

Abstract
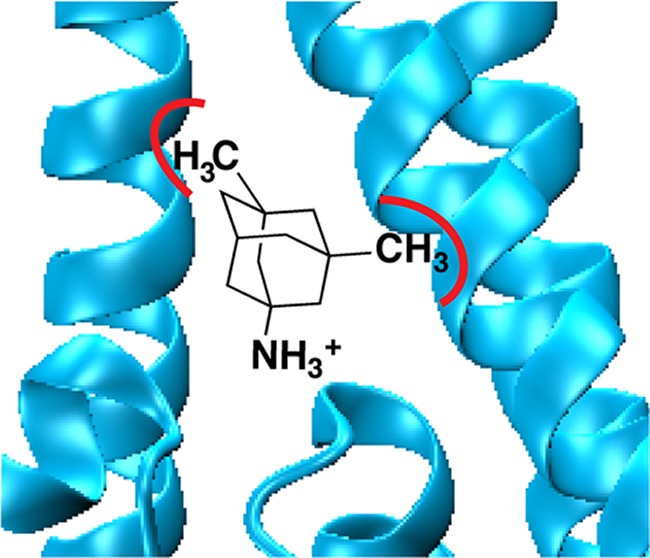
Memantine (Namenda) is prescribed as a treatment for moderate to severe Alzheimer’s Disease. Memantine functions by blocking the NMDA receptor, but the key binding interactions between drug and receptor are not fully elucidated. To determine key binding interactions of memantine, we made side-by-side comparisons of IC50 for memantine and amantadine, a structurally related drug, in the GluN1/GluN2B NMDA receptor. We identified hydrophobic binding pockets for the two methyl groups on memantine formed by the residues A645 and A644 on the third transmembrane helices of GluN1 and GluN2B, respectively. Moreover, we found that while adding two methyl groups to amantadine to produce memantine greatly improves affinity, adding a third methyl group to produce the symmetrical trimethylamantadine diminished affinity. Our results provide a better understanding of chemical-scale interactions between memantine and the NMDA channel, which will potentially benefit the development of new drugs for neurodegenerative diseases involving NMDA receptors.
Keywords: Memantine, open-channel blocker, amantadine, unnatural amino acid mutagenesis
N-Methyl-d-aspartate (NMDA) receptors are members of the ionotropic glutamate receptor (iGluR) family, which also includes AMPA and kainate receptors.1−3 These are fast, excitatory, ligand-gated ion channels activated by the agonist glutamate and, only in the case of NMDA receptors, a coagonist such as glycine or d-serine. The NMDA ion channel is highly permeable to Ca2+ and is blocked by Mg2+ in a voltage-dependent manner. The NMDA receptor is thought to play a central role in learning and memory and is essential to the normal function of the central nervous system. Overactivation of the receptor has been implicated in many neurological disorders, such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis, schizophrenia, epilepsy, and neurodegeneration following stroke.4 Several neuroprotective drugs have been developed to block the NMDA receptor, preventing overactivation. However, most of them cause debilitating side effects due to the critical roles that NMDA receptors play in brain function.
Memantine is unique among NMDA blockers and is currently approved for use in moderate to severe Alzheimer’s and is in a late-stage clinical trial for Parkinson’s disease.5−7 Memantine is thought to function by preferentially blocking open NMDA channels (an uncompetitive antagonist), and hence, a balance between open and closed channels can be achieved by adjusting dosage.8−10 The interaction between NMDA receptors and memantine is reversible, and the mechanism of block has not been fully elucidated. A better understanding of the chemical-scale interactions between the NMDA receptor and memantine will yield insight into the mechanism of memantine blockade that underlies its high clinical potential.

The structure of the transmembrane domain of the NMDA receptor is not currently available. It was proposed some time ago that the transmembrane domain of iGluRs is homologous to the pore region of potassium channels, but with the opposite orientation with respect to the membrane.11,12 This has been confirmed by a crystal structure of a full-length AMPA receptor,13 but unfortunately, the image is of a closed channel and is missing a significant number of residues in the pore loop. Therefore, we created a homology model of GluN1 and GluN2B transmembrane domains, based on the crystal structure of the Kv1.2 chimaera potassium channel (PDB ID 2R9R)14 in the open conformation (Figure 1).
