

Abstract
Rationale
Adenylyl cyclase (AC) represents one of the principal molecules in the β-adrenergic receptor (βAR) signaling pathway, responsible for the conversion of ATP to the second messenger, cAMP. AC type 5 (ACV) and 6 (ACVI) are the two main isoforms in the heart. While highly homologous in sequence, these two proteins nevertheless play different roles during the development of heart failure. Caveolin-3 is a scaffolding protein, integrating many intracellular signaling molecules in specialized areas called caveolae. In cardiomyocytes, caveolin is predominantly located along invaginations of the cell membrane known as t-tubules.
Objective
We take advantage of ACV and ACVI knockout mouse models to test the hypothesis that there is distinct compartmentalization of these two isoforms in ventricular myocytes.
Methods and Results
We demonstrate that ACV and ACVI isoforms exhibit distinct subcellular localization. ACVI isoform is localized in the plasma membrane outside of the t-tubular region, and is responsible for β1AR signaling-mediated enhancement of the L-type Ca2+ current (ICa,L) in ventricular myocytes. In contrast, ACV isoform is localized mainly in the t-tubular region where its influence on ICa,L is restricted by phosphodiesterase (PDE). We further demonstrate that the interaction between caveolin-3 with ACV and PDE is responsible for the compartmentalization of ACV signaling.
Conclusions
Our results provide new insights into the compartmentalization of the two AC isoforms in the regulation of ICa,L in ventricular myocytes. Since caveolae are found in most mammalian cells, the mechanism of βAR and AC compartmentalization may also be important for βAR signaling in other cell types.
Keywords: Adenylyl cyclase type 5, adenylyl cyclase type 6, L-type Ca2+ current, ventricular myocytes, calcium channel, adrenergic receptor
INTRODUCTION
Adenylyl cyclase (AC)1,2 represents one of the principal effector molecules in the β-adrenergic receptor (βAR) signaling pathway, responsible for the conversion of ATP to the second messenger, cAMP. cAMP activates protein kinase A (PKA) which phosphorylates a number of key Ca2+-cycling proteins in cardiac myocytes, including phospholamban, ryanodine receptors, troponin I, and L-type Ca2+ channel (Cav1.2). Phosphorylation of these target proteins increases cytoplasmic Ca2+, resulting in an increased contractile response of cardiomyocytes. Unlike other components of the βAR signaling pathway, over-expression of AC did not promote cardiac hypertrophy, making it an attractive therapeutic target for heart failure.3
The mammalian AC gene family contains at least ten members4 with a number of AC isoforms detected in the heart at the transcript level, however, only two isoforms (ACV and ACVI) are found in abundance.5,6 These two isoforms share approximately 65% protein sequence identity and both belong to a group of Ca2+-inhibitable AC, which can be inhibited by physiological concentrations of Ca2+.5,7 A fundamental difference between the two isoforms is that ACV appears to cause detrimental effects in the aging model of cardiomyopathy, whereas over-expression of the ACVI isoform confers beneficial effects in models of cardiac hypertrophy.8,9
Subcellular compartmentalization of the β-AR signaling proteins allows a variety of biological functions to be regulated at the same time by only a few β-AR subtypes.10,11 Signaling proteins that are located close to each other can form a working unit, also termed signalosome. Different subcellular localizations have been shown to occur through a number of scaffolding proteins. Previous studies by Ostrom and Insel have provided insights into the roles of caveolin-rich domain in subcellular localization of AC and β-ARs in cardiac myocytes.12,13 Additionally, it has been documented that the interactions between different compartments of the βAR signaling are limited by phosphodiesterase (PDE)-dependent cAMP degradation.14-16
Caveolin is an essential molecule for the formation of caveolae, or membrane invaginations, which are found in most cell types.17 Caveolae increase the cell surface area, which is important for vesicular trafficking, signal transduction, and macromolecular transport. In the heart, the most abundant isoform of caveolin is caveolin-3, which is associated with long membrane invaginations known as t-tubules in addition to the surface membrane.18
Compartmentalization contributes to the complexity of subcellular organization and alterations in a compartment’s signaling pathway could lead or contribute to cardiac pathology. For example, an increase in β2AR reactivity or redistribution of β2AR from t-tubule could contribute to cardiac toxicity during the progression of cardiomyopathy.19 Even though the concepts of compartmentalization are well documented, there is still limited understanding of how different compartments are organized and some of the key effectors in these specialized regions. Specifically, it is not known whether there is similar subcellular compartmentalization of the key effector molecules, ACV and ACVI, in the βAR signaling pathways. Moreover, it has not been established whether different βAR subtypes, namely β1AR and β2AR, mediate their downstream effects via different isoforms of ACs in the heart. Indeed, new insights into the distinct localization and function of the two main AC isoforms will help to shed light on these critical effector molecules during the development and progression of cardiac hypertrophy and failure, possibly provide new therapeutic rationales for these common cardiac conditions.
In order to accomplish these goals, we took advantage of ACV, ACVI, and ACV/ACVI knockout models to test the central hypothesis that there is subcellular compartmentalization of the two main AC isoforms in ventricular myocytes. ICa,L was recorded as a robust readout of the βAR signaling. The effects of β1AR vs. β2AR signaling via the two AC isoforms were tested. Moreover, we provide new mechanistic insights into the pivotal roles of caveolin-3 as a scaffolding protein which links the specific isoform of AC and PDE leading to the specialized subcellular organization, forming a separate functional compartment. Finally, we provide evidence that caveolin-3 represents a critical molecule for the compartmentalization of the βAR signaling cascade in cardiac myocytes.
METHODS
Detailed materials and methods are provided in the Online Supplement.
Animals
All animal care and use procedures were approved by the University of California, Davis Institutional Animal Care and Use Committee. Experiments were performed in accordance with National Institutes of Health and institutional guidelines. Genetically targeted mouse models including ACV knockout (ACV KO)20 and ACVI knockout (ACVI KO)7 were used for the studies. The double knockout (ACV/ACVI KO) mice were generated by crossing the homozygous single KO mice.
Patch-clamp recordings
Single left ventricular myocytes were isolated from 10-12 week old animals. All experiments were performed using the conventional whole-cell patch-clamp technique at room temperature. In some experiments, a detubulation technique consisting of an osmotic shock to internalize the t-tubules was used.21
Cell culture and transfection
Human Embryonic Kidney (HEK) 293 cells were transfected with ACV-HA, ACVI-AU1, and Caveolin3 constructs using Lipofectamine, LTX and PLUS reagents (Invitrogen).
Western blot analyses
Western blot analyses were performed as previously described.22 Primary antibodies used in the study include anti-β-tubulin (Abcam ab6046, 1:4,000 dilution) and anti-Cav1.2 (Sigma C1603, 1:200 dilution) antibodies.
