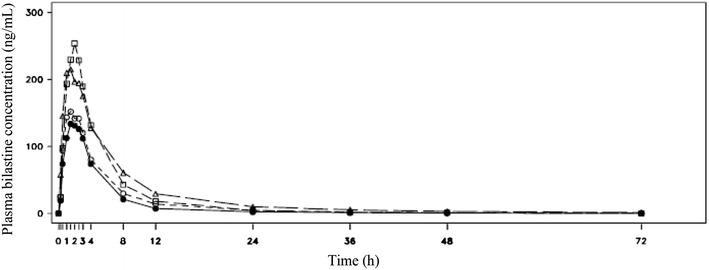
Mean plasma concentration–time profile of bilastine. Filled circles group 1-normal, GFR >80 mL/min/1.73 m2; open circles group 2-mild renal impairment, GFR 50–80 mL/min/1.73 m2; open squares group 3-moderate renal impairment, GFR 30–50 mL/min/1.73 m2; open triangles group 4-severe renal impairment, GFR <30 mL/min/1.73 m2. GFR glomerular filtration rate
The concentration–time profile was essentially parallel in all groups; however, mean plasma bilastine concentrations in subjects with renal impairment generally exceeded those in the control group during the first 12 h after dose administration. There was a clear separation in mean bilastine concentrations between the moderately and severely impaired groups relative to the healthy subjects.
Pharmacokinetic parameters are listed in Table 2 and the urinary concentration data in Table 3.
Table 2.
Pharmacokinetic parametersa,b
| Parameter (units) | Group 1: healthy (n = 6) | Group 2: mild (n = 6) | Group 3: moderate (n = 6) | Group 4: severe (n = 6) |
|---|---|---|---|---|
| AUClast (ng·h/mL) | 732.8 ± 260.0 | 946.9 ± 147.7 | 1,378.6 ± 264.4 | 1,654.6 ± 664.8 |
| AUC∞ (ng·h/mL) | 737.4 ± 260.8 | 967.4 ± 140.2 | 1,384.2 ± 263.2 | 1,708.5 ± 699.0 |
| Cmax (ng/mL) | 144.0 ± 57.8 | 172.1 ± 45.0 | 271.1 ± 30.4 | 228.8 ± 81.8 |
| tmax (h)c | 1.5 (1.0–3.0) | 1.5 (0.5–3.0) | 2.25 (1.0–2.5) | 1.5 (0.5–3.0) |
| Clast (ng/mL) | 0.3 ± 0.1 | 0.8 ± 0.3 | 0.4 ± 0.1 | 1.5 ± 1.1 |
| λz (h−1) | 0.0796 ± 0.0194 | 0.0593 ± 0.0326 | 0.0691 ± 0.0155 | 0.0499 ± 0.0254 |
| CLP/F (L/h) | 29.67 ± 8.86 | 21.05 ± 3.18 | 14.88 ± 2.75 | 16.24 ± 14.20 |
| Vz/F (L) | 382.9 ± 122.10 | 461.7 ± 244.27 | 222.6 ± 55.68 | 351.1 ± 193.42 |
| t½β (h) | 9.26 ± 2.788 | 15.08 ± 7.655 | 10.46 ± 2.340 | 18.38 ± 11.39 |
| CLR (L/h) | 8.72 ± 2.21 | 6.94 ± 0.80 | 5.49 ± 2.08 | 4.03 ± 3.76 |
| (CLP/F)/CLR | 3.40 | 3.03 | 2.71 | 4.03 |
aGroups categorized according to degree of renal impairment
bValues are mean ± standard deviation except where otherwise stated
cValues are median (range)
AUC, area under the plasma concentration–time curve; AUClast, AUC from time zero to time of last measurable plasma concentration; AUC∞, AUC from time zero to infinity; Clast, last measurable plasma concentration; CLP/F, plasma clearance after oral administration; CLR renal clearance; Cmax, maximum plasma concentration; t½β, terminal elimination half-life; tmax, time to reach Cmax; Vz/F, apparent volume of distribution during terminal phase after non-intravenous administration; λz, terminal disposition rate constant
Table 3.
