

Abstract
Influenza infection predisposes patients to secondary bacterial pneumonia that contributes significantly to morbidity and mortality. While this association is well documented, the mechanisms that govern this synergism are poorly understood. A window of hyporesponsiveness following influenza infection has been associated with a substantial increase in local and systemic IFNγ concentrations. Recent data suggests that the oxazolidinone antibiotic linezolid decreases IFNγ and TNFα production in vitro from stimulated peripheral blood mononuclear cells. We therefore sought to determine whether linezolid would reverse immune hyporesponsiveness after influenza infection in mice through its effects on IFNγ. In vivo dose response studies demonstrated that oral linezolid administration sufficiently decreased bronchoalveolar lavage fluid levels of IFNγ at day 7 post-influenza infection in a dose-dependent manner. The drug also decreased morbidity as measured by weight loss compared to vehicle-treated controls. When mice were challenged intranasally with S. pneumoniae 7 days after infection with influenza, linezolid pre-treatment led to decreased IFNγ and TNFα production, decreased weight loss, and lower bacterial burdens at 24 hours post bacterial infection in comparison to vehicle-treated controls. To determine whether these effects were due to suppression of IFNγ, linezolid-treated animals were given intranasal instillations of recombinant IFNγ before challenge with S. pneumoniae. This partially reversed the protective effects observed in the linezolid-treated mice, suggesting that the modulatory effects of linezolid are mediated partially by its ability to blunt IFNγ production. These results suggest that IFNγ, and potentially TNFα, may be useful drug targets for prophylaxis against secondary bacterial pneumonia following influenza infection.
INTRODUCTION
While primary influenza infection causes significant morbidity and mortality worldwide, a greater appreciation for the contribution of secondary bacterial pneumonias to these statistics is starting to emerge. Studies suggest that the majority of deaths associated with the 1918 pandemic influenza strain can be attributed to secondary bacterial infections of the upper respiratory tract (1, 2). The bacterial species most often encountered during recovery from influenza infection include S. pneumoniae, S. aureus, and H. influenzae (2–5). Clinical correlates for the association between influenza and pneumococcal infections are available from studies that examined the efficacy of pneumococcal vaccines. Studies show that the pneumococcal conjugate vaccine has 45% efficacy in the prevention of secondary pneumonia in pediatric patients with confirmed influenza infection suggesting that prophylaxis against secondary pneumococcal disease may greatly reduce the morbidity and mortality associated with influenza infection (6, 7).
A thorough understanding of the mechanisms that promote immune hyporesponsiveness is critical to advancing preventative measures to decrease the incidence of secondary infection. Studies have identified several factors that may contribute to increased susceptibility as a result of influenza infection, including physical damage to the lungs (8), enhanced bacterial binding via increased platelet-activating factor (9, 10), a dysfunctional neutrophil response (11, 12), and elevated levels of IL-10 (13, 14). Other studies suggest that excess IFNγ produced during the latter stages of influenza infection may mediate susceptibility to S. pneumoniae infection. Sun and Metzger (15) demonstrated that high concentrations of IFNγ that result from the immune response are sufficient to inhibit alveolar macrophage phagocytosis of S. pneumoniae, both in vitro and in vivo during the first 24 hours of infection. In this dual infection model, normal clearance of bacteria was restored in animals lacking IFNγ signaling and then impaired once again when alveolar macrophages or flu-infected mice were treated with exogenous IFNγ.
These studies suggest that the inhibition of influenza-associated IFNγ production may be beneficial in reducing the incidence of secondary S. pneumoniae infection. Pharmacologic inhibition of this cytokine response could provide a means to prevent this window of hyporesponsiveness. The oxazolidinone antibiotic linezolid has been shown to have inhibitory activity against proinflammatory cytokines. Linezolid’s pharmacological effects lie in its ability to bind to the 50S ribosomal subunit, thus preventing initiation of the 70S complex and inhibiting the initiation of protein synthesis (16). Linezolid treatment of whole human blood cells stimulated with LPS was shown to inhibit IFNγ and TNFα production in a dose-dependent manner (17). Further, linezolid reduced concentrations of IFNγ and TNFα in human PBMCs without altering endotoxin production in vitro (18). We therefore hypothesized that prophylactic treatment of influenza-infected mice with linezolid would reduce harmful proinflammatory IFNγ production and limit outgrowth of a secondary pulmonary challenge with S. pneumoniae. Indeed, we observed that linezolid treatment of influenza-infected mice resulted in decreased weight loss, inhibition of IFNγ and TNFα responses, and attenuation of bacterial outgrowth following S. pneumoniae challenge, while not adversely affecting the adequate response to and clearance of the virus. These studies provide further rationale for the exploration of IFNγ-targeting therapies to control and limit dangerous secondary bacterial infections.
MATERIALS AND METHODS
Mouse model of viral and bacterial infection
Eight- to 10-wk old female C57BL/6 mice were purchased from Taconic (Bar Harbor, ME). The A/Puerto Rico/8/34 (PR8) strain of influenza virus was grown in the allantoic fluid of 10-day-old embryonated specific pathogen-free chicken eggs as previously described (19). Viral stocks were tested for common mouse pathogens and were shown to contain only influenza virus. Viral yield was quantified by titration in eggs to determine the 50% egg-infectious dose (EID50). Mice were infected intranasally (i.n.) under isoflurane anesthesia with 1–2 EID50 /g bodyweight in a 50μl volume of HBSS. Following infection, mice were monitored daily for weight loss. When animals reached 10% body weight loss, daily s.c. injections of warmed HBSS were administered for rehydration purposes. Frozen stocks of S. pneumoniae strain A66.1 were thawed and immediately resuspended to appropriate inocula in 50μL of HBSS. On the morning of day 7 post-influenza infection, animals were challenged i.n. under isoflurane anesthesia with 6x104 to 1.4x105 CFU of bacteria, depending on the experimental replicate. Resultant inocula were confirmed following serial plating on 5% sheep’s blood agar and overnight incubation to determine CFU counts. Each experimental replicate included 4–5 mice per treatment group per timepoint, and data was combined for multiple replicates as noted in each figure legend.