Computer modeling
Rosetta-Membrane de novo method23 was used for structure prediction of caveolin. We first generated 10,000 caveolin models of the transmembrane regions of caveolin followed by model clustering as described previously.23 The center model of the largest cluster was then used for modeling of N- and Ctermini of caveolin using Rosetta loop modeling method.24
The ACV and mutated ACV molecule were modeled using the PHYRE database.25 Only the peptide segment before the first transmembrane segment was included in the modeling. After both ACV models were created, the peptides were truncated to 20 amino acids to mimic our experiments (amino acids 84- 104 of the mouse ACV sequence).
Molecular graphics and analyses were performed using the UCSF Chimera package.26 The Dock 6.4 was used for the docking of the peptides.27,28 The docking of the peptides used a rigid algorithm (the individual parts of the peptide were kept in the same orientation). The caveolin-3 peptide was treated as a receptor and the ACV wild type and mutant peptide was treated as a ligand. The model was run six times with a range of grid from 0.1 to 10 Å to confirm the results.
RESULTS
ACVI isoform is critical for the observed β-AR enhancement of ICa,L in mouse ventricular myocytes
We first evaluated the distinct role of ACV and ACVI in βAR stimulation of ICa,L using whole-cell patch-clamp technique (Figure 1). Ventricular myocytes were isolated from four different groups of animals including WT, ACV KO, ACVI KO, and the double knockout (ACV/ACVI KO). Application of isoproterenol (ISO) at a concentration of 1 μM significantly increased ICa,L in WT ventricular myocytes (Figure 1a). Similar results were observed in the ACV KO group (Figure 1a, middle panels). Remarkably, ISO had no effects on ICa,L in myocytes isolated from ACVI KO (Figure 1a, lower panels) or ACV/ACVI KO myocytes (Online Figure I). In addition, the basal current was significantly decreased (-1.6±0.4 pA/pF at 0 mV) in ACVI KO myocytes (Figure 1a, lower panels). There was no statistical difference in ICa,L between ACVI KO and the double KO myocytes. Summary data for the current density elicited at 0 mV are shown in Figure 1b.
Figure 1. β-AR enhances ICa,L in mouse ventricular cardiomyocytes via the activation of ACVI.
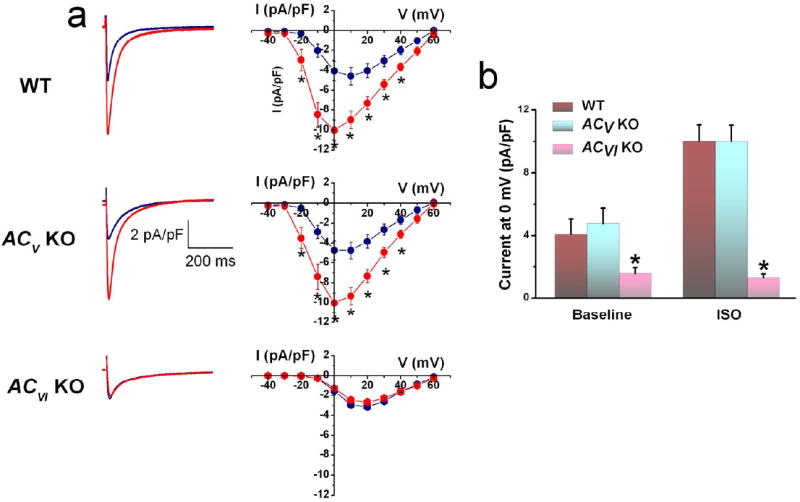
(a) Representative ICa,L recorded from ventricular myocytes isolated from WT, ACV KO, and ACVI KO, respectively. Current traces were elicited using a voltage step of 0 mV for 500 ms from a holding potential of -55 mV at baseline (blue) and after 20 minutes of ISO (red). The corresponding currentvoltage (IV) relations elicited using a family of voltage steps from -40 to +60 mV from a holding potential of -55 mV are shown to the right. (b) Schematic representation of the known regulation of ICa,L by β-AR stimulation. (c) Summary data of ICa,L density (in pA/pF) from the three groups of mice (n=8 for each group, *P<0.05).
ICa,L was recorded 10 minutes after patch rupture for the current to stabilize. Online Figure IIa shows the time course of the current at baseline and up to 30 minutes after ISO demonstrating a relatively stable current in our recording conditions. Indeed, in all our patch-clamp experiments, we have ensured that current recordings were stable with good access before the application of ISO or blockers.
Taken together, these results suggest that the observed βAR enhancement of ICa,L occur mainly via the ACVI isoform. There are three main possibilities as to why ACV does not contribute to the observed ICa,L enhancement. First, ACV isoform may become degraded in the absence of ACVI.7 Second, ACV isoform may not couple directly to βAR. Third, it is possible that the stimulatory effect of the ACV isoform may be compartmentalized and masked by the action of PDE. Indeed, suppression of β2AR stimulation by PDE was previously documented.15 To directly test the latter possibility, we investigated the role of ACV and ACVI separately in the β1AR and β2AR signaling pathways.
β2AR-mediated enhancement of ICa,L signals mainly via ACV isoform
Application of a selective β1AR blocker, CGP-20712A, completely abolished the effect of ISO in WT cardiomyocytes (Figure 2a, upper panel). However, the β2AR-dependent enhancement of ICa,L can be revealed by the application of selective PDE3 and PDE4 blockers, rolipram and cilostamide (Figure 2b & d). Similar to WT ventricular myocytes, application of CGP-20712A also abolished the effect of ISO in the ACV KO (Figure 2b, middle panel). In contrast, additional application of PDE blockers failed to reveal the β2AR-dependent increase of ICa,L (Figure 2b & d). Administration of CGP-20712A in ACVI KO cardiomyocytes did not change the outcome of ISO stimulation, as there was no increase in ICa,L (Figure 2a, lower panel). More importantly, the addition of PDE blockers in ACVI KO mice revealed the β2AR-dependent enhancement of ICa,L (Figure 2b & d) that was not statistically different from the WT group. Taken together, these results suggest that β2AR regulation of ICa,L may signal mainly via the ACV isoform and is under the strong influence of PDE (Schematic representation in Figure 2c).
Figure 2. ACV isoform is critical for β2AR enhancement of ICa,L in the mouse ventricular myocytes.
