

Abstract
Mass spectrometry has evolved at an exponential rate over the last 100 years. Innovations in the development of mass spectrometers have created powerful instruments capable of analyzing a wide range of targets, from rare atoms and molecules to very large molecules such as a proteins, protein complexes and DNA. These performance gains have been driven by sustaining innovations, punctuated by the occasional disruptive innovation. The use of mass spectrometry for proteome analysis was driven by disruptive innovations that created a capability for large-scale analysis of proteins and modifications.
Proteins are linear polymers created from a set of 20 amino acids encoded in DNA. If the amino acid sequence of a protein was sufficient to explain its role in biological processes, then there would be no need for protein analysis methods since sequences can be determined quite efficiently with DNA sequencing techniques. In fact, in 1978 Malcolm predicted that DNA sequencing methods would result in the “decline and fall of protein chemistry”, a prophecy that has not come true, in part, because a protein's function or role must be determined in its individual context and in the context of molecular and cellular systems1. The function of a protein can be dictated by its molecular interactions, by its location in the cell, by its time or level of expression or by its modification state. Malcolm, in a sense, was correct that DNA sequencing methods would lead to transformational changes in the biological sciences, but he, like everybody else at the time, did not envision a global effort to sequence the human genome and the genomes of model organisms, or the far reaching consequences of such an effort. When the Human Genome Project was proposed it was expected to benefit research in genetics and medicine and to accelerate the discovery of the causes of disease, but no one anticipated that protein analysis would also benefit from genome data. Despite the promise of the Human Genome Project, it quickly became clear that genetic data alone does not provide sufficient insight into the mechanisms of diseases to effect cures, and that even simple genetic mutations such as the deleted Phe at position 508 (ΔF508) in the Cystic Fibrosis Transport Regulator (CFTR) protein create complicated biology that has taken 20+ years to unravel2. Nevertheless, genome sequences unexpectedly created a resource for mass spectrometry that has accelerated the pace of biological research.
The Evolution of Shotgun Proteomics (From Amino Acids to Proteomes)
Mass spectrometry has evolved at an exponential rate over the last 100 years.3 Some of this evolution has been driven by innovations in the machining, electronic and computer industries which created higher performance components for mass spectrometers, and these improvements have resulted in steady performance gains. However, bigger gains have come from the occasional disruptive innovations- technological innovations which are transformational- that created entirely new levels of scale and capability. Large-scale analysis of proteins or proteomics was made possible by a collection of disruptive innovations driving the field at a fast moving pace.
After mass spectrometers were shown to be capable of analyzing organic molecules, it was natural to look to amino acids and small peptides as the next target. Amino acid and peptide analysis was complicated by the lack of volatility of these zwitterionic and polar molecules and by the mass range of early mass spectrometers. To overcome this problem, clever use of derivatization allowed evaporation of the modified amino acids and small peptides off a solids probe into an EI source where fragmentation patterns permitted determination of the peptide sequence4,5. As high resolution, accurate mass instruments emerged, accurate mass was used as a tool for sequence analysis of small peptides6. The ability to analyze small peptides led to the analysis of proteins using enzymatic digestion and acid hydrolysis of the intact protein to produce peptides small enough to be analyzed by the mass spectrometer6. By generating overlapping peptide fragments, the sequence of the protein could be reconstructed. Clearly, this strategy produced complicated mixtures of peptides that would require advances in separation technology and fortuitously concurrent innovations in gas chromatography (GC) provided a means to separate peptides using the same derivatization chemistry employed for mass spectrometry. It wasn't long before GC was interfaced with MS to allow simultaneous separation and structural analysis7 and using this strategy some impressive protein sequencing results were achieved8. Alternative strategies also emerged that made use of derivatization chemistries such as permethylation, enabling the analysis of longer peptides, which were often too involatile to be separated by GC, but could be fractionally distilled off a solids probe9. As DNA sequencing methods emerged, GCMS analysis of peptides was used to check the accuracy of DNA derived sequences and to establish the correct reading frame10. Errors in the middle of the DNA sequence could shift the reading frame, making parts of the sequence incorrect. The greatest challenge of the time was the ionization of peptides without the laborious derivatization steps since these steps meant that applications were limited to abundant proteins.
A major disruptive innovation occurred in 1981 with the development of Fast Atom Bombardment (FAB)11,12. For the first time peptides could be robustly ionized without modification and very large peptides (>1-2K Da) could be ionized. This innovation set off a drive to increase the mass range of mass spectrometers. FAB ionization was also energetic enough to activate some peptide ions to dissociate and produce low abundance fragmentation, often generating enough information to determine the amino acid sequence of a pure peptide12. Proteins could be sequenced using this approach by first purifying peptides from proteolytic digestion and then subjecting them to FAB-MS to derive sequence ions13. Some peptides would fragment well during FAB, but many would not. Within a few months of the introduction of FAB, Hunt et al integrated this new ionization technique with tandem mass spectrometry to create a robust method to sequence peptides14. This method circumvented several problems: it eliminated the need to purify peptides to homogeneity, it generalized fragmentation through collision induced dissociation (CID), and it improved signal to noise in the product ion spectrum by eliminating the high level of chemical noise created by FAB. Hunt also integrated the use of HPLC to separate proteolytically digested proteins by collecting fractions with off-line HPLC and then analyzing peptides by FAB-MSMS15. Tandem mass spectrometry data for peptides was then manually interpreted, which limited the throughput of the approach. Data could be collected very quickly, but interpretation was slow and complicated. The approach developed by Hunt et al which comprises digesting intact proteins, separating peptides by HPLC and then sequencing the peptides by tandem mass spectrometry is essentially the strategy used today for bottom-up proteomics.