Linezolid preparation and dosing
Based on the weight of the mice, animals were dosed with either 20mg/kg, 40mg/kg, or 80mg/kg of linezolid twice daily via oral gavage. Linezolid was prepared using either pure powder, or by crushing linezolid tablets (Pfizer, New York, NY). The drug was suspended in 2% methylcellulose based on a weight to weight calculation such that each 100μl of vehicle contained the desired amount of active drug to be administered. This dosing range was chosen based upon studies that examined the pharmacokinetic profile of linezolid in mice (20, 21). It was determined that 60mg/kg twice daily dosing in infected mice would likely mimic human plasma levels of the drug. Because Takahashi et. al. showed that linezolid blocked cytokine secretion down to a concentration of 2μg/ml, we predicted that this dosing range would prove effective (17).
Cell isolation
Mice were euthanized by exsanguination under deep anesthesia at post-infection timepoints. The lungs were lavaged with 1ml aliquots of HBSS containing 3mM EDTA to isolate alveolar cells, with the first aliquot being separated from the cellular fraction by centrifugation (deemed “first wash”). For experiments that did not include bacterial challenge, blood was removed from the lungs by perfusion of HBSS into the right side of the heart. Lungs were then excised and minced and incubated with 50 U/ml DNase (Sigma-Aldrich) and 1 mg/ml collagenase A (Sigma-Aldrich) in RPMI 1640 media containing 5% FCS. For experiments that included bacterial challenge, whole lungs were minced with a mechanical homogenizer. TBLN and digested lung tissue were pushed through cell strainers to obtain single cell suspensions. Subsequently, RBCs were lysed by exposure to a hypotonic solution, and the remaining cells were washed, counted, and resuspended in HBSS.
Flow cytometry
BAL, lung digest, and TBLN cells were washed in PBS containing 0.1% BSA and 0.02% sodium azide and incubated with appropriate concentrations of fluorochrome-conjugated antibodies specific for murine CD4, CD8, CD44, or CD62L. Antibodies were purchased from eBioscience or BD Biosciences. For intracellular cytokine staining, 2x106 cells from each sample were incubated for 4 hours with 50ng/ml PMA and 500ng/ml ionomycin, and in the presence of 10μg/ml brefeldin A for the final 2 hours. Cells were surface stained for CD4 and CD8 and fixed in 4% formalin. Cells were then permeabilized in 0.05% saponin, exposed to anti-CD16/32 Ab to block Fc binding, and incubated with fluorescently labeled anti-TNF-α, anti-IFNγ, and anti-IL-10 antibodies. Cells were washed thoroughly and resuspended in PBS for analysis. Labeled cells were analyzed using a BD LSRII Flow Cytometer System (BD Biosciences) as compared to isotype and one-color controls. Greater than 50,000 events per sample were routinely acquired. For all analyses, viable lymphocyte populations were defined on forward vs. side scatter plots and further examined on dot plots examining CD4 vs CD8 expression.
Cytokine analysis by ELISA
Lung lavage cells were separated from lavage fluid in the first 1mL lung lavage washout. ELISAs for IFNγ and TNFα were performed on cell-free lavage fluid using ELISA kits according to manufacturers’ instructions (eBioscience). Concentrations were normalized to total protein content in the lavage fluid as measured using the BCA Protein Quantification Kit (Bio-Rad, Hercules, CA).
Administration of exogenous IFNγ
In select experiments, mice were administered 2ng of recombinant (r)IFNγ (Peprotech) or PBS/0.1BSA vehicle in 25μL i.n. under isoflurane anesthesia on days 5 and 6 post-influenza infection. Mice were subsequently infected on day 7 with S. pneumoniae as described above.
Enumeration of bacterial loads
Following mechanical homogenization, aliquots of lung were isolated before RBC lysis. The aliquots were then serially diluted in sterile water and spread plated onto 5% sheep’s blood agar. After 24 hours, CFU were counted and calculated to represent entire bacterial contents of the lungs.
Determination of lung viral burdens
Lungs were sterilely isolated from infected mice and frozen at 80°C until analysis. Viral burdens were determined by plaque assay on Madin Darby Canine Kidney cells (ATCC, Manassas, VA) as described previously (22). Briefly, cells were grown to confluency in 6-well plates in DMEM media supplemented with non-essential amino acids and 10% heat inactivated FCS (Atlanta Biologicals, Lawrenceville, GA). Ten-fold dilutions of lung homogenate were incubated with the cells for 1 hour at 37°C. The cells were then washed and overlaid with DMEM media in 1% Bacto Agar with 1% trypsin (Sigma-Aldrich, St. Louis, MO). Three days later the cells were fixed with 20% acetic acid and the overlay removed. Plaques were visualized by staining with crystal violet and counted.
Statistics
Measurements were determined for each individual mouse, and the means and SD were generated for each treatment group. Differences between experimental groups were determined using t tests, 1-way, or 2-way ANOVA where appropriate. The Student Newman-Keuls post hoc test was used to discriminate the differences between individual groups at each time point when detected. Differences were considered statistically significant when p<0.05. Analyses were performed using GraphPad Prism software package (GraphPad Software, Inc., La Jolla, CA).