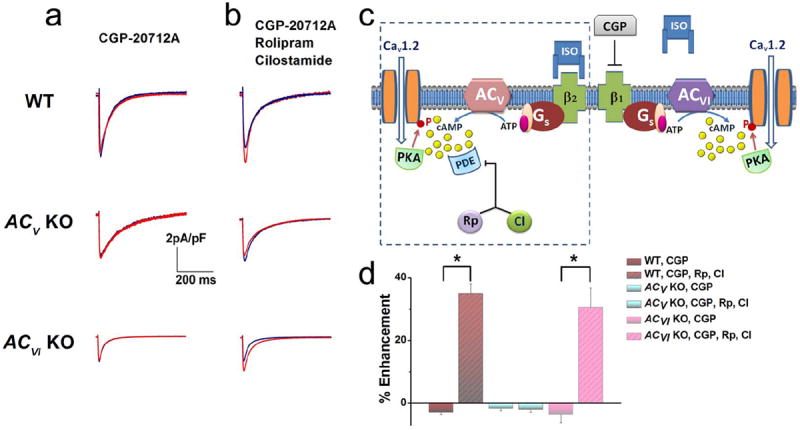
(a) Representative ICa,L recorded from ventricular myocytes isolated from WT, ACV KO, and ACVI KO, respectively. Current traces were elicited using a voltage step of 0 mV for 500 ms from a holding potential of -55 mV at baseline (blue) and after 20 minutes of ISO (red). β1ARs were blocked by CGP-20712A (CGP). No significant effect of β2AR stimulation was observed. (b) Application of a PDE3 blocker (rolipram, Rp) and a PDE4 blocker (cilostamide, CI) revealed the effect of β2AR stimulation only in WT and ACVI KO animals. (c) Schematic representation of the experimental results suggesting that ACV is localized within the same compartment as β2ARs and PDE shown in the box as outlined, separated from β1ARs. (d) Summary data of the percentages of ICa,L enhancement by β2AR stimulation in the three groups of mice with and without PDE blockers (n=7-8 for each group, *P<0.05).
For experiments using PDE blockers, cells were first treated with the blockers for 30 minutes before the addition of ISO. Online Figure IIb shows the time course of the current at baseline, after PDE blockers and then ISO in the presence of the blockers.
The significant decrease in the basal ICa,L in ACVI KO and double KO myocytes was not associated with significant changes in the expression at the protein or transcript levels (Online Figure III). Moreover, application of PDE3 and PDE4 blockers alone did not alter ICa,L (Online Figure IIb) suggesting minimal basal phosphorylation of ICa,L. Future experiments are required to further explore the mechanisms underlying the decrease in ICa,L in ACVI KO mice. Nonetheless, the data so far support distinct functional roles of the two isoforms of AC in ventricular myocytes.
Two distinct populations of β1ARs in ventricular myocytes
To directly examine the downstream signaling pathway of β1ARs, we tested the effect of ISO in the presence of ICI-118,551 (a specific β2AR blocker). In this condition, ISO increased ICa,L in the WT myocytes by 145.8±18.8% at 0 mV (Figure 3a, upper panel). Application of PDE blockers in this condition further potentiated the effect of ISO by 392±63% (Figure 3b & d). A similar enhancement of ICa,L in ACV KO myocytes was observed by the application of ISO in the presence of ICI-118,551 (108±16.3% increase in the current). However, in contrast to the WT myocytes, PDE blockers failed to further potentiate the effects of ISO in ACV KO myocytes (Figure 3b & d).
Figure 3. β1ARs mediate the enhancement effects on ICa,L via both ACV and ACVI isoforms.
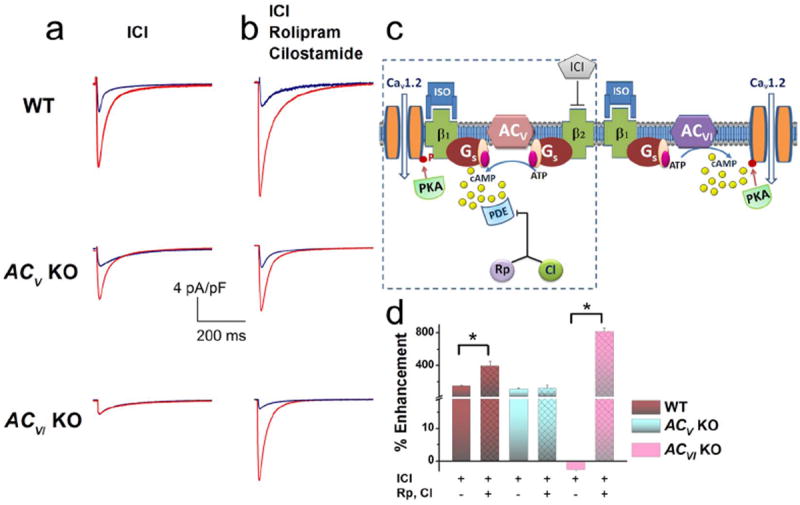
(a) Representative ICa,L recorded from ventricular myocytes isolated from WT, ACV KO, and ACVI KO, respectively. Current traces were elicited using a voltage step of 0 mV for 500 ms from a holding potential of -55 mV at baseline (blue) and after 20 minutes of ISO (red). β2-ARs were blocked by ICI- 118,551 (ICI). No β1AR stimulation of the ICa,L was observed in the ACVI KO group. (b) Application of a PDE3 blocker (Rp) and a PDE4 blocker (CI) revealed the stimulatory effect of β1AR in the ACVI KO mice and further enhanced the effects of β1AR in the WT mice. (c) Schematic representation of the experimental results which suggest that β1ARs mediate the enhancement effects on ICa,L via both ACV and ACVI isoforms. Moreover, only the effect through the ACV isoform is restricted by PDE. (d) Summary data of the percentages of ICa,L enhancement by β1AR stimulation in the three groups of mice with and without PDE blockers (n=6-8 for each group, *P<0.05).
As predicted from the results in Figure 1a (lower panels), there was no enhancement of ICa,L by ISO in the presence of ICI-118,551 in ACVI KO myocytes. However, the application of PDE blockers revealed a marked increase of ICa,L by 814.2±46% (Figure 3b & d). These results suggest that there may exist two populations of β1ARs; one that is associated with ACVI and another that is associated with ACV and is masked by PDE under basal conditions (see diagram in Figure 3c). However, PDE inhibition is known to have effects that are independent of ßAR stimulation which may confound the interpretations. Therefore, additional experiments were performed to further test our hypothesis (Figure 4-6).
Figure 4. Detubulation abolished the enhancement effects of β2AR stimulation on ICa,L via ACV isoform.
