

Abstract
Background
Levodopa remains the most effective drug for the treatment of Parkinson’s disease (PD). However, long-term levodopa treatment is associated with the emergence of levodopa-induced dyskinesia (LID), which has hampered its use for PD treatment. The mechanisms of LID are only partially understood. A previous study showed that KN-93, a Ca2+/calmodulin-dependent protein kinase II (CaMKII) inhibitor, could be used to ameliorate LID in rats. However, the precise mechanisms by which KN-93 acts as an antidyskinetic are not fully understood.
Methods
In the present study, a rat model of PD was induced by 6-hydroxydopamine (OHDA) injections. Then, the successfully lesioned rats were intrastriatally administered with a different dose of KN-93 (1 μg, 2 μg, or 5 μg) prior to levodopa treatment. Abnormal involuntary movements (AIMs) scores and apomorphine-induced rotations were measured in PD rats. Phosphorylated levels of GluR1 at Serine-845 (pGluR1S845) levels were determined by western blot. Arc and Penk levels were measured by real-time polymerase chain reaction (PCR).
Results
We found that both 2 μg and 5 μg KN-93 treatment lowered AIMs scores in levodopa priming PD rats without affecting the antiparkinsonian effect of levodopa. In agreement with behavioral analysis, KN-93 treatment (2 μg) reduced pGluR1S845 levels in PD rats. Moreover, KN-93 treatment (2 μg) reduced the expression of Gad1 and Nur77 in PD rats.
Conclusion
These data indicated that intrastriatal injections of KN-93 were beneficial in reducing the expression of LID by lowering the expression of pGluR1S845 via suppressing the activation of CaMKII in PD rats. Decreased expression of pGluR1S845 further reduced the expression of Gad1 and Nur77 in PD rats.
Keywords: Parkinson’s disease, levodopa-induced dyskinesia, KN-93
Introduction
Parkinson’s disease (PD) is a common neurodegenerative disease in elderly patients, which is characterized by loss of dopaminergic neurons in the substantia nigra.1 Although levodopa remains the best drug for the treatment of PD, 30%–50% of PD patients suffer from levodopa-induced dyskinesia (LID) after administration of levodopa for 5–10 years.2,3 At present, the mechanisms of LID are not fully understood; however, the main hypothesis for LID is that the responsiveness of the dopamine D1 receptor in the direct pathway is enhanced after repeated administration of levodopa.4,5 In addition to D1 receptor enhancement, it has been demonstrated that nondopaminergic receptors, especially GluR1, are also involved in LID.6 A previous study showed that phosphorylated GluR1 at Serine-831 (pGluR1S831), activated by Ca2+/calmodulin-dependent protein kinase II (CaMKII) contributed to the expression of LID expression in PD rats.7 Intrastriatal administration of KN-93 (2 μg), a CaMKII inhibitor, ameliorated LID by reducing the degree of pGluR1 S831 in a rat model of PD. As we know, GluR1 can be phosphorylated at Serine-831 and Serine-845. Whether phosphorylated GluR1 at Serine-845 (pGluR1 S845) contributes to the development of LID is still unknown. Numerous studies have indicated that the long-term effect of persistent activation of cyclic adenosine monophosphate (cAMP) signaling induces expression of many genes, such as Gad1 and Nur77,8,9 which are involved in the expression of LID. Studies also suggested that, in most cases, these genes specifically affect the striatonigral neurons of the direct pathway.10 But whether pGluR1 induces LID by changing the expression of genes associated with LID is also unknown. Thus, in the present study, we investigated the effects of intrastriatal administration of KN-93 at different doses (1 μg, 2 μg, or 5 μg) on the expression of LID in a rat model of PD. The effects of KN-93 on the expression of pGluR1S845 and other genes, including Gad1 and Nur77, were also investigated.