While FAB-MS and MSMS were big breakthroughs for peptide and protein analysis, they were limited by difficulty interfacing them directly with liquid separations. A method called continuous flow FAB was developed as an interface to HPLC, but the method was not robust and was never widely used16,17. In 1989 Fenn et al demonstrated electrospray ionization on proteins18. An extraordinary feature of the method was the ability to ionize large proteins and measure m/z ratios accurately in the mass spectrometer, but another valuable feature of the method was ionization at atmospheric pressure, which simplified interfacing liquid separations to the mass spectrometer. Smith et al very quickly implemented ESI to interface capillary electrophoresis to MS to demonstrate this feature19. ESI was clearly a disruptive innovation as FAB literally disappeared from mainstream use within a few years.
A clear benefit of ESI could be a more efficient sequencing strategy for peptides and proteins, although initial enthusiasm revolved around measuring the masses of intact proteins20. Several groups devised strategies for liquid chromatography of peptides, but a powerful synergy was found between ESI and the microscale packed capillary columns of Novotny and Jorgenson21,22. ESI is a concentration sensitive ionization method, so the low flow rate associated with capillary chromatography resulted in big gains in signal. Hunt et al devised a nanoLC ESI tandem mass spectrometry method to sequence peptides isolated from MHC proteins based on the previously developed FAB-MSMS strategy 23. A drawback to this approach was the need to manually select ions for MSMS, necessitating two analyses; one to identify peptide ion m/z values and a second to select ions for MSMS. This approach limited the number of ions that could be selected in a single analysis. To improve on this process a “peak parking” technique was used that slowed the flow rate of the HPLC effluent to allow more time to collect MSMS.23,24 Clearly, automating the process to collect tandem mass spectra would increase the efficiency of the process.
Instrument Control Language (ICL) was a unique and disruptive innovation which was developed by Sokolow et al at Finnigan MAT and first appeared on the Finnigan MAT TSQ70.25 ICL could be used to create computer programs to interact with data and control operation of the instrument based on that data in real time.26,27 ICL allowed automated data acquisition and imbued it with a level of crude “intelligence”. Davis et al created methods to automatically collect tandem mass spectra of peptides by first surveying the m/z values present in a scan and then selecting m/z values for MSMS based on their abundance levels27. The program would quickly set up the instrument for an MSMS experiment, collect 2 or 3 MS/MS and then resume MS1 scans to find more m/z values. Other ICL applications quickly followed. For instance, Yates et al designed an experiment to collect MSMS data on phosphopeptides based on a neutral loss scan to detect the loss of phosphoric acid from phospho-Ser and –Thr and then collecting MSMS data on those peptide ions exhibiting the neutral loss.26 ICL proved to be an enabling and disruptive innovation which increased the efficiency of MSMS and other experiments and made large-scale proteomics possible. It is now standard technology on all mass spectrometers used for proteomics.
In 1990 the DOE and NIH presented a joint plan to the US Congress to sequence the human genome28. By design, the collection of new human genome sequences was slow in the initial phases of the project while technology development and the collection of DNA sequence from several model organisms, notably E. coli, S. cerevisiae, D. melanogaster and C. elegans, and mouse, were pursued29. Databases started to fill with DNA sequence information and bioinformatic algorithms for mining the data proliferated. In 1994 a seminal method for automated analysis of peptide tandem mass spectrometry data emerged that involved searching MSMS data using the sequences being generated by genome sequencing projects30. This method solved the long standing problem of rapid and accurate interpretation of tandem mass spectrometry peptide data. Further work showed the method could be used to identify modifications to peptides that are not represented in databases and to search nucleotide databases employing 6 frame translations to identify new open reading frames26,31. Automated control of data acquisition and large-scale methods for data interpretation dovetailed beautifully with Hunt's methods for LC/MSMS analysis of peptides to create a strategy for the analysis of complex peptide mixtures26,32. This new strategy for data interpretation converted a protein sequence analysis strategy into one of protein identification (Figure 1). Even though the full complement of proteins in an organism are known after genome is sequencing (individuals within a species will certainly have sequence variations), biochemical experiments still require tools to identify and characterize the proteins involved in specific processes. Protein identification with tandem mass spectrometry facilitated that process and was critical to the creation of “shotgun” proteomics by enabling large-scale, high throughput data analysis- a process that was disruptive to traditional protein analysis methods.
Figure 1.
