

Abstract
Chronic hypoxia causes pulmonary hypertension with smooth muscle cell proliferation and matrix deposition in the wall of the pulmonary arterioles. We demonstrate here that hypoxia also induces a pronounced inflammation in the lung before the structural changes of the vessel wall. The proinflammatory action of hypoxia is mediated by the induction of distinct cytokines and chemokines and is independent of tumor necrosis factor-α signaling. We have previously proposed a crucial role for heme oxygenase-1 (HO-1) in protecting cardiomyocytes from hypoxic stress, and potent anti-inflammatory properties of HO-1 have been reported in models of tissue injury. We thus established transgenic mice that constitutively express HO-1 in the lung and exposed them to chronic hypoxia. HO-1 transgenic mice were protected from the development of both pulmonary inflammation as well as hypertension and vessel wall hypertrophy induced by hypoxia. Significantly, the hypoxic induction of proinflammatory cytokines and chemokines was suppressed in HO-1 transgenic mice. Our findings suggest an important protective function of enzymatic products of HO-1 activity as inhibitors of hypoxia-induced vasoconstrictive and proinflammatory pathways.
Acute hypoxia in the lung causes arteriolar vasoconstriction whereas prolonged hypoxia promotes proliferation and migration of vascular smooth muscle cells (VSMC) and extracellular matrix deposition in the arterial wall, a process known as vascular remodeling (1). These abnormalities are characteristic of pulmonary hypertension (2). Several clinical conditions characterized by lung inflammation have been linked to the development of chronic pulmonary hypertension (3). Interestingly, perivascular inflammatory cell infiltration as well as increased serum levels of proinflammatory cytokines, such as IL-1β and IL-6, have been reported in clinical cases of primary pulmonary hypertension (4, 5). However, little attention has been given up to now to the role of pulmonary inflammation in the pathogenesis of pulmonary hypertension induced by hypoxia.
Heme oxygenase (HO; EC 1.14.99.3) catalyzes the oxidation of heme to carbon monoxide (CO) and biliverdin, which is then converted to bilirubin by biliverdin reductase. Three isoforms of HO have been identified: the inducible HO-1 and the constitutively expressed HO-2 and HO-3 (6, 7). Our previous in vitro data suggest that CO released by HO-1 confers protection against vasoconstriction and vascular remodeling induced by hypoxia (8–10). More recently, Soares et al. have suggested anti-inflammatory properties of HO-1 in a cardiac transplantation model, although the molecular mechanisms have not been fully elucidated (11). Our recent in vivo data using an HO-1 null mouse model suggest that HO-1 plays a central role in protecting the right ventricle from hypoxic pulmonary pressure-induced injury (12).
In the present study, we established transgenic mice that overexpress HO-1 in the lung and exposed them to hypoxia to investigate the effects of HO-1 activity on the development of pulmonary hypertension. We report here that, in wild-type animals, hypoxia caused pulmonary hypertension with vascular remodeling as well as a striking inflammatory response in the lung parenchyma. In contrast, overexpression of HO-1 protected against the development of pulmonary hypertension as well as the hypoxia-induced inflammatory cell infiltration. Significantly, overexpression of HO-1 attenuated the hypoxic induction of proinflammatory cytokines and chemokines in the lung. These findings point to novel molecular mechanisms underlying the anti-inflammatory properties of HO-1 that may be crucial for the body's adaptive responses to hypoxia.
Methods
Transgenic Mice.
A plasmid containing a 3.7-kb genomic region encompassing the promoter of the human surfactant protein C (SP-C) gene was a kind gift of Jeffrey A. Whitsett (Children's Hospital Medical Center, Cincinnati). The SP-C promoter–human HO-1 cDNA (13) transgene was constructed by standard cloning methods, and microinjection was performed in FVB/N strain pronuclei by the Brigham & Women's Hospital Core Transgenic Mouse Facility (Boston). Founder mice were identified by Southern blot analysis of genomic DNA isolated from mouse-tail biopsies.
Hypoxic Exposure.
Six- to 8-week-old HO-1 transgenic mice and their nontransgenic littermates or age-matched nontransgenic mice were exposed to normobaric hypoxia (8–10% oxygen) or normoxia for the indicated time as described previously (14). Animals were killed by lethal injection of sodium pentobarbital. Excised lungs and other organs were frozen in liquid nitrogen and stored at −80°C for RNA and protein analysis. Some animals were also subjected to hemodynamic measurements, bronchoalveolar lavage fluid analysis, and histological analysis.
RNA Analysis.
Total RNA was extracted using RNAzol B (Tel-Test, Friendswood, TX) according to manufacturer's instructions. Twenty to 30 μg of RNA were separated on 1.2% agarose/6% formaldehyde gel, transferred to a nylon membrane (MSSI, Westboro, MA), and hybridized with radiolabeled HO-1 or HO-2 cDNA probes as described previously (8). RNase protection assay (RiboQuant, PharMingen) was performed according to manufacturer's instructions, and the quality of RNA was assessed by agarose gel electrophoresis followed by ethidium bromide staining before each assay. Expression levels of cytokines were quantified using an image analyzer (Molecular Dynamics).