Methods
Experimental design
As shown in Figure 1, 6-hydroxydopamine (OHDA) injections were used to produce a rat model of PD. After 3 weeks of injections, rats showing stable apomorphine-induced rotations (>7 turns/minutes) were selected as valid PD rats. Forty valid PD rats were treated with levodopa plus benserazide twice daily for 21 days. Abnormal involuntary movements (AIMs) scores were measured on days 1, 7, 14, and 21 of treatment. In addition, one group of PD rats (PD group, n = 10) and another group of sham-operated rats (sham group, n = 10) were treated with vehicle twice daily for 21 days. On day 22, 40 levodopa-treated PD rats were randomly divided into four groups: levodopa + KN-93 (1 μg) group, levodopa + KN-93 (2 μg) group, levodopa + KN-93 (5 μg) group, and levodopa + vehicle group. These rats were intrastriatally treated with different doses of KN-93 (1 μg, 2 μg, or 5 μg) or vehicle before levodopa treatment, respectively. As a control, the sham group and PD group rats received intrastriatal administration of vehicle before vehicle treatment. Apomorphine-induced rotations were measured to observe the antiparkinsonian effect of KN-93 on day 1 and day 22. AIMs scores were measured to observe the antidyskinetic effect of KN-93 in rats on days of 1, 7, 14, 21, and 22. By the end of the experiment, rats were sacrificed by deep anesthesia using 3% phenobarbital. Western blot was used to determine levels of GluR1 and pGluR1S845 in rats. Real-time polymerase chain reaction (PCR) was used to measure levels of Gad1 and Nurr77 in rats.
Figure 1.
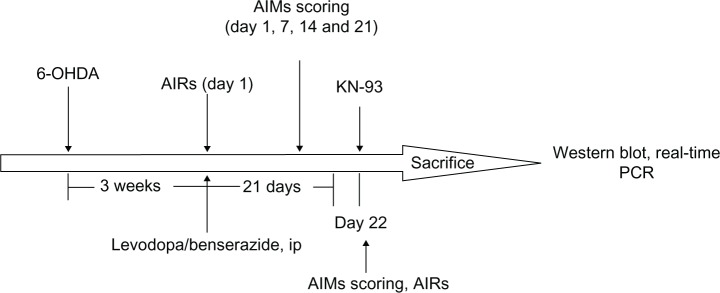
The experiment protocol.
Abbreviations: AIMs, abnormal involuntary movements; AIRs, apomorphine-induced rotations; ip, intraperitoneal; OHDA, hydroxydopamine; PCR, polymerase chain reaction.
Animals
Ninety adult rats (Sprague Dawley females weighing 180–220 g) were used in this study. All protocols involving the animals were approved by the Institutional Review Board of Xinhua Hospital and were performed according to the guidelines of the National Institutes of Health for the care and use of laboratory animals (NIH publication number 80–23).
Lesions
All animals received injections of a total of 8 μg of 6-OHDA (dissolved in 4 μL of 0.9% physiological saline containing 0.02% ascorbic acid; Sigma-Aldrich Corporation, St Louis, Missouri, USA). The coordinates were calculated using the rat brain atlas11 as follows: anterior-posterior (AP): −4.4 mm; medial-lateral (ML): −1.2 mm; and dorsal-ventral (DV): −7.8 mm. The tooth bar was set to −2.4 mm. One week after injections, the rats that exhibited a stable apomorphine-induced rotational asymmetry of at least seven full turns per minute away from the lesioned side were selected for the next experiment.
Treatment
Forty PD rats were treated with levodopa (25 mg/kg plus benserazide 12.5 mg/kg, subcutaneously), twice daily for 21 days. On day 22, 40 PD rats were randomly divided into four groups (n = 10) and intrastriatally administrated with KN-93 (1 μg, 2 μg, or 5 μg) or same volume of vehicle before levodopa administration. As controls, 10 sham-lesioned rats and 10 PD rats were treated with vehicle twice daily for 21 days. On day 22, these vehicle-treated rats were intrastriatally administrated with vehicle before vehicle treatment.
Behavioral test
For apomorphine-induced rotation, rats were injected with apomorphine (0.5 mg/kg, intraperitoneally). Ten minutes after injections, apomorphine-induced rotations were measured for a period of 30 minutes.