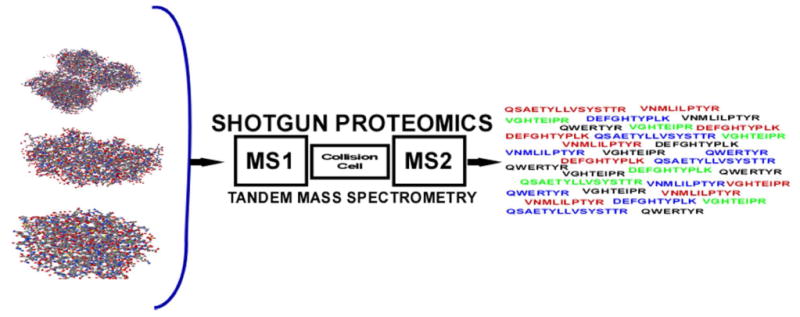
Protein sequencing becomes protein identification. In both strategies the intact protein is digested with a protease, typically trypsin which cleaves after arginine or lysine, to produce a collection of peptides. By using liquid separations coupled to a tandem mass spectrometer, peptide ions are fragmented as they elute into the instrument. In protein sequencing the tandem mass spectrum is interpreted to determine the amino acid sequence de novo. In protein identification, the tandem mass spectrum is searched through a collection of protein sequences to find the best amino acid sequence match to the spectrum.
A powerful capability of tandem mass spectrometry is that of mixture analysis.33 When many molecules co-ionize, a tandem mass spectrometer can select individual ions for fragmentation. The development of large-scale data analysis enabled protein mixture analysis because the increased efficiency of the process made it feasible to collect and interpret thousands of tandem mass spectra in a timely fashion. This was first realized by Eng et al and McCormack et al when they showed the intentional digestion of protein mixtures for the purpose of protein identification using LC/MSMS and database searching.30,34 Direct analysis of protein mixtures has obvious advantages over methods which require purification and sequential analysis. It's highly efficient and sensitive and by limiting sample manipulation, sample losses are minimized. This is particularly important for low abundance proteins where constant exposure to new, active surfaces can result in substantial losses. Furthermore, highly abundant proteins in a mixture can act as carriers to help protect low abundance proteins from active surfaces. Direct, solution based analysis of proteins also provides better opportunities to proteolytically digest proteins and to enrich peptides. The technological developments that allowed direct analysis of digested protein mixtures have revolutionized the analysis of proteins and proved to be a disruptive innovation for 2D gel electrophoresis (2DGE), the best method for proteomics at the time.
This new strategy for protein analysis was quickly used to develop new types of analyses for molecular and cellular biology (Figure 2). McCormack et al identified proteins involved in protein-protein interactions using three different methods to enrich interacting proteins, with subsequent analysis by direct solution digestion of the proteins and LC/MSMS with database searching.32 Cells compartmentalize activities into discrete sections or locations. Membrane proteins have long been difficult to isolate because of their hydrophobicity and this new strategy allowed digestion of the more soluble portions of the protein for easier analysis and identification35. Identifying the proteins residing in subcellular compartments assists in understanding the various activities that take place there as well as providing information about a protein's function or role36. Link et al used tandem mass spectrometry to identify the contents of the periplasmic space in E. coli.37 These studies demonstrated for the first time the interplay between molecular and cellular biological techniques and a new strategy to identify proteins using “shotgun protein analysis”, a term coined in 1998.38 A number of interesting applications have been performed using shotgun proteomics, including correlation profiling to identify the components of an organelle, subtractive analysis to identify those proteins enriched in the nuclear envelope versus the endoplasmic reticulum and the large-scale identification of protein complexes.39,40,32,41
Figure 2.
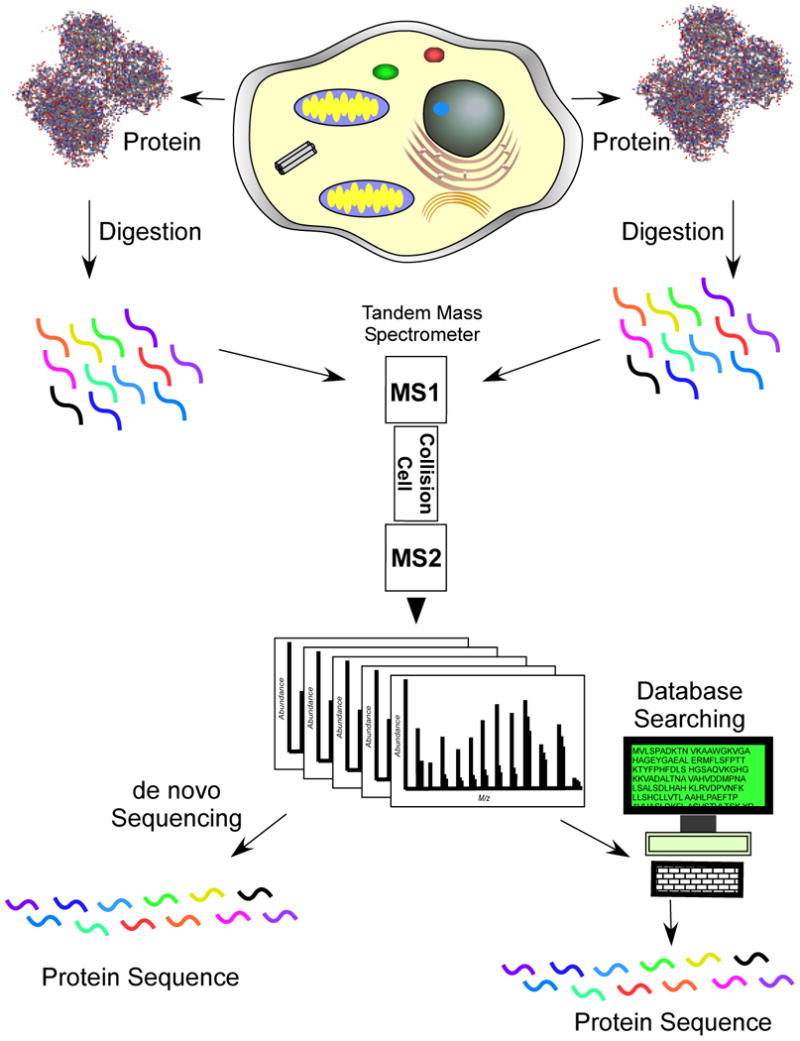
Shotgun proteomics can be used to directly identify the components of cells, organelles, protein complexes and proteins. Shotgun proteomics also simplifies the analysis of membrane proteins since the proteins can be digested directly in a lipid bilayer rather than trying to enrich the proteins. Membrane proteins are hydrophobic and difficult to manipulate in aqueous buffers.
