

Abstract
Background
Identification of QTL with large phenotypic effects conserved across genetic backgrounds and environments is one of the prerequisites for crop improvement using marker assisted selection (MAS). The objectives of this study were to identify meta-QTL (mQTL) for grain yield (GY) and anthesis silking interval (ASI) across 18 bi-parental maize populations evaluated in the same conditions across 2-4 managed water stressed and 3-4 well watered environments.
Results
The meta-analyses identified 68 mQTL (9 QTL specific to ASI, 15 specific to GY, and 44 for both GY and ASI). Mean phenotypic variance explained by each mQTL varied from 1.2 to 13.1% and the overall average was 6.5%. Few QTL were detected under both environmental treatments and/or multiple (>4 populations) genetic backgrounds. The number and 95% genetic and physical confidence intervals of the mQTL were highly reduced compared to the QTL identified in the original studies. Each physical interval of the mQTL consisted of 5 to 926 candidate genes.
Conclusions
Meta-analyses reduced the number of QTL by 68% and narrowed the confidence intervals up to 12-fold. At least the 4 mQTL (mQTL2.2, mQTL6.1, mQTL7.5 and mQTL9.2) associated with GY under both water-stressed and well-watered environments and detected up to 6 populations may be considered for fine mapping and validation to confirm effects in different genetic backgrounds and pyramid them into new drought resistant breeding lines. This is the first extensive report on meta-analysis of data from over 3100 individuals genotyped using the same SNP platform and evaluated in the same conditions across a wide range of managed water-stressed and well-watered environments.
Keywords: Breeding, Drought, Heritability, Maize, Managed water stress, Meta analysis, SNP
Background
Globally, maize (Zea mays ssp. mays L.) is an important source of food and nutritional security for millions of people in the developing world, especially in sub-Saharan Africa (SSA) and Latin America [1]. Maize is a staple food in many of the SSA countries and is commonly grown by resource poor, small-scale farmers in rural areas. It covers 25 million hectares in SSA that produce 38 million metric tons [1] but the average maize yield in the region is estimated at 1.4 tons per hectare, which is about 20%, 37% and 56% of the average maize yield in developed countries, Brazil and Philippines, respectively [2]. Several factors, including high frequency of drought stress, scarcity and high cost of irrigation, and farmers’ inability to obtain quality seeds and fertilizers, contribute to such low productivity in the region. Given the unpredictable nature of drought and climate variability over years, breeders must develop improved maize hybrids that are able to withstand drought stress without significant yield penalty under optimal rainfall conditions [3-5]. For developing drought tolerant maize, selection can be done directly under water stress, indirectly under well-watered (optimal) conditions, or under both optimal and stress conditions [6]. However, heritability of grain yield under water stress has been reported to be lower than yield under optimal environments [7]. Hence, physiologists and breeders have devoted significant efforts in identifying relevant secondary traits correlated to grain yield for indirect selection. These include anthesis silking interval (ASI) between male and female flowering and several other morpho-physiological traits [8,9].
The ability to transfer target genomic regions associated with trait(s) of interest using molecular markers resulted in extensive QTL mapping experiments in most economically important crops. Such studies aimed at the identification of molecular markers for marker assisted backcrossing (MABC), marker assisted recurrent selection (MARS) and QTL cloning [10]. Using MABC, Ribaut and Ragot [4] introgressed 5 QTL associated with yield components and flowering in maize from a donor parent into a drought susceptible recurrent parent. The authors reported increased grain yield and reduced ASI under water-limited conditions. The best MABC progeny outperformed the recurrent parent by two to four times under severe drought conditions, with no yield reduction under optimal conditions. However, drought is a complex trait influenced by genetic background and other environmental factors; thus, relying on a few QTL for MABC is unlikely to create optimally drought tolerant lines for target population of environments. Individual drought associated QTL generally explain a very small proportion of the phenotypic variance for grain yield, ASI or barrenness. QTL for drought related traits are also often cross-specific and remain undetected in crosses from different genetic backgrounds. Most QTL are detected under either drought stress or optimal conditions (not both), and there is no assurance that QTL detected from inbred lines will function in the same manner in hybrids. Thus, they must be fully validated in several environmental conditions and hybrid combinations before deployment in a large breeding program.
MARS is another marker based breeding technology that seeks to accumulate favorable alleles from several genomic regions within a single population [11]. In maize, the MARS protocol involves (a) development and evaluation of testcross performance of bi-parental populations in multi-location experiments; (b) genotyping of the F2:3 population (Cycle 0); (c) undertaking an ad hoc significance test to identify a subset of markers that are significantly associated with the target trait; and (d) one generation (cycle) of selection of the best Cycle 0 families based on phenotypic index derived from testcross performance, followed by 2-3 cycles of selection based solely on markers with significant effects [12-15]. Currently, the International Maize and Wheat Improvement Center (CIMMYT), in collaboration with the national agricultural research systems (NARS) from 14 countries in Africa, the International Institute of Tropical Agriculture (IITA), the African Agricultural Technology Foundation (AATF), the Monsanto Company, and several regional and national seed companies in Africa, is working in large scale projects that aim to develop and disseminate drought tolerant maize for SSA using conventional breeding, MARS, and/or transgenic technology. These include the drought tolerant maize for Africa (DTMA) and the water efficient maize for Africa (WEMA) projects. For the MARS component of the WEMA project, CIMMYT developed and evaluated 18 bi-parental mapping populations, which formed the base for this study. All these populations have been phenotyped with common protocols and genotyped under a common single nucleotide polymorphism (SNP) platform.
Comparisons among independent QTL mapping projects usually attempt to determine if loci identified in each are the same by comparing the chromosomal position of a common subset of markers across different studies and/or indirectly by comparing each mapping population to a reference map [16]. Co-localized QTL may not be identical, however, especially when they are associated with large confidence intervals. Meta QTL analysis [17] is a better method for combining data from independent studies to detect consensus QTL and to shrink the QTL confidence intervals. Meta-analyses have been used in maize, wheat, rice, rapeseed, potato, cotton, soybean, barley, cocoa and apricot [18,19]. In maize, meta QTL (mQTL) for drought tolerance [20], flowering time [21], grain yield components [22], ear rot resistance [23,24] and silage quality [19] have been reported. Hao and colleagues [20] collected published QTL results and data related to drought tolerance for 12 mapping populations from the MaizeGDB website (http://www.maizegdb.org) and conducted meta-analyses on a total of 239 and 160 QTL detected under water stressed and well watered conditions, respectively. The authors reported 39 consensus mQTL for drought-tolerance related traits under water stress and 36 mQTL under well watered conditions. In most QTL meta-analyses published so far [19,20,23,24], authors compiled published linkage maps and QTL results from independent studies using different phenotyping protocols, constructed consensus linkage maps using a subset of markers common to the different studies, and projected mQTL positions and their confidence intervals onto the consensus map. Limitations of those studies are caused by the use of different phenotyping protocols, different QTL mapping methods, too few common markers, or by too few populations, causing lower confidence in the mQTL and the delimited intervals. The objectives of the present study were to identify mQTL for grain yield and ASI across 18 bi-parental maize populations genotyped with a common SNP platform and phenotyped with a common protocol in multi-location experiments both under water stressed and well watered environments.
Results
Phenotypic distribution, heritability and correlations
The mean phenotypic distribution for GY and ASI for all 18 bi-parental populations under water stressed and optimum environments was either normal or approximately normal (data not shown). Broad-sense heritability for GY (Table 1) varied from 0 to 0.40 under water stressed and from 0.23 to 0.58 under optimum environments. For ASI, heritability under water-stressed and optimum environments varied from 0 to 0.37 and from 0.08 to 0.54, respectively (Table 2). Populations with heritability < 0.1 under stressed (6 populations for each trait, of which 2 were common between the two traits) and/or < 0.20 under optimum (only 2 pops for ASI) environments were excluded from QTL analysis. In the dataset used for QTL mapping, therefore, broad sense heritability for GY and ASI varied from 0.13 to 0.40 under water stressed and from 0.21 to 0.58 under optimum conditions. There was significant but low to moderate negative correlation between GY and ASI under stressed (-0.09 to -0.51; p <0.001) and optimum (-0.08 to -0.23; p <0.001) conditions (data not shown).
Table 1.