Figure 1.
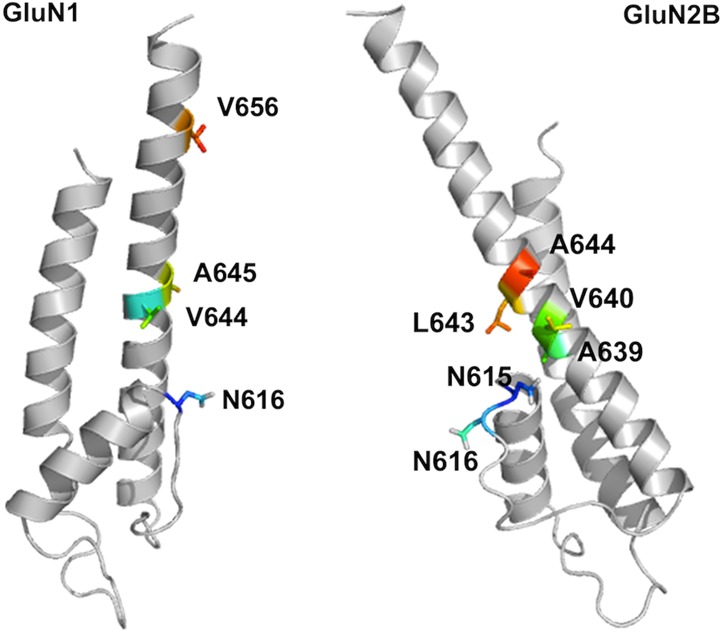
Homology model of the transmembrane region of GluN1 (left) and GluN2B (right) subunits of NMDA receptor based on the potassium channel open conformation (PDB ID 2R9R) without optimization. The relative position of the two subunits is currently unknown.
The primary binding site of memantine (the one with the highest affinity or lowest IC50) involves an interaction between the ammonium group of memantine and the side chain of an Asn residue (residue 616, the N/Q site) in the GluN1 subunit (Figure 1).8,15 This residue is located at the tip of the pore loop, which forms the narrowest constriction of the NMDA pore. Kashiwagi et al. suggested that mutations at residues on the third transmembrane (TM3) and post-TM3 regions of GluN1 had a considerable impact on memantine IC50.16 When we map these residues onto our homology model, we found some of them to be distant from the Asn residue that anchors the ammonium of memantine. It seemed very unlikely that a small molecule like memantine would interact directly with all these residues.
In the present study, we sought to define the scope of the memantine primary binding site by identifying the residues that directly contact the two methyl groups. We considered mutations in both GluN1 and GluN2B, and we will adopt a simplified notation for mutations, exemplified by N1-N616Q or N2-N615D. We assume that the ammonium of memantine is indeed interacting with the N1-N616 residue in its primary blocking site. Our preliminary results suggested that no point mutation deeper in the pore than residue N616 had a significant effect on memantine blockade (Supporting Information Figure S1), consistent with a previous report17 that memantine cannot block NMDA receptors from the intracellular site. Therefore, the methyl group binding pockets would serve as the upper boundary of the memantine primary binding site.
To identify the methyl group binding pockets, we considered amantadine, a common antiviral agent that is known to block the channel of NMDA receptors, but with a lower affinity than memantine.18−21 Amantadine has the same basic core structure as memantine, the only difference being that amantadine lacks the two methyl groups present on memantine. In spite of the small structural difference, the affinity of memantine is 75-fold higher than amantadine in the wild type receptor (Table 1; Figure 2), indicating that the two additional methyl groups play an important role in antagonism. If these two antagonists bind at the same site in the NMDA pore, mutations at residues that interact with the methyl groups are expected to have a larger effect on memantine affinity than amantadine, while other mutations should affect binding of the two antagonists in a similar way.