With the establishment of methods to analyze protein complexes and subcellular compartments, the obvious next step was to develop methods for the analysis of intact cells.35,41 Whole cell analysis is a complicated endeavor as cells contain a mixture of different compartments, many proteins are bound in complexes or inserted in lipid bilayers, and protein isoforms and modified forms can increase complexity. So the challenge to developing methods for whole cell analysis is twofold. First, good strategies to digest protein mixtures must be created, and then good methods must be developed to separate the complex peptide mixture. Good separation techniques allow you to obtain high levels of sequence coverage which can reveal data on isoforms and protein modifications or the “proteoforms” of proteins.
The digestion of proteins has long been a first step in the analysis of protein sequence or structure. Protein digestion was traditionally performed on a homogenous protein so it was a simple task to ensure the protein was denatured and digested. However, in protein mixtures proteolysis can be limited because of steric or chemical inhibition. When proteins are tightly bound in complexes or to DNA, a protease may be unable to access sites of cleavage. Additionally, membrane proteins may be protected by their folding through the lipid bilayer and by modifications frequently found on membrane proteins, such as glycosylation. Link et al overcame these issues by employing a two-step digestion procedure that employed endoproteinase Lys-C digestion in 8M urea followed by dilution of the solution to 2 M urea and trypsin digestion.41 The first step in this process uses a higher concentration of chaotrope to better denature the proteins. Because endoproteinase Lys-C is still active at that concentration, it is used to initiate digestion and then a lower concentration of chaotrope is used for a final trypsin digestion. Washburn et al improved on the process and captured more membrane proteins by segregating insoluble from soluble proteins using high salt and sodium carbonate washes and then subjecting the membrane fraction to cyanogen bromide digestion followed by trypsin.35 This process worked well and led to a very high coverage of the yeast membrane proteome. Blonder et al used a high concentration of alcohol in buffer to perform a trypsin digest on membrane proteins with good success.42 Wu et al used a non-specific protease to better cleave the exposed regions of membrane proteins.43 This process had an added advantage of providing information about how proteins were folded through the lipid bilayer, a strategy exploited by Blackler et al to study protein channel function.44 Liebler et al employed a filter aided digestion process to employ better chaotropes to denature proteins that were not mass spectrometry compatible.45 Wiśniewski, et al used the same method, initially denaturing proteins in SDS and then swapping the SDS for urea.46 The initial digestion of proteins in solution is a key starting point in whole proteome analysis and efficient and complete digestion is essential to high proteome coverage.
A critical aspect of large-scale proteomics is the ability to separate incredibly complex mixtures of peptides.47,48 The challenges of these separations have invigorated the mature field of liquid chromatographic separations with many new developments in HPLC pumps, chromatographic supports and separation strategies. In particular, HPLC pumps capable of driving very high pressure separations have proven to be important in order to allow the use of much smaller chromatographic supports which improve separation efficiency and increase peak capacity.49-51 Jorgenson's pioneering work in this area has driven the development of new pumps capable of sustaining pressures of 12-20K psi.52 To address the complexity of proteomic samples, there has been a resurgence in multi-dimensional separations with an emphasis on strategies which can be coupled to the mass spectrometer. Recent reviews of LC/LC provide a more in-depth treatment of this topic, but briefly, configurations in use frequently combine ion exchange methods such as Strong Cation Exchange or Strong Anion Exchange with reversed-phase (RP) separations or combine RP-RP with separations based on different pH in the RP columns.47,53 Regardless of configuration, RP is frequently the last step before the mass spectrometer so peptides can be desalted prior to ionization in order to avoid formation of salt adducts. An interesting development has been the use of porous layer open tubular columns (PLOT) for proteomics.54,55 Long used in gas chromatography to achieve high efficiency separations, PLOT columns are now seeing increased use as mass spectrometers have reached a level of sensitivity that is more compatible with the low capacity of these columns. A goal of proteomic separations is to increase peak capacity and separation efficiency in the shortest time possible, which is a very difficult proposition. If peak widths become too narrow, the mass spectrometer may not be able to scan fast enough to sample peptides, so peaks get missed; consequently the efficiency of separations has to be matched to the scan speed of the mass spectrometer employed. Good separations are critical to reducing ion suppression and increasing dynamic range, which will also advance protein sequence coverage during an analysis.
The Drive to Determine Protein Function
Genome projects provide information about the functional coding elements present in the DNA sequence. From DNA sequences bioinformatic algorithms can postulate the function of such coding elements through sequence similarity. Proteins can have multiple biological functions and simply knowing that a protein sequence is similar to that of known proteins does not provide proof of its activity or when the protein is active. Thus, an important task for proteomics is to determine the functions and roles of all proteins encoded in a genome sequence. Shotgun proteomic technologies have enabled new strategies to rapidly get at this information.