Western Blot Analysis and Immunoassay.
Protein samples were prepared by homogenization of frozen tissues in lysis buffer (10 mM Tris⋅HCl, pH 8/140 mM NaCl/5 mM EDTA/0.025% NaN3/1% Triton X-100/1% deoxycholate/0.1% SDS/0.5 mM PMSF/1 μg/μl leupeptin/1 μg/μl aprotinin). The samples (30 μg) were resolved on 12% SDS/PAGE, transferred onto a PVDF membrane (Millipore), and incubated with anti-HO-1 antibody (1:1,000, SPA-896, StressGen Biotechnologies, Victoria, Canada), followed by an anti-rabbit IgG horseradish peroxidase antibody (1:12,500, Jackson ImmunoResearch). Specific proteins were detected using ECL (Amersham Pharmacia). For quantification of cytokines, protein samples were prepared by homogenization of frozen tissues in the same lysis buffer for Western analysis but without SDS. They were analyzed in quadruplicate using a commercially available sandwich enzyme immunoassay for macrophage inflammatory protein-2 (MIP-2) peptide (Quantikine, R & D Systems). Values were expressed in picograms per milliliter, and the sensitivity of the assay was 1.5 pg/ml. The concentration of MIP-2 was reported in pg/mg total protein to normalize for protein recovered between the different animals and conditions.
HO Activity Measurement.
HO enzymatic activity was measured by bilirubin generation as described previously (8). Briefly, frozen tissues were homogenized in lysis buffer (250 mM Tris⋅HCl, pH 7.4/150 mM NaCl/250 mM sucrose/0.5 mM PMSF/1 μg/μl leupeptin/1 μg/μl aprotinin). Microsomal fraction was obtained by successive centrifugations (800 × g for 10 min, 8,800 × g for 10 min, and 105,000 × g for 30 min) and washed with 0.15 M KCl followed by centrifugation (105,000 × g for 30 min). The pellet was solubilized in 0.1 M potassium phosphate by sonication and stored at −80°C. The reaction was carried out in the mixture containing 2–3 mg/ml protein microsomal fraction/1 mM glucose-6-phosphate/0.2 unit/ml glucose-6-phosphate dehydrogenase/0.8 mM NADP/0.025 mg/ml hemin at 37°C for 45 min. After chloroform extraction, bilirubin was quantitated with a double-beam spectrophotometer.
Hemodynamic and Ventricular Weight Measurements.
Hemodynamic and ventricular weight measurements were performed as described (12). Briefly, mice were anesthetized with chloral hydrate (0.54 mg/g body weight). The trachea was cannulated, and mice were ventilated at room air (tidal volume 0.01 ml/g). The thoracic cavity was opened, PE-50 polyethylene tubing was inserted into the right ventricle, and right ventricular systolic pressure (RVSP) was measured with MacLab monitoring equipment. For ventricular weight measurement, hearts were excised and atria were removed. The right ventricular free wall was dissected, and each chamber was weighed. The ratio of right ventricular weight to left ventricular weight plus septum [RV/(LV + S)] was used as an index of right ventricular hypertrophy.
Bronchoalveolar Lavage (BAL) Fluid.
Animals were anesthetized with sodium pentobarbital after 48 h of hypoxia. BAL was performed with PBS (1 ml). Total white cell counts were determined by manual counting. Samples were cytocentrifuged and stained with DiffQuick (Fisher Scientific) for differential cell counts.
Lung Histology.
Lungs were inflated, and the pulmonary artery was perfused with 4% paraformaldehyde. Excised lungs were fixed in 4% paraformaldehyde overnight at 4°C and embedded in paraffin. For frozen sections, fixed samples were washed with PBS containing 5–20% sucrose and embedded in OCT compound. Frozen sections (12 μm) were used for immunostaining with neutrophil and macrophage-specific antibodies. Immunostaining for HO-1 was performed in the paraffin-embedded sections (6 μm). Sections were treated with 0.3% hydrogen peroxide in methanol for 20 min, preincubated with 5% goat serum, and treated with the anti-Ly-6G antibody (1:500, PharMingen) for neutrophils, anti-Mac-3 antibody for macrophages (1: 500, PharMingen), or anti-HO-1 antibody (1:200, SPA 896, StressGen Biotechnologies) for 1 h at 37°C. Next, the sections were incubated with a biotinylated goat secondary antibody, treated with the avidin–biotin complex (Elite ABC kit, Vector Laboratories), and stained with diaminobenzidine tetrahydrochloride and hydrogen peroxide. For pulmonary vascular morphometry, paraffin-embedded lung sections were stained with hematoxylin and eosin. Images for arterioles were captured with a microscope digital camera system (Olympus), and arterial area was measured using an image analysis program (NIH IMAGE 1.55). Percent wall thickness was calculated by the following formula: Wall thickness (%) = areaext − areaint/areaext × 100, where areaext and areaint are the area bounded by external and internal elastic lamina, respectively. Approximately 130 vessels of comparable size from the lungs of 25 different mice were estimated (n = 7 for nontransgenic hypoxic, n = 5 for transgenic hypoxic, n = 6 for nontransgenic normoxic, and n = 7 for transgenic normoxic).