Table 1. Memantine and Amantadine IC50, (Amantadine IC50)/(Memantine IC50) Ratio, Ω, and ΔΔG° for Wild Type and Mutant NMDA Receptorsa.
| memantine IC50 (μM) | N | amantadine IC50 (μM) | N | IC50 ratio | Ω | ΔΔG° | |
|---|---|---|---|---|---|---|---|
| WT | 0.54 ± 0.03 | 18 | 41 ± 5.6 | 6 | 75 | ||
| GluN1 mutants | |||||||
| N616Q | 5.9 ± 0.5 | 6 | 490 ± 73 | 10 | 83 | 1.1 | –0.06 |
| N616D | 14 ± 1.3 | 7 | 590 ± 21 | 5 | 41 | 0.55 | 0.35 |
| V644N | 12 ± 3.2 | 7 | 150 ± 27 | 8 | 12 | 0.16 | 1.1 |
| V644T | 0.26 ± 0.06 | 13 | 44 ± 15 | 13 | 170 | 2.2 | –0.48 |
| V644L | 0.26 ± 0.09 | 15 | 26 ± 6.5 | 11 | 100 | 1.3 | –0.17 |
| A645N | 240 ± 16 | 11 | 1000 ± 68 | 11 | 4.4 | 0.06 | 1.7 |
| A645L | 4.8 ± 0.4 | 9 | 53 ± 4.4 | 9 | 11 | 0.15 | 1.1 |
| A645V | 0.60 ± 0.06 | 9 | 21 ± 0.8 | 14 | 34 | 0.46 | 0.47 |
| V656N | 2.4 ± 0.2 | 8 | 94 ± 14 | 7 | 40 | 0.53 | 0.38 |
| GluN2B mutants | |||||||
| N615D | 1.3 ± 0.3 | 7 | 300 ± 17 | 9 | 230 | 3.1 | –0.67 |
| N616D | 1.0 ± 0.1 | 6 | 56 ± 5.3 | 10 | 55 | 0.74 | 0.18 |
| A639N | 3.6 ± 0.4 | 10 | 140 ± 26 | 11 | 39 | 0.51 | 0.39 |
| V640N | 0.29 ± 0.03 | 11 | 12 ± 2.6 | 8 | 42 | 0.55 | 0.35 |
| L643N | 34 ± 2.7 | 10 | 750 ± 130 | 13 | 22 | 0.29 | 0.73 |
| A644N | 90 ± 2.0 | 9 | 340 ± 29 | 10 | 3.7 | 0.05 | 1.8 |
| A644L | 0.29 ± 0.07 | 12 | 3.6 ± 0.4 | 12 | 12 | 0.17 | 1.0 |
| A644V | 0.41 ± 0.05 | 14 | 10. ± 0.7 | 11 | 25 | 0.33 | 0.66 |
N= number of cells. IC50 ratio = IC50(amantadine)/IC50(memantine). Ω = [(wild type memantine IC50)(mutant amantadine IC50)]/[wild type amantadine IC50)/(mutant memantine IC50)]. ΔΔG0 = RT ln(Ω), where R = 1.987 kcal·mol–1·K–1 and T = 298 K.
Figure 2.

Memantine and amantadine dose–response curves for the wild-type and the N1-N616Q mutant NMDA receptors.
Probing wild type versus a mutant receptor with two different antagonists sets up an opportunity for a mutant cycle analysis as a way to evaluate meaningful interactions. The basic scheme is shown in Figure 3. The coupling parameter, Ω, defines the deviation from additivity of the two “mutations”: the change to the receptor and the removal of the methyl groups of memantine to make amantadine. Significant coupling suggests an important interaction between the protein side chain being mutated and the methyl groups. The coupling parameter can be converted to a free energy by the equation ΔΔG° = −RT ln(Ω). We consider meaningful interactions to have values of Ω ≥ 3 (or ≤ 1/3), corresponding to |ΔΔG°| values > 0.6 kcal/mol.
Figure 3.