Urine concentrations of bilastine (ng/mL)a,b
| Time (h) | Group 1: healthy (n = 6) | Group 2: mild (n = 6) | Group 3: moderate (n = 6) | Group 4: severe (n = 6) |
|---|---|---|---|---|
| 0–12 | 4,387.5 ± 3,358.4 | 4,824.1 ± 2,073.0 | 7,184.3 ± 3,843.4 | 3,469.8 ± 2,066.2 |
| 12–24 | 531.7 ± 342.0 | 597.7 ± 159.2 | 472.9 ± 221.9 | 813.0 ± 567.5 |
| 24–48 | 103.4 ± 38.6 | 159.3 ± 43.3 | 126.7 ± 81.1 | 260.4 ± 212.1 |
| 48–72 | 20.9 ± 10.0 | 49.3 ± 40.7 | 30.3 ± 7.4 | 97.8 ± 69.3 |
aGroups categorized according to degree of renal impairment
bValues are mean ± standard deviation
There was a clear trend of increasing AUC and Cmax through the groups 1–4.
The subjects with renal impairment had higher mean bilastine AUC and Cmax values than the healthy subjects. The mean AUC∞ ranged from 737.4 to 1,708.5 ng·h/mL in healthy subjects and severely impaired subjects, respectively. There were no substantial differences between groups in median tmax or in mean ke for bilastine.
Urinary excretion of bilastine was rapid, most being excreted in the first 12 h, and was essentially complete by 72 h.
The relationship between CLP/F and CLR was maintained between groups independently of the degree of renal impairment, the mean values being 3.4, 3.0, 2.7, and 4.0 for healthy, mildly, moderately, and severely renally impaired subjects, respectively.
A reduction in CLP/F that was parallel to the decrease in GFR was found. This can be observed in Fig. 2, which shows a linear correlation between these two parameters. It is also important to point out that the regression line in Fig. 2, calculated by statistical regression analysis, tends to cross the origin, confirming that glomerular filtration was the main excretion pathway. An outlier can be observed with high CLP/F and low GFR values. It is likely that the apparently elevated CLP/F value was due to a low bilastine bioavailability. The regression analysis was repeated with and without this outlier and similar results were obtained. It was therefore decided to keep these data in the analysis dataset.
Fig. 2.
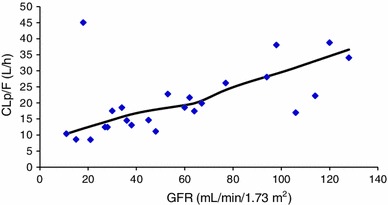
Relationship between CLP/F and GFR. The line represents regression calculated by statistical regression analysis. An outlier can be observed with high CLP/F and low GFR values. It is likely that the apparently elevated CLP/F value was due to a low bilastine bioavailability. The regression analysis was repeated with and without this outlier and similar results were obtained. r = 0.515; p < 0.01. CLP/F bilastine plasma clearance after oral administration, GFR glomerular filtration rate, p probability of a 22 degrees of freedom that it is due to chance, r Pearson’s r between non-compartmental analysis estimated using CLP/F and GFR
The results of the statistical analysis are shown in Table 4.
Table 4.