Summary of the QTL for grain yield detected in 18 bi-parental populations
| Population code |
Water stressed |
Well watered |
Heritability |
Genetic variance per QTL** |
||||
|---|---|---|---|---|---|---|---|---|
| No. of QTL | R2 (%)* | No. of QTL | R2 (%)* | Water stressed | Well watered | Water stressed (%) | Well watered (%) | |
| 6x1008 |
2 |
6.10 |
4 |
21.60 |
0.24 |
0.42 |
12.76 |
12.92 |
| 6x1015 |
0 |
0.00 |
7 |
42.70 |
0.26 |
0.35 |
0.00 |
17.48 |
| 6x1016 |
- |
- |
5 |
37.90 |
0.00 |
0.57 |
0.00 |
13.37 |
| 6x1017 |
- |
- |
7 |
65.70 |
0.00 |
0.47 |
0.00 |
19.97 |
| 6x1018 |
1 |
7.70 |
5 |
34.00 |
0.26 |
0.52 |
29.96 |
13.13 |
| 6x1019 |
- |
- |
6 |
35.20 |
0.00 |
0.38 |
0.00 |
15.60 |
| 6x1020 |
2 |
6.10 |
5 |
24.00 |
0.33 |
0.55 |
9.27 |
8.66 |
| 6x1021 |
9 |
48.80 |
7 |
71.70 |
0.21 |
0.58 |
25.34 |
17.69 |
| 6x1023 |
- |
- |
3 |
28.50 |
0.08 |
0.38 |
0.00 |
25.13 |
| 6x1024 |
- |
- |
5 |
43.70 |
0.02 |
0.44 |
0.00 |
19.73 |
| 6x1028 |
- |
- |
7 |
50.00 |
0.00 |
0.40 |
0.00 |
17.86 |
| 6x1115 |
0 |
0.00 |
3 |
11.10 |
0.22 |
0.29 |
0.00 |
12.76 |
| 6x1116 |
3 |
9.30 |
5 |
62.40 |
0.24 |
0.49 |
12.92 |
25.47 |
| 6x1117 |
0 |
0.00 |
2 |
13.70 |
0.27 |
0.48 |
0.00 |
14.27 |
| 6x1118 |
0 |
0.00 |
2 |
11.70 |
0.40 |
0.26 |
0.00 |
22.50 |
| 6x1120 |
0 |
0.00 |
3 |
26.30 |
0.11 |
0.51 |
0.00 |
17.19 |
| 6x1121 |
1 |
3.90 |
6 |
60.60 |
0.32 |
0.23 |
12.19 |
43.91 |
| 6x1122 |
0 |
0.00 |
1 |
1.50 |
0.36 |
0.37 |
0.00 |
4.05 |
| Total |
18.00 |
81.90 |
83.00 |
642.30 |
3.32 |
7.68 |
102.43 |
321.69 |
| Mean | 1.50 | 6.83 | 4.61 | 35.68 | 0.18 | 0.43 | 5.69 | 17.87 |
*R2 = the total phenotypic variance explained by all QTL.
** The genetic variance is the percent phenotypic variance explained by a single QTL divided by heritability of a trait.
Maize populations and environments are described in detail in Table 3.
Table 2.
Summary of the QTL for ASI detected in bi-parental populations
| Population code |
Water stressed |
Well watered |
Heritability |
Genetic variance per QTL** |
||||
|---|---|---|---|---|---|---|---|---|
| No. of QTL | R2 (%)* | No. of QTL | R2 (%)* | Water stressed | Well watered | Water stressed | Well watered | |
| 6x1008 |
3 |
13.40 |
3 |
21.80 |
0.27 |
0.40 |
16.54 |
18.17 |
| 6x1015 |
- |
- |
4 |
26.10 |
0.00 |
0.21 |
0.00 |
31.07 |
| 6x1016 |
1 |
1.80 |
1 |
4.70 |
0.13 |
0.36 |
13.85 |
13.06 |
| 6x1017 |
1 |
8.20 |
1 |
12.30 |
0.19 |
0.41 |
43.16 |
30.00 |
| 6x1018 |
- |
- |
- |
- |
0.00 |
0.08 |
0.00 |
0.00 |
| 6x1019 |
0 |
0.00 |
2 |
14.40 |
0.03 |
0.33 |
0.00 |
21.82 |
| 6x1020 |
5 |
30.60 |
4 |
28.40 |
0.26 |
0.33 |
23.54 |
21.52 |
| 6x1021 |
2 |
5.40 |
4 |
17.20 |
0.20 |
0.34 |
13.50 |
12.65 |
| 6x1023 |
3 |
18.20 |
5 |
44.30 |
0.29 |
0.45 |
20.92 |
19.69 |
| 6x1024 |
- |
- |
5 |
19.80 |
0.05 |
0.51 |
0.00 |
7.76 |
| 6x1028 |
6 |
23.20 |
5 |
43.20 |
0.23 |
0.37 |
16.81 |
23.35 |
| 6x1115 |
- |
- |
- |
- |
- |
- |
- |
- |
| 6x1116 |
- |
- |
7 |
38.70 |
0.00 |
0.46 |
0.00 |
12.02 |
| 6x1117 |
3 |
16.70 |
0 |
0.00 |
0.14 |
0.24 |
39.76 |
0.00 |
| 6x1118 |
- |
- |
0 |
0.00 |
0.00 |
0.23 |
0.00 |
0.00 |
| 6x1120 |
3 |
17.20 |
3 |
17.10 |
0.21 |
0.47 |
27.30 |
12.13 |
| 6x1121 |
2 |
12.90 |
0 |
0.00 |
0.37 |
0.54 |
17.43 |
0.00 |
| 6x1122 |
4 |
16.60 |
5 |
40.80 |
0.24 |
0.42 |
17.29 |
19.43 |
| Total |
33.00 |
164.20 |
49.00 |
328.80 |
2.61 |
6.15 |
250.10 |
242.65 |
| Mean | 2.75 | 13.68 | 3.06 | 20.55 | 0.15 | 0.36 | 14.71 | 14.27 |
*R2 = the total phenotypic variance explained by all QTL.
** The genetic variance is the percent phenotypic variance explained by a single QTL divided by heritability of a trait.
Maize populations and environments are described in detail in Table 3.
Linkage and consensus mapping
The map length in the population specific linkage maps varied from 426 to 1418 cM, with a mean of 1075 cM (Table 3). The total number of mapped SNPs per population varied from 118 to 202 with an average of 172.3. The average number of SNPs mapped per chromosome in the population specific maps was 17.1 (data not shown). Chromosome 10 had fewer markers (range 4-13; average 9) compared to all other chromosomes due to low marker polymorphism in the initial polymorphism screening between parents (Figure 1). The mean map distance between markers ranged from 2.8 to 7.8 cM and the overall mean across all 18 populations was 6.1 cM. The final consensus map consisted of 430 SNPs with a total map length of 1471 cM. As shown in Figure 1, the number of markers per chromosome in the consensus map ranged from 27 to 66 SNPs (with a mean of 43); map length per chromosome ranged from 126 to 207 cM, with a mean of 147 cM. The map distance between markers in the final consensus map ranged from 1.0 to 27.8 cM and the overall mean was 3.5 cM, which is much smaller than the overall mean distance (6.1 cM) of the population specific maps. All except 9 intervals had a map distance < 10 cM (Figure 2).
Table 3.