The concept of “guilt by association” is a powerful approach to develop initial clues about what proteins might do in biological systems. This is most easily done by enriching protein complexes and identifying the components. If something is known about the function of the “bait” protein, then the function of all proteins interacting with the bait can be inferred. For example, Carney et al identified Nbs1, a protein involved in double strand DNA repair and responsible for the disease Nijmegen breakage syndrome, by isolating a protein complex containing two other “bait” proteins, Mre11 and Rad5056, which were known to be involved in this repair process. This complex forms to repair damaged DNA after X-radiation of cells. Hazbun et al combined the analysis of protein complexes with several other techniques to identify the functions of 100 essential, hypothetical genes in S. cerevisiae.57 In this study they were able to assign functions to 77 of the 100 proteins. Sato et al and Conaway et al were able to find the long missing components of the mammalian Mediator complex using shotgun proteomics of the complex.58,59 Large-scale protein-protein interaction studies are now common and provide rich insights into the biology of organisms.60-65
A more traditional biochemical strategy to associate a function to a protein is to enrich proteins based on activity and then identify the protein involved. However, protein activity can be lost before a protein is enriched to homogeneity (or the activity can originate from a collection of proteins). Shotgun proteomics can be used to identify the proteins present in the enriched fractions before homogeneity is reached or the activity is lost. Sauerwald et al used this approach to identify O-phosphoseryl-tRNA synthetase as the enzyme involved in the formation of Cys-tRNACys in organisms lacking cysteinyl-tRNA synthetase.66 A more global approach to discover protein activities was developed by Liu et al using an activity based profiling method to identify proteins with a particular enzymatic activity.67 This approach uses a “suicide substrate” inhibitor or other types of tightly bound active site inhibitors tethered to a solid support to pull out enzymes that are in an active state. Most importantly, new enzymes of a class can be found using this method. A striking result in a search for hydrolases was the identification of many proteins whose sequences would not have classified them as hydrolases, but whose activity very clearly did.67 Activity-based methods combined with shotgun proteomics create precise methods to identify proteins with specific enzymatic activities.
Whole cell or organelle analyses can also provide functional insight into proteins. Proteins that co-localize in organelles such as the mitochondria share a common role and since the vast majority of proteins that localize in the mitochondria are derived from the nuclear genome, their presence in the mitochondria may be reflective of the tissue type and the needs of the tissue. Kislinger et al and Mootha et al identified proteins in the mitochondria from 6 different organs of the mouse and they found the distribution of proteins varied depending on the tissue type.68,69 Tissue dependent differences in mitochondrial proteins may reflect the needs of the tissues, as heart tissue may have very different energy needs from brain tissue. The presence of proteins at particular times in a cell or organism's lifecycle can reveal information about a protein's role. For example, Florens et al used proteomic methods to measure protein expression in different morphological states of Plasmodium falciparum (Pf), which, when combined with studies in other Plasmodium species, revealed keen insights into stage specific proteins.70,71Plasmodium undergoes a complicated lifecycle between two species of hosts and is very evasive of the human immune system.71,72 Comparing transcript expression with protein expression for each stage revealed how transcripts are prepared in one stage for translation in the next stage.73 Expression profiling, although complicated to interpret, can be a useful shotgun strategy to identify proteins with important functions, particularly when combined with genetic perturbations.74
Qualitative proteomic analysis has focused primarily on acquiring more information about a sample, such as cell tissue or protein complex, with the ultimate goal of comprehensive coverage of all proteins present. What technological advances are needed to achieve this goal? Sample preparation methods to ensure all proteins are properly digested and soluble have made good progress. Sequence coverage has been increased by using multi-protease digestions to ensure peptides are within the acquisition range of the mass spectrometer.75-77 In addition, new peptide dissociation methods such as electron transfer dissociation (ETD) have increased the size of peptides that can be efficiently fragmented.78 High sequence coverage increases the chance of observing modified peptides and peptides that may represent a splice junction which helps to differentiate protein isoforms. Two major challenges exist for complex mixtures such as a cell or tissue lysate: ion suppression and dynamic range. Ion suppression occurs whenever a complex mixture is ionized, as some molecules ionize preferentially, suppressing the ionization of others. This issue has been summarized by Cech and Enke.79 Anderson observed this problem in an attempt to perform targeted mass spectrometry of peptides in digested serum, where he found big increases in signal and success by pre-enriching the peptides with antibodies prior to analysis.80 Eliminating the bulk of the matrix background reduced ion suppression and improved signals for the target peptides. Wolters et al observed a similar effect and improved analyses by using LC/LC to increase fractionation of the complex peptide mixtures to reduce the complexity during ionization.81 If methods can be developed to better reduce or eliminate ion suppression, more uniform ionization could be created for peptides, which will improve both qualitative and quantitative analysis. The challenge of measuring the large dynamic range of peptide and protein abundances is linked to ion suppression, but is primarily associated with limits of detection in the mass spectrometer. Both ion suppression and dynamic range can be partially overcome by increasing fractionation of peptides, but this increases time of analysis and limits its usefulness as an experimental strategy (e.g. long analysis times). Fonslow et al recently described two strategies to selectively remove more abundant proteins from complex protein mixtures.82,83 One method uses differential affinity for a hexapeptide library to remove more abundant proteins and another harnesses Michelis-Menton enzyme kinetics to preferentially digest abundant proteins in complex mixtures. In addition to ion suppression and dynamic range, a third issue is one of MS peak capacity. Typically, precursor ions are selected with a 3 amu wide window which increases the chance of precursor ion “contamination” with other precursors and results in poor search results. A workaround is to purposely try to multiplex MSMS and then deconvolute spectra for searching.84,85 This strategy is called Data Independent Acquisition (DIA). It has been proposed by several groups and is commercially available (MSe).84-88 As mass spectrometers scan faster it becomes more feasible to use DIA for identification and to improve quantitation.85,86 Tandem mass spectrometers have improved significantly with each generation, moving shotgun proteomics closer to achieving a complete proteome. Determining when a complete proteome is achieved, however, is a difficult proposition as proteomes are dynamic and it's not clear exactly how many proteins should be present. In addition, it is critical that a complete proteome be achieved with a reasonable experimental strategy and not through a time-consuming brute-force strategy (e.g. extensive fractionation).