Statistical Analysis.
All values were expressed as mean ± SEM. Comparison of results between different groups was performed by one-way analysis of variance or Mann–Whitney U test using statview, Version 4.5 (Abacus Concepts, Berkeley, CA).
Results
Generation of HO-1 Transgenic Mice.
Two founder lines of transgenic mice harboring the human HO-1 gene under the control of the human SP-C promoter were obtained, and the inheritance of the transgene was confirmed by Southern blotting of genomic DNA. Expression levels of HO-1 mRNA and protein in the lungs from one line of transgenic mice were 4–6-fold higher than their nontransgenic littermates, whereas no difference in HO-2 expression was detected in the lungs between transgenic and nontransgenic control mice (Fig. 1 a and b). Furthermore, a 2.5-fold increase in HO activity was observed in the lungs of this line as compared with nontransgenic control mice (163.3 ± 15.1 vs. 60.3 ± 9.4 pmol/h per mg protein, P < 0.05, six to seven mice for each group, Mann–Whitney U test). A less significant increase in levels of HO-1 protein and enzymatic activity (<2-fold) was detected in the lungs of the second line compared with wild-type animals. Both lines of mice were exposed to hypoxia to determine the development of pulmonary hypertension compared with their nontransgenic littermates, as described below.
Figure 1.

Expression of HO-1 in transgenic mice. (a) Northern blot analysis for HO-1 and HO-2. HO-1 (Top) and HO-2 mRNA levels (Middle) were examined in total RNA (30 μg) extracted from the lungs of HO-1 transgenic mice (Tg) or nontransgenic littermates (non-Tg). β-actin mRNA levels were examined to confirm equal loading (Bottom). This blot is representative of three independent experiments, six mice per group. (b) Western blot analysis for HO-1 in the lung. Lung whole-cell lysates (30 μg) from five to six HO-1 Tg and a corresponding number of non-Tg littermates were analyzed for HO-1 protein levels using an anti-HO-1 antibody. The same blot was reprobed with an anti-β-actin antibody to verify equal loading (Lower). Note that the anti-HO-1 antibody used here is raised against the N-terminal amino acid sequence of the protein, a region conserved between mouse and human HO-1. (c) Western blot analysis for HO-1 in nonlung tissues. Tissue distribution of HO-1 was analyzed in whole cell lysates (30 μg) of heart, liver, spleen, and kidney from Tg mice (+) or non-Tg littermates (−). This blot represents identical patterns of expression observed in three Tg and three non-Tg animals.
Lung-Specific Expression of HO-1 Transgene.
High levels of HO-1 protein were observed in the spleen, moderate levels in the liver, and low levels in the heart and kidney by Western analysis (Fig. 1c). In all tissues, HO-1 levels were equal in both transgenic (Tg+) and nontransgenic (Tg−) control mice, verifying that overexpression of HO-1 was successfully targeted to the lung. Because of the tissue specificity of the SP-C promoter (15, 16), immunostaining for HO-1 was observed in the alveolar cells of the lungs of transgenic mice, shown here for the high expressor line (Fig. 2 a and b), whereas no immunostaining was detected in the sections treated with control preimmune rabbit serum (Fig. 2c). In contrast, little or no HO-1 specific immunostaining was observed in the lungs of nontransgenic control mice (Fig. 2d), consistent with the low levels of endogenous HO-1 expression detected through Western blot analysis (Fig. 1b). No structural abnormalities were noted in the lungs of transgenic mice up to the age of 2 years (data not shown).
Figure 2.
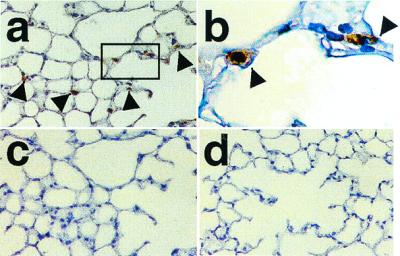
Immunostaining for HO-1 in the lung of Tg mice. Paraffin-embedded sections of the lungs were analyzed for HO-1 expression by immunohistochemistry. (a) High levels of HO-1 expression are evident in the lung of Tg mice (brown, arrow). (b) High power photograph of the rectangle area indicated in a. (c) The section adjacent to that in a incubated with preimmune rabbit serum. (d) Absence of detectable HO-1 immunoreactivity in the lung of non-Tg mice. Original magnification: ×100 for a, c, and d and ×400 for b. Staining was performed in lung sections of four Tg and four non-Tg control mice.
Development of Hypoxia-Induced Pulmonary Hypertension: Time Course and Dose-Dependent Protection by HO-1.
