

Abstract
Axonal transport of synaptic vesicle proteins is required to maintain neurons’ ability to communicate via synaptic transmission. Neurotransmitter-containing synaptic vesicles are assembled at synaptic terminals via highly regulated endocytosis of membrane proteins. These synaptic vesicle membrane proteins are synthesized in the cell body and transported to synapses in carrier vesicles that make their way down axons via microtubule-based transport utilizing kinesin molecular motors. Identifying the cargos that each kinesin motor protein carries from the cell bodies to the synapse is key to understanding both diseases caused by motor protein dysfunction and how synaptic vesicles are assembled. However, obtaining a bulk sample of axonal transport complexes from central nervous system (CNS) neurons to use for identification of their contents has posed a challenge to researchers. To obtain axonal carrier vesicles from primary cultured neurons, we fabricated a microfluidic chip designed to physically isolate axons from dendrites and cell bodies and developed a method to remove bulk axonal samples and label their contents. Synaptic vesicle protein carrier vesicles in these samples were labeled with antibodies to the synaptic vesicle proteins p38, SV2A, and VAMP2, and the anterograde axonal transport motor KIF1A, after which antibody overlap was evaluated using single-organelle TIRF microscopy. This work confirms a previously discovered association between KIF1A and p38 and shows that KIF1A also transports SV2A- and VAMP2-containing carrier vesicles.
Keywords: microfluidics, TIRF microscopy, axonal transport, single organelles, synaptic vesicle proteins, kinesin motor proteins
Axonal transport is critical for maintaining neurons’ unique highly polarized structure. Synaptic proteins are synthesized in the cell body and transported down the axon in carrier vesicles and protein complexes bound to kinesin motors moving on microtubule tracks to the synapse. After arrival at the synapse, these proteins are incorporated into synaptic vesicles via exocytosis of the carrier vesicles followed by endocytotic assembly of synaptic vesicles containing the correct protein composition. Several kinesin superfamily motors (KIFs) have been implicated in the anterograde transport of synaptic vesicle proteins, specifically KIF1A, KIF1Bβ, and KIF5B.1−6 Despite some redundancy in their cargos, all three of these motors are required for neuronal function, and the absence of any one of them is a fatal phenotype.2,3 Much of what we know about these motors and their cargos comes from studies of whole brain3,5,7,8 or peripheral nervous system1 carrier vesicles purified by immunoisolation, or from small-scale studies of fluorescently labeled cells.2,4−10 In spinal axons obtained from the cauda equina, synaptic vesicle proteins are carried in distinct carriers by at least two different molecular motors,1 consistent with the idea that some synaptic vesicle proteins stay segregated until formation of synaptic vesicles. To explore the specificity and variance of protein transport in central nervous system axons, we developed a microfluidic method to collect axonal carriers from CNS neurons and a protocol to label and analyze their cargos at the single organelle level.
Microfluidic devices for neuronal cell culture and analysis have developed rapidly over the past decade.11−19 They offer a number of advantages over traditional coverslip-based cultures, including the ability to control the locations of cells or cell processes with a high degree of precision, and to easily create and manipulate microenvironments within a single culture. Poly(dimethylsiloxane) (PDMS)-based microfluidic systems in particular offer a number of features particularly conducive to cell culture. These include oxygen permeability, a highly biocompatible surface composition, ease of fabrication, and the low cost of rapidly producing many culture devices. This ease of use has motivated the development of microfluidic devices designed to permit axons to isolated from dendrites and cell bodies.11,15,16,19 However, these devices do not allow for the removal of axonal material without the use of detergents or time-consuming machining and surface treatments. Therefore, we refined this approach to obtain axonal material from cultures in microfluidic devices to combine physical isolation with the ability to selectively remove axonal material through mechanical means.
This paper describes our technique and its application to the analysis of axonal transport carriers for synaptic vesicle proteins isolated from cultured mouse hippocampal neurons. Single vesicles were labeled using immunofluorescence and imaged to assess the coexistence of the synaptic vesicle proteins p38, SV2A, and VAMP2 with the anterograde transport motor KIF1A. We found that all three proteins are carried by this motor, unlike in the PNS where only p38 and VAMP2 are transported by KIF1A.1 While low variability was found in the populations of protein examined in this study, this method would allow for variability to be observed. Future studies of other proteins may find there are subpopulations in carrier vesicle contents and that these contents differ over the lifespan of the culture.
Results and Discussion
Axonal Sample Preparation
We designed and fabricated a microfluidic device, based on previous designs,11,15 that allows for the physical isolation and removal of the axons from 14 to 17 days in vitro (d.i.v.) hippocampal neurons in culture (Figures 1 and 2). This device allows neurons to be cultured on-chip with their axons physically isolated from the dendrites and cell bodies. By increasing the density of the connecting channels between the somal and axonal chambers compared to previous designs, enough axons grow into the axonal chamber to allow for significant quantities of axonal material to be collected for further analysis. Dense, healthy cultures of neurons maintained in this device yielded over a microgram of purified axonal material. These microfluidic devices are constructed of PDMS and poly-d-lysine-coated glass, with the PDMS slabs molded to contain the desired microfluidic channel pattern using soft lithography.20 Freshly dissociated hippocampal neurons from newly born mice are plated directly into the somal chambers and the connecting channels and axonal chamber are filled with fresh media. The channel layout takes advantage of the faster growth rate and length of axons, so that after 14 d.i.v. only axons have grown longer than the 500 μm long connecting channels between the somal and axonal compartments (Figure 1b–d).
Figure 1.