RESULTS
Linezolid inhibits IFNγ production in a dose-dependent manner
Studies in mice demonstrate that the window of immune hyporesponsiveness following influenza infection, as defined by a secondary bacterial challenge, is associated with heightened IFNγ production in the lungs ranging from day 7 to day 10 post-infection (14, 15). Using these same timepoints, we therefore hypothesized that use of linezolid following influenza infection would be sufficient to limit IFNγ responses. Animals were orally gavaged with 20, 40, or 80mg/kg linezolid or vehicle control twice daily starting 24h after influenza challenge. While all influenza-infected animals experienced substantial weight loss over the course of the study, we consistently observed that animals treated with a dose of 80mg/kg linezolid lost significantly less weight than vehicle-treated controls (Fig. 1A). As is typical in models of influenza infection, weight loss began on day 4 post-infection and progressed to 15–20% loss from baseline by day 10. In the group that received the highest linezolid dose, mean losses only dropped by 12–14% for each experimental replicate. Alveolar concentrations of IFNγ were observed to be low on day 4 post-infection, but dramatically elevated on day 7 post-infection, a timing that corresponds to the hyporesponsive window (Fig. 1B) (15). Linezolid treatment decreased IFNγ concentrations in the BAL fluid in a dose-dependent manner in comparison to vehicle-treated controls at 7 days post-infection, the difference becoming significant in the 80mg/kg dose group. By day 10 post-infection, IFNγ concentrations had decreased, but were still 100- to 1000-fold higher than those observed on day 4 post-infection. These data suggest that twice daily dosing of mice with linezolid at a dose of 80mg/kg is optimal for inhibition of IFNγ production at the time of peak production. While 7 days post-infection was found to be the time of maximal TNFα production, no statistical difference was observed in TNFα production between animals being treated with any dosage of linezolid in comparison to vehicle-treated controls at any time point examined (Fig. 1C).
FIGURE 1.

Dose-dependent suppression of IFNγ by linezolid following influenza infection. C57BL/6 mice were infected intranasally with 2 EID50/g influenza strain PR8. Animals were orally gavaged with linezolid at 20mg/kg, 40mg/kg, or 80mg/kg or with 2% methylcellulose vehicle control twice a day. Animals were weighed daily and sacrificed at given timepoints. (A) Weight loss is represented as percentage of weight loss in comparison to day 0. (B and C) Bronchoalveolar lavage fluid was collected and analyzed for expression of IFNγ (B) and TNFα (C) protein by ELISA. Data are a compilation of 3 independent experiments (12–16 mice per treatment group per timepoint). Mean values ± SD are depicted and compared using one-way ANOVA (and 2-way ANOVA for weight curve data) along with the Student Newman-Keuls post hoc test. *p<0.05
Linezolid does not significantly alter other immune responses following influenza infection
We sought to evaluate the effects of linezolid on the activation status of T cells and the production of cytokines following influenza infection to determine whether blunting IFNγ production had an impact on other responses to the virus. As shown in Fig. 2A (top), linezolid did not alter the total numbers of activated CD4+ T cells found in the lung lavage (left), lung digest (middle), or in TBLN (right). As is typical of antiviral immune responses, increases in T cell activation can initially be detected in the draining lymph nodes (here on day 7 post-infection), with subsequent activated T cell infiltration of pulmonary tissues and alveolar spaces. Similarly, linezolid did not modulate the activation of CD8 T cells over the course of study (Fig. 2A, bottom panel). Production of cytokines was measured by intracellular cytokine staining of cells isolated from lung lavage, digest, and TBLN. Linezolid treatment did not significantly alter the absolute number of CD4 T cells producing IFNγ (Fig. 2B), TNFα (Fig. 2C), or IL-10 (Fig. 2D). No statistical differences were observed in CD8 T cell production of IFNγ (Fig. 2B, bottom), TNFα (Fig. 2C, bottom), or IL-10 (Fig. 2D, bottom). Despite the lack of significance, we consistently observed a trend towards linezolid-mediated suppression of IL-10 and TNFα in the cells of both the lung lavage and TBLN. These data are consistent with the observation of reduced IFNγ concentrations in the first wash of linezolid treated mice (Fig. 1B). This also corresponds to the in vitro data reported by Takahashi et al that shows a blunting of TNFα production by the drug in peripheral blood monocytes (17). In addition, we observed no differences between linezolid-treated and control animals in the percentages of CD4+ or CD8+ cells that stained positive for activation or cytokine production observed over time (data not shown). To confirm these findings, we performed cell differential counts on lung digest cells, which demonstrated no statistical difference in the percentages of lymphocytes, monocyte/macrophages, or polymorphonuclear cells in the lungs of influenza-infected animals (data not shown).
FIGURE 2.

Linezolid does not alter other lymphocyte responses following influenza infection. C57BL/6 mice were infected intranasally with influenza strain PR8 and dosed twice daily with linezolid (80mg/kg) or vehicle. Animals were sacrificed at given times and BAL, lung digest, and TBLN were processed for flow cytometry. (A) Graphs represent absolute numbers of CD4 (top panels) and CD8 (bottom panels) cells expressing activation markers CD44hiCD62Llo. (B–D) Intracellular cytokine staining yielded absolute numbers of CD4 and CD8 cells producing IFNγ (B), TNFα (C), and IL-10 (D) in BAL, lung digest, and TBLN compartments. Data are representative of 2 independent experiments, with 4–5 mice per group per timepoint in each experimental replicate. Mean ± SD are depicted with comparison using one-way ANOVA along with the Student Newman-Keuls post hoc test. No statistically significant differences were observed.