Right ventricular hypertrophy (RVH) is a hallmark of pulmonary hypertension resulting from RV pressure overload. The ratio of right ventricle weight to left ventricle plus septum weight [RV/(LV + S)] was measured to assess the development of RVH upon exposure of nontransgenic mice to hypoxia (8–10% O2 environment) for varying periods of time. RV/(LV + S) was significantly increased as early as 48 h after hypoxia, and RVH was developed in a time-dependent manner peaking at 2 weeks (Fig. 3a). Under these conditions, endogenous HO-1 in nontransgenic mouse lungs was minimally increased after 6 h of hypoxia and remained significantly lower compared with lungs of transgenic mice for up to 2 weeks of hypoxic exposure (data not shown). HO-1 protein levels in transgenic mouse lungs were not affected by the hypoxic exposure and remained constitutively elevated (4.6-fold above nontransgenic controls).
Figure 3.

Time course of hypoxic right ventricular hypertrophy: protection by HO-1 overexpression. (a) Non-Tg mice were exposed to hypoxia (8–10% O2) for 18 h, 5 days (d), or 2–3 weeks (w). Development of RVH after hypoxic exposure was determined by the RV/(LV + S) ratio as described in Materials and Methods. *, P < 0.01, **, P < 0.001 vs. time point 0 (n = 6 for each group, one-way ANOVA). (b) Tg homozygous, heterozygous, and non-Tg control mice from founder 1 and homozygous Tg and non-Tg mice from line 2 were exposed to normoxia or hypoxia for 3 weeks. Under normoxia, line 1 homozygous Tg (n = 19), heterozygous Tg (n = 6), and non-Tg controls (n = 14) as well as line 2 homozygous Tg (n = 5) or non-Tg (n = 5) did not differ in baseline RV/(LV + S) (P > 0.05). After hypoxic exposure, line 1 homozygous Tg (n = 26) and heterozygous Tg mice (n = 8) show significant reduction in RV/(LV + S) as compared with non-Tg controls (n = 22). Similarly, homozygous line 2 Tg mice (n = 5) manifest significantly lower RV/(LV + S) values as compared with their non-Tg controls (n = 5) exposed to hypoxia. (*, P < 0.05, **, P < 0.005 vs. hypoxic controls; †P < 0.01 vs. normoxic controls, Mann–Whitney U test). Data are mean ± SEM.
To determine whether the overexpression of HO-1 in the lung protects against the development of pulmonary hypertension, transgenic and nontransgenic mice from both founder lines were exposed to hypoxia (8–10%) for 3 weeks, and RV/(LV + S) was measured. Under normoxia, there was no difference in RV/(LV + S) between transgenic and nontransgenic control mice (Fig. 3b). After hypoxic exposure, a significant increase in RV/(LV + S) was observed in nontransgenic mice, which was suppressed in transgenic mice. Heterozygous mice from line 1 and homozygous mice from line 2 whose lung HO-1 transgene levels were comparable (≤2-fold above controls) demonstrated intermediate protection with mild RVH. RV/(LV + S) was higher than normoxic mice but significantly lower than nontransgenic hypoxic control mice (Fig. 3b). This protection against pulmonary hypertension and secondary RVH persisted in transgenic mice up to 8 weeks of hypoxic exposure (data not shown).
RVSP was measured after 2 and 3 weeks of hypoxia and compared between transgenic and nontransgenic controls. Under normoxia, there was no difference in RVSP measurements; however, after hypoxic exposure, a significant increase in RVSP was observed in nontransgenic mice compared with normoxic mice or with transgenic hypoxic mice (Fig. 4a). Although intermediate pressures were noted in transgenic hypoxic mice, they remained significantly elevated above the normoxic controls (P < 0.01). Morphometric analysis of the pulmonary vasculature revealed no difference in vessel wall architecture between transgenic and nontransgenic animals under normoxia (23% vs. 26% medial wall thickness, respectively, P > 0.05). Two weeks after hypoxic exposure, nontransgenic mice demonstrated a significant increase in the wall thickness of pulmonary arterioles, which was completely inhibited in hypoxic transgenic mice (39% vs. 26%, P < 0.05) (Fig. 4b). Combined, these findings indicate that overexpression of HO-1 decreases active vasoconstriction and prevents pulmonary vascular remodeling with RVH. It is noted that transgenic and nontransgenic control mice did not differ in hematocrits either at baseline or after hypoxic exposure. Hematocrits were significantly elevated in both groups of mice after hypoxic exposure (data not shown).
Figure 4.