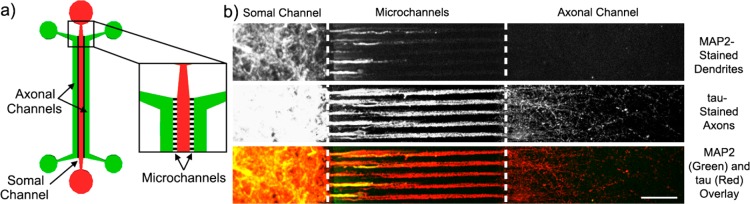
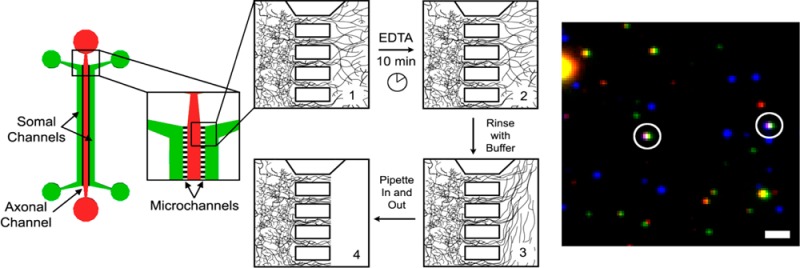
The axonal sample isolation device allows for axons to be physically isolated from other neuronal components. (a) A schematic of the microfluidic chip for neuronal growth and axonal isolation. Three large channels, 1.5 mm wide and ∼300 μm high, are spaced 500 μm apart. The inside channel is designated the somal channel (shown in red) is where freshly dissociated neurons are plated. The outside channels are designated the axonal channels (shown in green) and are only filled with media. The three channels are all connected by small microchannels that are 10 μm wide, 3 μm high, 500 μm lonh, and spaced 20 μm apart (shown in black in the inset, not to scale). These microchannels are too small to allow for cell bodies to enter, so only dendrites and axons grow into them. Dendrites do not grow to the same length or as quickly as axons grow, so at 14 d.i.v. they are still significantly shorter than the 500 μm microchannels connecting the somal chambers to the axonal chamber (see MAP2-labeled dendrites in b, top panel). Axons, however, grow long enough and quickly enough to fill the axonal chambers with a dense mat of processes (see tau-labeled axons in b, middle panel). Two-color overlay between the MAP2-labeled (green) dendrites tau-labeled (red) axons at 14 d.i.v. is shown in b, bottom panel. Scale bar is 100 μm.
Figure 2.
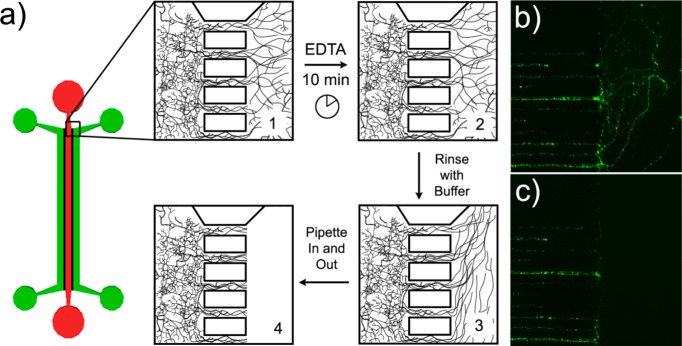
Samples of pure axonal material can be mechanically isolated using the axonal sample isolation device. (a) Axonal sample isolation device and method. Step 1: Plate neurons in the somal (red) channels and wait until 14 d.i.v. for complete axonal growth. Step 2: Fill axonal (green) channel with EDTA to deadhere axons from coverglass. After 10 min, remove the EDTA and rinse the axons with sample buffer. Step 3: Using fresh sample buffer, pipet in and out at the entrance to the channel to pull and break axons. Step 4: Rinse axonal channels with sample buffer to remove any remaining axonal material. Dendrites in the small connecting channels remain undisturbed. (b) EGFP expressing axons in the connecting channels and axonal chamber prior to and (c) just after removal. Connecting channels are 10 μm wide and spaced 20 μm apart.
At 14–17 d.i.v., axons are lifted off of the glass surface using EDTA and then removed through using trituration (see Figure 2a). Due to the high fluidic resistance of the small channels connecting the somal and axonal chambers, the axons break off where the channels begin, leaving both the dendrites in the small connecting channels and cell bodies in the somal chambers (see Figure 2b) undisturbed. In contrast to other methods used to obtain samples of axonal proteins from cultured CNS neurons, no detergents are used so that a wider variety of downstream assays can be performed,15 and no special stamping or machining of each glass culture surface is required prior to use.19 The resulting bulk sample of pure axonal material is free of dendritic and somal contamination, which is critical for any analysis of axonal transport. The absence of dendrites in the sample minimizes the number of mature synapses as most synapses are between axons and dendrites.
Axonal Sample Labeling and Imaging
To isolate carriers from within axons, samples were flash-frozen and thawed to disrupt the axonal plasma membrane, homogenized, and unbroken axons and debris cleared by centrifugation. The remaining supernatant, containing the light membrane fraction, was immunolabeled for proteins and motors of interest (see Figure 3). Briefly, samples were first incubated in two primary antibodies, one against a vesicle cargo protein and another against a motor protein. Excess primary antibody was removed with anti-mouse and anti-rabbit IgG agarose beads, which have been shown to preferentially remove free antibodies over antibodies bound to organelles.21 Antibodies bound to motor proteins were labeled with a fluorescent secondary antibody. Antibodies bound to vesicle proteins were labeled with two secondary antibodies, conjugated to fluorescent dyes with different emission wavelengths. Carrier vesicles are expected to have multiple copies of each type of cargo protein; thus, double-labeling cargo proteins provides additional assurance that fluorescent entities are bona fide carrier vesicle–motor complexes and not errant free antibodies.
Figure 3.

Axonal carrier vesicles can be labeled with primary and secondary antibodies for facile visualization. Schematic: Step 1: Incubate sample overnight with primary antibodies, two for a synaptic vesicle protein of interest and one for the motor of interest. Step 2: Incubate sample with anti-mouse and anti-rabbit IgG agarose beads to remove excess primary antibody. Step 3: Incubate sample with dye-conjugated secondary antibodies, one color for each primary, for 3–6 h. Step 4: Incubate sample with anti-goat IgG agarose beads to remove excess secondary antibody.