Analysis of variance of pharmacokinetic parametersa
| Group | PK parameter | Geometric LS means | Geometric mean ratiob | 90 % CIc | |
|---|---|---|---|---|---|
| Test group | Reference (group 1: healthy) | ||||
| 2: Mild | AUC∞ | 958.8 | 703.3 | 1.36 | 0.94–1.97 |
| AUClast | 937.0 | 698.6 | 1.34 | 0.93–1.94 | |
| Cmax | 167.0 | 134.6 | 1.24 | 0.89–1.72 | |
| t max d | 1.50 | 1.50 | 0.00 | ||
| 3: Moderate | AUC∞ | 1,363.9 | 703.3 | 1.94 | 1.34–2.81 |
| AUClast | 1,358.0 | 698.6 | 1.94 | 1.35–2.81 | |
| Cmax | 269.7 | 134.6 | 2.00 | 1.45–2.78 | |
| tmax d | 2.25 | 1.50 | 0.75 | ||
| 4: Severe | AUC∞ | 1,513.9 | 703.3 | 2.15 | 1.49–3.12 |
| AUClast | 1,472.3 | 698.6 | 2.11 | 1.46–3.04 | |
| Cmax | 214.0 | 134.6 | 1.59 | 1.15–2.20 | |
| tmax d | 1.50 | 1.50 | 0.00 | ||
aGroups categorized according to degree of renal impairment
bRatio of geometric means of test and control groups
cA 90 % CI that falls within the interval 0.8–1.25 leads to the conclusion that the two groups are bioequivalent
dThe median values in the first two columns and the difference of median values in the column of geometric mean ratio are presented, respectively
AUC, area under plasma concentration–time curve; AUClast, AUC from time zero to time of last measurable plasma concentration; AUC∞, AUC from time zero to infinity; Cmax, maximum plasma concentration; LS, least squares; PK, pharmacokinetic; tmax, time to reach Cmax
Generally, when compared with healthy subjects, the geometric mean ratios of AUC∞ and AUClast were higher as the degree of renal insufficiency progressed, being highest in those subjects with severe insufficiency. In all groups of renally impaired subjects, the corresponding 90 % CIs of these parameters were not enclosed within the 0.8–1.25 interval, indicating that the bioequivalence between cohorts was not demonstrated based on these results.
Plasma protein binding was quantified in renally impaired subjects and comparable values were found among the renal impairment groups [25].
Safety
No serious AEs or deaths were reported.
Six subjects, one each in the healthy, mildly, and moderately impaired groups and three in the severely renally impaired group experienced mild AEs during the study. All were considered possibly treatment related; these were diarrhea (in three subjects), with single occurrences of anorexia, abdominal discomfort, and tension headache.
In the healthy and moderately impaired groups, all of the mean results for all hematology, blood chemistry, and urinalysis parameters were within reference ranges throughout the study. In the mildly impaired group, only the mean blood glucose concentration at the end of study was outside the normal range 120.8 mg/dL (reference range 60–115 mg/dL).
In the severely impaired group, some parameters were outside the reference range at the start of the study: hemoglobin, hematocrit, and red blood count were lower while blood urea nitrogen and creatinine levels were higher. There were no notable changes in these parameters throughout the study. Lactate dehydrogenase was initially above the reference range but normalized during the study.
No clinically meaningful changes were observed in vital signs, and no apparent trends were observed over time.
Of the 24 subjects enrolled and dosed in the study, a total of five (20.8 %) had outlier ECG results. Of these five subjects, two (33.3%) were in the healthy group and one (16.7%) each were in the mild, moderate, and severe impairment groups. These subjects had increases in QT duration from baseline >30 ms or QT >450 ms for males, >430 ms for females. One subject in the mild and one in the moderate group also had change in QTcF from baseline >30 ms or QTcF >450 ms for males, >430 ms for females. None of the abnormal ECGs were considered clinically significant.
Discussion
In this open-label, single-centre, single-dose, parallel-group, non-randomized study, the pharmacokinetics and safety of bilastine were evaluated in subjects with various degrees of renal insufficiency. A single oral dose of 20 mg of bilastine was given to each subject.
As bilastine is renally excreted, renal insufficiency was expected to lead to a greater exposure to the drug. This was borne out by the progressive increase in mean Cmax and AUC with increasing renal insufficiency, such that both parameters in group 4 (severe renal insufficiency) were approximately double their value in group 1 (healthy). There were no substantial differences in median tmax or in the mean terminal rate constants for bilastine.