Linezolid does not alter influenza viral replication
Targeting IFNγ production as a prophylaxis against secondary bacterial infection is only a reasonable approach if administration of the drug does not exacerbate influenza viral replication. Influenza-infected mice were dosed twice daily with 80mg/kg linezolid or vehicle control and were euthanized on day 7 post-infection. Plaque assays were used to determine total viral burdens in the lungs of infected mice. The data in Fig. 3 demonstrates that there was no significant difference in influenza viral loads between vehicle and linezolid-treated mice. This data, combined with our results showing that linezolid treatment decreases the weight loss after infection with influenza and the data presented in Fig. 2, supports that linezolid’s effects are primarily upon IFNγ production which does not impact the ability of the mice to clear the infecting virus.
FIGURE 3.
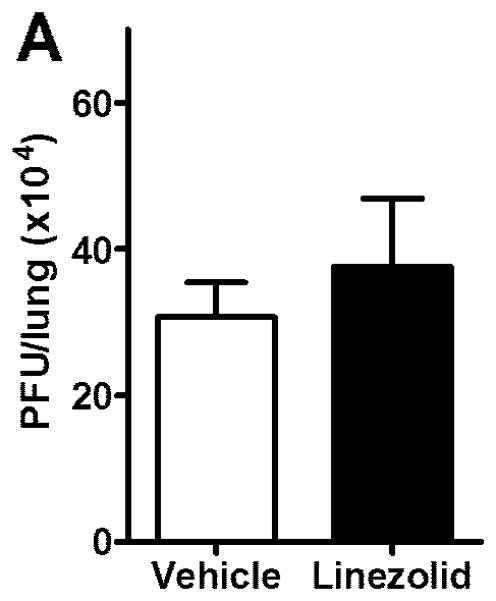
Linezolid does not alter influenza viral burdens. Influenza-infected mice were dosed twice daily with 80mg/kg linezolid or vehicle control. (A) On day 7 post-infection, lungs were harvested and homogenized and viral burden was assessed using the plaque assay. Data are represented as PFU per lung. Mean ± SD are depicted (5 mice per group) and compared by t test. Results are representative of 2 replicated experiments. No statistically significant differences were observed.
Linezolid inhibits IFNγ responses and reduces bacterial loads following secondary S. pneumoniae infection
To model secondary bacterial pneumonia, influenza-infected animals were intranasally challenged with a sublethal dose of S. pneumoniae on day 7 post-infection. Mice received twice daily dosing of linezolid starting on day 1 post-influenza infection, with the last dose being administered 18h before S. pneumoniae challenge (Fig. 4A). In these studies, mice began to lose weight following day 3 of influenza infection, an effect that was blunted by linezolid treatment to a significant degree by day 4 post-infection (Fig. 4B). Following challenge with S. pneumoniae on day 7, the control group on average lost an additional 3.4% body weight, in comparison to a weight loss of only 2.8% in linezolid treated animals. Mice were humanely euthanized at 4 hours and 24 hours after secondary bacterial infection. Here in the co-infection experiments, IFNγ concentrations 4 hours after bacterial challenges were not affected (Fig. 4C). However, linezolid treatment significantly reduced IFNγ concentrations in the BAL at the 24 hours timepoint (p=0.017). Similarly, TNFα concentrations were also significantly diminished at 24h following bacterial challenge in the presence of linezolid (p=0.007; Fig. 4D). The bacterial burdens within the lungs of the animals were determined, and while there was a trend towards suppression of bacterial outgrowth in linezolid-treated animals at 4 hours post-Pneumococcal challenge, the drug significantly decreased bacterial burdens in the lungs of influenza-infected animals at 24 hour post-infection (Fig. 4E). Linezolid decreased bacterial burdens by nearly 1000-fold in comparison to vehicle-treated animals at this time point (p<0.001), a level comparable with animals that were not primarily infected with influenza. We were concerned that levels of residual linezolid remaining in the lungs at the time of challenge with S. pneumoniae might have been sufficient to limit bacterial outgrowth. To rule this out, flu-naïve animals were dosed twice daily with 80mg/kg linezolid or with vehicle for 6 days, with the last dose given 18h before bacterial challenge. When bacterial outgrowth was determined following 24h of S. pneumoniae infection, we observed no differences in the levels of viable Pneumococci recovered from the lungs of either treatment group (Fig. 4F). These data suggest that linezolid is capable of reversing the influenza-dependent immunosuppressive phenotype observed in our studies, without having any direct effect on the growth of S. pneumoniae.
FIGURE 4.