Summary of the 18 bi-parental mapping populations used in the present study
| Population code | Population type | Cross | Managed water stressed evaluation sites | Well watered evaluation sites | Population size | No. of SNPs used for genotyping | Total number of SNPs mapped | Total map length (cM) |
|---|---|---|---|---|---|---|---|---|
| 6x1008 |
F2:3 |
CZL00009/CML505 |
Chisumaban, Isinya, Kibokooko and Nanga |
Embu, Kakamega, Kiboko and Mtwapa |
165 |
201 |
195 |
1400 |
| 6x1015 |
F2:3 |
CZL04003/CZL00009 |
Isinya, Kibokooko and Nanga |
Embu, Kakamega, Kiboko and Mtwapa |
162 |
190 |
179 |
1227 |
| 6x1016 |
F2:3 |
CZL00009/CZL99017 |
Isinya, Kibokooko and Nanga |
Embu, Kakamega, Kiboko and Mtwapa |
148 |
191 |
171 |
1342 |
| 6x1017 |
F2:3 |
CZL00009/CML539 |
Isinya, Kibokooko and Nanga |
Embu, Kakamega, Kiboko and Mtwapa |
184 |
210 |
199 |
1305 |
| 6x1018 |
F2:3 |
CML505/CZL99017 |
Kibokooko and Nanga |
Embu, Kakamega, Kiboko and Mtwapa |
184 |
212 |
177 |
1333 |
| 6x1019 |
F2:3 |
CZL04008/CZL0719 |
Kibokooko and Nanga |
Embu, Kakamega, Kiboko and Mtwapa |
173 |
202 |
182 |
1344 |
| 6x1020 |
F2:3 |
CZL0723/CZL0724 |
Kibokooko and Nanga |
Embu, Kakamega, Kiboko and Mtwapa |
181 |
218 |
196 |
1418 |
| 6x1021 |
F2:3 |
CZL0723/CZL0719 |
Isinya, Kibokooko and Nanga |
Embu, Kakamega, Kiboko and Mtwapa |
184 |
217 |
202 |
1376 |
| 6x1023 |
F2:3 |
CZL0618/VL062655 |
Chisumaban, Isinya, Kibokooko and Nanga |
Embu, Kakamega, Kiboko and Mtwapa |
184 |
225 |
200 |
1351 |
| 6x1024 |
F2:3 |
CZL02001/VL062590 |
Chisumanje, Kiboko and Nanga |
Embu, Kakamega, Kiboko and Mtwapa |
181 |
204 |
176 |
1246 |
| 6x1028 |
F2:3 |
CZL074/VL062645 |
Chisumabans and Kibokooko |
Embu, Kakamega, Kiboko and Mtwapa |
174 |
205 |
184 |
1166 |
| 6x1115 |
BC1F3 |
CKL09004/CZL00003//CKL09004 |
Isinya, Chiredzi, Chisumanje, Kiboko and Mtwapa |
Kiboko, Kti and Kakamega |
184 |
166 |
144 |
1034 |
| 6x1116 |
BC1F3 |
CKL09007/CML395//CML395 |
Isinya, Chiredzi, Chisumanje, Kiboko and Mtwapa |
Kiboko, Kti and Kakamega |
184 |
185 |
145 |
426 |
| 6x1117 |
BC1F3 |
CKL09007/CML444//CML444 |
Isinya, Chiredzi, Chisumanje, Kiboko and Mtwapa |
Kiboko, Kti and Kakamega |
160 |
163 |
152 |
459 |
| 6x1118 |
BC1F3 |
CKL09001/CML444//CML444 |
Isinya, Chiredzi, Chisumanje, Kiboko and Mtwapa |
Kiboko, Kti and Kakamega |
178 |
177 |
164 |
502 |
| 6x1120 |
F2:3 |
CKL09008/CML395 |
Isinya, Chiredzi, Chisumanje, Kiboko and Mtwapa |
Kiboko, Kti and Kakamega |
173 |
166 |
164 |
1180 |
| 6x1121 |
BC1F3 |
CKL09002/CZL03011//CKL09002 |
Isinya, Chiredzi, Chisumanje, Kiboko and Mtwapa |
Kiboko, Kti and Kakamega |
176 |
173 |
118 |
656 |
| 6x1122 | BC1F3 | CKL09006/CZL03011//CKL09006 | Isinya, Chiredzi, Chisumanje, Kiboko and Mtwapa | Kiboko, Kti and Kakamega | 155 | 185 | 153 | 581 |
Figure 1.

Summary of the consensus map of 10 maize chromosomes, showing map length, initial number of SNPs integrated in the consensus maps and final number of SNPs retained for meta analyses after excluding all markers with map distance of <1 cM.
Figure 2.
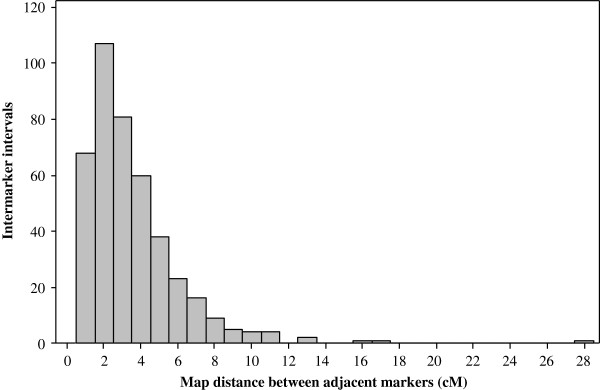
Frequency distribution of the 430 SNPs that were used for generating the final consensus linkage maps.
QTL in individual populations
From the 18 studies, composite interval mapping (CIM) uncovered a total of 101 QTL for GY and 82 QTL for ASI (Tables 1, 2 and Additional file 1). Figures 3 and 4 show the frequency distribution of the QTL for GY and ASI by LOD score and phenotypic variance explained by each QTL and chromosome. Under stressed environments, 18 QTL for GY were uncovered (Table 1) in 6 of the 12 populations with heritability > 0.10 (range: 0-9; average = 1.5 QTL per population). The proportion of phenotypic variance explained by each GY QTL under stress environments varied from 1.3 to 8.4%, and there were between 1 and 4 QTL per chromosome (except chromosome 10). The average phenotypic and genotypic variance explained by each GY QTL under stressed environments was 4.6% and 17.1%, respectively. In the optimum environments, a total of 83 QTL for GY were detected across all 18 populations. The number of GY QTL per population under optimum environments varied from 1 to 7 with an average of 4.6 QTL, and each QTL explained 1.2 to 19.1% of the phenotypic variance. The QTL were distributed across all chromosomes with the number of QTL per chromosome ranging from 4 to 14, with a mean of 4.8 QTL. The mean phenotypic and genotypic variance explained by each GY QTL under optimum environments was 7.7% and 18.1%, respectively.
Figure 3.

Frequency distribution of the 183 the 183 GY and ASI QTL by a) LOD score and b) R2.
Figure 4.

Frequency distribution of (a) population–specific QTL used for projection and total number of mQTL, and (b) the number of mQTL per chromosome.
For ASI, a total of 33 QTL with heritability > 0.10 were uncovered in 11 populations under stressed environments (Table 2 and Additional file 1). Each QTL for ASI explained 0.1 to 12.1% of the phenotypic variance under water stress, and they were distributed across all chromosomes with each chromosome containing from 1 to 6 QTL, (an average of 3.3 QTL per population). The mean phenotypic and genotypic variance explained by each QTL for ASI under stress was 5.0% and 22.7%, respectively. QTL for ASI with heritability > 0.20 were detected in 13 of the 16 populations under optimum environments, with the number of QTL varying from 1 to 7 and an average of 3.8 QTL per population. Each QTL for ASI explained 1.1 to 17.3% of the phenotypic variance under optimum environments. The QTL for ASI under optimum environments were distributed across all chromosomes with the number of QTL per chromosome ranging from 2 to 8 and an average of 5.1 per chromosome. The average phenotypic and genotypic variance explained by each QTL for ASI under optimum environments was 6.7% and 18.7%, respectively. Several QTL for GY and ASI had overlapping confidence intervals, and they appeared in clusters in the linkage maps (Additional files 1 and 2).
Meta-analyses
All QTL identified in individual populations were projected on the consensus map separately for GY and ASI first, and then for the combined QTL results of both traits (Additional file 2). The analysis of the combined traits increased the number of QTL per chromosome from a range of 4-17 to a range of 8-27. The statistical power via single trait-analysis and combined traits analyses was the same (data not shown). The meta-analysis sharply reduced the total number of QTL from 183 to 68 mQTL, compared to individual populations (Figure 4). Nine of these mQTL were specific to ASI, 15 to GY, and the remaining 44 were common to both GY and ASI (Figures 4 and 5). Table 4 and Additional file 3 present information about each mQTL, including chromosomal position, genetic and physical confidence interval, R2, flanking markers, and number of candidate genes in the interval. Eight of the 68 mQTL were associated either with ASI (5 mQTL) or both ASI and GY (3 mQTL) under water stressed environments only. The other 28 mQTL were detected both under stressed and optimum environments and the remaining 32 mQTL were associated with GY, ASI or both traits under optimum environments only.
Figure 5.

The positions of the 68 mQTL for grain yield and anthesis silking interval. The 95% genetic confidence interval of mQTL for grain yield and anthesis silking interval are shaded in green and pink colors, respectively, while those shaded in black coincided for both traits. The mQTL detected in >4 populations are indicated on the right side of each chromosome.
Table 4.