For AIMs rating, similar to the method described previously,12 on test days, rats were individually placed in plastic trays 5 minutes before drug treatments. Following injections, each rat was assessed for exhibition of axial, limb, orolingual, and locomotor movements. At 20 minute intervals (20, 40, 60, 80 minutes, etc), AIMs were rated for 60 seconds for each rat for a total of 2 hours (120 minutes). During this time, a severity score of 0–4 was assigned for each AIMs category. For each AIMs category, the scores were summed for each time point.
Protein extraction
Total- and membrane-enriched protein were extracted using a method described previously7 The striatal tissue was homogenized by sonication. The homogenate was centrifuged at 800g for 10 minutes at 4°C, and the pellet containing mainly nuclei and large debris was discarded. Then, the supernatant was centrifuged again at 10,000g at 4°C for 30 minutes. After centrifugation, the pellet containing membrane and cytoplasmic compartments was used as total striatal protein homogenate. To pellet the synaptosomal membrane fraction, the total protein pellet was rehomogenized in ice-cold sample buffer containing 320 mM sucrose and centrifuged at 25,000g at 4°C for 30 minutes. After each centrifugation, the resulting pellet was rinsed briefly with ice-cold TEVP (10 mM Tris-Cl (pH 7.4), 5 mM NaF, 1 mM Na3VO4, 1 mM EDTA, 1 mM EGTA) buffer before subsequent fractionations to avoid possible crossover contamination. The total striatal protein homogenate pellet and synaptosomal membrane-enriched pellet were resuspended in ice-cold sample buffer. Protein concentrations were measured using a bicinchoninic acid (BCA) assay kit (Biyuntian, Jiangsu, People’s Republic of China).
Western blot
Striatal tissues were homogenized (1:10, weight:volume) in 20 mM Tris–HCl (pH 7.4), containing 1 mM NaF, 150 mM NaCl, 1% Triton-100, and freshly-added protease inhibitor cocktail (CalBiochem International, La Jolla, CA, USA), and 100 μM phenylmethylsulfonyl fluoride. Cytosols were prepared by centrifugation at 12,000 g for 10 minutes at 4°C. Total protein (40 μg) was separated on a 10% sodium dodecyl sulfate-polyacrylamide gel and electrophoretically transferred to polyvinylidene fluoride membranes in Tris-glycine transfer buffer. Then, membranes were blocked in 5% (weight/volume) instant non-fat dried milk for 2 hours at room temperature, and incubated with primary antibodies corresponding to GluR1 (1:1000; Cell Signaling Technology, Beverly, MA, USA), or pGluR1S845 (1:500; Cell Signaling) at 4°C overnight. The membranes were subsequently washed with (50 mM Tris–HCl [pH 7.5], 150 mM NaCl, 0.05% Tween 20) and incubated with secondary horseradish peroxidase conjugated IgG for 1 hour at room temperature. Immunoreactive proteins were visualized by LumiGLO (Cell Signaling Technology) chemiluminescent reagent and peroxide. The light-emitting bands were detected on X-ray film.
Real-time PCR
After rats were sacrificed by deep anesthesia, striatal tissues of rats were homogenized and total ribonucleic acid (RNA) was extracted by Trizol reagent (Life Technologies Corporation, Carlsbad CA, USA). The primer sequences used in this study were as follows: 5′-CTGCACAGCTTGGGTGTTGA-3′ (forward) and 5′-CAGACAGCTAGCAATGCGGTTC-3′ (reverse) for Nurr77; 5′-TCACAAGATGATGGGTGTGCTG-3′ (forward) and 5′-GTCATACTGCTTGTCTGGCTGGA-3′ (reverse) for Gad1. The PCR amplifications were performed with 50 cycles of denaturation at 95°C for 15 seconds, annealing at 60°C for 30 seconds, and extension at 72°C for 20 seconds using the ABI 7300 Real-Time PCR System (Applied Biosystems, Carlsbad CA, USA). Results were expressed as relative expression corrected to the housekeeping gene β-actin. The detector used in real-time PCR reaction is Taqman.