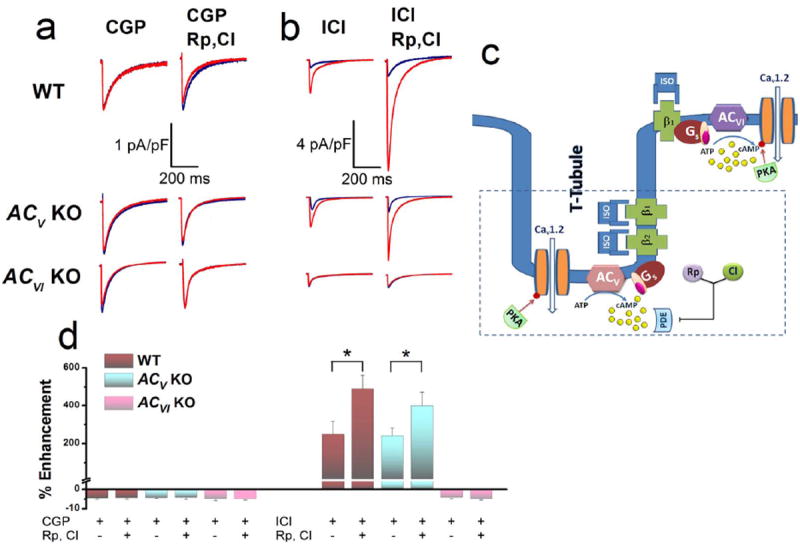
(a) Representative ICa,L recorded from ventricular myocytes after detubulation from WT, ACV KO, and ACVI KO, respectively. Current traces were elicited using a voltage step of 0 mV for 500 ms from a holding potential of -55 mV at baseline (blue) and after 20 minutes of ISO (red) in the presence of a β1AR blocker (CGP) without PDE blockers (left column) and with PDE blockers (right column). Compared to Figure 2, after detubulation, PDE blockers failed to enhance ICa,L in the WT or ACVI KO mice. (b) Similar experiments conducted in the presence of a β2AR blocker (ICI) without PDE blockers (left column) and with PDE blockers (right column). Compared to Figure 3, after detubulation, PDE blockers failed to enhance ICa,L in the ACVI KO group. (c) Schematic representation of the experimental results which support the compartmentalization of ACV signaling within the t-tubule. (d) Summary data of the percentages of ICa,L enhancement in the three groups of mice with and without PDE blockers after detubulation in the presence of β1AR or β2AR blockers (n=6-8 for each group, *P<0.05).
Figure 6. Cavelin 3 associates with PDE and localizes PDE to ACV compartment within t-tubules in ventricular myocytes.
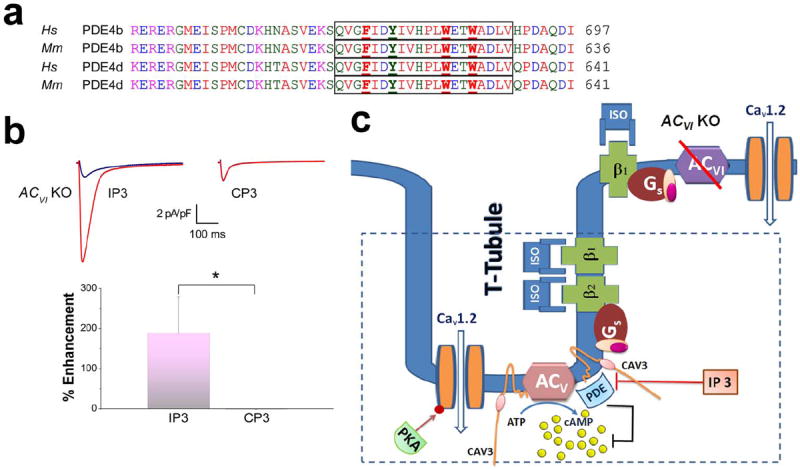
(a) Amino acid sequence alignment of human (Hs) and mouse (Mm) PDE4b and d. Both isoforms contain putative caveolin-binding domains. A 20-amino-acid inhibitory peptide was generated encompassing the putative caveolin-binding domain (outlined in the box, IP3). A corresponding control peptide (CP3) was generated by mutating the aromatic amino acids (shown in bold and underlined) to alanine. (b) Representative ICa,L recorded from ACVI KO cardiomyocytes in the presence of IP3 (left) and CP3 (right) in control (blue) and after 20 minutes ISO application (red). Lower panel shows the summary data of the percentages of ICa,L enhancement in ACVI KO cardiomyocytes in the presence IP3 compared to CP3 (n=6 for each group, *P<0.05). (c) Schematic representation of the experimental results showing the association between caveolin-3 and PDE and the compartmentalization of PDE and ACV isoform within the t-tubules in ventricular myocytes.
ACV and ACVI are the two key isoforms of AC in ventricular myocytes
In order to test the key isoforms of AC in ventricular myocytes, the effects of ISO on ICa,L was tested in ACV/ACVI KO myocytes in the absence and presence of PDE blockers. There were no observable effects of ISO in these conditions (Online Figure Ia-c), suggesting that ACV and ACVI represent the two main isoforms of AC in ventricular myocytes.
Parallel assessment of cAMP levels in ventricular myocytes isolated from ACV/ACV KO and WT mice was performed as previously described.29 The same protocol for patch-clamp recordings was used. Specifically, cells were first incubated with PDE inhibitors for 30 minutes prior to the addition of ß-adrenergic agonist plus PDE inhibitors (Online Figure Id). Results demonstrate a lack of cAMP stimulation in ACV/ACV KO consistent with our patch-clamp data.
ACV isoform is compartmentalized within the t-tubules under the strong influence of PDE while ACVI is localized outside of the t-tubules
Our findings thus far support the signaling of β2AR mainly via the ACV isoform. Since previous studies have provided evidence for the localization of β2AR within the t-tubules,19 we tested the hypothesis that the ACV is similarly compartmentalized within the t-tubules. After the detubulation procedure, there were no changes in the effects of ISO on ICa,L in the WT, ACV KO and ACVI KO myocytes (Online Figure IVa-b compared to Figure 1a). However, there was a dramatic decrease in the cell capacitance and the amplitude of ICa,L (Online Figure IVc, and e). In order to verify successful detubulation, DI-8-ANEPPS was used to demonstrate the loss of the striation pattern (Online Figure IVd).
More importantly, after detubulation, the application of PDE blockers failed to reveal the β2AR enhancement of ICa,L current in WT and ACVI KO cardiomyocytes (Figure 4a, right column compared to Figure 2b). In addition, detubulation led to the absence of β1AR enhancement of ICa,L in ACVI KO cardiomyocytes in the presence of PDE blockers (Figure 4b, lower panels compared to Figure 3b). In contrast, in WT and ACV KO cardiomyocytes, PDE blockers could potentiate the effect of β1AR stimulation after detubulation (Figure 4b, upper and middle panels). Taken together, the data support the notion that β2ARs signal mainly via the ACV isoform. ACV isoform is compartmentalized within the t-tubules and is under the strong influence of PDE. Moreover, ACVI isoform appears to be localized outside of the t-tubules and is under weaker influence of PDE.