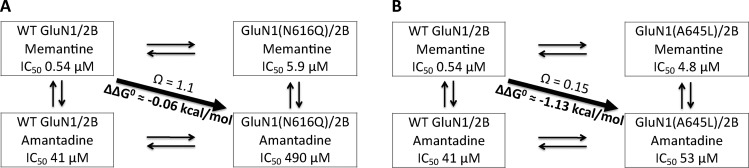
Examples of mutant-cycle analysis. (a) The GluN1-N616Q mutation showed no coupling at all to the methyl groups of memantine, producing Ω ≈ 1 and ΔΔG° ≈ 0 kcal/mol. (b) The GluN1-A645L mutation strongly coupled to the methyl groups as shown by the substantial coupling energy, ΔΔG°.
Memantine and amantadine show similar responses to the N1-N616Q and the N1-N616D mutations compared to the wild type, indicating that the two drugs block the channel at the same general location and with the same orientation (Table 1, Figure 2). Stated differently, these two mutations, which are thought to probe the ammonium group binding site, show no significant coupling to the memantine/amantadine pair (ΔΔG° < 0.4) (Table 1), which is a probe of methyl group binding. Therefore, comparison of the IC50 shifts between the two drugs is a valid strategy to probe for the memantine methyl groups’ binding site.
Preliminary mutational scanning suggested that only mutations at residues V644, A645, and V656 in GluN1 have a meaningful impact on memantine block (Supporting Information Figure S1). Since we are probing for a hydrophobic binding pocket for the methyl groups, our strategy was to make hydrophobic side chains more hydrophilic. Therefore, we mutated these three residues to Asn. The impact of mutations in the GluN1 subunit on IC50 values of both memantine and amantadine are shown in Figure 4 and Table 1. The highest memantine concentration used in all IC50 experiments was 100 μM to minimize complications involving the secondary (lower affinity) binding site and/or antagonist trapping.8,15 Though this choice prevented completion of full dose–response curves for some mutations, meaningful IC50 values (unlike EC50 values) can be obtained from such plots. The EC50 for glutamate was measured for all the mutant receptors to ensure that (i) the mutant receptors are functional and (ii) a saturating dose of glutamate (4 or 10 μM) was applied to activate the mutant receptors in the IC50 experiments (Supporting Information Table S1).
Figure 4.

Memantine IC50, amantadine IC50, and ΔΔG° for wild-type and mutant NMDA receptors containing mutations in GluN1. (a) Effect of introducing Asn mutations at select sites. (b) Aliphatic mutations at residue N1-645. The values for IC50 ± SEM are shown in Table 1. ΔΔG° values are shown above the corresponding columns.
The mutation N1-V644N impacted the binding of memantine significantly more than amantadine. The IC50 ratio between the two drugs decreased to 12-fold, compared to the 75-fold difference seen in the wild-type receptor, producing a ΔΔG° of 1.1 kcal/mol (Table 1, Figure 4a). The adjacent N1-A645N mutation showed an even larger effect, with only a 4.4-fold difference between memantine and amantadine IC50 and a ΔΔG° value of 1.7 kcal/mol (Table 1, Figure 4a). A previous study based on the substituted cysteine accessibility method (SCAM) showed that, in the related GluN1/GluN3 receptor, N1-A645 in TM3 is in close proximity to the N site, N1-N616,22 and the homology model in Figure 1 also supports this finding.
Although the Asn mutations at GluN1 residue 644 and 645 affected glutamate EC50 values much less than drug IC50's, the N1-A645N and N1-V656N mutations did show shifts in Glu EC50 of 6- and 10-fold, respectively (Supporting Information Table S1). The N1-V656N mutation did not affect drug binding significantly, and so the perturbation in glutamate EC50 was not a concern. However, in order to determine whether the data in Figure 4a resulted from an unwanted structural perturbation, we tested N1-V644T, N1-V644L, N1-A645V, and N1-A645L mutations. All of these mutations shift glutamate EC50 significantly less than N1-A645N (Supporting Information Table S1). The additional mutations at residue 644 did not have a considerable impact on memantine IC50, amantadine IC50, or the ratio between the two (Table 1).