Results
Intrastriatal KN-93 treatment had no effect on apomorphine-induced rotations in PD rats
Apomorphine-induced rotation was measured in PD rats treated with KN-93 (1 μg, 2 μg, or 5 μg) or vehicle. As shown in Figure 2, no differences in apomorphine-induced rotations were observed in PD rats treated with different dose of KN-93 or vehicle, suggesting that KN-93 had no anti-parkinsonism effect in PD rats.
Figure 2.

KN-93 had no antiparkinsonian effect on PD rats. Levodopa treatment decreased apomorphine-induced rotations in PD rats on day 22 compared to day 1.
Notes: There was no significance in apomorphine-induced rotations between rats treated with vehicle and different dose of KN-93. Data are presented as mean ± standard error of the mean (error bars). *P < 0.01, compared to day 1.
Abbreviation: PD, Parkinson’s disease.
Intrastriatal KN-93 treatment reduced levodopa-induced dyskinesia in PD rats
As shown in Figure 3, increased AIMs scores in PD rats were observed in PD rats after administration of levodopa. However, intrastriatal KN-93 treatment (1 μg, 2 μg, or 5 μg) reduced AIMs scores in PD rats compared to vehicle-treated animals (P < 0.05). Moreover, the AIMs scores of both 2 μg and 5 μg treatment groups were significantly decreased when compared to 1 μg treatment group (P < 0.01 for both). This indicates that 2 μg or 5 μg KN-93 treatment might offer better results than 1 μg KN-93 treatment. However, there was no difference between PD rats treated with 2 μg or 5 μg KN-93.
Figure 3.
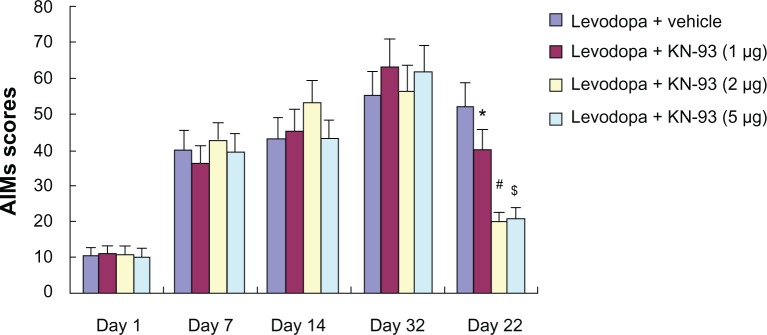
KN-93 treatment reduced levodopa-induced dyskinesia in PD rats. Intermittent levodopa treatment induced increased AIMs scores in PD rats. Intrastriatal KN-93 administration prior to levodopa treatment decreased AIMs scores in PD rats.
Notes: Data are presented as mean ± standard error of the mean (error bars). *P < 0.05 versus Levodopa + vehicle. #P < 0.01 and $P < 0.01 versus Levodopa + KN-93 (1 μg); n = 10.
Abbreviations: AIMs, abnormal involuntary movements; PD, Parkinson’s disease.
Intrastriatal KN-93 treatment reduced phosphorylated levels of GluR1 in the striatum of PD rats
Western blot was used to determine the levels of GluR1 in the total striatal homogenized protein and membrane as well as pGluR1S845 levels in the membrane. No significant difference in GluR1 levels in the total striatal homogenates was found in rats of different groups (data not shown). Similarly, there was no difference in GluR1 levels in the membranes of rats treated with different doses of KN-93 or vehicle (data not shown). Next we analyzed pGluR1S845 levels in the membrane. The results showed that 6-OHDA lesions induced decreased levels of striatal pGluR1S845 in the membrane of PD rats (Figure 4; P < 0.05). Subsequent levodopa treatment increased pGluR1S845 levels in PD rats compared to controls (P < 0.01). Reversely, intrastriatal KN-93 treatment (2 μg) reduced the increase of pGluR1S845 in PD rats (P < 0.05).