Control and regulation of Biological Systems
Proteins are modified by a dazzling array of molecular structures. Many are simply structural modifications that alter the chemical characteristics of the protein, such as lipid or carbohydrate modifications that seem to have no obvious regulatory function, but other modifications are keenly involved in regulation. A feature that distinguishes regulatory from non-regulatory modifications is often reversibility, with regulatory modifications typically being reversible and non-regulatory modifications typically being non-reversible. One exception is proteolytic processing which can convert enzymes from an inactive state to an active state and is irreversible.
Mass spectrometry has been used for the analysis of protein modifications for a long time. The measurement of mass in highly regular molecules such as proteins is a straightforward way to identify the addition of unexpected molecular structures. Early examples include Gerber et al, who discovered a pyroglutamic acid on the N-terminus of bacteriorhodopsin89 and Carr et al, who developed an elegant method to identify the presence of the labile modification gamma-carboxyglutamic acid.90 The development of ionization methods such as FAB improved the direct observation and analysis of post translational modifications such as pyroglutamic acid and C-terminal amides91. Mass spectrometry analysis of protein phosphorylation sites was developed using FAB-MS and was extended to the analysis of sulfation, which occurs on tyrosine and can be easily confused for tyrosine phosphorylation.92,93 Tandem mass spectrometry is more effective than FAB-MS for identifying sites of modification, and was implemented by Hunt et al, who used it to identify phosphopeptides which had been enriched by iron (III) affinity chromatography.94,95 In these pre-genome sequence era strategies, the protein sequence was known and the protein was purified to homogeneity before attempting to identify the sites of modification.
As genome sequences began to appear and protein identification methods were developed, the nature of the problem to identify modifications changed. Yates et al demonstrated the use of database searching methods to identify post translational modifications using tandem mass spectrometry data.26 The challenge of identifying the site of modification confidently is greater than simply identifying the amino acid sequence of the peptide, as there can be multiple sites of modification within a peptide with different types of modifications at each site. A search algorithm must assess all the possibilities and determine which amino acid is most likely modified. A good example of challenges associated with identifying modifications is provided by phosphorylation. Phosphorylation can occur on Ser, Thr and Tyr, to a lesser extent His and a few other amino acids. Even just considering the major sites of phosphorylation, if multiples of Ser, Thr or Tyr appear in a sequence, each one is a potential site of modification and thus increases the possibilities to be considered by a search algorithm. For example, if there are 3 sites within a sequence, then there are 23 possible ways the peptide can be modified by phosphorylation. The molecular weight of the measured peptide can rule out whether the peptide has 1, 2, or 3 phosphorylations, but determining the sites when the peptide is not fully modified requires having specific fragment ions that define the mass shifts associated with a modification site. An additional issue is that a peptide can be modified at different sites, giving the modified peptides the same molecular weight and often the same HPLC elution time causing co-elution and co-fragmentation in the mass spectrometer. When the tandem mass spectrometer collects a spectrum that represents two or more modified peptides, evidence for both possibilities exists in the tandem mass spectrum. If they are equimolar, then evidence will likely be strong for both possibilities, but if they are not equimolar, then evidence will be stronger for one possibility rather than the other. Most search programs will simply assign the match to the strongest match, but on visual inspection of the spectrum, “contamination” may be observed. No algorithms have appeared that can quantitate the amount of each modified site within a spectrum, although algorithms exist to determine the probability that the dominant site is correctly identified.96 Search algorithms have also appeared to search for modifications in a blind manner without regard to the type of modification.97 The ability to rapidly interpret modified tandem mass spectra and assign sites of modification created the ability to perform these analyses on a large-scale and has led to a better understanding of the biology of modifications.