HO-1 overexpressing mice are protected from pulmonary hypertension induced by hypoxia. (a) RVSP measurements were performed in Tg and non-Tg mice exposed to 2–3 weeks of hypoxia. RVSP did not differ between Tg (n = 13) and non-Tg (n = 8) mice at baseline normoxic conditions. Under hypoxia, Tg mice (n = 25) had significantly lower pressure measurements than non-Tg controls (n = 8) (*, P < 0.05, Mann–Whitney U test). Data are means ± SEM. (b) Hematoxylin and eosin staining of paraffin-embedded lung sections of Tg (Right) or non-Tg control mice (Left) exposed to hypoxia for 2–3 weeks. Original magnification: ×400. Percent wall thickness in arterioles of comparable size was estimated based on analysis of area as described in Materials and Methods (lower graph). Significant reduction in the wall thickness is observed in Tg mice as compared with non-Tg control. Data are expressed as mean ± SEM (n = 5–7). *, P < 0.05 vs. non-Tg control (Mann–Whitney U test).
Overexpression of HO-1 Protects Against Pulmonary Inflammation Induced by Hypoxia.
In addition to vascular remodeling, we particularly noted that hypoxic lungs exhibited hypercellularity compared with normoxic lungs of nontransgenic mice. This difference was more evident at early time points (48 h) of hypoxic exposure and was significantly diminished after 2 weeks of hypoxia. Interestingly, this cellular infiltration was suppressed in the hypoxic lungs of transgenic mice. To determine whether the protective effects of HO-1 involve anti-inflammatory mechanisms, we performed immunohistochemical staining for neutrophils and macrophages in lungs from mice exposed to hypoxia for 48 h. A prominent infiltration of neutrophils was observed in the hypoxic lungs of nontransgenic mice, whereas the minimal staining detected in hypoxic transgenic mice was comparable with that in normoxic control lungs (Fig. 5a, Left). Although macrophages were observed in the normoxic lung, hypoxia induced further accumulation of macrophages in the lungs of nontransgenic control mice but not in transgenic mice (Fig. 5a, Right). These observations were supported by BAL fluid analysis. The number of neutrophils and macrophages in BAL fluid was significantly increased after hypoxic exposure in nontransgenic mice, whereas this increase was suppressed in transgenic mice under hypoxia (Fig. 5b), suggesting that HO-1 overexpression protects against the pulmonary inflammation induced by hypoxia. We have previously reported that mice deficient in HO-1 had a maladaptive response to chronic hypoxia (5–7-week exposure to 8–10% O2) with right ventricular dilatation and infarction (12). Of interest, the lungs of these mice manifested sustained hypercellularity with inflammatory cell infiltrates even after several weeks of hypoxia compared with normoxic null mice or wild-type hypoxic mice (unpublished observations).
Figure 5.

HO-1 overexpressing mice are protected against pulmonary inflammation induced by hypoxia. (a) Immunostaining for neutrophils and macrophages. Frozen lung sections from Tg or non-Tg control mice that had been exposed to hypoxia for 48 h were stained with anti-Ly-6G antibody for neutrophils and anti-Mac-3 antibody for macrophages (brown staining). Original magnification: ×100. (b) The number of neutrophils and macrophages in BAL fluid after hypoxia. Differential cell counts for neutrophils (Neut, Upper) and macrophages (Mac, Lower) were performed in the BAL fluid of Tg (closed bars) and non-Tg control mice (open bars) after 48 h of hypoxia. Data are expressed as mean ± SEM (n = 4). *, P < 0.05 vs. normoxic non-Tg control. †, P < 0.05 vs. hypoxic non-Tg control (Mann–Whitney U test).
HO-1 Overexpression Inhibits Induction of Proinflammatory Cytokines and Chemokines Under Hypoxia.
The recruitment of leukocytes is regulated by the action of cytokines and chemokines (17). These mediators also function as potent mitogens and chemoattractants for VSMC, resulting in vascular remodeling (18, 19). To investigate mechanisms underlying the anti-inflammatory properties of HO-1, we examined cytokine expression in the hypoxic lung. We found that the expressions of IL-1β, IL-6, monocyte chemoattractant protein (MCP)-1, and macrophage inflammatory protein (MIP)-2, a functional murine homologue of human IL-8, were significantly elevated in the lungs of nontransgenic mice in response to hypoxia, with a peak at 48 h after exposure (Fig. 6, non-Tg). In contrast, other proinflammatory cytokines, such as tumor necrosis factor (TNF)α, IFN-γ, and IL-1α, and chemokines such as RANTES were unaffected or only minimally affected (Fig. 6 and data not shown). Anti-inflammatory mediators, such as IL-10, were also unaffected (data not shown). More importantly, the induction of IL-1β, IL-6, MCP-1, and MIP-2 were significantly suppressed in the hypoxic lungs of transgenic mice (Fig. 6, Tg), suggesting that inhibition of these cytokines is involved in the mechanisms mediating the anti-inflammatory effects of HO-1. Along with RNA analysis, we correlated protein levels of MIP-2 in the lungs of these transgenic and nontransgenic hypoxic and normoxic mice. MIP-2 protein was undetectable at baseline in all normoxic transgenic and nontransgenic lungs. After 48 h and 5 days of hypoxia, averages of 3.75 and 2.9 pg of MIP-2/mg total protein, respectively, were detected in nontransgenic lungs. MIP-2 protein levels remained undetectable in transgenic hypoxic lungs both at 48 h and 5 days.