Figure 4.

Intrastriatal KN-93 treatment reduced pGluR1S845 levels in PD rats. Total protein levels and membrane levels of GluR1 were decreased in PD rats. (A) Typical immunoblot images detected by an antibody against pGluR1S845. (B) pGluR1S845 levels in the membrane were decreased in PD rats compared to sham-operated rats. Repeated administration of levodopa increased the level of pGluR1S845 in PD rats. Conversely, intrastriatal KN-93 treatment reduced the increase in pGluR1S845 induced by repeated levodopa administration. Data are presented as mean ± standard error of the mean (error bars). *P < 0.05 versus sham; **P < 0.01 versus PD; #P < 0.05 versus Levodopa + vehicle; n = 5.
Abbreviation: PD, Parkinson’s disease.
Intrastriatal KN-93 treatment reduced the expression of Gad1 and Nur77 in PD rats
After extraction of total RNA in PD rats treated with KN-93 or vehicle, real-time PCR was used to determine the levels of Gad1 and Nur77 in rats. As shown in Figure 5, 6-OHDA induced increased levels of Gad1 and Nur77 in PD rats (all P < 0.05). Further increased levels of Gad1 and Nur77 were found in PD rats after repeated administration of levodopa (all P < 0.05), which indicates that Gad1 and Nur77 may be involved in the expression of LID. However, intrastriatal KN-93 treatment (2 μg or 5 μg) reduced the increase in Gad1 and Nur77 while reducing AIMs scores in PD rats (all P < 0.05).
Figure 5.

Intrastriatal KN-93 treatment reduced the expression of Gad1 (A) and Nur77 (B) in PD rats. 6-OHDA lesions induced increased Gad1 and Nur77 in PD rats. Levodopa treatment induced further increase in PD rats. However, intrastriatal KN-93 administration prior to levodopa treatment prevented the increase of Gad1 and Nur77 in PD rats.
Notes: Data are presented as mean ± standard error of the mean (error bars). In (A), *P < 0.05 versus sham. #P < 0.05 versus PD. ##P < 0.05 versus Levodopa + vehicle. In (B), *P < 0.05 versus sham. #P < 0.01 versus PD. ##P < 0.01 versus Levodopa + vehicle; n = 5.
Abbreviations: mRNA, messenger ribonucleic acid; OHDA, hydroxydopamine; PD, Parkinson’s disease.
Discussion
In the present study, we found that both 2 μg KN-93 and 5 μg KN-93 treatment could reduce the expression of LID in levodopa primed PD rats. One μg KN-93 treatment had no effect on LID expression in PD rats. Notably, there was no difference in AIM scores between 2 μg KN-93 and 5 μg KN-93-treated PD rats, suggesting that the 2 μg KN-93 treatment was sufficient to ameliorate LID in PD rats. More importantly, we found that KN-93-treated PD rats showed decreased apomorphine-induced rotations, which was comparable with vehicle-treated PD rats, suggesting that KN-93 had no antiparkinsonian effect on PD rats. We also found that the levels of pGluR1S845 were increased after 21 days of levodopa treatment. In contrast, intrastriatal administration of KN-93 (2 μg or 5 μg) reduced the increase in pGluRS845 levels while ameliorating the expression of LID. In addition, we found that Gad1 and Nur77 gene expression increased after chronic levodopa treatment in PD rats. However, intrastriatal administration of KN-93 (2 μg or 5 μg) reduced the increase of Gad1 and Nur77.