Summary of the 68 meta QTL (mQTL) for grain yield and anthesis-silking interval detected across 18 maize populations
| Chromosome | Meta QTL name | K | Predicted meta QTL position (cM) | Meta QTL 95% genetic confidence interval (cM) | Initial number of QTL | Mean initial confidence interval (cM) | Mean LOD score of the initial QTL | Mean R2 for the initial QTL | Δ in the 95% confidence interval (cM) | 95% physical confidence interval (kb) | Physical distance (kb) | No. of candidate genes within the physical interval | No. of populations where the meta QTL was uncovered | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
MQTL1.1 |
1 |
4.3 |
5.3 |
3 |
9.3 |
3.5 |
7.7 |
4.0 |
3639-4732 |
1,093.0 |
59 |
3 |
|
| 1 |
MQTL1.2 |
2 |
20.5 |
5.0 |
3 |
12.7 |
2.9 |
4.3 |
7.7 |
10061-14464 |
4,402.1 |
213 |
3 |
|
|
1 |
MQTL1.3 |
3 |
42.2 |
3.8 |
5 |
14.4 |
3.3 |
9.3 |
10.6 |
28552-30583 |
2,031.9 |
72 |
5 |
|
| 1 |
MQTL1.4 |
4 |
53.9 |
7.5 |
3 |
13.3 |
3.5 |
7.8 |
5.8 |
45277-46989 |
1,711.8 |
73 |
3 |
|
| 1 |
MQTL1.5 |
5 |
64.0 |
4.0 |
1 |
4.0 |
4.3 |
5.9 |
0.0 |
51515-58369 |
6,854.6 |
227 |
1 |
|
| 1 |
MQTL1.6 |
6 |
78.5 |
9.1 |
2 |
16.0 |
3.2 |
6.8 |
7.0 |
70862-79893 |
9,031.5 |
257 |
2 |
|
| 1 |
MQTL1.7 |
7 |
100.8 |
7.1 |
4 |
16.0 |
3.5 |
7.1 |
8.9 |
191405-198269 |
6,864.1 |
227 |
3 |
|
| 1 |
MQTL1.8 |
8 |
128.0 |
9.0 |
1 |
18.0 |
2.5 |
12.3 |
9.0 |
223836-229962 |
6,125.7 |
234 |
1 |
|
| 1 |
MQTL1.9 |
9 |
180.8 |
3.3 |
2 |
12.0 |
2.9 |
4.0 |
8.7 |
284698-288173 |
3,475.7 |
145 |
1 |
|
|
2 |
MQTL2.1 |
1 |
12.4 |
4.5 |
6 |
14.0 |
3.9 |
8.0 |
9.5 |
3534-3847 |
313.2 |
25 |
6 |
|
|
2 |
MQTL2.2 |
2 |
30.2 |
3.3 |
5 |
10.8 |
3.1 |
5.8 |
7.6 |
6141-6957 |
815.7 |
48 |
5 |
|
| 2 |
MQTL2.3 |
3 |
42.2 |
4.7 |
2 |
6.0 |
5.8 |
9.6 |
1.3 |
9969-11272 |
1,302.3 |
71 |
2 |
|
| 2 |
MQTL2.4 |
4 |
56.2 |
3.5 |
3 |
8.7 |
3.8 |
6.6 |
5.2 |
18063-19837 |
1,774.0 |
68 |
3 |
|
| 2 |
MQTL2.5 |
5 |
65.7 |
4.4 |
4 |
11.0 |
4.1 |
7.1 |
6.6 |
28812-37684 |
8,871.8 |
322 |
3 |
|
| 2 |
MQTL2.6 |
6 |
116.7 |
5.1 |
3 |
13.3 |
3.4 |
4.4 |
8.3 |
209512-214904 |
5,391.9 |
268 |
3 |
|
| 2 |
MQTL2.7 |
7 |
135.4 |
0.6 |
4 |
8.0 |
3.5 |
2.8 |
7.4 |
226386-230734 |
4,347.5 |
287 |
2 |
|
| 3 |
MQTL3.1 |
1 |
2.0 |
3.6 |
2 |
6.0 |
4.0 |
4.5 |
2.4 |
1384-1699 |
315.4 |
32 |
2 |
|
| 3 |
MQTL3.2 |
2 |
20.7 |
12.5 |
2 |
21.0 |
4.4 |
6.4 |
8.5 |
3266-5558 |
2,291.9 |
108 |
2 |
|
|
3 |
MQTL3.3 |
3 |
42.7 |
7.3 |
5 |
20.8 |
3.5 |
5.8 |
13.5 |
7218-10471 |
3,253.7 |
138 |
4 |
|
| 3 |
MQTL3.4 |
4 |
54.5 |
4.7 |
2 |
7.0 |
3.3 |
7.2 |
2.3 |
27986-53834 |
25,847.8 |
619 |
2 |
|
| 3 |
MQTL3.5 |
5 |
74.6 |
10.5 |
2 |
15.0 |
3.5 |
7.3 |
4.5 |
141780-165485 |
23,704.8 |
660 |
2 |
|
| 3 |
MQTL3.6 |
6 |
93.5 |
4.6 |
3 |
10.7 |
3.8 |
6.5 |
6.1 |
175055-197483 |
22,428.3 |
926 |
3 |
|
| 3 |
MQTL3.7 |
7 |
108.0 |
4.0 |
1 |
4.0 |
3.6 |
6.7 |
0.0 |
208947-211212 |
2,264.7 |
92 |
1 |
|
| 3 |
MQTL3.8 |
8 |
120.0 |
9.4 |
1 |
12.0 |
3.9 |
3.6 |
2.6 |
211133-218712 |
7,578.4 |
308 |
1 |
|
| 4 |
MQTL4.1 |
1 |
9.5 |
4.5 |
3 |
8.7 |
3.1 |
4.2 |
4.2 |
1613-4989 |
3,376.2 |
206 |
3 |
|
| 4 |
MQTL4.2 |
2 |
30.8 |
4.1 |
3 |
7.3 |
3.2 |
6.1 |
3.2 |
11272-13790 |
2,518.2 |
87 |
3 |
|
| 4 |
MQTL4.3 |
3 |
40.0 |
6.0 |
1 |
6.0 |
3.6 |
1.2 |
0.0 |
14511-18502 |
3,991.4 |
125 |
1 |
|
| 4 |
MQTL4.4 |
4 |
53.4 |
3.3 |
2 |
5.0 |
6.2 |
12.7 |
1.7 |
41714-81616 |
39,902.1 |
851 |
1 |
|
| 4 |
MQTL4.5 |
5 |
98.0 |
6.0 |
1 |
6.0 |
4.0 |
6.5 |
0.0 |
196386-203074 |
6,688.0 |
238 |
1 |
|
| 4 |
MQTL4.6 |
6 |
118.2 |
6.0 |
2 |
14.0 |
3.0 |
4.4 |
8.0 |
240863-242930 |
2,067.0 |
18 |
2 |
|
| 5 |
MQTL5.1 |
1 |
4.2 |
3.3 |
3 |
13.3 |
2.7 |
3.4 |
10.0 |
887-2799 |
1,911.3 |
189 |
2 |
|
|
5 |
MQTL5.2 |
2 |
28.9 |
3.1 |
4 |
8.0 |
3.9 |
8.5 |
5.0 |
6821-9201 |
2,379.9 |
128 |
4 |
|
| 5 |
MQTL5.3 |
3 |
42.0 |
6.0 |
1 |
6.0 |
3.9 |
5.3 |
0.0 |
11666-13316 |
1,650.1 |
83 |
1 |
|
| 5 |
MQTL5.4 |
4 |
52.0 |
5.7 |
2 |
8.0 |
2.6 |
5.9 |
2.3 |
21458-42985 |
21,527.0 |
645 |
2 |
|
| 5 |
MQTL5.5 |
5 |
60.0 |
4.0 |
1 |
4.0 |
4.9 |
9.9 |
0.0 |
46400-75946 |
29,545.5 |
863 |
1 |
|
| 5 |
MQTL5.6 |
6 |
76.4 |
6.3 |
3 |
11.3 |
3.7 |
7.7 |
5.0 |
158664-169720 |
11,056.0 |
324 |
3 |
|
| 5 |
MQTL5.7 |
7 |
116.4 |
4.7 |
3 |
9.3 |
3.4 |
5.0 |
4.6 |
199917-203728 |
3,810.3 |
191 |
3 |
|
| 5 |
MQTL5.8 |
8 |
128.6 |
3.2 |
5 |
20.5 |
4.3 |
5.5 |
17.3 |
204095-208963 |
4,867.9 |
305 |
3 |
|
|
6 |
MQTL6.1 |
1 |
0.4 |
1.9 |
4 |
8.0 |
3.1 |
4.0 |
6.1 |
1535-3654 |
2,119.7 |
66 |
4 |
|
| 6 |
MQTL6.2 |
2 |
16.0 |
10.0 |
1 |
10.0 |
3.7 |
6.0 |
0.0 |
7800-21710 |
13,909.7 |
301 |
1 |
|
| 6 |
MQTL6.3 |
3 |
45.8 |
1.9 |
3 |
8.0 |
4.2 |
10.5 |
6.1 |
109410-109818 |
407.2 |
5 |
3 |
|
| 6 |
MQTL6.4 |
4 |
58.0 |
12.0 |
1 |
12.0 |
2.7 |
5.2 |
0.0 |
115280-138426 |
23,145.6 |
829 |
1 |
|
| 6 |
MQTL6.5 |
5 |
80.6 |
4.7 |
4 |
12.5 |
4.6 |
8.8 |
7.8 |
153123-156645 |
3,521.2 |
201 |
3 |
|
| 6 |
MQTL6.6 |
6 |
94.2 |
5.4 |
4 |
11.0 |
4.6 |
9.6 |
5.6 |
160735-161977 |
1,241.9 |
77 |
2 |
|
| 6 |
MQTL6.7 |
7 |
116.0 |
6.5 |