Finally, from the detubulation experiments, the fraction of L-type Ca2+ channels which are localized in the t-tubule versus the sarcolemmal membrane are estimated to be ~ 86% (Online Figure IVe). In addition, we estimated the ratio of Ca2+ channels in the t-tubules which are regulated by β1 vs. β2ARs to be 2.4:1 (Online Figure IVe).
Caveolin-3 may anchor and localize ACV within t-tubules
In order to investigate the mechanisms for the proposed compartmentalization of ACV within the t-tubules, we tested the hypothesis that caveolin-3, a scaffolding protein, may interact with both ACV and PDE, thus, localizing ACV within t-tubules under the strong influence of PDE. Indeed, comparison of the amino acid sequence alignment between the N termini of ACV and ACVI isoforms reveals a putative caveolin-binding domain in ACV but not ACVI isoform (Figure 5a). The proposed caveolin-binding domain is found to be conserved in both mouse and human ACV isoform and contains four aromatic amino acids including phenylalanine (PHE) at positions 94, 96, and 98, and tryptophan (TRP) at position 104 in mouse ACV (Figure 5a, Online Figure Va).
Figure 5. Caveolin-3 interacts and anchors ACV isoform within t-tubules in ventricular myocytes.
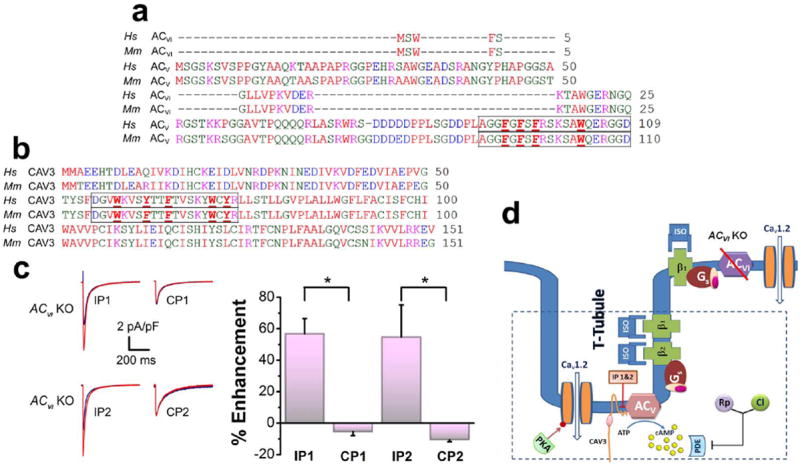
(a, b) Amino acid sequence alignment of the N-termini of human (Hs) and mouse (Mm) ACV and ACVI isoforms in (a) and the N-termini of human (Hs) and mouse (Mm) caveolin-3 (CAV3) in (b). Two inhibitory peptides were generated encompassing the conserved region of ACV (outlined in the box, IP1) and the scaffolding domain in CAV3 (outlined in the box, IP2), respectively. The corresponding control peptides (CP1, CP2) were generated by mutating the aromatic amino acids (shown in bold and underlined in a & b) into alanine. Note that the ACVI isoform does not contain the putative caveolin-binding sequence. (c) Representative ICa,L recorded from ACVI KO in the presence of the inhibitory and control peptides in control (blue) and after 20 minutes ISO application (red). Both inhibitory peptides (IP1 and IP2) revealed the significant stimulatory effects of ISO. Summary data of the percentages of ICa,L enhancement in ACVI KO cardiomyocytes in the presence of the inhibitory or control peptides (n=6 for each group, *P<0.05). (d) Schematic representation of the experimental results which suggest a caveolin binding domain on N-terminus of the ACV isoform. Caveolin-3 interacts and anchors the ACV isoform within the t-tubules of the cardiac myocytes. A pink ellipse represents the caveolin scaffolding domain.
To test the hypothesis that the putative caveolin-binding domain may interact with the previously documented caveolin-scaffolding domain in the N terminus of caveolin-3 (Figure 5b), we first modeled the caveolin-3 protein utilizing the Rosetta Stone algorithm with a modification for the transmembrane region (Online Figure Vb). Utilizing the docking algorithm, we observed the predicted interactions between the putative caveolin-binding domain and caveolin-scaffolding domain (Online Figure Vd & f). Changing each of the amino acids PHE 94, 96, 98, and TRP 104 in the putative caveolin-binding domain to alanine (Online Figure 5e) abolished the interactions in the docking procedure.
Inhibitory peptides derived from the putative caveolin-binding domain or caveolin-scaffolding domain unmask the stimulatory effects of ISO on ICa,L in ACVI KO mice
We next developed two inhibitory peptides (IPs) containing 20 amino acids derived from the N termini of ACV and caveolin-3, which encompass the putative caveolin-binding domain (IP1) and caveolin-scaffolding domain (IP2), respectively. Two control peptides (CPs) were developed by mutating the critical aromatic amino acids (shown in bold in the outlined boxes in Figure 5a & b) to alanine.
If caveolin-3 is indeed interacting with ACV and PDE to localize ACV under the strong influence of PDE, we predict that intracellular application of IPs may disrupt the interaction between caveolin-3 and ACV and unmask the stimulatory effects of ISO on ICa,L in ACVI KO myocytes. In fact, this is what we have observed (Figure 5c compared to Figure 1a, lower panel). In contrast, application of the two control peptides (CP1 or CP2) did not affect the response of ICa,L to ISO in ACVI KO myocytes (Figure 5c). Nonetheless, there are other possible interpretations of the results. It is possible that the IPs may result in the relief of the tonic caveolin-3 inhibition on ACV.
Similarly, we also predict a putative caveolin-binding domain in PDE4 isoforms which appears to be critical for the compartmentalization of ACV isoform (Figure 6a). This sequence is found to be conserved in mouse and human PDE4 across all splice variants (more then 20). An inhibitory peptide was developed using 20 amino acid sequences encompassing the putative caveolin-binding domain of PDE 4b and 4d (IP3, Figure 6a). A control peptide was developed by mutating the aromatic amino acid residues to alanine (CP3). Intracellular application of IP3 resulted in the enhancement of ISO on ICa.L in ACVI KO cardiomyocytes (Figure 6b compared to Figure 1a, lower panel). In contrast, intracellular application of CP3 did not alter the response of ACVI KO cardiomyocytes to ISO (Figure 6b).
The interactions between caveolin-3 and ACV may help to compartmentalize the β2AR signaling
To further test the hypothesis that the interactions between caveolin-3 and ACV may help to compartmentalize the β2AR signaling, we tested the effects of the two inhibitory peptides (IP1 or IP2) on the β2AR regulation of ICa,L (Online Figure VIa). Currents were recorded in control and after ISO stimulation in the presence of a selective β1AR blocker (CGP-20712A). Control experiments are shown to the right using control peptides (CP1 or CP2). In contrast to Figure 2a, there was a significant enhancement of ICa,L in the presence of the inhibitory peptides (~22-30%, Online Figure VIb and c). The enhancement of ICa,L was not observed in ACV KO ventricular myocytes consistent with data shown using PDE inhibitors (Figure 2b, Middle Panel).