In contrast, the N1-A645L mutation had a significant impact on memantine IC50 but not amantadine IC50, resulting in a ΔΔG° of 1.1 kcal/mol (Table 1, Figure 4b). Since Leu and Ala are both hydrophobic, this could be considered a steric effect, suggesting a steric clash with the methyl groups of memantine when Ala is mutated to Leu. With amantadine, however, essentially no effect is seen. To test this interpretation, we studied the smaller hydrophobic residue Val. The lesser impact of the Val mutation compared to Leu (Figure 4b) is consistent with this analysis. Leu can be considered to be isosteric to Asn, and so the additional perturbation for the Asn mutation (ΔΔG° 1.7 kcal/mol) relative to Leu (ΔΔG° 1.1 kcal/mol) can be considered a polarity effect (Figure 4b). Both results are consistent with the notion that N1-A645 contributes to a hydrophobic binding pocket for the methyl groups on memantine.
The mutation N1-V656N is not impactful, causing only a 4.4-fold shift in IC50 for memantine and a 2.3-fold shift for amantadine (Table 1, Figure 4a). This is perhaps not surprising given its distance from the proposed memantine binding site in the homology model (Figure 1). Other workers have also reported a modest shift in memantine IC50 when residue V656 in GluN1 is mutated.16 Interestingly, this mutation causes a nearly 10-fold shift in glutamate EC50 (Supporting Information Table S1), which may imply a strong perturbation to receptor gating.
Models of the NMDA receptor heterotetramer indicate that both GluN1 and GluN2 contribute to the channel region being probed here. However, it is not safe to assume that the residue N2-A644, which would typically be considered to align with N1-A645, also contributes to a methyl group binding site.12,23 A previous SCAM study of GluN1/GluN2C suggests that there may be an offset by four residues in the TM3 regions between the GluN1 and GluN2C.24 In contrast, a study of the binding to GluN1/GluN2B by felbamate, an anticonvulsant drug that is structurally dissimilar to those being considered here, supports a model in which the offset in the TM3 region between GluN1 and GluN2B is minimal.25
Asymmetry in the region of the memantine binding site is supported by our studies of N615 and N616 of GluN2B, residues that could be considered to align with N1-N616. Both N2-N615D and N2-N616D produce relatively modest effects. The N2-N615D mutation is unique in that it affects amantadine binding more than memantine. This suggests an asymmetry in the region of the ammonium group binding site, such that the GluN1 subunit plays a more important role in memantine block, in agreement with previous proposals.15
To probe for contributions to a methyl group binding site by GluN2B, we considered the aligning residues L643 and A644 as well as the residues A639 and V640 which are one helix turn lower in the structure (Figure 1). The N2-A639N and N2-V640N mutations had a negligible effect on memantine and amantadine binding (Figure 5a). In contrast, N2-L643N and N2-A644N substantially impacted blockade. Similar to what is seen with GluN1, N2-L643N shows a modest differentiation between memantine and amantadine, while N2-A644N shows a quite substantial effect (Figure 5a).
Figure 5.

Memantine IC50, amantadine IC50, and ΔΔG° for wild-type and mutant NMDA receptors mutations in GluN2B. (a) Effect of introducing Asn mutations at select sites. (b) Aliphatic mutations at residue N2-644. The values for IC50 ± SEM are shown in Table 1. ΔΔG° values are shown above the corresponding columns.
Parallel to the study in GluN1 subunit, we also mutated N2-A644 to the hydrophobic side chains Leu and Val. All these mutations resulted in minimal changes to glutamate EC50 (Supporting Information Table S1). The trends in ΔΔG° values very much parallel those seen for N1-A645 (Figure 5b). The large, polar residue Asn has the greatest effect; the isosteric but hydrophobic residue Leu has a smaller but still significant effect; the smaller hydrophobic residue Val has a small/negligible effect. These results suggest these two residues, N1-A645 and N2-A644, play similar roles in shaping the memantine methyl binding site. A subtle distinction between the two sites is that the N2-A644L mutation shows a meaningfully lower IC50 for amantadine than the wild type, while a comparable effect is not seen for memantine (Figure 5b). It could be the smaller size of amantadine is better able to accommodate the stabilizing hydrophobicity of the larger Leu side chain.