LID induced by long-term levodopa treatment has hampered the use of levodopa for the treatment of PD.13 The main cause of LID is that the responsiveness of striatal neurons in the direct striatonigral pathway is enhanced after intermittent administration of levodopa.14–16 Persistent hyper-activation of cAMP signaling produced via activation of the D1 receptor plays an important in the expression of LID.17 Numerous studies have indicated that long-term levodopa treatment easily induced LID in PD rats.18,19 In line with a previous study,20 in the present work we found that 21 days of levodopa treatment induced full LID expression including axial dyskinesia, forelimb dyskinesia, orolingual dyskinesia, and locomotive dyskinesia as demonstrated by increased AIMs in PD rats. Conversely, intrastriatal administration of KN-93 (2 μg or 5 μg) reduced AIMs scores in PD rats. We also found that there was no difference in AIMs scores in PD rats treated with 2 μg or 5 μg KN-93. This indicates that 2 μg KN-93 may be a suitable dose for reducing LID expression in PD rats.
A previous study indicated that during chronic treatment with levodopa, activation of several postsynaptic pathway downstream glutamate receptors are progressively dysregulated after chronic levodopa treatment, inducing a loss of control of phosphorylation cascades with increased phosphorylation of striatal substrates, such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor subunits.21 As we know, chronic levodopa treatment enhances the sensitivity of D1 receptor in PD rats.22 More importantly, D1 receptor stimulation enhances phosphorylation of the AMPA glutamate receptor subunit GluR1, increases surface expression of AMPA receptors, and facilitates their synaptic insertion in several brain areas.23 In addition, accumulating evidence suggests that AMPA antagonists can attenuate LID in animal models of PD, indicating that AMPA receptors play a crucial role to the induction of LID.24–26 GluR1, a subunit of the AMPA receptor, can be activated by phosphorylating at Serine-831 and Serine-845. It has previously been demonstrated that phosphorylated GluR1 is vital in changing synaptic plasticity.7 Increased levels of phosphorylated GluR1 can be found in the striatum of PD rats after repeated levodopa administration, which might disturb the biochemical machinery underlying long-term potentiation formation.7 KN-93, a competitive and selective inhibitor of CaMKII, has been shown to ameliorate LID by reducing levels of pGluR1S831.7 In the present study, we found that KN-93 reduced pGluR1S845 levels in PD rats treated with levodopa, which suggests that pGluR1S845 also contributes to the expression of LID.
Previous studies also show that long-term activation of the D1 receptor directly influenced gene expression.12,27 It has been reported that LID is accompanied by increased levels of Gad1 and Nur77, which aid the production of dynorphin, which is closely related to LID expression.28 In the present study, we found that Gad1 and Nur77 levels increased after intermittent administration of levodopa, which was consistent with previous studies. However, KN-93 treatment reduced the increase in Gad1 and Nur77 in PD rats treated with levodopa.
Taken together, we found that intrastriatal administration of KN-93 (2 μg or 5 μg) reduced the expression of LID in levodopa primed PD rats. In addition, KN-93 treatment reduced pGluR1S845 levels and the expression of Gad1 and Nur77. We assume that KN-93 can ameliorate LID expression by reducing the expression of Gad1 and Nur77 which subsequently lowers the levels of pGluR1S845 in PD rats.