We further test the effects of PDE4 inhibitor alone on the β2AR stimulation of WT ventricular myocytes (Online Figure VId). PDE4 inhibitor alone was sufficient in relieving the endogenous PDE inhibition on the ICa,L enhancement consistent with our data using IP3 (Figure 6).
Immunofluorescence confocal microscopic imaging of caveolin-3 after detubulation
In order to properly interpret the results of the detubulation shown in Figure 4, we further confirm that there were no significant changes in the localization of caveolin-3 after the detubulation process (see Online Figure VII and Online Movies). The data further support previous findings that caveolin-3 is localized predominantly in the t-tubules but also present on the surface membrane.12,13
DISCUSSION
βARs represent one of the most important signaling pathways in the control of heart rates and cardiac contractility. Interests in the βAR pathways stem from their seminal roles in both physiologic and pathologic conditions.30-32 Indeed, the use of βAR blockers has been shown to be one of the most beneficial therapies for the treatment of heart failure, hypertension, and cardiac arrhythmias.33
Stimulation of L-type Ca2+ channels by β1 and β2ARs is mediated by different isoforms of AC
Functional differences between ACV and ACVI isoforms have not been extensively investigated until recently. The lack of isoform-specific antibodies and the relatively small amount of AC available on the membrane presented technical challenges which have recently been overcome with the development of gene-targeted mouse models. Previous studies have provided evidence that ACV disruption increases longevity and protect against stress.34 In ACVI KO animals,7 there is a significant decrease in βAR reactivity (approximately 78% reduction). Double knockout animals were previously reported to be non viable, however, we found that double knockout animals could survive despite high mortality rates within the first 3 weeks after birth. After this period, the mice survive without gross abnormalities. This is similar to the limited survival rate of β1AR KO animals.35
Our data support the divergent roles of the two isoforms of AC in the heart. In contrast to previous studies, our data suggest the critical roles of both isoforms. There is a complete lack of enhancement of ICa,L by βAR stimulation using isoproterenol in ACVI KO and ACV/ACVI double knockout myocytes. This is in contrast to the WT and ACV KO myocytes. In order to explain this finding, we considered a possibility that the effects of βAR stimulation via ACV isoform may be masked by PDE. Without PDE blockers, the stimulation of ICa,L from isoproterenol was mostly mediated through β1AR activation (Figures 2a & 3a). Detailed investigation involving the separation of β1AR and β2AR suggest that the effects of β2AR stimulation via ACV isoform are masked by PDE (as shown in Figure 2b). Moreover, the enhancement of ICa,L via ACV isoform appears to be compartmentalized with both β1AR and β2AR, whereas the enhancement of ICa,L via ACVI is mediated mainly through β1AR. Using the detubulation technique, our data suggest that the enhancement effect of ICa,L via ACV is compartmentalized within the t-tubule while the effects of ACVI are coupled to L-type Ca2+ channels which are localized outside of the t-tubules. Indeed, the data is in good agreement with previous findings suggesting that both β1AR and β2AR are located within the t-tubules, whereas only β1AR is present outside the t-tubules.19 Nonetheless, it remains a possibility that ACV isoform is present in other cellular domains where it might not be coupled to the L-type Ca2+ channels directly. Likewise, ACVI isoform may be localized but does not couple to L-type Ca2+ channel in the t-tubules. Future experiments are required to develop antibodies which are isoform-specific to directly establish the subcellular localization of the two isoforms.
Mechanisms underlying the distinct subcellular organization and compartmentalization of AC isoforms
The different isoforms of AC have similar primary structure containing an N-terminus, a six transmembrane domain, a C1 portion of the catalytic domain, a second six transmembrane domain, and a C2 portion of the catalytic domain. The most striking difference between ACV and ACVI is in the N-termini. The N-terminus of ACV is much longer than that of ACVI, leading us to hypothesize that the specific localization of ACV within the t-tubule may be associated with the N-terminus. Since t-tubules are known to contain different lipid contents compared to the outer membrane, we investigated potential differences in the hydrophobicity of the N termini of the two isoforms. However, a hydrophobicity plot did not reveal significant differences between the N-termini of ACV and ACVI isoforms.
On the other hand, several isoforms of ACs have been shown to be suppressed by the addition of caveolin peptides.36 Since caveolin is a scaffolding protein localized within the t-tubules in addition to the surface sarcolemma, we first tested the hypothesis that the ACV isoform may possess a caveolin-binding domain. Indeed, caveolin-binding domain sequences vary greatly among different proteins, but all share a similar pattern of aromatic alternating with non aromatic amino acid residues.37 We observe a putative caveolin-binding sequence within the N terminus of ACV. This stretch of amino acids is absent in the ACVI isoform (Figure 5a). Inhibitory peptides generated from the putative binding sequences are able to disrupt the interaction between caveolin and ACV and unmask the effects of ACV in ACVI KO animals without PDE blockers. Taken together, the data support our notion that this region within the N terminus of ACV is responsible for the association between ACV and caveolin, thus, the specific compartmentalization of ACV within the t-tubules.
The location of PDE4 isoforms along the z-line region was previously reported.16 Two isoforms of PDE, PDE4b and 4d, have been shown to regulate ICa,L in response to βAR stimulation.16,38,39 We observe a putative caveolin-binding domain within the C termini of PDE4b and 4d isoforms which is highly conserved across species. In fact, the sequence is conserved in more than 20 different splice variants of PDE4b and 4d, suggesting its biological significance. Finally, inhibitory peptides derived from this putative binding domain unmask the effects of ACV isoform, suggesting that the binding domain is responsible for the interaction between caveolin and PDEs leading to the compartmentalization of PDE and ACV isoform within the t-tubules. The schematic representation is presented in Figure 6c.
Potential clinical importance and future directions
Our findings support the divergent subcellular localization of the two main isoforms of AC in ventricular myocytes. The data further suggest that the two isoforms represent distinct downstream effector molecules for β1AR vs. β2AR signaling pathways. Due to its specialized location, the effects of ACV isoform are masked by the action of PDE. Finally, the specific protein-protein interactions with the scaffolding protein, caveolin-3, is responsible for the compartmentalization of ACV isoform and PDE4b and 4d within the t-tubules of cardiomyocytes.