Linezolid blunts IFNγ responses and prevents outgrowth of bacteria following a post-influenza secondary bacterial infection. (A) Mice were intranasally infected (large arrow) with influenza virus and dosed twice daily with linezolid (80mg/kg) or vehicle (small arrows). At day 7, animals were intranasally challenged with S. pneumoniae A66.1 as discussed in the Methods section, and animals were sacrificed at 4h and 24h following bacterial challenge (large arrows). (B) Mean weight loss is represented as percentage of weight loss in comparison to day 0. (C and D) Bronchoalveolar lavage fluid was collected and analyzed for expression of IFNγ (C) and TNFα (D) by ELISA. (E) Lung digest samples were plated for CFU enumeration on blood agar plates and expressed per 0.1gm of lung, with the addition of a flu-naïve group for comparison. (F) To ensure that linezolid did not have a direct effect on bacterial growth, mice were left influenza-naïve, dosed with linezolid or vehicle twice daily, and infected with S. pneumoniae. Bacterial enumeration was performed at 24h post-infection and expressed per lung. Mean values for each group, denoted by horizontal lines (each point representative of one animal), were compared using t tests or one-way ANOVA along with the Student Newman-Keuls post hoc test where appropriate. Data are a compilation of two independent experiments. *p<0.05, **p<0.01, ***p<0.0001
Administration of exogenous rIFNγ reverses the linezolid-induced phenotype observed following secondary bacterial infection
To confirm whether the effects of linezolid are mediated through its impact on IFNγ, animals were infected with influenza, dosed with linezolid or vehicle, and then administered exogenous rIFNγ at 42h and 18h before S. pneumoniae challenge as depicted in Fig. 5A. Administration of rIFNγ to linezolid-treated mice resulted in levels of IFNγ in the BAL that were comparable to that of vehicle-treated controls, although the differences were not statistically significant (Fig. 5B, left panel). As shown in Fig. 4, linezolid treatment dramatically decreased the TNFα present in the alveolar space (Fig. 5B, right panel). The administration of rIFNγ did not reverse the drug’s ability to blunt the production of TNFα, as the mean concentration of the cytokine was not altered post-pneumococcal challenge. However, rIFNγ administration did reverse, in part, the effect of linezolid treatment on the growth of S. pneumoniae (Fig. 5C). While the mean bacterial burden in the mice that received both linezolid and rIFNγ was significantly higher than the group that received linezolid alone, the recombinant cytokine did not completely block the effect of the drug, as the mean CFU burden was significantly lower than that of the group treated with vehicle only. Finally, we performed intracellular cytokine staining on lung digest samples and analyzed the production of TNFα by non-lymphocytes based upon forward and side scatter characteristics as evaluated by flow cytometry. This analysis showed a decrease in the overall number of non-lymphocytic cells in the lungs that were producing TNFα in both the linezolid and linezolid plus rIFNγ groups. These data suggest that linezolid’s ability to decrease the infectivity of S. pneumoniae is at least partially dependent upon the impact on IFNγ.
FIGURE 5.

Linezolid-induced effects on post-influenza secondary bacterial infections is IFNγ-dependent. (A) Animals were intranasally infected with influenza virus and dosed twice daily with linezolid (80mg/kg) or vehicle as depicted. At days 5 and 6 post-infection, a subset of mice that were receiving linezolid were given intranasal instillations of 2ng rIFNγ, with the remainder of the mice receiving vehicle only. At 18h after the last IFNγ dose, animals were intranasally challenged with S. pneumoniae A66.1 as described in the Methods section and sacrificed at 24h following bacterial challenge. An additional group of mice were challenged with S. pneumoniae without prior influenza infection. (B) Bronchoalveolar lavage fluid was collected and analyzed for expression of IFNγ (left panel) and TNFα (right panel) by ELISA. (C) To assess bacterial burden at 24h post-infection, lung digest samples were plated for CFU enumeration on blood agar plates and expressed per 0.1gm of lung. (D) To compare the number of non-lymphocytes that were producing TNFα, intracellular cytokine staining was performed on lung digest samples, and the number of cells within the non-lymphocyte gate that fluoresced positive for TNFα as analyzed by flow cytometry are depicted. Horizontal lines represent mean values, and each symbol is representative of one animal. Data was pooled from multiple experimental replicates, and means were compared using 1-way ANOVA along with the Student Newman-Keuls post hoc test. *p<0.05, **p<0.01, ***p<0.001
DISCUSSION
In our studies, we confirmed that an intranasal challenge with influenza virus A, strain PR8, causes a window of hyporesponsiveness that leads to increased susceptibility to secondary Streptococcus pneumoniae infection, and that this period corresponds with a dramatic rise in IFNγ production. Our studies demonstrated that linezolid treatment of influenza-infected animals was sufficient to blunt both IFNγ and TNFα responses in a dose-dependent manner. Upon secondary infection with Streptococcus pneumoniae 7 days after viral infection, linezolid significantly attenuated the level of bacterial outgrowth at 24h post-bacterial infection and continued to both limit weight loss and suppress IFNγ and TNFα secretion in the alveolar space. To further elucidate the role of IFNγ, exogenous IFNγ was administered to animals receiving linezolid, which caused partial reversal of the drug’s ability to protect from the pathology associated with secondary infection. While these studies suggest that IFNγ contributes to increased susceptibility to secondary bacterial infections, the administration of exogenous IFNγ was insufficient to completely reverse the linezolid phenotype, suggesting that other mediators may be contributing to this phenomenon. While this work did not directly address the role that TNFα contributes to linezolid’s beneficial properties, this is an important consideration that should be a focus of further research.
Host defense against influenza virus is characterized by initial inflammation followed by an adaptive response mounted by CD4 and CD8 T cells, resulting in clearance of the organism along with effective humoral immunity that provides protection against subsequent challenge. An initial influx of neutrophils and macrophages are responsible for limiting viral replication and for the elimination of dying cells. These cells release high levels of proinflammatory cytokines, including IFNγ, IL-1, IL-6, and TNFα. As the adaptive response begins to develop, CD8 T cells are directly responsible for lysis of influenza-infected cells and contribute to resolution of infection with production of the anti-inflammatory cytokine IL-10 (23). The excessive proinflammatory cytokine production is thought to be a potential mediator of the increased susceptibility to bacterial infection. IFNγ has been shown to not be required for clearance of influenza infection (24), but its production by CD8+ T cells may lessen the severity of inflammation (25).
The period of hyporesponsiveness has been linked to several factors, including influenza-induced lung inflammation and cell damage (8), dysfunctional neutrophil responses (11, 12), and increased IL-10 concentrations (13, 14). While initial responses immediately following acute influenza exposure may reveal additional binding sites on lung epithelia for bacterial pathogens (8), the majority of inflammation is resolved between days 4–6 of infection (26, 27), before the time of bacterial infection in our model. Moreover, in a similar time-frame, between days 3 and 6 post-influenza challenge, neutrophils have been demonstrated to be impaired in their ability to phagocytose and generate toxic reactive oxygen species (12). These investigators further showed that in addition to decreased neutrophil functionality, there were neutrophil-independent mechanisms contributing to post-influenza susceptibility to bacterial infection, including that induced by high levels of IL-10 production.