1 |
10.0 |
2.9 |
1.5 |
3.5 |
163973-165726 |
1,753.2 |
116 |
1 |
|
| 7 |
MQTL7.1 |
1 |
11.6 |
7.2 |
2 |
12.0 |
6.4 |
11.5 |
4.8 |
1977-3000 |
1,023.8 |
71 |
2 |
|
| 7 |
MQTL7.2 |
2 |
33.9 |
3.7 |
4 |
15.0 |
3.2 |
5.3 |
11.3 |
7344-8456 |
1,112.2 |
51 |
3 |
|
| 7 |
MQTL7.3 |
3 |
45.6 |
4.0 |
3 |
8.0 |
4.1 |
8.7 |
4.0 |
11348-17346 |
5,998.1 |
177 |
2 |
|
| 7 |
MQTL7.4 |
4 |
51.5 |
3.3 |
2 |
5.0 |
3.1 |
5.8 |
1.7 |
92209-111874 |
19,664.7 |
387 |
2 |
|
|
7 |
MQTL7.5 |
5 |
63.4 |
8.2 |
4 |
16.5 |
3.4 |
6.8 |
8.3 |
119475-128209 |
8,734.7 |
289 |
4 |
|
| 7 |
MQTL7.6 |
6 |
86.4 |
9.6 |
2 |
14.0 |
3.4 |
4.4 |
4.4 |
155968-156660 |
692.0 |
28 |
1 |
|
| 7 |
MQTL7.7 |
7 |
102.0 |
2.3 |
1 |
6.0 |
3.6 |
4.2 |
3.7 |
162969-163528 |
559.5 |
23 |
1 |
|
| 8 |
MQTL8.1 |
1 |
14.0 |
4.0 |
1 |
4.0 |
2.6 |
2.2 |
0.0 |
8255-12884 |
4,628.6 |
174 |
1 |
|
| 8 |
MQTL8.2 |
2 |
28.2 |
8.7 |
2 |
14.0 |
7.0 |
12.9 |
5.3 |
12884-16036 |
3,152.4 |
118 |
2 |
|
|
8 |
MQTL8.3 |
3 |
45.8 |
4.0 |
5 |
11.6 |
4.0 |
7.8 |
7.7 |
19226-22594 |
3,368.0 |
112 |
4 |
|
| 8 |
MQTL8.4 |
4 |
56.6 |
4.9 |
3 |
11.3 |
3.4 |
7.3 |
6.4 |
94597-114908 |
20,310.3 |
572 |
3 |
|
| 8 |
MQTL8.5 |
5 |
68.0 |
4.2 |
2 |
6.0 |
4.1 |
11.2 |
1.8 |
130389-136848 |
6,459.5 |
238 |
2 |
|
| 8 |
MQTL8.6 |
6 |
90.5 |
10.1 |
2 |
15.0 |
4.2 |
7.9 |
4.9 |
161536-162181 |
645.2 |
39 |
2 |
|
| 8 |
MQTL8.7 |
7 |
109.2 |
3.7 |
3 |
14.7 |
3.2 |
4.7 |
11.0 |
165872-166992 |
1,119.5 |
67 |
3 |
|
| 9 |
MQTL9.1 |
1 |
4.9 |
6.7 |
3 |
12.0 |
3.0 |
4.1 |
5.4 |
558-1039 |
480.9 |
17 |
2 |
|
|
9 |
MQTL9.2 |
2 |
28.6 |
2.5 |
6 |
10.7 |
3.4 |
3.5 |
8.3 |
12863-15939 |
3,076.3 |
107 |
6 |
|
| 9 |
MQTL9.3 |
3 |
48.9 |
10.7 |
2 |
16.0 |
3.3 |
4.7 |
5.3 |
78943-107127 |
28,183.8 |
676 |
2 |
|
| 9 |
MQTL9.4 |
4 |
64.9 |
8.3 |
3 |
14.7 |
4.2 |
7.1 |
6.4 |
122950-134502 |
11,552.0 |
424 |
3 |
|
| 9 |
MQTL9.5 |
5 |
85.6 |
2.5 |
4 |
13.0 |
3.8 |
5.2 |
10.5 |
139422-142013 |
2,590.7 |
118 |
3 |
|
| 10 |
MQTL10.1 |
1 |
0.0 |
4.0 |
1 |
4.0 |
2.9 |
1.5 |
0.0 |
5116-5692 |
576.2 |
27 |
1 |
|
| 10 |
MQTL10.2 |
2 |
12.0 |
2.7 |
3 |
5.3 |
4.3 |
9.4 |
2.6 |
7133-13608 |
6,475.7 |
192 |
2 |
|
| 10 |
MQTL10.3 |
3 |
24.7 |
3.5 |
3 |
6.0 |
7.0 |
13.1 |
2.5 |
15276-62174 |
46,897.5 |
857 |
3 |
|
| 10 | MQTL10.4 | 4 | 46.0 | 4.2 | 1 | 8.0 | 4.0 | 6.5 | 3.8 | 127259-131505 | 4,245.7 | 160 | 1 |
See Additional file 3 for more information.
The number of mQTL identified on each chromosome varied from 4 on chromosome 10 to 9 on chromosome 1, with an average of 6.8 mQTL per chromosome. The low marker density on chromosome 10 may have reduced the number of mQTL uncovered on this chromosome. The mean phenotypic variance explained by each mQTL varied from 1.2 to 13.1% and the overall average was 6.5%. The 95% genetic confidence intervals for the mQTL varied between 0.6 and 12.5 cM, with an average of 5.4 cM, which is half the sizes of their respective original QTL (range = 4.0-21.0 cM; average = 10.7 cM). The 95% physical confidence intervals ranged from 313 to 46,898 kb with an average of 7,574 kb. The total number of candidate genes within the physical intervals varied from 5 to 926, with an average of 239 candidate genes per mQTL (Table 4). The physical to genetic distance ratio varied from 64 to 13,554 kb/cM, and the average was 7,574 kb/cM.
Eighteen of the 68 mQTL were detected in only a single population, 20 mQTL in 2 populations, 21 mQTL in 3 populations, 5 mQTL in 4 populations, 2 mQTL in 5 populations, and 2 mQTL in 6 populations (Table 4). No mQTL was detected in more than 6 of the 18 populations. Among the 9 mQTL mapped in 4-6 populations, four mQTL (mQTL2.2, mQTL6.1, mQTL7.5 and mQTL9.2) were associated with GY under both stress and optimum conditions; 2 mQTL (mQTL1.3 and mQTL5.2) were associated with GY only under optimum conditions; and the remaining 3 mQTL (mQTL2.1, mQTL3.3 and mQTL8.3) were associated with GY under optimum and ASI under water stressed and/or optimum environments. MQTL2.2 is located on chromosome 2 between 28.6 and 31.8 cM and has a physical interval of 816 kb. This QTL explains on average 5.8% of the phenotypic variance for GY both under water stress and optimum environments and encompassed 48 candidate genes. MQTL6.1 is located at the proximal end of chromosome 6 and has a physical interval of 2,120 kb; it accounted on average for 4.0% of the phenotypic variance for GY both under water stress and optimum environments and encompassed 66 candidate genes. MQTL7.5 is located on chromosome 7 between 59.4 and 67.5 cM and has a physical interval of 1,069 kb; this QTL explains on average 6.8% of the phenotypic variance for GY both under water stress and optimum environments and encompassed 289 candidate genes. MQTL9.2 is located on chromosome 9 between 27.4 and 29.8 cM and has a physical interval of 12,569 kb. The latter mQTL explains on average 3.5% of the phenotypic variance for GY both under water stress and optimum environments and encompassed 107 candidate genes.