Antibody-labeled carriers were imaged in microfluidic chips using TIRF microscopy.21,22 TIRF microscopy was chosen over epifluorescence imaging for single-organelle imaging due to its reduced background fluorescence when imaging fluorescently labeled single organelles.21 A schematic of our imaging setup is shown in Figure 4a. Sample was introduced into a straight microfluidic channel 1 mm wide and 300 μm high and allowed to flow through the channel. The channel was made of PDMS bonded to a glass coverslip (see Methods). Previous studies revealed that membranous organelles stick to the glass surface when loaded in isotonic buffers.21 Excess volume was then removed from the wells at both ends of the microchannel, and the adhered organelles were rinsed with fresh AC buffer prior to imaging. Images were taken sequentially of each field-of-view on the channel’s glass surface using lasers emitting 488 nm, 561 nm, and 638 nm light with emission filters limited to the emission ranges of AlexaFluor-488, AlexaFluor-568, and AlexaFluor-647 (see Figure 4b–e). This procedure was repeated with samples containing only the primary antibodies to p38 and KIF1A and the AlexaFluor-488, AlexaFluor-568, and AlexaFluor-647 secondary antibodies as a control.
Figure 4.
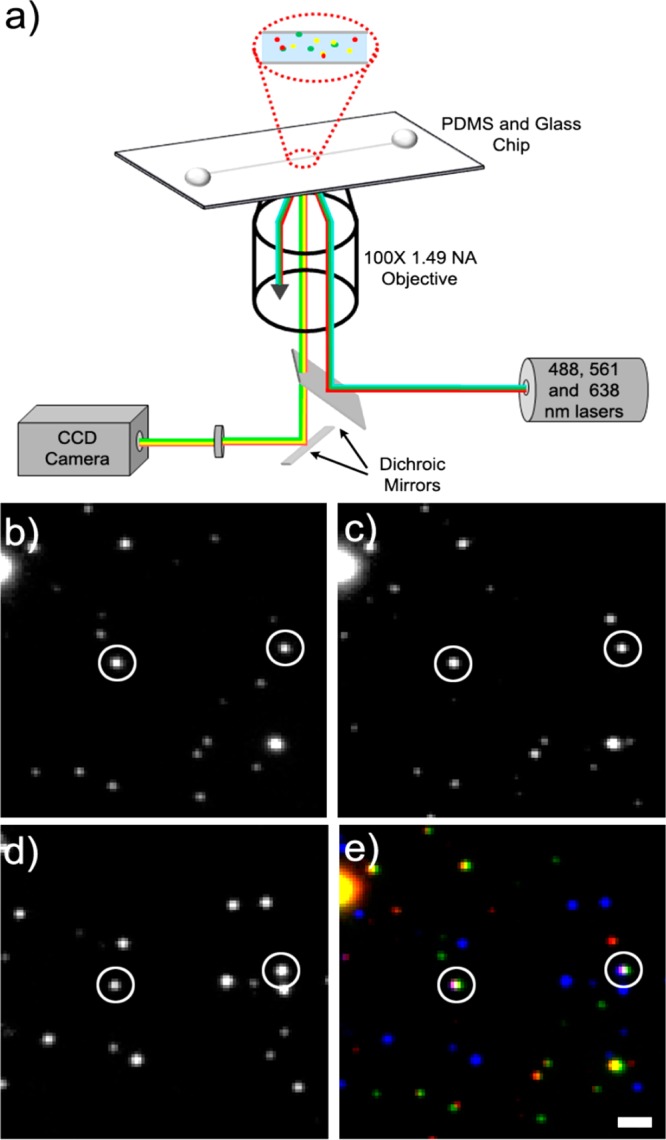
Fluorescently labeled axonal carrier samples can be viewed in three colors using TIRF microscopy. (a) TIRF imaging setup schematic. (b) Alexa Fluor 488 puncta (p38 rabbit polyclonal primary antibody), (c) Alexa Fluor 568 puncta (p38 rabbit polyclonal primary antibody), and (d) Alexa Fluor 647 puncta (KIF1A mouse monoclonal primary antibody) are (e) checked for colocalization (Alexa Fluor 488 shown in green, Alexa Fluor 568 shown in red, and Alexa Fluor 647 shown in blue). Puncta that fit the three-color colocalization criteria are circled in white. Scale bar is 1.6 μm.
Axonal Sample Data Analysis
Images were analyzed by first selecting spots that contained both colors used to label the synaptic vesicle protein of interest, and then for overlap with the color used to label the motor of interest. Samples that had protein/motor overlap percentages comparable to antibody complexes were discarded as labeling failures. If a motor–protein pair sample continually failed this test, the motor and protein were considered to be in different carrier complexes. Colocalization between spots was defined as having fluorescent centroids calculated to be within 160 nm of each other.
Because the small size of the carrier vesicles that transport synaptic vesicle proteins render them below the diffraction limits of light microscopy, one of the most important considerations when using light microscopy to analyze colocalization of fluorescent labels is the acceptance distance permitted between the centroids of the differently colored fluorescent puncta. While modulating the permitted acceptance distance can allow for larger organelles or aggregates to be included in the data set, it can also increase the number of false positives. Figure 5 illustrates how choosing the permitted acceptance distance helps distinguish between errant free antibody complexes/aggregates and actual single transport vesicles. At small acceptance distances, larger organelles will be excluded and the puncta must be very bright to ensure the accuracy of the perceived centroids of the puncta. As the acceptance distance is increased, the amount of random matching with fluorescent puncta that are near and not part of the organelle increases for very small organelles such as these. KIF1A anterograde transport vesicles are estimated to be in the sub-150 nm range,6 so we limited our acceptance distance to 160 nm to exclude larger vesicles and protein aggregates. Note that this level of distinction requires that the optics do not introduce a shift between the images in each spectral range and that the fluorophores used are bright enough to accurately calculate the centroid of the fluorescent puncta. For larger organelles, this is not an issue, but for the smaller transport organelles analyzed here the ability to distinguish centroids within 100 nm was critical.
Figure 5.