While some studies support the notion that elevated IL-10 concentrations may play a role in increasing susceptibility to infection (13, 14), other investigators have challenged these results. Studies have shown that IL-10−/− mice infected with influenza do not differ from wild type mice in the ability to clear S. pneumoniae in the first four hours following bacterial challenge and that IL-10−/− and wild type mice are equally protected by IFNγ neutralization in the dual infection model (15). In addition, some authors have suggested that the induction and sustained maintenance of alternatively activated macrophages during this period of influenza recovery may play a role in susceptibility to infection with S. pneumoniae, although further evidence is needed to support these claims (28). In our infection model, while the differences did not reach statistical significance, the number of T cells producing IL-10 in the alveolar spaces was consistently decreased, and therefore linezolid’s activity may also be partially related to this.
The inability of exogenous rIFNγ to completely reverse the linezolid-induced phenotype suggests that other mediators may be contributing to this phenotype. Indeed, we found that linezolid causes a trend toward suppression of TNFα production in the alveolar spaces of infected mice. Other investigators have examined the role of TNFα in resolution of influenza infection in the absence of secondary bacterial infection. These studies show that while TNFα is not necessary for the resolution of acute viremia following influenza infection, TNFα−/− mice demonstrate increased lung immunopathology on days 7 through 21 post-influenza infection. TNFα−/− mice had increased IFNγ production and cellular infiltration, in addition to worsened lung histopathology in comparison to wild type mice (23). Our data demonstrates that linezolid is able to blunt TNFα production at day 7 post-influenza infection without altering the percentages of immune cells infiltrating the lung or increasing viral replication. As for the dual infection effect on TNFα, the decreased number of cell producing TNFα and the concentration of TNFα in the BAL are likely due to the inhibiting of the bacterial infection. This is confirmed by the fact that rIFNγ administration partially reversed the bacterial growth but had no effect on restoring TNFα production.
To further examine other potential mechanisms governing the impact on IFNγ levels observed in our studies, we investigated the production of IL-12 and IL-18 in our experimental samples. IL-12 and IL-18 have been shown to augment IFNγ levels following influenza infection, in an antigen-independent manner (29, 30). We examined production of IL-12 and IL-18 protein in BAL samples from animals infected with influenza and treated with 80mg/kg linezolid or with vehicle control and observed no differences in protein production between treatment groups (data not shown). Work from other investigators has shown that compensatory mechanisms exist to govern IFNγ production in the absence of IL-12 or IL-18 (31). Indeed, in our experimental system it appears that the effect of linezolid on IFNγ production is independent of IL-12 and IL-18. Future work will focus on examining further mechanisms governing the observed suppression of IFNγ production by linezolid.
Influenza A infection preferentially increases susceptibility to some bacterial pathogens over others (32, 33). In investigating this discrepancy, authors noted the increase in IFNγ concentrations following influenza A infection, and found that macrophage phagocytosis of varied bacterial strains was differentially affected by the presence of IFNγ. IFNγ inhibited macrophage phagocytosis of the gram-positive bacterium Staphylococcus aureus but failed to inhibit phagocytosis of the gram-negative pathogen E. coli (34). In accordance with other studies (15), this suggests that high IFNγ concentrations following influenza infection may contribute to hyper-susceptibility to S. pneumoniae infection by inhibiting bacterial phagocytosis by macrophages. Other studies have shown that ultra-low doses of anti-IFNγ antibodies are effective in limiting influenza-induced pathology and morbidity (35), which may suggest that attenuation of IFNγ levels may limit susceptibility to secondary infection. The most convincing argument comes from the laboratory of Metzger et al, who demonstrate that animals that lack IFNγ signaling, via genetic deletion or antibody-neutralization, are less susceptible to secondary bacterial infection following influenza, in comparison to animals with intact IFNγ signaling (15).
Despite our results, the use of linezolid as a prophylaxis against secondary pneumonia is unlikely. Linezolid is commonly used to treat resistant gram positive infections (36). Induction of antimicrobial resistance, along with the drug’s high cost, make the use of linezolid in this manner impractical (37). Our results show however that IFNγ could serve as a significant drug target for specific populations at high risk for secondary infections due to influenza.
In our studies, we demonstrate that use of the antimicrobial agent linezolid is sufficient to limit IFNγ production in the lungs of mice, which leads to decreased bacterial outgrowth following challenge with S. pneumoniae. Since linezolid did not alter viral replication or bacterial survival on its own, we can conclude that linezolid’s effects on IFNγ production are important in limiting secondary pneumonia in our model. Further work will focus on IFNγ as a therapeutic target for drug development. Ideal drug candidates should have little to no outside effects on the host responses that may compromise immunity to other common infectious agents. Further work will focus on deeper mechanistic understanding of the role of IFNγ in promoting these infections in order to develop better drug targets for future therapeutics.
Acknowledgments
We thank Jennifer Strange and Greg Bauman of the UK Flow Cytometry Core Facility, and in addiiton we thank Melissa Hollifield and Linda Schutzman for their excellent technical assistance.
This work was supported by the Pfizer ASPIRE grant WS795849 (to DJF) and by the National Institutes of Health grants R21 AI083528 (to BAG) and R01 AI088989 (to BAG).