The increase in exposure was due to the reduction of both plasma and renal clearances, which paralleled the decrease of GFR across the groups. The constancy of the (CLP/F)/CLR ratio across the groups, and the reduction in CLR from mild to severe renal insufficiency, consistent with the parallel reduction in the GFR, suggest that renal excretion is the main elimination route for bilastine, with no alternative elimination routes (hepatic, biliary, or others) being used even in severe renal insufficiency.
The relationship between CLP/F and GFR (linear and crossing the origin when correlated) suggests that glomerular filtration is the main renal excretion mechanism.
The measurement of the protein-bound fraction of bilastine has shown that the amount of unbound bilastine is not affected by progressive renal insufficiency, showing that protein binding of bilastine was independent of concentration [25]. It is important to point out that the degree of renal impairment is not affecting plasma protein binding: a change in plasma protein binding can be the consequence of higher drug concentration but also due to the disease itself which can produce several molecules/agents that can act to displace bound drugs. It has been confirmed that this is not the case for bilastine as there is a constant plasma protein binding; therefore, even in severely renally impaired subjects [25], with higher bilastine concentrations and high levels of urea and other substances, known displacers of drugs bound to albumin. As the percentage of plasma protein binding is constant among the different groups, independent of the disease status and bilastine concentration, the results from the total plasma concentrations obtained in this pharmacokinetic study can be more or less directly linked with efficacy and safety in order to conclude a dose adjustment is not needed.
The higher exposure observed in the renal impaired groups was below that seen in healthy subjects given bilastine 50 and 100 mg orally as a single dose or as a multiple 14-day dose administration, which appeared to be safe and well-tolerated [7].
The rapid urinary excretion of bilastine suggests daily doses would not lead to accumulation even in subjects with severe renal insufficiency, since the amount in the plasma after 24 h was less than 5 % of the Cmax in all the groups. Moreover overall exposure (AUC∞) in severely impaired subjects was within safety margins that corresponded to the mean estimated exposure and 95 % CI of a single 80 mg oral dose of bilastine (AUC∞ of 4,225.6 ng·h/mL, 95 % CI 3,174.7–6278.4) [6]. This was the dose at which the incidence of central nervous system events indicative of sedation (somnolence) showed a statistically significant difference versus placebo in a previous phase I study in healthy volunteers [13]. This indicates that even patients with severe renal impairment receiving 20 mg of bilastine once a day are unlikely to attain a bilastine exposure exceeding the estimated safety margin.
Bilastine given as a 20 mg oral dose was well-tolerated. The reported AEs were all mild. Clinical laboratory values were generally within reference ranges for all parameters in this study. Vital signs, ECGs, and physical measurements did not indicate any significant safety concerns.
Conclusions
It appears that a once-daily 20 mg dose of bilastine can be safely administered in the presence of various degrees of renal insufficiency, even if this causes higher exposure to bilastine. This is in accordance with the approved posology of bilastine that does not require dosage adjustments for renally impaired subjects.
Acknowledgments
FAES FARMA funded the conduct of this study at Clinical Pharmacology of Miami, the analysis and reporting of data and writing of the manuscript by inVentiv Health Clinical, and the pharmacokinetic modeling and analysis performed by Dr. Mónica Rodríguez (Dynakin SL). Ander Sologuren is an employee of FAES FARMA S.A., the sponsor of the study. The other authors have no potential conflicts of interest that are directly relevant to the content of this study.