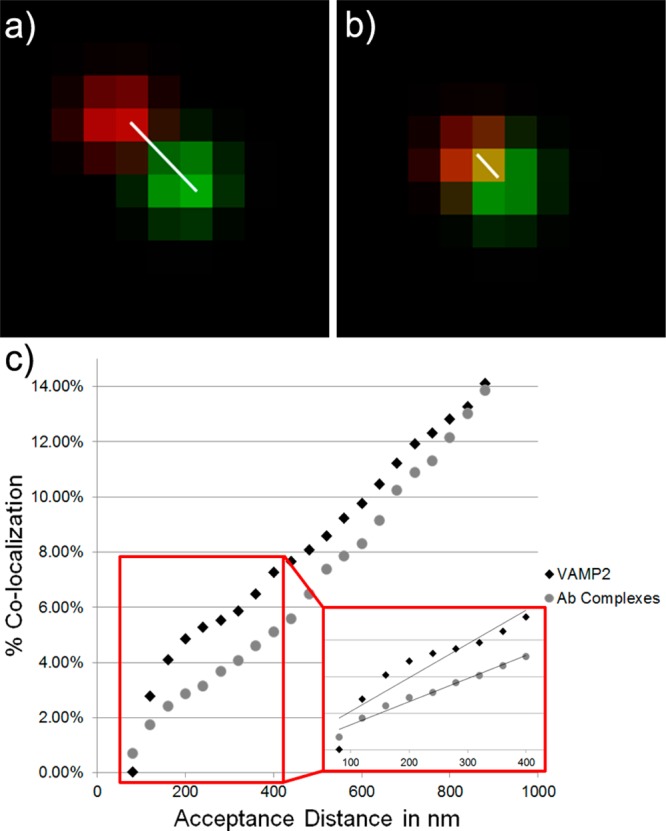
Acceptance distance variation affects colocalization. Example puncta where the distance between their fluorescence peaks is (a) 2.8 pixels or 448 nm apart and (b) 1 pixel or 160 nm apart. Setting a high acceptance distance, such as 3.0 pixels, would allow for fluorescent puncta notably farther apart than the size of a single organelle (such as those in (a)) to be considered colocalized and thus the proteins they label are incorrectly considered to be part of the same transport complex. By limiting the acceptance distance to 1.0 pixels, only fluorescent puncta that are labeling proteins on the same transport complex are accepted as colocalizing puncta. The white lines connect the fluorescent intensity peaks of the red and green puncta to aid in visualizing the distance between them. (c) Acceptance distance effects. The effects of modulating the permitted acceptance distance between fluorescent puncta is illustrated for a single sample labeled to identify colocalization between a single sample of VAMP2 and KIF1A and a control sample containing only primary–secondary antibody complexes and no axonal organelles. As the acceptance distance is increased beyond that of the estimated vesicle size, aggregates and other organelles or free antibody complexes near to the vesicle are falsely matched to it. A certain amount of false matching, considered to be the background level, is due to the sticking and aggregation of free antibody complexes and is controlled for by examining samples of free antibody complexes. Note that when the acceptance distance is limited to the size range axonal carrier vesicle-motor transport complexes are anticipated to be, the percentage of colocalization between VAMP2-labeled puncta and KIF1A-labeled puncta increases above the amount that would be anticipated with random matching of free antibody complex puncta (see inset). This indicates that actual axonal transport complexes are being visualized, not just free antibody complexes, and that the protein and motor are both present on these complexes. The inset graph in (c) shows the best linear fits to the data in the matching range in which these carrier vesicles and motor are expected to be found. The R2 value for the free antibody complex data is 0.979, while the R2 value for the VAMP2 data is 0.858.
Single Carrier Analysis Reveals That KIF1A Transports Synaptophysin (p38), VAMP2, and SV2 Down Axons in Hippocampal Neurons
We examined the colocalization of three synaptic vesicle proteins, p38, SV2A, and VAMP2, with KIF1A, the motor implicated in the transport of most synaptic vesicle proteins. While p38 has been found to be a cargo of KIF1A, previous work suggested that SV2 was carried exclusively by the motor protein KIF1Bβ3 and not by KIF1A.1 Additionally, VAMP2 and SV2 were previously shown to be in many of the same axonal transport packets,9 but no association between VAMP2 and KIF1A has been found.
In contrast to these findings, our analyses revealed that p38, SV2A, and VAMP2 are all carried by KIF1A, though to varying degrees (Figure 6). p38-labeled organelles repeatedly colocalized with KIF1A, confirming previous work done in PNS samples1 and indicating that p38 is carried primarily by KIF1A. SV2A- and VAMP2-labeled organelles also colocalized with KIF1A motors at a rate of more than twice background levels, consistent with their moving down axons via KIF1A some of the time. To provide additional assurance that the apparent colocalization of KIF1A with VAMP2 and SV2 was not due to overcrowded images, we assessed the effects of introducing an artificial shift of 320 nm horizontally between the KIF1A images and vesicle protein images. We found that the rate of colocalization between VAMP2/SV2 and KIF1A dropped below that of colocalization in control samples containing only antibody complexes. This suggests that the colocalization is likely to reflect true association of KIF1A with carriers containing these vesicle proteins.
Figure 6.