Abbreviations used in this paper
- BAL
bronchoalveolar lavage
- TBLN
tracheobronchoalveolar lymph nodes
References
- 1.Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. 2008;198:962–970. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sheng ZM, Chertow DS, Ambroggio X, McCall S, Przygodzki RM, Cunningham RE, Maximova OA, Kash JC, Morens DM, Taubenberger JK. Autopsy series of 68 cases dying before and during the 1918 influenza pandemic peak. Proc Natl Acad Sci U S A. 2011;108:16416–16421. doi: 10.1073/pnas.1111179108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwarzmann SW, Adler JL, Sullivan RJ, Jr, Marine WM. Bacterial pneumonia during the Hong Kong influenza epidemic of 1968–1969. Arch Intern Med. 1971;127:1037–1041. [PubMed] [Google Scholar]
- 4.Hageman JC, Uyeki TM, Francis JS, Jernigan DB, Wheeler JG, Bridges CB, Barenkamp SJ, Sievert DM, Srinivasan A, Doherty MC, McDougal LK, Killgore GE, Lopatin UA, Coffman R, MacDonald JK, McAllister SK, Fosheim GE, Patel JB, McDonald LC. Severe community-acquired pneumonia due to Staphylococcus aureus, 2003–04 influenza season. Emerg Infect Dis. 2006;12:894–899. doi: 10.3201/eid1206.051141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valiquette L, Cossette B, Garant MP, Diab H, Pepin J. Impact of a reduction in the use of high-risk antibiotics on the course of an epidemic of Clostridium difficile-associated disease caused by the hypervirulent NAP1/027 strain. Clin Infect Dis. 2007;45(Suppl 2):S112–121. doi: 10.1086/519258. [DOI] [PubMed] [Google Scholar]
- 6.Madhi SA, Klugman KP. A role for Streptococcus pneumoniae in virus-associated pneumonia. Nat Med. 2004;10:811–813. doi: 10.1038/nm1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klugman KP, Chien YW, Madhi SA. Pneumococcal pneumonia and influenza: a deadly combination. Vaccine. 2009;27(Suppl 3):C9–C14. doi: 10.1016/j.vaccine.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 8.Kash JC, Walters KA, Davis AS, Sandouk A, Schwartzman LM, Jagger BW, Chertow DS, Li Q, Kuestner RE, Ozinsky A, Taubenberger JK. Lethal synergism of 2009 pandemic H1N1 influenza virus and Streptococcus pneumoniae coinfection is associated with loss of murine lung repair responses. MBio. 2011;2:1–9. doi: 10.1128/mBio.00172-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCullers JA, Rehg JE. Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J Infect Dis. 2002;186:341–350. doi: 10.1086/341462. [DOI] [PubMed] [Google Scholar]
- 10.van der Sluijs KF, van Elden LJ, Nijhuis M, Schuurman R, Florquin S, Shimizu T, Ishii S, Jansen HM, Lutter R, van der Poll T. Involvement of the platelet-activating factor receptor in host defense against Streptococcus pneumoniae during postinfluenza pneumonia. Am J Physiol Lung Cell Mol Physiol. 2006;290:L194–199. doi: 10.1152/ajplung.00050.2005. [DOI] [PubMed] [Google Scholar]
- 11.Abramson JS, Giebink GS, Mills EL, Quie PG. Polymorphonuclear leukocyte dysfunction during influenza virus infection in chinchillas. J Infect Dis. 1981;143:836–845. doi: 10.1093/infdis/143.6.836. [DOI] [PubMed] [Google Scholar]
- 12.McNamee LA, Harmsen AG. Both Influenza-Induced Neutrophil Dysfunction and Neutrophil-Independent Mechanisms Contribute to Increased Susceptibility to a Secondary Streptococcus pneumoniae Infection. Infect Immun. 2006;74:6707–6721. doi: 10.1128/IAI.00789-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Sluijs KF, van Elden LJ, Nijhuis M, Schuurman R, Pater JM, Florquin S, Goldman M, Jansen HM, Lutter R, van der Poll T. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J Immunol. 2004;172:7603–7609. doi: 10.4049/jimmunol.172.12.7603. [DOI] [PubMed] [Google Scholar]
- 14.van der Sluijs KF, Nijhuis M, Levels JH, Florquin S, Mellor AL, Jansen HM, van der Poll T, Lutter R. Influenza-induced expression of indoleamine 2,3-dioxygenase enhances interleukin-10 production and bacterial outgrowth during secondary pneumococcal pneumonia. J Infect Dis. 2006;193:214–222. doi: 10.1086/498911. [DOI] [PubMed] [Google Scholar]
- 15.Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat Med. 2008;14:558–564. doi: 10.1038/nm1765. [DOI] [PubMed] [Google Scholar]
- 16.Shinabarger DL, Marotti KR, Murray RW, Lin AH, Melchior EP, Swaney SM, Dunyak DS, Demyan WF, Buysse JM. Mechanism of action of oxazolidinones: effects of linezolid and eperezolid on translation reactions. Antimicrob Agents Chemother. 1997;41:2132–2136. doi: 10.1128/aac.41.10.2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takahashi G, Sato N, Yaegashi Y, Kojika M, Matsumoto N, Kikkawa T, Shozushima T, Akitomi S, Aoki K, Ito N, Hoshikawa K, Suzuki Y, Inoue Y, Wakabayashi G, Endo S. Effect of linezolid on cytokine production capacity and plasma endotoxin levels in response to lipopolysaccharide stimulation of whole blood. J Infect Chemother. 2010;16:94–99. doi: 10.1007/s10156-009-0012-5. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Roca P, Mancilla-Ramirez J, Araceli SS, Fernandez-Aviles M, Calderon-Jaimes E. Linezolid Diminishes Inflammatory Cytokine Production from Human Peripheral Blood Mononuclear Cells. Archives of medical research. 2006;37:31–35. doi: 10.1016/j.arcmed.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 19.Cottey R, Rowe CA, Bender BS. Influenza virus. Curr Protoc Immunol. 2001;Chapter 19(Unit 19):11. doi: 10.1002/0471142735.im1911s42. [DOI] [PubMed] [Google Scholar]