Our data further provide prediction for the disparate function of the two isoforms of AC in the heart. Disruption of ACV and PDE4b and 4d could promote βAR signaling. Indeed, an increase in β2AR reactivity has been reported in heart failure, which was explained by the loss of t-tubules in ventricular cardiomyocytes and the redistribution of β2ARs.19 Based on our findings, the loss of t-tubules may also be predicted to disrupt the compartmentalization of the ACV/PDE interaction which may further result in β2AR’s contribution in cardiac toxicity. Finally, our findings help explain the domain organization of βARs in the ventricular cardiomyocytes and suggest that the ACV isoform and PDE4b and 4d isoforms work in concert. Since caveolin-3 represents common scaffolding protein essential for the formation of caveolae in different cell types, our findings may be important not only for cardiac physiology and pathophysiology, but also for the organization of cellular compartmentalization in other cell types.
Supplementary Material
Novelty and Significance.
What Is Known?
Adenylyl cyclase (AC) represents one of the principal effector molecules in the β-adrenergic receptor (βAR) signaling pathway.
AC type 5 (ACV) and type 6 (ACVI) are the two main isoforms in the heart.
Caveolin-3 is a scaffolding protein, integrating many intracellular signaling molecules in specialized areas called caveolae.
What New Information Does This Article Contribute?
The two AC isoforms involved in the regulation of L-type Ca2+ current (ICa,L) in ventricular myocytes show distinct compartmentalization.
ACV compartmentalized within the t-tubule enhances ICa, L via regulatory signaling restricted by phosphodiesterase (PDE).
ACVI, enhances Ica, L localized to the outside of t-tubules.
ACVI isoform is responsible for β1AR signaling-mediated enhancement of the L-type Ca2+ current (ICa,L).
The interaction between caveolin-3 with ACV and PDE is responsible for the compartmentalization of ACV signaling.
AC represents one of the principal effector molecules in the βAR signaling pathway. Even though ACV and ACVI have been identified as the two main isoforms in the heart, it is not known whether these two isoforms have distinct subcellular localization. Here we report that enhancement of ICa,L via ACV isoform is compartmentalized within the t-tubule and is mediated by both β1AR and β2AR. In contrast, the effects of ACVI are coupled to L-type Ca2+ channels which are localized outside of the t-tubules and are mediated mainly through β1AR. We also found that a specific protein-protein interaction with caveolin-3 is responsible for the compartmentalization of ACV isoform and PDE4b and 4d within the t-tubules of cardiomyocytes. Hence, loss of t-tubules, which has been reported in heart failure, could disrupt the compartmentalization of the ACV and PDE, resulting in an increase in the contribution of βAR to cardiac toxicity.
Acknowledgments
The authors acknowledge the UC Davis Health System (UCDHS) Confocal Microscopy Facility.
SOURCES OF FUNDING
Supported by the Department of Veteran Affairs Merit Review Grant (I01BX000576) and the National Institutes of Health Grants (HL085844, HL085727) to N.C., NIH grants (P01 HL066941, HL088426, and HL081741), and a VA Merit Review Award to H.K.H., R.E.M. and P.S. are trainees in NIH T32 HL86350 Training Grant in Basic and Translational Cardiovascular Science to UC Davis, and American Heart Association Grant-in-Aid to T.T.
Nonstandard Abbreviations
- AC
Adenylyl cyclase
- PDE
Phosphodiesterase
- AR
Adrenergic Receptor
- CAV3
Caveolin-3
- ICa,L
L-type Ca2+ current
Footnotes
DISCLOSURES
None.
References
- 1.Sutherland EW, Oye I, Butcher RW. The Action of Epinephrine and the Role of the Adenyl Cyclase System in Hormone Action. Recent Prog Horm Res. 1965;21:623–646. [PubMed] [Google Scholar]
- 2.Sutherland EW, Robison GA. The role of cyclic-3’,5’-AMP in responses to catecholamines and other hormones. Pharmacol Rev. 1966;18:145–161. [PubMed] [Google Scholar]
- 3.Tang T, Gao MH, Hammond HK. Prospects for gene transfer for clinical heart failure. Gene therapy. 2012;19:606–612. doi: 10.1038/gt.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sunahara RK, Dessauer CW, Gilman AG. Complexity and diversity of mammalian adenylyl cyclases. Annu Rev Pharmacol Toxicol. 1996;36:461–480. doi: 10.1146/annurev.pa.36.040196.002333. [DOI] [PubMed] [Google Scholar]
- 5.Beazely MA, Watts VJ. Regulatory properties of adenylate cyclases type 5 and 6: A progress report. European journal of pharmacology. 2006;535:1–12. doi: 10.1016/j.ejphar.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 6.Cooper DM. Molecular and cellular requirements for the regulation of adenylate cyclases by calcium. Biochemical Society transactions. 2003;31:912–915. doi: 10.1042/bst0310912. [DOI] [PubMed] [Google Scholar]
- 7.Tang T, Gao MH, Lai NC, Firth AL, Takahashi T, Guo T, Yuan JX, Roth DM, Hammond HK. Adenylyl cyclase type 6 deletion decreases left ventricular function via impaired calcium handling. Circulation. 2008;117:61–69. doi: 10.1161/CIRCULATIONAHA.107.730069. [DOI] [PubMed] [Google Scholar]
- 8.Roth DM, Bayat H, Drumm JD, Gao MH, Swaney JS, Ander A, Hammond HK. Adenylyl cyclase increases survival in cardiomyopathy. Circulation. 2002;105:1989–1994. doi: 10.1161/01.cir.0000014968.54967.d3. [DOI] [PubMed] [Google Scholar]
- 9.Vatner SF, Yan L, Ishikawa Y, Vatner DE, Sadoshima J. Adenylyl cyclase type 5 disruption prolongs longevity and protects the heart against stress. Circ J. 2009;73:195–200. doi: 10.1253/circj.cj-08-0957. [DOI] [PubMed] [Google Scholar]
- 10.Steinberg SF, Brunton LL. Compartmentation of G protein-coupled signaling pathways in cardiac myocytes. Annu Rev Pharmacol Toxicol. 2001;41:751–773. doi: 10.1146/annurev.pharmtox.41.1.751. [DOI] [PubMed] [Google Scholar]
- 11.Xiang YK. Compartmentalization of beta-adrenergic signals in cardiomyocytes. Circulation research. 2011;109:231–244. doi: 10.1161/CIRCRESAHA.110.231340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Insel PA, Head BP, Ostrom RS, Patel HH, Swaney JS, Tang CM, Roth DM. Caveolae and lipid rafts: G protein-coupled receptor signaling microdomains in cardiac myocytes. Ann N Y Acad Sci. 2005;1047:166–172. doi: 10.1196/annals.1341.015. [DOI] [PubMed] [Google Scholar]