References
- 1.Bachert C, Kuna P, Zuberbier T. Bilastine in allergic rhinoconjunctivitis and urticaria. Allergy. 2010;65(Suppl 93):1–13. doi: 10.1111/j.1398-9995.2010.02404.x. [DOI] [Google Scholar]
- 2.Corcóstegui R, Labeaga L, Innerárity A, Berisa A, Orjales A. Preclinical pharmacology of bilastine, a new selective histamine H1 receptor antagonist: receptor selectivity and in vitro antihistaminic activity. Drugs R&D. 2005;6(6):371–384. doi: 10.2165/00126839-200506060-00005. [DOI] [PubMed] [Google Scholar]
- 3.Corcóstegui R, Labeaga L, Innerárity A, Berisa A, Orjales A. In vivo pharmacological characterisation of bilastine, a potent and selective histamine H1 receptor antagonist. Drugs R&D. 2006;7(4):219–231. doi: 10.2165/00126839-200607040-00002. [DOI] [PubMed] [Google Scholar]
- 4.Crean C, Valiente R, Sologuren A, McLaverty D. Effect of grapefruit juice on the pharmacokinetics of bilastine [abstract] J Clin Pharmacol. 2007;47(9):1198. [Google Scholar]
- 5.Roupe K, Sologuren A, Crean C, Valiente R. Effect of age and gender on the pharmacokinetics and pharmacodynamics of bilastine [abstract] J Clin Pharmacol. 2007;47(9):1198. [Google Scholar]
- 6.Jauregizar N, de la Fuente L, Lucero ML, Sologuren A, Leal N, Rodríguez M. Pharmacokinetic-pharmacodynamic modelling of the antihistaminic H1 effect of bilastine. Clin Pharmacokinet. 2009;48(8):543–554. doi: 10.2165/11317180-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 7.Crean C, Roupe K, Sologuren A, Valiente R. The pharmacokinetics of bilastine after single and 14 days once daily administration [abstract] Basic Clin Pharmacol Toxicol. 2007;101(Suppl 1):148. [Google Scholar]
- 8.Crean C, Sologuren A, Valiente R, McLaverty D. The drug–drug interaction of ketoconazole on bilastine pharmacokinetics [abstract] Basic Clin Pharmacol Toxicol. 2007;101(Suppl 1):148–149. [Google Scholar]
- 9.Lucero ML, Gonzalo A, Ganza A, Leal N, Soengas I, Ioja E, Gedey S, Jahic M, Bednarczyk D. Interactions of bilastine, a new oral H1 antihistamine, with human transporter systems. Drug Chem Toxicol. 2012;35(Suppl 1):8–17. doi: 10.3109/01480545.2012.682653. [DOI] [PubMed] [Google Scholar]
- 10.Lucero ML, Gonzalo A, Mumford R, Betanzos M, Alejandro A. An overview of bilastine metabolism during preclinical investigations. Drug Chem Toxicol. 2012;35(Suppl 1):18–24. doi: 10.3109/01480545.2012.682651. [DOI] [PubMed] [Google Scholar]
- 11.Tyl B, Kabbaj M, Azzam S, Sologuren A, Valiente R, Reinbolt E, Roupe K, Blanco N, Wheeler W. Lack of significant effect of bilastine administered at therapeutic and supratherapeutic doses and concomitantly with ketoconazole on ventricular repolarization: results of a thorough QT study (TQTS) with QT-concentration analysis. J Clin Pharmacol. 2012;52(6):893–905. doi: 10.1177/0091270011407191. [DOI] [PubMed] [Google Scholar]
- 12.Graff C, Struijk JJ, Kanters JK, Andersen MP, Toft E, Tyl B. Effects of bilastine on T-wave morphology and the QTc interval: a randomized, double-blind, placebo-controlled, thorough QTc study. Clin Drug Investig. 2012;32(5):339–351. doi: 10.2165/11599270-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 13.García-Gea C, Martínez-Colomer J, Antonijoan RM, Valiente R, Barbanoj MJ. Comparison of peripheral and central effects of single and repeated oral dose administrations of bilastine, a new H1 antihistamine: a dose-range study in healthy volunteers with hydroxyzine and placebo as control treatments. J Clin Psychopharmacol. 2008;28(6):675–685. doi: 10.1097/JCP.0b013e31818b2091. [DOI] [PubMed] [Google Scholar]