Acknowledgments
The study was supported by the Projects of National Science Foundation of China (81071025, 81171203, 81171204 and 81200871), and Projects of the Shanghai Committee of Science and Technology, People’s Republic of China (11nm0503300, 11410708900 and 12XD1403800).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Devine MJ, Gwinn K, Singleton A, Hardy J. Parkinson’s disease and α-synuclein expression. Mov Disord. 2011;26(12):2160–2168. doi: 10.1002/mds.23948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iravani MM, Jenner P. Mechanisms underlying the onset and expression of levodopa-induced dyskinesia and their pharmacological manipulation. J Neural Transm. 2011;118(12):1661–1690. doi: 10.1007/s00702-011-0698-2. [DOI] [PubMed] [Google Scholar]
- 3.Carta M, Bezard E. Contribution of pre-synaptic mechanisms to L-DOPA-induced dyskinesia. Neuroscience. 2011;198:245–251. doi: 10.1016/j.neuroscience.2011.07.070. [DOI] [PubMed] [Google Scholar]
- 4.Mela F, Marti M, Bido S, Cenci MA, Morari M. In vivo evidence for a differential contribution of striatal and nigral D1 and D2 receptors to L-DOPA induced dyskinesia and the accompanying surge of nigral amino acid levels. Neurobiol Dis. 2012;45(1):573–582. doi: 10.1016/j.nbd.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 5.Kobylecki C, Hill MP, Crossman AR, Ravenscroft P. Synergistic antidyskinetic effects of topiramate and amantadine in animal models of Parkinson’s disease. Mov Disord. 2011;26(13):2354–2363. doi: 10.1002/mds.23867. [DOI] [PubMed] [Google Scholar]
- 6.Santini E, Sgambato-Faure V, Li Q, et al. Distinct changes in cAMP and extracellular signal-regulated protein kinase signalling in L-DOPA-induced dyskinesia. PLoS ONE. 2010;5(8):e12322. doi: 10.1371/journal.pone.0012322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ba M, Kong M, Yu G, Sun X, Liu Z, Wang X. GluR1 phosphorylation and persistent expression of levodopa-induced motor response alterations in the Hemi-Parkinsonian rat. Neurochem Res. 2011;36(6):1135–1144. doi: 10.1007/s11064-011-0461-y. [DOI] [PubMed] [Google Scholar]
- 8.Mahmoudi S, Samadi P, Gilbert F, et al. Nur77 mRNA levels and L-Dopa-induced dyskinesias in MPTP monkeys treated with docosahexaenoic acid. Neurobiol Dis. 2009;36(1):213–222. doi: 10.1016/j.nbd.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sgambato-Faure V, Buggia V, Gilbert F, Lévesque D, Benabid AL, Berger F. Coordinated and spatial upregulation of arc in striatonigral neurons correlates with L-dopa-induced behavioral sensitization in dyskinetic rats. J Neuropathol Exp Neurol. 2005;64(11):936–947. doi: 10.1097/01.jnen.0000186922.42592.b7. [DOI] [PubMed] [Google Scholar]
- 10.Darmopil S, Martín AB, De Diego IR, Ares S, Moratalla R. Genetic inactivation of dopamine D1 but not D2 receptors inhibits L-DOPA-induced dyskinesia and histone activation. Biol Psychiatry. 2009;66(6):603–613. doi: 10.1016/j.biopsych.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 11.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Sydney: Academic Press; 2007. [Google Scholar]
- 12.Morgese MG, Cassano T, Cuomo V, Giuffrida A. Anti-dyskinetic effects of cannabinoids in a rat model of Parkinson’s disease: role of CB(1) and TRPV1 receptors. Exp Neurol. 2007;208(1):110–119. doi: 10.1016/j.expneurol.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alcacer C, Santini E, Valjent E, Gaven F, Girault JA, Hervé D. Gα(olf) mutation allows parsing the role of cAMP-dependent and extracellular signal-regulated kinase-dependent signaling in L-3,4-dihydroxyphenylalanine-induced dyskinesia. J Neurosci. 2012;32(17):5900–5910. doi: 10.1523/JNEUROSCI.0837-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abuirmeileh A, Harkavyi A, Rampersaud N, et al. Exendin-4 treatment enhances L-DOPA evoked release of striatal dopamine and decreases dyskinetic movements in the 6-hydoxydopamine lesioned rat. J Pharm Pharmacol. 2012;64(5):637–643. doi: 10.1111/j.2042-7158.2011.01394.x. [DOI] [PubMed] [Google Scholar]