The two methyl groups on memantine are crucial for NMDA receptor blockade, increasing memantine affinity to the open NMDA receptor channel and making it a much better neuroprotective drug than amantadine. To further probe the possible role of methyl groups and asymmetry in the binding region, we considered the molecule trimethylamantadine (TMAm). The additional methyl group of TMAm introduces a 3-fold rotation axis that is absent in memantine. We found that this molecule blocks the NMDA receptor with an IC50 of 3.5 μM (Supporting Information Table S2), intermediate between the values for memantine and amantadine. However, the N1-N616Q mutation that displays a substantial shift in both memantine and amantadine IC50 does not have any effect on TMAm block. Similarly, Asp mutations at N2-N615 or N2-N616 do not shift the TMAm IC50 from the wild-type value (Supporting Information Table S2). These data imply that the TMAm molecule binds in a different orientation than memantine and amantadine. TMAm is sensitive to N1-A645N and N2-A644N mutations, but the mutations have a significantly smaller effect on IC50 shifts for TMAm compared to memantine. Altogether, our results suggest that the additional methyl group on TMAm prevents it from binding the receptor at the same location or orientation as memantine and amantadine. Therefore, the special property of memantine as an NMDA receptor blocker stems not only from the presence of the additional hydrophobicity gained from the two methyl groups on the amantadine core, but also proper shape-matching to the binding site.
Our results indicate that the primary binding interaction of the methyl groups of memantine is formed by N1-A645 and N2-A644. Mutation at these residues had a significantly larger effect on memantine block compared to amantadine block. When coupled with the interaction between the ammonium group and N1-N616, these results provide excellent guidance for efforts to model the binding of memantine to the receptor. Because these alanine residues are conserved in all the GluN2 subunits (GluN2A/B/C/D), it is possible that the methyl group binding pockets are the same for other GluN1/GluN2 receptor subtypes. These alanine residues are located immediately upstream to the SYTANLAAF motif, which has been implicated to play a crucial role in gating of the NMDA receptor.26−28 These results provide further insight into the chemical-scale interactions between the NMDA receptor and memantine, hopefully contributing to efforts to understand the drug’s high clinical potential and accelerate the development of other therapeutic NMDA receptor antagonists.
Methods
NMDAR Clones and Mutagenesis
The rat GluN1-1a and rat GluN2B cDNA clones were in the pAMV vector. Mutant GluN1 and GluN2B subunits were prepared by site-directed mutagenesis using the standard Stratagene QuikChange protocol and verified through sequencing. All cDNA was linearized with NotI, and mRNA was synthesized by in vitro runoff transcription using the T7 mMESSAGE mMACHINE kit (Ambion).
Electrophysiological Recordings
Amantadine was purchased from Aldrich, and memantine from Tocris Bioscience. Glycine and l-glutamic acid hydrochloride were purchased from Aldrich.
Stage V–VI Xenopuslaevis oocytes (Nasco) were injected with 4–75 ng of mRNA in a total volume of 50 nL per oocyte. Oocytes were incubated in ND96+ solution for 18–48 h. Macroscopic current recordings were made in two-electrode voltage clamp mode using the OpusXpress 6000A instrument (Molecular Devices). Oocytes were evaluated in a Mg2+- and Ca2+-free saline solution (96 mM NaCl, 5 mM HEPES, 2 mM KCl, and 1 mM BaCl2, pH 7.5). Eight oocytes were simultaneously clamped at −80 mV. The receptors were first activated in a Mg2+- and Ca2+-free solution containing 10 μM glycine and 20 μM glutamate. In the cases of GluN1(A645N) and GluN1(V656N) mutations, 10 μM glycine and 4 μM glutamate were used to activate the receptors to avoid overly saturated glutamate concentrations. Dose–response relationships were obtained by delivery of various concentrations of antagonists in 1 mL aliquots during the NMDA receptor activation. Representative traces are shown in the Supporting Information.