Discussion
The projection of many QTL on a consensus map for meta-analysis allows to ascertain whether the QTL detected under water stressed conditions are a subset of those detected under optimal conditions, and if the QTL are common across different mapping populations (genetic backgrounds). Our study clearly demonstrated four times as many mQTL expressed under optimum conditions than under stressed environments. There was lower broad-sense heritability both for GY and ASI under stressed (0.13 to 0.40) than under optimum (0.21 to 0.58) environments, which may be an indication of a larger environmental component to the variance associated with stressed compared to optimum conditions. Although heritability under stress is sometimes comparable with heritability under optimum conditions, many studies [25-27] have also reported lower heritability under water stress than under optimum conditions. The mQTL were very specific to genetic background, but 8 of the 9 mQTL (MQTL1.3, MQTL2.1, MQTL2.2, MQTL3.3, MQTL5.2, MQTL6.1, MQTL8.3 and MQTL9.2; Table 4) found in 4 to 6 populations had small to medium genetic (1.9-8.2 cM) and physical (313-3368 kb) intervals and may be important regions for marker assisted backcrossing, QTL cloning for transformation, and/or functional analysis. Four of these mQTL (mQTL2.2, mQTL6.1, mQTL7.5 and mQTL9.2) seem the most suitable for future studies and eventual incorporation into breeding lines because (a) they were associated with GY under both water stressed and optimum environments; (b) they were detected up to 6 genetic backgrounds; (c) they accounted on average 3.5 to 6.8% of the phenotypic variance for GY under stress and optimum conditions, and (d) they encompassed lower number (48 to 289) of candidate genes.
Candidate genes can be identified through positional cloning using QTL confidence intervals [10], but confidence intervals need to be as small as possible. Combining results from several genome-wide surveys [28] and/or by merging QTL data from different studies [29] can help accomplish this. Our results from meta-analysis clearly demonstrated a gain in precision, reaching up to 12-fold smaller confidence interval in the mQTL, as compared with the population specific maps (Table 4). Similar results have been reported in other studies [20,23,24,30,31]. As shown in Table 4, the number of candidate genes within the 4 most conserved mQTL associated with GY both under water stressed and optimum environments varied from 48 to 289, and will thus require a further shrinking by fine mapping. This can most easily be done by increasing marker density evenly in the target regions, for example via genotyping-by-sequencing (GBS), which will generate nearly a million SNPs per sample at a cost of about $22 to $38 (http://igd.cornell.edu/index.cfm/page/GBS/GBSpricing.htm). At least 10% of these SNPs are expected to be polymorphic between parents in a given cross, and will thus generate at least 100 thousand polymorphic SNPs for fine mapping. The mapping populations presented in this study have been submitted for GBS, which may narrow down the physical confidence interval of the mQTL. Further fine mapping and/or QTL validation can also be done by increasing the size of the mapping populations. The four mQTL for GY detected in multiple populations under both stressed and optimum environments were also associated with ASI under stress and/or optimum conditions. However, we are unsure if this was due to the pleiotropic action of a single gene or multiple linked genes [16,32]. If the cause is tight linkage of multiple genes, fine mapping of large numbers of recombinants will break up the linkage. Although this is a labor and time-consuming process, it will be proposed for the conserved mQTL of large phenotypic effect.
Some of the mQTL detected in this study explained up to 13% of the phenotypic variance for GY and ASI under stress and/or optimum conditions. Because each mapping populations had an average of 174 progenies, it is possible that some of the mQTL of large effect may showed upward biased estimation (Beavis effect) of the phenotypic effects [33,34]. MQTL with large physical intervals may also contain several linked genes influencing the same trait. This has been reported even in cases where QTL effects have been fine mapped to more than one specific gene [32]. As far as we are aware, this is the first study that reports extensive mQTL results using over 3100 individuals that were genotyped using the same SNP platform and phenotyped in the same way across a wide range of managed water stressed and well watered environments. Future investigations may involve fine mapping and/or verification of some of the mQTL regions detected across 4-6 populations using large population size and high marker density. The results from this study provide highly valuable information for researchers working on QTL mapping for possible use in marker assisted selection and/or QTL cloning.
Conclusions
Meta-analyses reduced the number of QTL by 68% and narrowed the confidence intervals up to 12-fold, but none of the mQTL were detected in more than 6 populations, confirming the uniqueness of QTL from different populations. Nevertheless, at least 4 of the 68 mQTL were detected at least in 4 populations and may be considered for fine mapping and validation using large population sizes and high marker density, such as GBS. These four mQTL were located on chromosomes 2 (mQTL2.2), chromosome 6 (mQTL6.1), chromosome 7 (mQTL7.5) and chromosome 9 (mQTL9.2). About 65% of the mQTL uncovered under water stressed and/or optimum environments coincided between grain yield and ASI but it is unclear whether such large number of coincident mQTL was due to pleiotropic effect or tight linkage.
Methods
Population development, phenotyping and genotyping
A total of 25 MARS populations were initiated in 2008 and 2009. Quality control (QC) genotyping [35] of F1s and their parents with 100 SNP markers identified all F1s with true-to-type parental alleles for ≥ 95% of the polymorphic SNPs for advancement either to F2:3 or BC1F3, while those with >5% non-parental alleles were discarded. Seven of the 25 MARS populations either failed to pass the quality control genotyping criteria or had broad sense heritability < 0.10 and/or < 0.20 in the combined analyses of all the stressed and optimum environments, respectively, and were excluded from analyses. Phenotypic evaluations were performed on testcrosses derived by crossing either the F2:3 or BC1F3 families with one single cross tester from opposite heterotic group. The parents crossed with the same tester, and selected commercial checks were included in each of the trials. Each population was planted using an alpha lattice design, with 2 replications per location, and evaluated in 2-4 managed water stressed and 3-4 well watered locations (Table 3). Each entry was planted in a 5 m long row with spacing of 0.75 m between rows and 0.25 m between plants. In maize, it is well known that grain yield is often reduced 2-3 times more when water deficits coincide with flowering, compared with other growth stages [36]. Therefore, water stress evaluation was conducted during the dry (rain free) season in Kenya, Zimbabwe and Zambia by withdrawing irrigation two weeks before flowering. Irrigation was resumed at the end of the flowering stage, corresponding to the end of silk emergence, and maintained until harvest to allow grain filling. Evaluation under optimum conditions in the 3 countries was carried out during the long rainy season.
Each population was evaluated for 12-17 different traits, including grain yield, anthesis date, number of ears per plant, and leaf senescence, which are commonly associated with drought tolerance. Only grain yield and ASI were selected as the main target traits in the present study. ASI was computed as the difference between days to silking and anthesis. Each trial was harvested when all leaves had senesced. Ears were dried and shelled, grain was weighed, and grain moisture determined by a capacitance meter. SAS program v9.2 was used for phenotypic data analyses, including calculating Best Linear Unbiased Predictor (BLUP), variance components and heritability under stressed and optimum environments.
Linkage and QTL mapping in individual populations
All mapping populations were genotyped by the Monsanto Company using a TaqMan assay (http://www.appliedbiosystems.com). For each segregating SNP, a χ2 goodness-of-fit analysis was performed to test for deviation from the expected segregation ratio. The chromosomal position and locus order of all SNPs used in the present study was provided by the Monsanto Company and this a priori information was used as a reference for determining locus order in our mapping populations. Linkage groups were established using LOD scores ranging between 3 and 15, and recombination frequency of 0.30. The order of the SNPs on each chromosome was determined as described elsewhere [37] using the Kosambi mapping function. χ2 analyses and linkage mapping were performed using JoinMap version 4.0 [38]. The number of polymorphic SNPs used for genotyping the populations varied from 163 to 225 (Table 3). Final linkage maps were constructed after excluding a total of 389 non-informative SNPs (an average of 22 SNPs per population) because they i) did not meet the threshold value for goodness-of-fit, ii) contributed to negative distance in the final map, iii) changed the expected marker order, or iv) mapped to unexpected chromosomal locations compared to the a priori information. QTL mapping was performed with BLUP values obtained across the combined analyses of all the stressed and optimum environments for each population. Composite interval mapping (CIM) was conducted as described elsewhere [39] using a minimum LOD score of 2.5 and the PLABQTL software, version 1.2 [40,41].