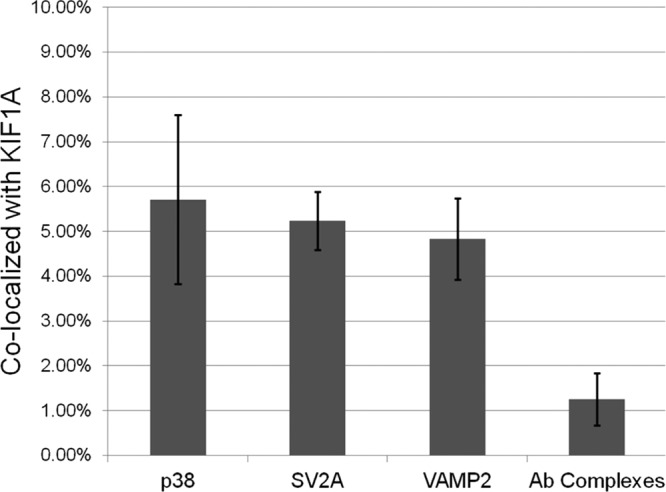
Fluorescently labeled synaptic vesicle proteins colocalize with fluorescently labeled KIF1A more frequently than free antibody complexes colocalize with one another. (a) Percent of colocalization between protein spots (defined as puncta where the two colors that label the synaptic vesicle protein of interest are colocalized) and motor spots (defined as puncta of the color that labels the KIF1A). Percentage colocalization for antibody complexes is defined as the colocalization between the two-color spots of the colors used to label a protein and the color used to label a motor in a sample containing primary and secondary antibodies but no axonal material. Error bars represent the standard error (SE). Specifically, 5.71% p38 colocalizes with KIF1A (SE: 1.89%), 5.23% of SV2A colocalizes with KIF1A (SE: 0.65%), and 4.83% of VAMP2 colocalizes with KIF1A (SE: 0.91%). 1.25% of primary–secondary antibody complexes labeled with the two colors of secondary antibody used to label the synaptic vesicle protein colocalize with primary–secondary antibody complexes labeled with the color used to label the KIF1A motor (SE: 0.59%).
We note that the coexistence of all three labels was low in all samples, suggesting that the majority represent a combination of primary–secondary antibody complexes, transport organelles that have dissociated from their motors, and transport organelles containing the protein but associated with another motor such as KIF1Bβ or KIF5B. The lower coexistence of VAMP2 and SV2 with KIF1A suggests that they may also be carried by motors other than KIF1A.
Method Advantages and Considerations
The method we report here offers a number of advantages over traditional biochemical analysis and in situ analysis of axonal transport complexes. First, the sample we obtain is much cleaner than a sample of axonal material directly taken from the PNS. Unlike a sample purified directly from an adult animal, the amount of material obtained using the axonal isolation device is dependent on culture age and health. For a dense and healthy culture of hippocampal neurons, the current device can yield over a microgram of purified axonal material in the light membrane fraction after removal from the device and centrifugation to remove large pieces of membrane and debris.
By looking at individual carriers instead of the entire sample in bulk, variability and subpopulations within a sample can be identified. Additionally, this technique allows for the use of samples containing significantly less material than required for analysis by Western blotting. As with a Western blot of immunoisolated material, it is possible to rapidly discern the molecular composition of the axons as has previously been shown1,6 while still allowing for subtleties in molecular composition and subpopulations of complexes to still be observable. This is not possible with in situ microscopy methods.
The analyses reported here, unlike previous studies of single synaptic vesicles,21 revealed sufficient free primary–secondary antibody complexes bound to the glass surface, necessitating a method to filter out false positive signals. This appears to be due to the fact that the samples analyzed here were more dilute. In more highly concentrated samples, less volume (1–3 μL) is loaded into the imaging channels and the amount of free antibody complexes constitute a much smaller portion of the total sample. This is likely why Mutch et al. saw no free antibody complexes in their analyses of brain synaptic vesicles. In more dilute samples where larger sample volumes (20–80 μL) of sample are loaded, the ratio of free antibody to sample is higher and the amount of free antibody loaded into the channel is higher. By using two secondary antibodies to label one of the target proteins and requiring colocalization of all three labels, we can control for the fact that free antibody complexes account for a higher proportion of the spots in our images. Working with dilute samples also requires that all reagents used be carefully screened before use to ensure that no fluorescent contamination is present. For example, we found that several commercially available protease inhibitor cocktails contain diffraction-limited particles that were fluorescent throughout the visible spectrum. Future work will focus on attaching antibodies to the glass surface of channels to increase the likelihood that only organelles stick to the channel surface. A further consideration is the use of agarose beads to reduce the amount of unbound antibodies in the samples. While the beads selectively bind to free antibodies remaining in the sample, which is in excess to antibody bound to protein, it is possible they may strip weakly bound antibodies from their antigens. However, given that we observed previously unreported associations, we do not believe this to be a significant issue.
In summary, we have developed a technique that allows for bulk samples of cultured axons to be obtained. The resulting sample is free of detergents and leaves subcellular organelles intact for analysis by a variety of methods, including fluorescent analysis of individual carrier vesicle–motor complexes. Using this technique to look at three synaptic vesicle proteins and a kinesin implicated in their anterograde transport, we were able to illuminate a previously unknown association between the motor protein KIF1A and the synaptic vesicle proteins SV2A and VAMP2. In addition to single-organelle imaging, other analysis techniques such as mass spectrometry could also be used to analyze these samples. We anticipate this technique will become a valuable part of the neurobiologist’s toolbox for probing the molecular contents of axons.
Methods
Antibodies
Anti-KIF1A monoclonal antibody (KIF1A mAb) was obtained from BD Biosciences, and this antibody has been shown to be specific to KIF1A using RNAi studies.23,24 Anti-VAMP2 polyclonal antibody grown in rabbit against an epitope near the N-terminus of the protein was obtained from Synaptic Systems and does not label any protein in VAMP2 knockout mice.25 Anti-synaptophysin 1 polyclonal antibody grown in rabbit against an epitope near the C-terminus of the protein was also obtained from Synaptic Systems. Anti-SV2A polyclonal antibody (SV2A pAb) recognizes an epitope in the cytoplasmic domain of the protein26 and does not label any protein in cells deficient in SV2A.27 Fluorescently labeled secondary antibodies, specifically goat anti-rabbit (GAR) Alexa Fluor 488, GAR Alexa Fluor 568, and goat anti-mouse (GAM) Alexa Fluor 647 were obtained from Invitrogen. Agarose beads conjugated to GAM, GAR, or rabbit anti-goat IgG, anti-MAP2, and anti-tau antibody were obtained from Sigma-Aldrich.