- 14.Conen S, Theunissen EL, Van Oers AC, Valiente R, Ramaekers JG. Acute and subchronic effects of bilastine (20 and 40 mg) and hydroxyzine (50 mg) on actual driving performance in healthy volunteers. J Psychopharmacol. 2011;25(11):1527–1533. doi: 10.1177/0269881110382467. [DOI] [PubMed] [Google Scholar]
- 15.Sastre J, Mullol J, Valero A, Valiente R, on behalf of Bilastine Study Group Efficacy and safety of bilastine 20 mg compared with cetirizine 10 mg and placebo in the treatment of perennial allergic rhinitis. Curr Med Res Opin. 2012;28(1):121–130. doi: 10.1185/03007995.2011.640667. [DOI] [PubMed] [Google Scholar]
- 16.Bachert C, Kuna P, Sanquer F, Ivan P, Dimitrov V, Gorina MM, van de Heyning P, Loureiro A. Comparison of the efficacy and safety of bilastine 20 mg vs desloratadine 5 mg in seasonal allergic rhinitis patients. Allergy. 2009;64(1):158–165. doi: 10.1111/j.1398-9995.2008.01813.x. [DOI] [PubMed] [Google Scholar]
- 17.Church MK. Safety and efficacy of bilastine: a new H(1)-antihistamine for the treatment of allergic rhinoconjunctivitis and urticaria. Expert Opin Drug Saf. 2011;10(5):779–793. doi: 10.1517/14740338.2011.604029. [DOI] [PubMed] [Google Scholar]
- 18.Kuna P, Bachert C, Nowacki Z, van Cauwenberge P, Agache I, Fouquert L, Roger A, Sologuren A, Valiente R. Efficacy and safety of bilastine 20 mg compared with cetirizine 10 mg and placebo for the symptomatic treatment of seasonal allergic rhinitis: a randomized, double-blind, parallel-group study. Clin Exp Allergy. 2009;39(9):1338–1347. doi: 10.1111/j.1365-2222.2009.03257.x. [DOI] [PubMed] [Google Scholar]
- 19.Zuberbier T, Oanta A, Bogacka E, Medina I, Wesel F, Uhl P, Antepara I, Jáuregui I, Valiente R. Comparison of the efficacy and safety of bilastine 20 mg vs levocetirizine 5 mg for the treatment of chronic idiopathic urticaria: a multi-centre, double-blind, randomized, placebo-controlled study. Allergy. 2010;65(4):516–528. doi: 10.1111/j.1398-9995.2009.02217.x. [DOI] [PubMed] [Google Scholar]
- 20.European Medicines Agency. Note for guidance on the evaluation of the pharmacokinetics of medicinal products in patients with impaired renal function. CHMP/EWP/225/02, London, 23 Jun 2004. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003123.pdf. Accessed 10 Jul 2013
- 21.Wilson DM, Bergert JH, Larson TS, Liedtke BS. GFR determined by nonradiolabeled iothalamate using capillary electrophoresis. Am J Kidney Dis. 1997;30(5):646–652. doi: 10.1016/S0272-6386(97)90488-1. [DOI] [PubMed] [Google Scholar]
- 22.Piquet N, Montminy V, Gonzalo A, Lucero ML. A rapid simple validated LC-MS/MS method for analysis of bilastine in human plasma and urine [abstract no. R6359]. Pharmaceutical Sciences World Congress; 14–18 Nov 2010; New Orleans. Congress Abstracts Book; 2010:233
- 23.European Medicines Agency. E 14—the clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. CHMP/ICH/2/2004, November 2005. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002879.pdf. Accessed 10 Jul 2013.
- 24.Cao L, Mathew T. A simple numerical approach towards improving the two one-sided test for average bioequivalence. Biom J. 2008;50(2):205–211. doi: 10.1002/bimj.200710407. [DOI] [PubMed] [Google Scholar]
- 25.Rodríguez M. In vitro determination of bilastine unbound fraction in patients with different degrees of renal impairment. Derio: Dynakin S.L.; 2009 (data on file).