Map projection and QTL meta-analyses
For the same chromosome across multiple populations, a consensus linkage map of all SNPs was constructed from the population specific maps using BioMercator version 2.1 as described by Arcade et al. [42]. Markers that showed inversions in the consensus map were discarded. The initial consensus map consisted of 961 markers but about 55% of the SNPs (531 of the 961 SNPs) had a map distance < 1 cM to adjacent markers, so they were excluded from the final consensus map. All QTL identified in individual populations using PlabQTL were projected on the consensus map separately for GY and ASI first, and then for the combined QTL results of both traits. The information on the original chromosomal position, LOD score, confidence interval (CI) and proportion of phenotypic variance (R2) explained by each QTL (as summarized in Additional file 1) were used for the projection. For each chromosome, meta-analysis was used to estimate the numbers, positions, and 95% confidence interval of the mQTL using BioMercator version 3.0 software (http://moulon.inra.fr/index.php/en/scientific-output/software/doc_details/15-biomercator-v-3) [43]. The meta-analysis first determines the best model based on model choice criteria of the following: AIC (Akaike information criterion), AICc, AIC3, BIC (Bayesian information criterion) and AWE (average weight of evidence). The best QTL model was selected when values of the model selectin criteria were the lowest at least in 3 of the 5 models (Additional file 4). The best model was then used in the MQTLView method. QTL with probability of membership in a given mQTL > 60% were assigned to the same mQTL. The 95% confidence intervals of the mQTL were drawn using the MapChart program, version 2.1 [44].
Candidate genes
Flanking markers of each mQTL were used to search for candidate genes within each mQTL interval. The genetic map of all proprietary SNPs used in this study, along with over 52,000 public markers, was provided by the Monsanto Company. The map was created using the company’s proprietary mapping population. For each mQTL, the public markers with known physical positions that were closest to the two flanking SNPs found in this study were chosen to define the interval. The physical positions of these flanking public markers were then used to search for candidate genes using the Maize Sequence database (http://www.maizesequence.org/index.html). This browser provides the latest sequence and annotation of the Zea mays ssp. mays genome from the Maize Genome Sequencing Project.
Competing interest
The authors declare no competing financial interests.
Authors’ contributions
KS was responsible for data analyses and writing the manuscript; YB was responsible in population development, experimental design and overall multi-location phenotyping of the populations; AT coordinated phenotyping of the populations both in Zambia and Zimbabwe; SM contributed in the project planning and overall coordination; BM and PS were responsible for coordinating sample management, polymorphic markers selection, DNA extraction and genotyping, and IT requirements at the Monsanto company; both MLW and BMP contributed to the data analyses and edited the manuscript. All authors have made their contribution in editing the manuscript and approved the final version.
Supplementary Material
Summary of the population-specific QTL detected by Composite Interval Mapping for grain yield (GY) and anthesis-silking interval (ASI) for 18 maize populations evaluated under managed water stressed (WS) and well-watered (WW) environments.
Summary of the projected position of the 183 QTL for grain yield (GY) and anthesis-silking interval (ASI) using BioMercator version 3.0.
Additional information to the meta QTL described in Table 4.
Summarizes the model selection criteria in the meta-analyses.
Contributor Information
Kassa Semagn, Email: k.semagn@cgiar.org.
Yoseph Beyene, Email: y.beyene@cgiar.org.
Marilyn L Warburton, Email: Marilyn.Warburton@ARS.USDA.GOV.
Amsal Tarekegne, Email: a.tarekegne@cgiar.org.
Stephen Mugo, Email: s.mugo@cgiar.org.
Barbara Meisel, Email: barbara.meisel@monsanto.com.
Pierre Sehabiague, Email: pierre.sehabiague@monsanto.com.
Boddupalli M Prasanna, Email: b.m.prasanna@cgiar.org.
Acknowledgements
We are grateful to J. Crossa for the initial phenotype data analyses, CIMMYT Field Technicians at the different stations in Kenya, Zimbabwe and Zambia for the phenotypic evaluation, and the Monsanto Company for genotyping the populations and also providing marker database that links the genetic positions of the proprietary SNP markers with public markers. This work was carried as part of the WEMA project that has been funded by the Bill & Melinda Gates Foundation and Howard Buffet Foundation.
References
- Shiferaw B, Prasanna BM, Hellin J, Bänziger M. Crops that feed the world 6. Past successes and future challenges to the role played by maize in global food security. Food Security. 2011;3:307–327. doi: 10.1007/s12571-011-0140-5. [DOI] [Google Scholar]
- Smale M, Byerlee D, Jayne T. Maize revolutions in Sub-Saharan Africa: World Bank Policy Research Working Paper. 2011. p. 34. http://econ.worldbank.org/external/default/main?pagePK=64165259&theSitePK=544849&piPK=64165421&menuPK=64166093&entityID=000158349_20110511144001.
- Campos H, Cooper M, Habben JE, Edmeades GO, Schussler JR. Improving drought tolerance in maize: a view from industry. Field Crops Research. 2004;90:19–34. doi: 10.1016/j.fcr.2004.07.003. [DOI] [Google Scholar]
- Ribaut JM, Ragot M. Marker-assisted selection to improve drought adaptation in maize: the backcross approach, perspectives, limitations, and alternatives. J Exp Bot. 2007;58:351–360. doi: 10.1093/jxb/erl214. [DOI] [PubMed] [Google Scholar]
- Sambatti JBM, Caylor KK. When is breeding for drought tolerance optimal if drought is random? New Phytol. 2007;175:70–80. doi: 10.1111/j.1469-8137.2007.02067.x. [DOI] [PubMed] [Google Scholar]
- Byrne PF, Bolaños J, Edmeades GO, Eaton DL. Gains from selection under drought versus multilocation testing in related tropical maize populations. Crop Sci. 1995;35:63–69. doi: 10.2135/cropsci1995.0011183X003500010011x. [DOI] [Google Scholar]
- Venuprasad R, Lafitte HR, Atlin GN. Response to direct selection for grain yield under drought stress in rice. Crop Sci. 2007;47:285–293. doi: 10.2135/cropsci2006.03.0181. [DOI] [Google Scholar]
- Bolaños J, Edmeades GO. The importance of the anthesis-silking interval in breeding for drought tolerance in tropical maize. Field Crops Research. 1996;48:65–80. doi: 10.1016/0378-4290(96)00036-6. [DOI] [Google Scholar]
- Ribaut JM, Hoisington DA, Deutsch JA, Jiang C. González de León D: Identification of quantitative trait loci under drought conditions in tropical maize. 1. Flowering parameters and the anthesis-silking interval. Theor Appl Genet. 1996;92:905–914. doi: 10.1007/BF00221905. [DOI] [PubMed] [Google Scholar]
- Salvi S, Tuberosa R. To clone or not to clone plant QTLs: present and future challenges. Trends Plant Sci. 2005;10:297–304. doi: 10.1016/j.tplants.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Edwards M, Johnson L. Analysis of molecular marker data American Society for Horticultural Science, Crop Science Society of America. Corvallis, Oregon; 1994. RFLPs for rapid recurrent selection; pp. 33–40. (Joint Plant Breeding Symposia Series). [Google Scholar]
- Bernardo R, Charcosset A. Usefulness of gene information in marker-assisted recurrent selection: a simulation appraisal. Crop Sci. 2006;46:614–621. doi: 10.2135/cropsci2005.05-0088. [DOI] [Google Scholar]
- Mayor PJ, Bernardo R. Doubled haploids in commercial maize breeding: one-stage and two-stage phenotypic selection versus marker-assisted recurrent selection. Maydica. 2009;54:439–448. [Google Scholar]
- Mayor PJ, Bernardo R. Genomewide selection and marker-assisted recurrent selection in doubled haploid versus F2 populations. Crop Science. 2009;49:1719–1725. doi: 10.2135/cropsci2008.10.0587. [DOI] [Google Scholar]
- Massman JM, Jung H-JG, Bernardo R. Genomewide Selection versus Marker-assisted Recurrent Selection to Improve Grain Yield and Stover-quality Traits for Cellulosic Ethanol in Maize. Crop Sci. 2013;53:58–66. [Google Scholar]