Microfluidic Chip Fabrication and Preparation
Microfluidic devices for both neuronal culture and TIRF imaging of carrier vesicle–motor complexes were fabricated using standard photolithography to generate the silicon masters and poly(dimethylsiloxane) (PDMS) replica molding techniques to generate the final microfluidic devices.20,28,29 For the neuron culture device masters, silicon wafers were coated with a 3 μm thick layer of SU-8 2002 photoresist (MicroChem) and exposed to UV light through an exposure mask with the pattern for the small connecting channels between the cell culture (somal) and axonal channels. Following development and a 30 min hard bake at 150 °C, a 300–400 μm thick layer of SU-8-2150 photoresist was spun on the same wafer, and the exposure mask with the 1.5 mm wide somal and axonal channels was aligned to the existing features and then again exposed to UV light. Following development of the second layer of features and another 30 min hard bake at 150 °C, the master was exposed overnight to tridecafluoro-1,1,2,2-tetrahydrooctyl-1-trichlorosilane vapor to aid in removing the PDMS slabs from the master. For the TIRF imaging device masters, the same procedure was followed but only one 300 μm thick layer of SU-8-2150 photoresist was spun prior to a single UV exposure through the feature mask with the 1 mm wide straight channels.
The microfluidic straight channels were formed out of PDMS and glass coverslips. A 10:1 ratio of prepolymer to catalyst was mixed, degassed under a vacuum, poured onto the master wafer, and allowed to bake for 2–4 h at 65 °C for the imaging devices and 1 h at 115 °C for the cell culture devices. After being cut out and removed from the master, access holes were drilled into the PDMS slabs using a ∼5 mm punch and then the slabs were plasma bonded to borosilicate glass coverslips. Prior to use, all coverslips were cleaned by boiling for 1–2 h in a 3:2:1 mixture of Milli-Q H2O, 30% hydrogen peroxide, and ammonium hydroxide and then rinsed in Milli-Q H2O. Following plasma bonding, culture devices were sterilized under a UV lamp, filled with 100 μg/mL poly-d-lysine solution, and incubated at 37 °C for 2 h. Prior to use, the devices were rinsed with Milli-Q H2O and then filled with hippocampal culture medium. Immediately after plasma bonding, imaging devices were filled with AC buffer (35 mM PIPES, 5 mM MgSO4, 5 mM EGTA, 0.5 mM EDTA, 1 mM DTT, 2 mM Mg-ATP, 2 mM potassium phosphoenolpyruvate, 5 U/mL pyruvate kinase, and complete protease inhibitor (EDTA-free tablets, Roche, pH 7.4)). All straight channels for imaging were sealed within 6 h of use.
Hippocampal Cultures
Cultures of primary mouse hippocampal neurons were prepared using previously established procedures.30 In brief, hippocampi from newly born (Postnatal Day 0 or 1) mice were isolated and incubated in papain for 30 min at 37 °C. Following incubation, they were washed, mechanically dissociated, filtered through a 70 μm cell filter, and centrifuged to concentrate the cells in 500–900 μL of culture medium. The somal channels of the device were then loaded with 100–200 μL of densely suspended cells. After allowing the cells to adhere to the channel for 30–60 min, the wells at either end of the somal and axonal chambers were filled with additional culture medium and left to grow for 14–17 days. Additional medium was added to the wells as needed. Hippocampal culture medium contained minimum essential medium supplemented with 10% fetal horse serum, 25 mM HEPES, 20 mM glucose, 50 U/mL penicillin, 50 μg/mL streptomycin, 1:200 N2 supplement, 1:100 B27 supplement, and 0.1 mM sodium pyruvate.
Axonal Sample Preparation
At 14–17 days in vitro, the axonal and somal channels were rinsed with 1× PBS, and then the axonal channel was filled with 10 mM EDTA and incubated for 10 min at 37 °C to help lift the axons off the culture substrate. After 10 min, the EDTA was removed from the axonal channel and the axons were gently rinsed with AC Buffer. Using 200 μL of fresh AC buffer per axonal chamber, a micropipet placed at the entrance to one end of the axonal chamber was vigorously pipetted in and out for 30–45 s to sever the axons at the edges of the axonal channel. This axonal material was placed on ice and then flash-frozen in dry ice cooled ethanol prior to storage at −80 °C. Previously collected axonal samples were thawed on ice and homogenized using a Dounce homogenizer. All steps were either performed at 4 °C or on ice. The resulting homogenate was spun for 10 min at 18 000g to remove large cellular debris, and the pellet was discarded. Supernatant containing axonal organelles was diluted 1–4-fold and aliquoted into 100 μL samples for the labeling process.
Axonal Transport Carrier Complex Labeling
Axonal organelles were incubated overnight with primary antibodies, two for the synaptic vesicle protein of interest and one for the motor protein of interest, diluted at 1:000. Following incubation, samples were incubated for 30 min with 20 μL each of anti-mouse and anti-rabbit IgG agarose beads to remove excess primary antibody. Dye-conjugated secondary antibodies from goat with Alexa Fluor 488, Alexa Fluor 568, and Alexa Fluor 647 were added to the samples at 1:500 and incubated for 3–6 h, followed by removal of excess secondary antibody with anti-goat IgG agarose beads. Samples were then stored on ice in the dark until imaging within 6 h after secondary antibody removal.
Axonal Carrier Complex Imaging
Total internal reflection fluorescence was generated as previously described,21 generating a ∼300 nm thick evanescent field that excited molecules and organelles immobilized on the surface of the imaging chip. Specifically, samples were introduced into the imaging channels using gravity-driven flow. Excess AC buffer was removed from the inlet and outlet, followed by the addition of 20–80 μL of sample. Samples were allowed to flow through the channel and absorb for up to 5 min before excess sample was removed and 50–100 μL of fresh AC buffer was added to the channel to wash away any unadhered organelles. Images were acquired on a Nikon Ti-E inverted microscope outfitted with a motorized XY stage for automated scanning, a Nikon Perfect Focus System, and a 100× TIRF Apochromat 1.49 NA oil objective lens using an Andor iXon DU-897 EMCCD camera. Each area of interest was imaged three times, using a 488 nm 50 mW argon laser, a 561 nm 20 mW DPSS laser, and a 638 nm 100 mW diode laser. Image acquisition and stage movement were controlled by Nikon’s NIS-Elements software.