Figure 6.

HO-1 overexpression inhibits hypoxic induction of cytokines in the lung. HO-1 Tg and non-Tg control mice were exposed to hypoxia (8–10% O2) for the times indicated [48 h, 5 days (d), or 2 weeks (w)]. After exposure, total lung RNA (10 μg) was examined for cytokine expression by RNase protection assay. Levels of L32, a ribosomal protein mRNA, were unaffected by hypoxia and served as an internal control. Expression of TNFα 3 h after hypoxia in non-Tg mice is also shown (Lower). Similar results were observed in four independent experiments.
Discussion
Chronic hypoxia is well known to cause hypertension and vascular remodeling in the pulmonary vasculature in various animal models of human pathophysiology. Shorter, more pronounced hypoxic exposure (6% O2 for 4–8 h) leads to induction of tissue factor and results in fibrin deposition and thrombosis in the pulmonary vasculature of mice (20). In the present study, we observed that less severe hypoxia (8–10% O2 for 48 h to 5 days) lead to a marked induction of proinflammatory cytokines and chemokines in association with pulmonary inflammation that preceded the development of pulmonary hypertension. Moreover, we demonstrated that overexpression of HO-1 in the lung inhibited cytokine gene expression and protected against both pulmonary inflammation as well as pulmonary hypertension induced by hypoxia.
In the process of pulmonary inflammation, cytokines are produced and amplified manyfold by target cells, such as monocytes, endothelial cells, and epithelial cells, leading to the recruitment of leukocytes that release a variety of pro-oxidants and proteolytic enzymes capable of causing vasoconstriction and lung injury (17, 21). In addition, proinflammatory cytokines and chemokines directly induce changes in vascular permeability and thrombogenicity (22, 23), processes implicated in the pathogenesis of pulmonary hypertension. These cytokines were also reported to promote proliferation and migration of VSMC in vitro (18, 19), and disruption of the CCR2 chemokine receptor gene decreased vascular remodeling in a model of atherosclerosis in vivo (24). Moreover, it was demonstrated that administration of an IL-1 receptor antagonist or anti-MCP-1 antibody protected against monocrotaline-induced pulmonary hypertension (25, 26). Together with our data of pronounced cytokine and chemokine induction in hypoxic pulmonary hypertension, these findings suggest a crucial role for proinflammatory cytokines and chemokines in the development of pulmonary vascular remodeling. Cytokine induction and cellular infiltration peak at 48 h and decrease by 2 weeks of hypoxia, yet RVH and vascular remodeling persist for several weeks of continued hypoxia. It is possible that these cytokines initiate a cascade of events leading to VSMC proliferation that is later amplified by the release of vasoactive mediators and growth factors that alter vessel wall structure and function. For example, the initial injury and loss of endothelial barrier function can lead to extravasation of serum and activation of endogenous elastolytic activity from vascular smooth muscle cells (27, 28). Increased serine elastase activity results in proteolytic degradation of extracellular matrix and the release of matrix-bound growth factors amplifying smooth muscle cell proliferation and remodeling of the pulmonary arterioles (27, 29). Indeed, treatment of rats with elastase inhibitors completely reversed malignant pulmonary hypertension induced by monocrotaline (30). In this report, we demonstrate a pronounced neutrophil and macrophage infiltration in the lungs of hypoxic mice that is linked with pulmonary hypertension. It is possible that neutrophil elastase as well as other proteolytic enzymes may trigger similar molecular events in the hypoxic setting. Future studies will determine whether inhibition of inflammation can prevent hypoxia-induced pulmonary hypertension.
The mechanism of protection provided by HO-1 may depend on its enzymatic product, CO, to function as a vasodilator against vasoconstriction and bilirubin as an antioxidant against the reactive oxygen species that are generated under hypoxic conditions (31). Because the recruitment of leukocytes was greatly suppressed in the hypoxic lungs of HO-1 transgenic mice, inhibition of cytokine induction may be another important mechanism by which HO-1 activity imparts protection against the development of pulmonary hypertension induced by hypoxia. In two recent studies, HO-1 activity and, in particular, CO, were reported to have potent anti-inflammatory properties in models of vascular injury (11, 32). The anti-inflammatory properties of CO were mediated by the inhibition of proinflammatory cytokines, TNFα, IL1-β, and MIP-1β, in the model of lipopolysaccharide-induced systemic inflammation (32). HO-1 was also shown to have protective effects in hyperoxic lung injury as well as ischemia–reperfusion in the liver, two well known models of inflammation-mediated injury (33, 34). In this report, we demonstrate a previously unrecognized inflammatory response induced by hypoxia. The mechanisms underlying the induction of cytokines by hypoxia are most likely different from other models of systemic or pulmonary inflammation. For example, in endotoxin-induced pulmonary inflammation models, early response cytokines, such as TNFα, are rapidly induced (within 6 h), and this early response leads to the activation of cytokine networks including the induction of IL-6, MCP-1, and MIP-2 (21). In contrast, induction of TNFα was not detected in the lungs 3, 6, and 18 h after hypoxia (Fig. 6 and data not shown), indicating that the hypoxic induction of IL-6, MCP-1, and MIP-2 is not secondary to that of this early response cytokine. Similarly, IL-6 has been suggested to function as an anti-inflammatory cytokine in endotoxin-induced pulmonary inflammation because IL-6 deficiency resulted in an increase in proinflammatory cytokines, including TNFα and IL-1β (35). However, despite reduced IL-6 expression in the hypoxic lungs of transgenic mice, we did not observe increased expression of other proinflammatory cytokines. In addition, treatment with lipopolysaccharide induced expression of the anti-inflammatory cytokine IL-10, whereas we did not find increased IL-10 expression under hypoxia. Hypoxia, therefore, induces a network of cytokines and chemokines that is distinct from other inflammatory pathways such as those induced by lipopolysaccharide.