- Tuberosa R, Salvi S, Sanguineti MC, Landi P, Maccaferri M, Conti S. Mapping QTLs regulating morpho-physiological traits and yield: case studies, shortcomings and perspectives in drought-stressed maize. Annals of Botany. 2002;89:941–963. doi: 10.1093/aob/mcf134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffinet B, Gerber S. Quantitative Trait Loci: A Meta-analysis. Genetics. 2000. pp. 463–473. [DOI] [PMC free article] [PubMed]
- Danan S, Veyrieras JB, Lefebvre V. Construction of a potato consensus map and QTL meta-analysis offer new insights into the genetic architecture of late blight resistance and plant maturity traits. BMC Plant Biology. 2011;11:19. doi: 10.1186/1471-2229-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truntzler M, BarriA"re Y, Sawkins MC, Lespinasse D, Betran J, Charcosset A, Moreau L. Meta-analysis of QTL involved in silage quality of maize and comparison with the position of candidate genes. Theoretical and Applied Genetics. 2010;121:1465–1482. doi: 10.1007/s00122-010-1402-x. [DOI] [PubMed] [Google Scholar]
- Hao Z, Li X, Liu X, Xie C, Li M, Zhang D, Zhang S. Meta-analysis of constitutive and adaptive QTL for drought tolerance in maize. Euphytica. 2010;174:165–177. doi: 10.1007/s10681-009-0091-5. [DOI] [Google Scholar]
- Chardon F, Virlon B, Moreau L, Falque M, Joets J, Decousset L, Murigneux A, Charcosset A. Genetic architecture of flowering time in maize as inferred from quantitative trait loci meta-analysis and synteny conservation with the rice genome. Genetics. 2004;168:2169–2185. doi: 10.1534/genetics.104.032375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JZ, Zhang ZW, Li YL, Wang QL, Zhou YG. QTL consistency and meta-analysis for grain yield components in three generations in maize. TAG Theoretical and Applied Genetics. 2011;122:771–782. doi: 10.1007/s00122-010-1485-4. [DOI] [PubMed] [Google Scholar]
- Xiang K, Reid LM, Zhang Z, Zhu X, Pan G. Characterization of correlation between grain moisture and ear rot resistance in maize by QTL meta-analysis. Euphytica. 2012;183:185–195. doi: 10.1007/s10681-011-0440-z. [DOI] [Google Scholar]
- Xiang K, Zhang ZM, Reid LM, Zhu XY, Yuan GS, Pan GT. A meta-analysis of QTL associated with ear rot resistance in maize. Maydica. 2010;55:281–290. [Google Scholar]
- Weber VS, Melchinger AE, Magorokosho C, Makumbi D, Bänziger M, Atlin GN. Efficiency of managed-stress screening of elite maize hybrids under drought and low nitrogen for yield under rainfed conditions in Southern Africa. Crop Science. 2012;52:1011–1020. doi: 10.2135/cropsci2011.09.0486. [DOI] [Google Scholar]
- Edmeades GO, Bolaños J, Chapman SC, Lafitte HR, Bänziger M. Selection improves drought tolerance in tropical maize populations: I. Gains in biomass, grain yield, and harvest index. Crop Science. 1999;39:1306–1315. doi: 10.2135/cropsci1999.3951306x. [DOI] [Google Scholar]
- Fukai S, Cooper M. Development of drought-resistant cultivars using physio-morphological traits in rice. Field Crops Research. 1995;40:67–86. doi: 10.1016/0378-4290(94)00096-U. [DOI] [Google Scholar]
- Keightley PD. Inference of genome-wide mutation rates and distributions of mutation effects for fitness traits: a simulation study. Genetics. 1998;150:1283–1293. doi: 10.1093/genetics/150.3.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quraishi UM, Murat F, Abrouk M, Pont C, Confolent C, Oury FX, Ward J, Boros D, Gebruers K, Delcour JA. Combined meta-genomics analyses unravel candidate genes for the grain dietary fiber content in bread wheat (Triticum aestivum L.) Functional & Integrative Genomics. 2011;11:71–83. doi: 10.1007/s10142-010-0183-2. [DOI] [PubMed] [Google Scholar]
- Swamy BPM, Vikram P, Dixit S, Ahmed HU, Kumar A. Meta-analysis of grain yield qtl identified during agricultural drought in grasses showed consensus. BMC Genomics. 2011;12:16. doi: 10.1186/1471-2164-12-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truntzler M, Barrière Y, Sawkins MC, Lespinasse D, Betran J, Charcosset A, Moreau L. Meta-analysis of QTL involved in silage quality of maize and comparison with the position of candidate genes. TAG Theoretical and Applied Genetics. 2010;121:1465–1482. doi: 10.1007/s00122-010-1402-x. [DOI] [PubMed] [Google Scholar]
- Studer AJ, Doebley JF. Do large effect QTL fractionate? A case study at the maize domestication QTL teosinte branched1. Genetics. 2011;188:673–681. doi: 10.1534/genetics.111.126508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchinger AE, Utz HF, Schon CC. Quantitative trait locus (QTL) mapping using different testers and independent population samples in maize reveals low power of QTL detection and large bias in estimates of QTL effects. Genetics. 1998;149:383–403. doi: 10.1093/genetics/149.1.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon CC, Utz HF, Groh S, Truberg B, Openshaw S, Melchinger AE. Quantitative trait locus mapping based on resampling in a vast maize testcross experiment and its relevance to quantitative genetics for complex traits. Genetics. 2004;167:485–498. doi: 10.1534/genetics.167.1.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semagn K, Beyene Y, Makumbi D, Mugo S, Prasanna BM, Magorokosho C, Atlin G. Quality control genotyping for assessment of genetic identity and purity in diverse tropical maize inbred lines. Theoretical and Applied Genetics. 2012;125:1487–1501. doi: 10.1007/s00122-012-1928-1. [DOI] [PubMed] [Google Scholar]
- Grant RF, Jackson BS, Kiniry JR, Arkin GF. Water deficit timing effects on yield components in maize. Agronomy Journal. 1989;81:61–65. doi: 10.2134/agronj1989.00021962008100010011x. [DOI] [Google Scholar]
- Semagn K, Bjornstad A, Skinnes H, Maroy AG, Tarkegne Y, William M. Distribution of DArT, AFLP, and SSR markers in a genetic linkage map of a doubled-haploid hexaploid wheat population. Genome. 2006;49:545–555. doi: 10.1139/G06-002. [DOI] [PubMed] [Google Scholar]
- van Ooijen JW. JoinMap 4: Software for the calculation of genetic linkage maps in experimental populations of diploid species. Wageningen, the Netherlands: Plant Research International B.V. and Kyazma B.V; 2006. [Google Scholar]
- Semagn K. Skinnes H, rnstad A, Mar<o>y AG, Tarkegne Y: Quantitative trait loci controlling Fusarium head blight resistance and low deoxynivalenol content in hexaploid wheat population from 'Arina' and NK93604. Crop Science. 2007;47:294–303. doi: 10.2135/cropsci2006.02.0095. [DOI] [Google Scholar]
- Utz HF, Melchinger AE, SchAn CC. Bias and sampling error of the estimated proportion of genotypic variance explained by quantitative trait loci determined from experimental data in maize using cross validation and validation with independent samples. Genetics. 2000;154:1839–1849. [PMC free article] [PubMed] [Google Scholar]
- Utz HF, Melchinger AE. PLABQTL: A computer program to map QTL, Version 1.2. Stuttgart, Germany: Institute of Plant Breeding, Seed Science, and Population Genetics, University of Hohenheim; 2003. [Google Scholar]
- Arcade A, Labourdette A, Falque M, Mangin B, Chardon F, Charcosset A, Joets J. BioMercator: integrating genetic maps and QTL towards discovery of candidate genes. Bioinformatics. 2004;20:2324–2326. doi: 10.1093/bioinformatics/bth230. [DOI] [PubMed] [Google Scholar]
- Sosnowski O, Charcosset A, Joets J. BioMercator V3: an upgrade of genetic map compilation and QTL meta-analysis algorithms. Bioinformatics. 2012;28:2082–2083. doi: 10.1093/bioinformatics/bts313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorrips RE. MapChart: Software for the Graphical Presentation of Linkage Maps and QTLs. Journal of Heredity. 2002;93:77–78. doi: 10.1093/jhered/93.1.77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of the population-specific QTL detected by Composite Interval Mapping for grain yield (GY) and anthesis-silking interval (ASI) for 18 maize populations evaluated under managed water stressed (WS) and well-watered (WW) environments.
Summary of the projected position of the 183 QTL for grain yield (GY) and anthesis-silking interval (ASI) using BioMercator version 3.0.
Additional information to the meta QTL described in Table 4.
Summarizes the model selection criteria in the meta-analyses.