- 15.Maranis S, Stamatis D, Tsironis C, Konitsiotis S. Investigation of the antidyskinetic site of action of metabotropic and ionotropic glutamate receptor antagonists. Intracerebral infusions in 6-hydroxydopamine-lesioned rats with levodopa-induced dyskinesia. Eur J Pharmacol. 2012;683(1–3):71–77. doi: 10.1016/j.ejphar.2012.02.036. [DOI] [PubMed] [Google Scholar]
- 16.Cheshire PA, Williams DR. Serotonergic involvement in levodopa-induced dyskinesias in Parkinson’s disease. J Clin Neurosci. 2012;19(3):343–348. doi: 10.1016/j.jocn.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 17.Santini E, Valjent E, Usiello A, et al. Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J Neurosci. 2007;27(26):6995–7005. doi: 10.1523/JNEUROSCI.0852-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quintana A, Sgambato-Faure V, Savasta M. Effects of L-DOPA and STN-HFS dyskinesiogenic treatments on NR2B regulation in basal ganglia in the rat model of Parkinson’s disease. Neurobiol Dis. 2012;48(3):379–390. doi: 10.1016/j.nbd.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 19.Iderberg H, Francardo V, Pioli EY. Animal models of L-DOPA-induced dyskinesia: an update on the current options. Neuroscience. 2012;211:13–27. doi: 10.1016/j.neuroscience.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 20.Kong M, Ba M, Song L, Liu Z. Comparative effects of acute or chronic administration of levodopa to 6-OHDA-lesioned rats on the expression and phosphorylation of N-methyl-D-aspartate receptor NR1 subunits in the striatum. Neurochem Res. 2009;34(8):1513–1521. doi: 10.1007/s11064-009-9939-2. [DOI] [PubMed] [Google Scholar]
- 21.Takuma K, Tanaka T, Takahashi T, et al. Neuronal nitric oxide synthase inhibition attenuates the development of L-DOPA-induced dyskinesia in hemi-Parkinsonian rats. Eur J Pharmacol. 2012;683(1–3):166–173. doi: 10.1016/j.ejphar.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 22.Maranis S, Stamatis D, Tsironis C, Konitsiotis S. Investigation of the antidyskinetic site of action of metabotropic and ionotropic glutamate receptor antagonists. Intracerebral infusions in 6-hydroxydopamine-lesioned rats with levodopa-induced dyskinesia. Eur J Pharmacol. 2012;683(1–3):71–77. doi: 10.1016/j.ejphar.2012.02.036. [DOI] [PubMed] [Google Scholar]
- 23.Lees A, Fahn S, Eggert KM, et al. Perampanel, an AMPA antagonist, found to have no benefit in reducing “off” time in Parkinson’s disease. Mov Disord. 2012;27(2):284–288. doi: 10.1002/mds.23983. [DOI] [PubMed] [Google Scholar]
- 24.Kobylecki C, Cenci MA, Crossman AR, Ravenscroft P. Calcium-permeable AMPA receptors are involved in the induction and expression of l-DOPA-induced dyskinesia in Parkinson’s disease. J Neurochem. 2010;114(2):499–511. doi: 10.1111/j.1471-4159.2010.06776.x. [DOI] [PubMed] [Google Scholar]
- 25.Ouattara B, Hoyer D, Grégoire L, et al. Changes of AMPA receptors in MPTP monkeys with levodopa-induced dyskinesias. Neuroscience. 2010;167(4):1160–1167. doi: 10.1016/j.neuroscience.2010.03.022. [DOI] [PubMed] [Google Scholar]
- 26.Silverdale MA, Kobylecki C, Hallett PJ, et al. Synaptic recruitment of AMPA glutamate receptor subunits in levodopa-induced dyskinesia in the MPTP-lesioned nonhuman primate. Synapse. 2010;64(2):177–180. doi: 10.1002/syn.20739. [DOI] [PubMed] [Google Scholar]
- 27.Tamim MK, Samadi P, Morissette M, et al. Effect of non-dopaminergic drug treatment on Levodopa induced dyskinesias in MPTP monkeys: common implication of striatal neuropeptides. Neuropharmacology. 2010;58(1):286–296. doi: 10.1016/j.neuropharm.2009.06.030. [DOI] [PubMed] [Google Scholar]
- 28.Westin JE, Vercammen L, Strome EM, Konradi C, Cenci MA. Spatiotemporal pattern of striatal ERK1/2 phosphorylation in a rat model of L-DOPA-induced dyskinesia and the role of dopamine D1 receptors. Biol Psychiatry. 2007;62(7):800–810. doi: 10.1016/j.biopsych.2006.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]