Data Analysis
A MATLAB image-processing program based on previously published algorithms22 was developed to identify puncta and check for two- and three-color overlay. Puncta smaller than 4 pixels were rejected as noise and puncta larger than the resolution limit of the imaging setup (∼2.3 pixels), allowing for slight deviations in the focal plane, were rejected as aggregates. All accepted puncta were required to have a signal-to-noise ratio greater than 3.0.
Immunocytochemistry
Axonal isolation devices that had been conformally sealed, not plasma bonded, and filled with neurons were allowed to grow for 14 d.i.v. At this time, the PDMS slabs were removed without disturbing the channel contents, leaving only the neurons on the coverslip for fixation. These coverslips were rinsed with 1× PBS, fixed in 4% paraformaldehyde for 20 min, and then blocked in blocking solution (2% normal goat serum, 0.4% saponin, and 1.0% BSA in 1× PBS) for 1 h. After blocking, coverslips were incubated with anti-MAP2 and anti-tau antibodies at a ratio of 1:500 in blocking solution for 2 h, rinsed, and incubated with Alexa Fluor 488 and Alexa Fluor 568 secondary antibodies at a ratio of 1:1000 in blocking solution for 1 h. Following a 10 min incubation in Hoechst stain (1:10 000, Invitrogen), coverslips were rinsed and mounted on slides using Fluoromount-G (Electron Microscopy Sciences) for fluorescence imaging.
Lentiviral Constructs, Production, and Infection
cDNA encoding enhanced green fluorescent protein was subcloned into the carrier vector pRRL. To generate virons carrying the EGFP cDNA, human embryonic kidney 239T (HEK293T) fibroblasts were cotransfected with packaging plasmids pLP1, pLP2, and pLP VSV-G from the Virapower packaging mix (Invitrogen) and the EGFP-pRRL carrying vector using calcium phosphate. After incubating overnight, the medium (Dulbecco’s modified Eagle's medium supplemented with 10% fetal bovine serum, 50 U/mL penicillin, and 50 μg/mL streptomycin) was replaced with fresh medium, and subsequently viron-containing media was harvested twice at 24 h intervals. Viron-containing media was then filtered through 0.22 μm pore-size PVDF membrane, and then virons were concentrated by centrifuging at 6700g for a minimum of 16 h at 4 °C. Pellets were resuspended in media and stored at −80 °C until use. Cultures were infected with these virons at 1–3 d.i.v. for imaging axonal removal from the axonal channel (Figure 2).
Fixed and Live Cell Imaging
Fluorescence images of fixed cells were taken on the same Nikon TIRF setup described in the axonal carrier complex imaging section. Fluorescence images of EGFP-expressing cells were taken on a Deltavision deconvolution microscope.
Acknowledgments
The authors thank Lisa Baldwin for animal husbandry and Dr. Bryant Fujimoto for assistance with Matlab and image analysis.
Author Contributions
A.E.S. designed the technique, performed the experiments and data analysis, and wrote the manuscript. S.M.B. and D.T. C. provided lab space, equipment, supervised, and provided input to the overall project and final manuscript.
This work was supported by Grant NIH R01 NS052637 from the National Institute of Neurological Disease and Stroke to S.M.B. and D.T.C.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Okada Y.; Yamazaki H.; Sekine-Aizawa Y.; Hirokawa N. (1995) The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell 81, 769–780. [DOI] [PubMed] [Google Scholar]
- Yonekawa Y.; Harada A.; Okada Y.; Funakoshi T.; Kanai Y.; Takei Y.; Terada S.; Noda T.; Hirokawa N. (1998) Defect in Synaptic Vesicle Precursor Transport and Neuronal Cell Death in KIF1A Motor Protein-deficient Mice. J. Cell Biol. 141, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C.; Takita J.; Tanaka Y.; Setou M.; Nakagawa T.; Takeda S.; Yang H. W.; Terada S.; Nakata T.; Takei Y.; Saito M.; Tsuji S.; Hayashi Y.; Hirokawa N. (2001) Charcot-Marie-Tooth Disease Type 2A Caused by Mutation in a Microtubule Motor KIF1B[β]. Cell 105, 587–597. [DOI] [PubMed] [Google Scholar]
- Nakata T.; Hirokawa N. (2003) Microtubules provide directional cues for polarized axonal transport through interaction with kinesin motor head. J. Cell Biol. 162, 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q.; Pan P.-Y.; Sheng Z.-H. (2007) Syntabulin-Kinesin-1 Family Member 5B-Mediated Axonal Transport Contributes to Activity-Dependent Presynaptic Assembly. J. Neurosci. 27, 7284–7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa S.; Tanaka Y.; Hirokawa N. (2008) KIF1B[β]- and KIF1A-mediated axonal transport of presynaptic regulator Rab3 occurs in a GTP-dependent manner through DENN/MADD. Nat. Cell Biol. 10, 1269–1279. [DOI] [PubMed] [Google Scholar]
- Zhai R. G.; Vardinon-Friedman H.; Cases-Langhoff C.; Becker B.; Gundelfinger E. D.; Ziv N. E.; Garner C. C. (2001) Assembling the Presynaptic Active Zone: A Characterization of an Active Zone Precursor Vesicle. Neuron 29, 131–143. [DOI] [PubMed] [Google Scholar]
- Shapira M.; Zhai R. G.; Dresbach T.; Bresler T.; Torres V. I.; Gundelfinger E. D.; Ziv N. E.; Garner C. C. (2003) Unitary Assembly of Presynaptic Active Zones from Piccolo-Bassoon Transport Vesicles. Neuron 38, 237–252. [DOI] [PubMed] [Google Scholar]