Data Analyses
All data were analyzed using the Clampfit 9.0 software (Axon). To determine IC50, the fraction of remaining current during the block (I/Imax) was determined for each test dose of antagonist, where I is the current response to an antagonist application when the receptor is already activated by the coagonists and Imax is the maximal current response to agonist activation for that given dose of the antagonist. Then the I/Imax values were averaged for each antagonist concentration across different cells, and the averages were fitted to the Hill equation. All dose–response data were obtained from at least five cells and at least two batches of oocytes. Dose responses of individual oocytes were also examined and used to determine outliers.
Although we performed our experiments in a magnesium-free environment, it is worth noting that a decrease in the potencies of both memantine and amantadine has been reported in the presence of physiological concentrations of magnesium ion.29,30 This observation suggests a competitive behavior between memantine and magnesium, consistent with the notion that they share a common blocking location at the tip of the pore loop.
Acknowledgments
We thank Dr. Kathryn McMenimen for helpful discussions.
Glossary
Abbreviations
- TM3
third transmembrane domain
- SCAM
scanning cysteine accessibility mutagenesis
- TMAm
trimethylamantadine
Supporting Information Available
Additonal tables and figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
Contributed to research design: W.L., E.B., W.Y., H.A.L., and D.A.D. conducted experiments: W.L., E.B., and W.Y. Performed data analysis: W.L., E.B., W.Y., H.A.L., and D.A.D. Writing of the manuscript: W.L., W.Y., H.A.L., and D.A.D.
This work was supported by the NIH (NS 34407 to D.A.D.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Dingledine R.; Borges K.; Bowie D.; Traynelis S. F. (1999) The glutamate receptor ion channels. Pharmacol. Rev. 51(1), 7–61. [PubMed] [Google Scholar]
- Madden D. R. (2002) The structure and function of glutamate receptor ion channels. Nat. Rev. Neurosci. 3(2), 91–101. [DOI] [PubMed] [Google Scholar]
- Traynelis S. F.; Wollmuth L. P.; McBain C. J.; Menniti F. S.; Vance K. M.; Ogden K. K.; Hansen K. B.; Yuan H.; Myers S. J.; Dingledine R. (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62(3), 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine D., Jiang R., Taly A., Chataigneau T., Specht A., and Grutter T. (2012) Ligand-Gated Ion Channels: New Insights into Neurological Disorders and Ligand Recognition. Chem. Rev. published online Sept 18, 2012. DOI: 10.1021/cr3000829. [DOI] [PubMed] [Google Scholar]
- Bormann J. (1989) Memantine is a potent blocker of N-methyl-d-aspartate (NMDA) receptor channels. Eur. J. Pharmacol. 166(3), 591–592. [DOI] [PubMed] [Google Scholar]
- Wilkinson D. (2012) A review of the effects of memantine on clinical progression in Alzheimer's disease. Int. J. Geriatr. Psychiatry 27(8), 769–776. [DOI] [PubMed] [Google Scholar]
- Witt A.; Macdonald N.; Kirkpatrick P. (2004) Memantine hydrochloride. Nat. Rev. Drug Discovery 109–110. [DOI] [PubMed] [Google Scholar]
- Chen H.-S. V.; Lipton S. A. (2006) The chemical biology of clinically tolerated NMDA receptor antagonists. J. Neurochem. 97(6), 1611–1626. [DOI] [PubMed] [Google Scholar]
- Johnson J. W.; Kotermanski S. E. (2006) Mechanism of action of memantine. Curr. Opin. Pharmacol. 6(1), 61–67. [DOI] [PubMed] [Google Scholar]
- Parsons C. G.; Stöffler A.; Danysz W. (2007) Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system--too little activation is bad, too much is even worse. Neuropharmacology 53(6), 699–723. [DOI] [PubMed] [Google Scholar]
- Kuner T.; Seeburg P. H.; Guy H. R. (2003) A common architecture for K+ channels and ionotropic glutamate receptors?. Trends Neurosci. 26(1), 27–32. [DOI] [PubMed] [Google Scholar]