- 20.Andes D, van Ogtrop ML, Peng J, Craig WA. In vivo pharmacodynamics of a new oxazolidinone (linezolid) Antimicrob Agents Chemother. 2002;46:3484–3489. doi: 10.1128/AAC.46.11.3484-3489.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keel RA, Crandon JL, Nicolau DP. Pharmacokinetics and pulmonary disposition of tedizolid and linezolid in a murine pneumonia model under variable conditions. Antimicrob Agents Chemother. 2012;56:3420–3422. doi: 10.1128/AAC.06121-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lines JL, Hoskins S, Hollifield M, Cauley LS, Garvy BA. The migration of T cells in response to influenza virus is altered in neonatal mice. J Immunol. 2010;185:2980–2988. doi: 10.4049/jimmunol.0903075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Damjanovic D, Divangahi M, Kugathasan K, Small CL, Zganiacz A, Brown EG, Hogaboam CM, Gauldie J, Xing Z. Negative regulation of lung inflammation and immunopathology by TNF-alpha during acute influenza infection. Am J Pathol. 2011;179:2963–2976. doi: 10.1016/j.ajpath.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Price GE, Gaszewska-Mastarlarz A, Moskophidis D. The role of alpha/beta and gamma interferons in development of immunity to influenza A virus in mice. J Virol. 2000;74:3996–4003. doi: 10.1128/jvi.74.9.3996-4003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiley JA, Cerwenka A, Harkema JR, Dutton RW, Harmsen AG. Production of Interferon-gamma by Influenza Hemagglutinin-Specific CD8 Effector T Cells Influences the Development of Pulmonary Immunopathology. Am J Pathol. 2001;158:119–130. doi: 10.1016/s0002-9440(10)63950-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayden FG, Fritz R, Lobo MC, Alvord W, Strober W, Straus SE. Local and systemic cytokine responses during experimental human influenza A virus infection. Relation to symptom formation and host defense. J Clin Invest. 1998;101:643–649. doi: 10.1172/JCI1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaiser L, Fritz RS, Straus SE, Gubareva L, Hayden FG. Symptom pathogenesis during acute influenza: interleukin-6 and other cytokine responses. J Med Virol. 2001;64:262–268. doi: 10.1002/jmv.1045. [DOI] [PubMed] [Google Scholar]
- 28.Chen WH, Toapanta FR, Shirey KA, Zhang L, Giannelou A, Page C, Frieman MB, Vogel SN, Cross AS. Potential role for alternatively activated macrophages in the secondary bacterial infection during recovery from influenza. Immunol Lett. 2012;141:227–234. doi: 10.1016/j.imlet.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Denton AE, Doherty PC, Turner SJ, La Gruta NL. IL-18, but not IL-12, is required for optimal cytokine production by influenza virus-specific CD8+ T cells. Eur J Immunol. 2007;37:368–375. doi: 10.1002/eji.200636766. [DOI] [PubMed] [Google Scholar]
- 30.Liu B, Mori I, Hossain MJ, Dong L, Takeda K, Kimura Y. Interleukin-18 improves the early defence system against influenza virus infection by augmenting natural killer cell-mediated cytotoxicity. J Gen Virol. 2004;85:423–428. doi: 10.1099/vir.0.19596-0. [DOI] [PubMed] [Google Scholar]
- 31.Zhang JH, Wei HM, Tian ZG. Analysis tissue expression of IFN-gamma in IL-12 and/or IL-18 gene ablated naive mice. Cellular & molecular immunology. 2005;2:68–72. [PubMed] [Google Scholar]
- 32.Brundage JF. Interactions between influenza and bacterial respiratory pathogens: implications for pandemic preparedness. Lancet Infect Dis. 2006;6:303–312. doi: 10.1016/S1473-3099(06)70466-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rothberg MB, Haessler SD, Brown RB. Complications of viral influenza. Am J Med. 2008;121:258–264. doi: 10.1016/j.amjmed.2007.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hang do TT, Choi EJ, Song JY, Kim SE, Kwak J, Shin YK. Differential effect of prior influenza infection on alveolar macrophage phagocytosis of Staphylococcus aureus and Escherichia coli: involvement of interferon-gamma production. Microbiol Immunol. 2011;55:751–759. doi: 10.1111/j.1348-0421.2011.00383.x. [DOI] [PubMed] [Google Scholar]
- 35.Tarasov SA, V, Zarubaev V, Gorbunov EA, Sergeeva SA, Epstein OI. Activity of ultra-low doses of antibodies to gamma-interferon against lethal influenza A(H1N1)2009 virus infection in mice. Antiviral Res. 2012;93:219–224. doi: 10.1016/j.antiviral.2011.11.018. [DOI] [PubMed] [Google Scholar]
- 36.Chien JW, Kucia ML, Salata RA. Use of linezolid, an oxazolidinone, in the treatment of multidrug-resistant gram-positive bacterial infections. Clin Infect Dis. 2000;30:146–151. doi: 10.1086/313597. [DOI] [PubMed] [Google Scholar]
- 37.Ager S, Gould K. Clinical update on linezolid in the treatment of Gram-positive bacterial infections. Infect Drug Resist. 2012;5:87–102. doi: 10.2147/IDR.S25890. [DOI] [PMC free article] [PubMed] [Google Scholar]