- Ahmari S. E.; Buchanan J.; Smith S. J. (2000) Assembly of presynaptic active zones from cytoplasmic transport packets. Nat. Neurosci 3, 445–451. [DOI] [PubMed] [Google Scholar]
- Kaether C.; Skehel P.; Dotti C. G. (2000) Axonal Membrane Proteins Are Transported in Distinct Carriers: A Two-Color Video Microscopy Study in Cultured Hippocampal Neurons. Mol. Biol. Cell 11, 1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A. M.; Blurton-Jones M.; Rhee S. W.; Cribbs D. H.; Cotman C. W.; Jeon N. L. (2005) A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat. Methods 2, 599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin F.; Nishimura N.; Griscom L.; LePioufle B.; Fujita H.; Takamura Y.; Tamiya E. (2006) Constraining the connectivity of neuronal networks cultured on microelectrode arrays with microfluidic techniques: A step towards neuron-based functional chips. Biosens. Bioelectron. 21, 1093–1100. [DOI] [PubMed] [Google Scholar]
- Millet L. J.; Stewart M. E.; Sweedler J. V.; Nuzzo R. G.; Gillette M. U. (2007) Microfluidic devices for culturing primary mammalian neurons at low densities. Lab Chip 7, 987–994. [DOI] [PubMed] [Google Scholar]
- Sato K.; Mawatari K.; Kitamori T. (2008) Microchip-based cell analysis and clinical diagnosis system. Lab Chip 8, 1992–1998. [DOI] [PubMed] [Google Scholar]
- Park J.; Kim H.; Byun J.; Ryu H.; Jeon N. (2009) Novel microfluidic platform for culturing neurons: culturing and biochemical analysis of neuronal components. Biotechnol. J. 4, 1573–1577. [DOI] [PubMed] [Google Scholar]
- Kim Y.-t.; Karthikeyan K.; Chirvi S.; Dave D. P. (2009) Neuro-optical microfluidic platform to study injury and regeneration of single axons. Lab Chip 9, 2576–2581. [DOI] [PubMed] [Google Scholar]
- Lovchik R. D.; Bianco F.; Tonna N.; Ruiz A.; Matteoli M.; Delamarche E. (2010) Overflow Microfluidic Networks for Open and Closed Cell Cultures on Chip. Anal. Chem. 82, 3936–3942. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Osakada Y.; Vrljic M.; Chen L.; Mudrakola H. V.; Cui B. (2010) Single-molecule imaging of NGF axonal transport in microfluidic devices. Lab Chip 10, 2566–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.-I.; Cheng G.-H.; Wong Y.-Y.; Lin C.-M.; Fang W.; Chow W.-Y.; Chang Y.-C. (2010) A lab-on-a-chip platform for studying the subcellular functional proteome of neuronal axons. Lab Chip 10, 647–653. [DOI] [PubMed] [Google Scholar]
- Xia Y.; Whitesides G. M. (1998) Soft Lithography. Annu. Rev. Mater. Sci. 28, 153–184. [Google Scholar]
- Mutch S. A.; Kensel-Hammes P.; Gadd J. C.; Fujimoto B. S.; Allen R. W.; Schiro P. G.; Lorenz R. M.; Kuyper C. L.; Kuo J. S.; Bajjalieh S. M.; Chiu D. T. (2011) Protein Quantification at the Single Vesicle Level Reveals That a Subset of Synaptic Vesicle Proteins Are Trafficked with High Precision. J. Neurosci. 31, 1461–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutch S. A.; Fujimoto B. S.; Kensel-Hammes P.; Gadd J. C.; Schiro P. G.; Bajjalieh S. M.; Chiu D. T. (2011) Determining the number of specific proteins in cellular compartments by quantitative microscopy. Nat. Protoc. 6, 1953–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo K. Y.; Kuzmin A.; Unger S. M.; Petersen J. D.; Silverman M. A. (2011) KIF1A is the primary anterograde motor protein required for the axonal transport of dense-core vesicles in cultured hippocampal neurons. Neurosci. Lett. 491, 168–173. [DOI] [PubMed] [Google Scholar]
- Rivière J.-B.; Ramalingam S.; Lavastre V.; Shekarabi M.; Holbert S.; Lafontaine J.; Srour M.; Merner N.; Rochefort D.; Hince P.; Gaudet R.; Mes-Masson A.-M.; Baets J.; Houlden H.; Brais B.; Nicholson G. A.; Van Esch H.; Nafissi S.; De Jonghe P.; Reilly M. M.; Timmerman V.; Dion P. A.; Rouleau Guy A. (2011) KIF1A, an Axonal Transporter of Synaptic Vesicles, Is Mutated in Hereditary Sensory and Autonomic Neuropathy Type 2. Am. J. Hum. Genet. 89, 219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch S.; Deák F.; Königstorfer A.; Mozhayeva M.; Sara Y.; Südhof T. C.; Kavalali E. T. (2001) SNARE Function Analyzed in Synaptobrevin/VAMP Knockout Mice. Science 294, 1117–1122. [DOI] [PubMed] [Google Scholar]
- Bajjalieh S.; Frantz G.; Weimann J.; McConnell S.; Scheller R. (1994) Differential expression of synaptic vesicle protein 2 (SV2) isoforms. J. Neurosci. 14, 5223–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowder K. M.; Gunther J. M.; Jones T. A.; Hale B. D.; Zhang H. Z.; Peterson M. R.; Scheller R. H.; Chavkin C.; Bajjalieh S. M. (1999) Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A). Proc. Natl. Acad. Sci. U.S.A. 96, 15268–15273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. R.; Chiu D. T.; Jackman R. J.; Cherniavskaya O.; McDonald J. C.; Wu H.; Whitesides S. H.; Whitesides G. M. (2000) Fabrication of Topologically Complex Three-Dimensional Microfluidic Systems in PDMS by Rapid Prototyping. Anal. Chem. 72, 3158–3164. [DOI] [PubMed] [Google Scholar]
- Fiorini G. S.; Chiu D. T. (2005) Disposable microfluidic devices: fabrication, function, and application. BioTechniques 38, 429–446. [DOI] [PubMed] [Google Scholar]
- Banker G., and Goslin K., Eds. (1998) Culturing Nerve Cells, 2nd ed., MIT Press, Cambridge, MA. [Google Scholar]