- Wollmuth L. P.; Sobolevsky A. I. (2004) Structure and gating of the glutamate receptor ion channel. Trends Neurosci. 27(6), 321–328. [DOI] [PubMed] [Google Scholar]
- Sobolevsky A. I.; Rosconi M. P.; Gouaux E. (2009) X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 462(7274), 745–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S. B.; Tao X.; Campbell E. B.; Mackinnon R. (2007) Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450(7168), 376–382. [DOI] [PubMed] [Google Scholar]
- Chen H. S.; Lipton S. A. (2005) Pharmacological implications of two distinct mechanisms of interaction of memantine with N-methyl-d-aspartate-gated channels. J. Pharmacol. Exp. Ther. 314(3), 961–971. [DOI] [PubMed] [Google Scholar]
- Kashiwagi K.; Masuko T.; Nguyen C. D.; Kuno T.; Tanaka I.; Igarashi K.; Williams K. (2002) Channel blockers acting at N-methyl-d-aspartate receptors: differential effects of mutations in the vestibule and ion channel pore. Mol. Pharmacol. 61(3), 533–545. [DOI] [PubMed] [Google Scholar]
- Parsons C. G.; Gilling K. E.; Jatzke C. (2008) Memantine does not show intracellular block of the NMDA receptor channel. Eur. J. Pharmacol. 587(1–3), 99–103. [DOI] [PubMed] [Google Scholar]
- Antonov S. M.; Johnson J. W. (1996) Voltage-dependent interaction of open-channel blocking molecules with gating of NMDA receptors in rat cortical neurons. J. Physiol. 493(Pt 2), 425–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpied T. A.; Boeckman F. A.; Aizenman E.; Johnson J. W. (1997) Trapping channel block of NMDA-activated responses by amantadine and memantine. J. Neurophysiol. 77(1), 309–323. [DOI] [PubMed] [Google Scholar]
- Blanpied T. A.; Clarke R. J.; Johnson J. W. (2005) Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J. Neurosci. 25(13), 3312–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky A. I.; Koshelev S. G.; Khodorov B. I. (1998) Interaction of memantine and amantadine with agonist-unbound NMDA-receptor channels in acutely isolated rat hippocampal neurons. J. Physiol. 512(Pt 1), 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada A.; Takahashi H.; Lipton S. A.; Chen H.-S. V. (2006) NR3A modulates the outer vestibule of the NMDA receptor channel. J. Neurosci. 26(51), 13156–13166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky A. I.; Prodromou M. L.; Yelshansky M. V.; Wollmuth L. P. (2007) Subunit-specific contribution of pore-forming domains to NMDA receptor channel structure and gating. J. Gen. Physiol. 129(6), 509–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky A. I.; Rooney L.; Wollmuth L. P. (2002) Staggering of subunits in NMDAR channels. Biophys. J. 83(6), 3304–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H.-R.; Kuo C.-C. (2008) Molecular determinants of the anticonvulsant felbamate binding site in the N-methyl-d-aspartate receptor. J. Med. Chem. 51(6), 1534–1545. [DOI] [PubMed] [Google Scholar]
- Blanke M. L.; VanDongen A. M. J. (2008) The NR1 M3 domain mediates allosteric coupling in the N-methyl-d-aspartate receptor. Mol. Pharmacol. 74(2), 454–465. [DOI] [PubMed] [Google Scholar]
- Chang H.-R.; Kuo C.-C. (2008) The activation gate and gating mechanism of the NMDA receptor. J. Neurosci. 28(7), 1546–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H.; Erreger K.; Dravid S. M.; Traynelis S. F. (2005) Conserved structural and functional control of N-methyl-d-aspartate receptor gating by transmembrane domain M3. J. Biol. Chem. 280(33), 29708–29716. [DOI] [PubMed] [Google Scholar]
- Otton H. J.; Mclean A. L.; Pannozzo M. A.; Davies C. H.; Wyllie D. J. A. (2011) Quantification of the Mg2+-induced potency shift of amantadine and memantine voltage-dependent block in human recombinant GluN1/GluN2A NMDARs. Neuropharmacology 60(2–3), 388–396. [DOI] [PubMed] [Google Scholar]
- Kotermanski S. E.; Johnson J. W. (2009) Mg2+ imparts NMDA receptor subtype selectivity to the Alzheimer's drug memantine. J. Neurosci. 29(9), 2774–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.