In summary, we demonstrate that hypoxia induced an early inflammatory response in the lung before the development of pulmonary hypertension. HO-1 overexpression effectively inhibited pulmonary inflammation and prevented pulmonary hypertension induced by hypoxia. HO-1 may be an important therapeutic target to inhibit hypoxia-induced cytokine signaling and prevent the inflammatory pathways that may lead to pulmonary hypertension.
Acknowledgments
This work is dedicated to the memory of Dr. Arthur Mu-En Lee, an insightful scientist and warm friend. He will remain an inspirational role model to all of us. We greatly thank Laura Lynch for her expert technical assistance. We also thank Jessica Johnson for her expert assistance in the preparation of this manuscript. H.C. was supported by National Institutes of Health Grant KO8 HL03917. M.A.P. was supported by National Institutes of Health Grants RO1s HL60788 and GM53249. This work was supported by the American Heart Association and by National Institutes of Health Grants RO1 HL55454 and SCOR 1P50 HL56398 (to S.K.).
Abbreviations
- HO
heme oxygenase
- RVH
right ventricular hypertrophy
- RVSP
right ventricular systolic pressure
- VSMC
vascular smooth muscle cell
- BAL
bronchoalveolar lavage
- MCP
monocyte chemoattractant protein
- TNF
tumor necrosis factor
References
- 1.Kourembanas S, Morita T, Liu Y, Christou H. Kidney Int. 1997;51:438–443. doi: 10.1038/ki.1997.58. [DOI] [PubMed] [Google Scholar]
- 2.Mecham R P, Whitehouse L A, Wrenn D S, Parks W C, Griffin G L, Senior R M, Crouch E C, Stenmark K R, Voelkel N F. Science. 1987;237:423–426. doi: 10.1126/science.3603030. [DOI] [PubMed] [Google Scholar]
- 3.Zapol W M, Snider M T. N Engl J Med. 1977;3:476–480. doi: 10.1056/NEJM197703032960903. [DOI] [PubMed] [Google Scholar]
- 4.Tuder R M, Groves B, Badesch D B, Voelkel N F. Am J Pathol. 1994;144:275–285. [PMC free article] [PubMed] [Google Scholar]
- 5.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Am J Respir Crit Care Med. 1995;151:1628–1631. doi: 10.1164/ajrccm.151.5.7735624. [DOI] [PubMed] [Google Scholar]
- 6.Abraham N G, Drummond G S, Lutton J D, Kappas A. Cell Physiol Biochem. 1996;6:129–168. [Google Scholar]
- 7.McCoubrey W K, Huang T J, Maines M D. Eur J Biochem. 1997;247:725–732. doi: 10.1111/j.1432-1033.1997.00725.x. [DOI] [PubMed] [Google Scholar]
- 8.Morita T, Perrella M A, Lee M-E, Kourembanas S. Proc Natl Acad Sci USA. 1995;92:1475–1479. doi: 10.1073/pnas.92.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morita T, Kourembanas S. J Clin Invest. 1995;96:2676–2682. doi: 10.1172/JCI118334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morita T, Mitsialis S A, Koike H, Liu Y, Kourembanas S. J Biol Chem. 1997;272:32804–32809. doi: 10.1074/jbc.272.52.32804. [DOI] [PubMed] [Google Scholar]
- 11.Soares M P, Lin Y, Anrather J, Csizmadia E, Takigami K, Sato K, Grey S T, Colvin R B, Choi A M, Poss K D, Bach F H. Nat Med. 1998;4:1073–1077. doi: 10.1038/2063. [DOI] [PubMed] [Google Scholar]
- 12.Yet S-F, Perrella M A, Layne M D, Hsieh C-M, Maemura K, Kobzik L, Wiesel P, Christou H, Kourembanas S, Lee M-E. J Clin Invest. 1999;103:R23–R29. doi: 10.1172/JCI6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abraham N G, Lavrovsky Y, Schwartzman M L, Stoltz R A, Levere R D, Gerritsen M E, Shibahara S, Kappas A. Proc Natl Acad Sci USA. 1995;92:6798–6802. doi: 10.1073/pnas.92.15.6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christou H, Yoshida A, Arthur V, Morita T, Kourembanas S. Am J Respir Cell Mol Biol. 1998;18:768–776. doi: 10.1165/ajrcmb.18.6.2980. [DOI] [PubMed] [Google Scholar]