- 13.Ostrom RS, Gregorian C, Drenan RM, Xiang Y, Regan JW, Insel PA. Receptor number and caveolar co-localization determine receptor coupling efficiency to adenylyl cyclase. J Biol Chem. 2001;276:42063–42069. doi: 10.1074/jbc.M105348200. [DOI] [PubMed] [Google Scholar]
- 14.Fischmeister R, Castro LR, Abi-Gerges A, Rochais F, Jurevicius J, Leroy J, Vandecasteele G. Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circulation research. 2006;99:816–828. doi: 10.1161/01.RES.0000246118.98832.04. [DOI] [PubMed] [Google Scholar]
- 15.Rochais F, Abi-Gerges A, Horner K, Lefebvre F, Cooper DM, Conti M, Fischmeister R, Vandecasteele G. A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circulation research. 2006;98:1081–1088. doi: 10.1161/01.RES.0000218493.09370.8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leroy J, Richter W, Mika D, Castro LR, Abi-Gerges A, Xie M, Scheitrum C, Lefebvre F, Schittl J, Mateo P, Westenbroek R, Catterall WA, Charpentier F, Conti M, Fischmeister R, Vandecasteele G. Phosphodiesterase 4B in the cardiac L-type Ca2+ channel complex regulates Ca2+ current and protects against ventricular arrhythmias in mice. The Journal of clinical investigation. 2011;121:2651–2661. doi: 10.1172/JCI44747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams TM, Lisanti MP. The caveolin proteins. Genome Biol. 2004;5:214. doi: 10.1186/gb-2004-5-3-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Head BP, Patel HH, Roth DM, Lai NC, Niesman IR, Farquhar MG, Insel PA. G-protein-coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin-3-associated microdomains in adult cardiac myocytes. J Biol Chem. 2005;280:31036–31044. doi: 10.1074/jbc.M502540200. [DOI] [PubMed] [Google Scholar]
- 19.Nikolaev VO, Moshkov A, Lyon AR, Miragoli M, Novak P, Paur H, Lohse MJ, Korchev YE, Harding SE, Gorelik J. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science. 2010;327:1653–1657. doi: 10.1126/science.1185988. [DOI] [PubMed] [Google Scholar]
- 20.Lee KW, Hong JH, Choi IY, Che Y, Lee JK, Yang SD, Song CW, Kang HS, Lee JH, Noh JS, Shin HS, Han PL. Impaired D2 dopamine receptor function in mice lacking type 5 adenylyl cyclase. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22:7931–7940. doi: 10.1523/JNEUROSCI.22-18-07931.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brette F, Komukai K, Orchard CH. Validation of formamide as a detubulation agent in isolated rat cardiac cells. Am J Physiol Heart Circ Physiol. 2002;283:H1720–1728. doi: 10.1152/ajpheart.00347.2002. [DOI] [PubMed] [Google Scholar]
- 22.Lu L, Zhang Q, Timofeyev V, Zhang Z, Young JN, Shin HS, Knowlton AA, Chiamvimonvat N. Molecular coupling of a Ca2+-activated K+ channel to L-type Ca2+ channels via alpha-actinin2. Circ Res. 2007;100:112–120. doi: 10.1161/01.RES.0000253095.44186.72. [DOI] [PubMed] [Google Scholar]
- 23.Bonneau R, Strauss CE, Baker D. Improving the performance of Rosetta using multiple sequence alignment information and global measures of hydrophobic core formation. Proteins. 2001;43:1–11. doi: 10.1002/1097-0134(20010401)43:1<1::aid-prot1012>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 24.Wang C, Bradley P, Baker D. Protein-protein docking with backbone flexibility. J Mol Biol. 2007;373:503–519. doi: 10.1016/j.jmb.2007.07.050. [DOI] [PubMed] [Google Scholar]
- 25.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 26.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 27.Moustakas DT, Lang PT, Pegg S, Pettersen E, Kuntz ID, Brooijmans N, Rizzo RC. Development and validation of a modular, extensible docking program: DOCK 5. J Comput Aided Mol Des. 2006;20:601–619. doi: 10.1007/s10822-006-9060-4. [DOI] [PubMed] [Google Scholar]
- 28.Graves AP, Shivakumar DM, Boyce SE, Jacobson MP, Case DA, Shoichet BK. Rescoring docking hit lists for model cavity sites: predictions and experimental testing. J Mol Biol. 2008;377:914–934. doi: 10.1016/j.jmb.2008.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Timofeyev V, Porter CA, Tuteja D, Qiu H, Li N, Tang T, Singapuri A, Han PL, Lopez JE, Hammond HK, Chiamvimonvat N. Disruption of adenylyl cyclase type V does not rescue the phenotype of cardiac-specific overexpression of Gaq protein-induced cardiomyopathy. Am J Physiol Heart Circ Physiol. 2010;299:H1459–1467. doi: 10.1152/ajpheart.01208.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brodde OE, Bruck H, Leineweber K. Cardiac adrenoceptors: physiological and pathophysiological relevance. Journal of pharmacological sciences. 2006;100:323–337. doi: 10.1254/jphs.crj06001x. [DOI] [PubMed] [Google Scholar]
- 31.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 32.Dorn GW, 2nd, Liggett SB. Pharmacogenomics of beta-adrenergic receptors and their accessory signaling proteins in heart failure. Clin Transl Sci. 2008;1:255–262. doi: 10.1111/j.1752-8062.2008.00059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bristow MR. Treatment of chronic heart failure with beta-adrenergic receptor antagonists: a convergence of receptor pharmacology and clinical cardiology. Circulation research. 109:1176–1194. doi: 10.1161/CIRCRESAHA.111.245092. [DOI] [PubMed] [Google Scholar]
- 34.Yan L, Vatner DE, O’Connor JP, Ivessa A, Ge H, Chen W, Hirotani S, Ishikawa Y, Sadoshima J, Vatner SF. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell. 2007;130:247–258. doi: 10.1016/j.cell.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 35.Rohrer DK, Desai KH, Jasper JR, Stevens ME, Regula DP, Jr, Barsh GS, Bernstein D, Kobilka BK. Targeted disruption of the mouse beta1-adrenergic receptor gene: developmental and cardiovascular effects. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:7375–7380. doi: 10.1073/pnas.93.14.7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toya Y, Schwencke C, Couet J, Lisanti MP, Ishikawa Y. Inhibition of adenylyl cyclase by caveolin peptides. Endocrinology. 1998;139:2025–2031. doi: 10.1210/endo.139.4.5957. [DOI] [PubMed] [Google Scholar]
- 37.Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolaeassociated proteins. J Biol Chem. 1997;272:6525–6533. doi: 10.1074/jbc.272.10.6525. [DOI] [PubMed] [Google Scholar]
- 38.Maltsev VA, Ji GJ, Wobus AM, Fleischmann BK, Hescheler J. Establishment of beta-adrenergic modulation of L-type Ca2+ current in the early stages of cardiomyocyte development. Circulation research. 1999;84:136–145. doi: 10.1161/01.res.84.2.136. [DOI] [PubMed] [Google Scholar]
- 39.Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.