The large-scale acquisition of peptide data by tandem mass spectrometry, together with high throughput data analysis enabled Ficaro et al to combine a phosphopeptide enrichment strategy for a large-scale analysis of phosphopeptides in S. cerevisiae.98 Phosphorylation can be difficult to observe because it often occurs in hydrophilic regions of proteins, exists at substoichiometric levels, and modifies lower abundance proteins, so Ficarro et al incorporated the use of phosphopeptide enrichment into their analysis to improve detection of phosphopeptides.98 Iron (III) affinity chromatography, first used in conjunction with mass spectrometry by Michel et al, had been widely employed to enrich phosphopeptides from single proteins, but Ficarro et al was the first to use it in a large-scale “shotgun” format.94,98 This work triggered a “space race” to collect as many phosphopeptide sites as possible from different cell types and tissues. Beauloslil et al identified over 5000 phosphopeptides in HeLa cells and the numbers from other studies continue to grow.99-102 While existing studies have done a tremendous job of cataloging protein phosphorylation sites, the next step is establishing the context of phosphorylation at sites that may be regulating processes or function. Such studies are more exacting and not nearly as high throughput. Kunz et al and Kubota et al have developed a method, “KAYAK”, to study the regulatory roles of specific kinase phosphorylation sites.103,104 Large-scale analysis of modification sites has expanded to include any modification that can be enriched, which has also fueled the development of new enrichment methods.105-107 These methods and studies will help bring into focus the role of modifications in controlling and regulating biological processes.
Protein Quantitation
The idea of quantitating molecules using mass spectrometry dates back to the origins of mass analysis when Aston discovered the existence of stable isotopes.108 With Nier's enrichment of the 13C stable isotope, the ability to trace molecules specifically labeled with stable isotopes based on mass differences in the mass spectrometer became possible.109 In early mass spectrometry peptide sequencing strategies, stable isotope-labeled reagents were also used to shift fragment ion series or to differentiate amino acids whose masses become isobaric (equal molecular weight) during derivatization chemistry8. Furthermore, stable isotope labeling was a sine qua non of quantitative mass spectrometry for in vivo studies of metabolism such as determining amino acid essentiality.
Early quantitative studies in proteomics involved the use of 2-D Gel Electrophoresis (2-DGE) with measurement of changes based on protein coloration through a protein staining method or radioactivity. The density of the stain reflected the amount of protein present, but care had to be taken to stain proteins in precisely the same manner, as development time could alter the density of staining. The advent of mass spectrometry based protein identification techniques allowed the identity of proteins on 2-DGE to be readily determined, providing a huge boon to the use of 2-DGE for biological studies. Mass Spectrometry protein identification methods reduced the time and labor needed to determine what protein was in a spot and allowed identification to be combined with quantitation.
To create more accurate methods of quantitation, stable isotope labeling methods were combined with mass spectrometry for the analysis of intact proteins (Figure 3). Several approaches have emerged which employ stable isotope metabolic labeling methods or covalent tagging with reagents containing labels. In 1998 Langen et al presented a method employing 15N and 13C labeled amino acids to metabolically label proteins for quantitation.110 In 1999 three papers were published on the use of stable isotope labeling to measure expression changes.111-113 Oda et al used 15N stable isotope metabolic labeling in S. cerevisiae to identify expression changes and to quantitate a phosphorylated peptide.112 Pasa-Tolic et al used isotope depleted media (13C, 15N, 2H) to create differences from normal media to measure changes in intact proteins of E. coli.113 Patents were filed by Chait et al and Franza and Rochon in 1999 to cover various aspects of metabolic labeling for protein quantitation including the use stable isotope labeled amino acids such as heavy Lys and Arg.114,115 In 1999 a different approach was also published by Gygi et al as they described the use of a stable isotope labeled reagent to modify Cys residues and introduce a mass differential tag.111 The reagent also allowed affinity isolation of the labeled peptide. The concept and design of the tag was based on the reagent developed by Gerber et al to measure changes in metabolites in urine.116 While the ICAT method was elegant in concept, it had a number of drawbacks including that identification and quantification were often based on one peptide per protein, which limits statistical analysis. There was also difficulty with peptide recovery from the avidin based system used for enrichment. Munchbach et al created an N-terminal labeling method that introduced a stable isotope label and helped direct fragmentation during CID, presaging the isobaric tagging methods to be introduced a few years later.117 Conrads et al used 15N labeling together with a Cys affinity capture system to create a method similar to ICAT.118 Zhang et al explored the use of deuterium labeled derivatizing agents, eventually culminating in an isotope coding strategy.119 Washburn et al used shotgun proteomics together with 15N labeling to measure protein expression changes on a large-scale.120 Rather than attempt to enrich for peptides or to separate by gel electrophoresis, the Washburn et al approach used a large-scale separation method for shotgun proteomics of labeled peptides. Shu et al and Ong et al used the addition of stable isotope labeled amino acids to media as a way to label proteins that were then separated by gel electrophoresis. 121,122 Following in the work of Munchbach et al, two methods to label the N-terminus of peptides were developed by Thompson et al and Ross et al that had an unusual twist.123,124 Both methods had a set of labels that were isobaric until peptides were fragmented and then they revealed a unique mass tag. This method allows experiments to be multiplexed and, surprisingly, produces fairly good measurement accuracy. Dephoure and Gygi recently demonstrated hyperplexing with 18 channels using isobaric tags and stable isotope labeling.125
Figure 3.