- 15.Korfhagen T R, Glasser S W, Wert S E, Bruno M D, Daugherty C C, McNeish J D, Stock J L, Potter S S, Whitsett J A. Proc Natl Acad Sci USA. 1990;87:6122–6126. doi: 10.1073/pnas.87.16.6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glasser S W, Korfhagen T R, Wert S E, Bruno M D, McWilliams K M, Vorbroker D K, Whitsett J A. Am. Phys. Soc. 1991. L349–L356. [DOI] [PubMed] [Google Scholar]
- 17.Xing A, Jordana M, Gauldie J, Wang J. Histol Histopathol. 1999;14:185–201. doi: 10.14670/HH-14.185. [DOI] [PubMed] [Google Scholar]
- 18.Roth M, Nauck M, Tamm M, Perruchoud A P, Ziesche R, Blck L H. Proc Natl Acad Sci USA. 1995;92:1312–1316. doi: 10.1073/pnas.92.5.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yue T L, Wang X, Sung C P, Olson B, McKenna P J, Gu J L, Feuerstein G Z. Circ Res. 1994;75:1–7. doi: 10.1161/01.res.75.1.1. [DOI] [PubMed] [Google Scholar]
- 20.Lawson C A, Yan S D, Yan S F, Liao H, Zhou Y S, Sobel J, Kisiel W, Stern D M, Pinsky D J. J Clin Invest. 1997;99:1729–1738. doi: 10.1172/JCI119337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strieter R M, Kunkel S L, Keane M P, Standford T J. Chest. 1999;116:103S–110S. doi: 10.1378/chest.116.suppl_1.103s. [DOI] [PubMed] [Google Scholar]
- 22.Ali M H, Schlidt S A, Chandel N S, Hynes K L, Schumacker P T, Gewertz B L. Am J Physiol. 1999;277:L1057–L1065. doi: 10.1152/ajplung.1999.277.5.L1057. [DOI] [PubMed] [Google Scholar]
- 23.Schecter A D, Rollins B J, Zhang Y J, Charo I F, Fallon J T, Rossikhina M, Giesen P L A, Nemerson Y, Taubman M B. J Biol Chem. 1997;272:28568–28573. doi: 10.1074/jbc.272.45.28568. [DOI] [PubMed] [Google Scholar]
- 24.Boring L, Gosling J, Cleary M, Charo I F. Nature (London) 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 25.Voelkel N F, Tuder R M, Bridges J, Arend W P. Am J Respir Cell Mol Biol. 1994;11:664–675. doi: 10.1165/ajrcmb.11.6.7946395. [DOI] [PubMed] [Google Scholar]
- 26.Kimura H, Kasahara Y, Kurosu K, Sugito K, Takiguchi Y, Terai M, Mikata A, Natsume M, Mukaida N, Matsushima K, Kuriyama T. Lab Invest. 1998;78:571–581. [PubMed] [Google Scholar]
- 27.Thompson K, Rabinovitch M. J Cell Physiol. 1996;166:495–505. doi: 10.1002/(SICI)1097-4652(199603)166:3<495::AID-JCP4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 28.Maruyama K, Ye C, Woo M, Venkatacharya H, Lines L D, Silver M M, Rabinovitch M. Am J Physiol. 1991;261:H1716–H1726. doi: 10.1152/ajpheart.1991.261.6.H1716. [DOI] [PubMed] [Google Scholar]
- 29.Jones P L, Cowan K N, Rabinovitch M. Am J Pathol. 1997;150:1349–1360. [PMC free article] [PubMed] [Google Scholar]
- 30.Cowan K N, Heilbut A, Humpl T, Lam C, Ito S, Rabinovitch M. Nat Med. 2000;6:698–702. doi: 10.1038/76282. [DOI] [PubMed] [Google Scholar]
- 31.Chandel N S, Maltepe E, Goldwasser E, Mathieu C E, Simon M C, Schumacker P T. Proc Natl Acad Sci USA. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otterbein L E, Bach F H, Alam J, Soares M, Lu H T, Wysk M, Davis R J, Flavell R A, Choi A M K. Nat Med. 2000;6:422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 33.Otterbein L E, Kolls J K, Mantell L L, Cook J L, Alam J, Choi A M K. J Clin Invest. 1999;103:1047–1054. doi: 10.1172/JCI5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amersi F, Beulow R, Kato H, Ke B, Coito A J, Shen X-D, Zhao D, Zaky J, Melinek J, Lassman C R, et al. J Clin Invest. 1999;104:1631–1639. doi: 10.1172/JCI7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei Z-F, Achong M K. J Clin Invest. 1998;101:311–320. doi: 10.1172/JCI1368. [DOI] [PMC free article] [PubMed] [Google Scholar]