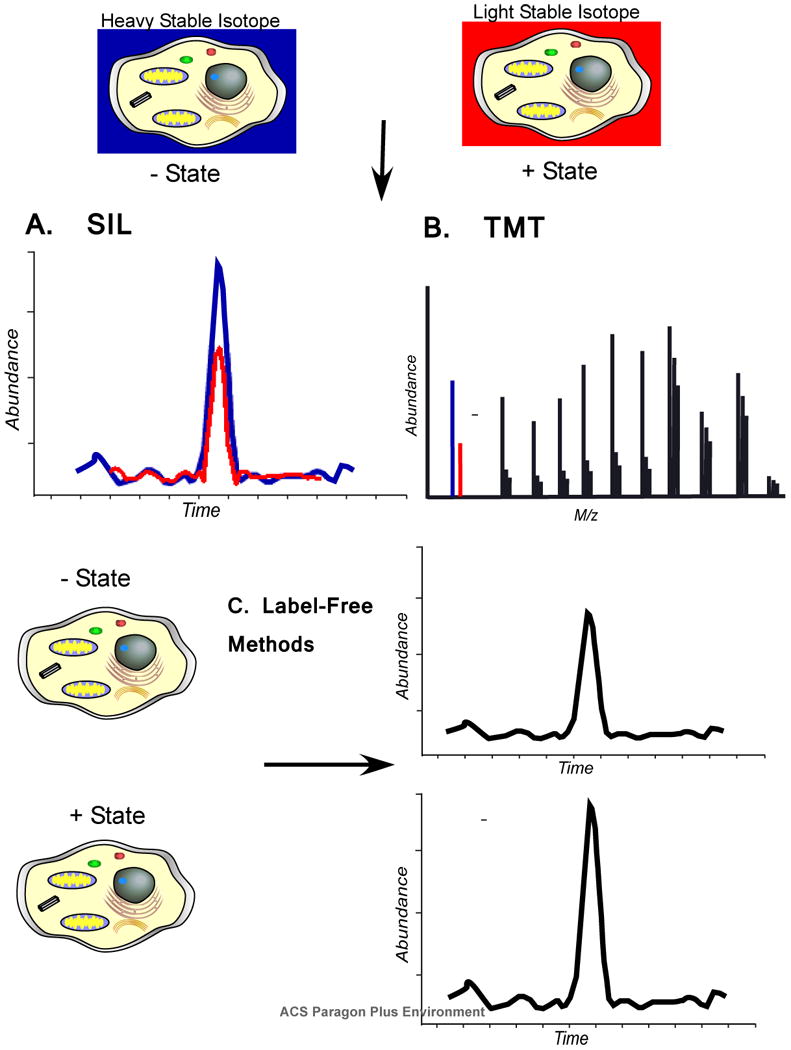
Quantitation of proteins can be performed using stable isotope labels, covalent tags, or label free methods. A) In stable isotope labeling (SIL) methods, a heavy stable isotope label allows peptide masses to be distinguished in the mass spectrometer between different experimental states. Abundance differences can be visualized by selected ion chromatograms. B) Isobaric tags add a mass to the peptides of each state that is isobaric until the peptide ions are fragmented which then reveals a mass difference. The difference in abundance is quantified from the reporter ions in the tandem mass spectrum. C) Two different experimental states can be compared using “label-free” methods. Ion intensity can be measured and compared by selected ion chromatograms as with SIL methods, but these measurements are taken from two different analyses. Another method uses “spectral counting” as a surrogate for abundance based on the observation that proteins which are more abundant have more peptide ions acquired. Label free methods are typically not as accurate as other methods, but can often provide sufficient information to prioritize follow up experiments.
In vivo Labeling Whole Animals
The introduction of stable isotope labels in humans and animals was used to measure metabolic fates of molecules.126 Metabolic analyses were then performed with trace levels of stable isotope labeled amino acid and very specific and sensitive mass spectrometers such as isotope ratio mass spectrometers (IRMS). Animal labeling with heavy isotopes was studied for safety and was thought to be safe for use at tracer levels, but the high levels necessary for proteomics were never studied. For this reason a study by Krijgsveld et al that labeled C. elegans and D. melanogaster with 15N was received with much enthusiasm.127 These metazoans, while not as complex as mammals, are still multicellular model systems used for the study of complex biology and thus would be useful for proteomic studies. Wu et al developed methods to label rats with 15N to very high levels of atomic percent enrichment using a method similar to Krijgsveld et al and (Figure 4)128 these rats were used to study brain development.129-131 A similar method to label mice with only stable isotope labeled lysine added to the diet was published by Kruger et al. but this method required labeling several generations of animals to achieve sufficient enrichment for proteomic studies. 132 Metabolic stable isotope labeling has been shown to be a very powerful method to study animal biology. Recently, Savas et al showed that the nuclear pore complex (NPC) is extremely long lived in post mitotic cells of tissues like the brain.133 By labeling an animal with 15N to 95% enrichment and then shifting the animals back to an 14N diet the proteins that are long lived can be identified at various time points in the animal's life. At 1 year, some of the proteins of the neuronal NPC are still labeled with 15N demonstrating turnover of proteins in the complex is very slow. Whole animal labeling enables questions that involve more complicated systems than cell lines and may better reflect organismic biology. For example, McClatchy et al measured protein expression changes in mitochondria and synaptosomes in rat brains as a function of developmental time points and location in the brain.134 New proteins were clearly observed that followed expression programs similar to proteins involved in specific developmentally based diseases. McClatchy also measured phosphorylation differences in rat livers and brain by mixing heavy labeled liver into light labeled brain in a strategy that allows a direct view of how tissues might be different.135 Similarly, Gygi et al measured phosphorylation analysis of mouse tissues using label free quantitation methods. This study demonstrated the ability to compare phosphorylation of proteins and peptides across different types of tissues.102 Ishihama et al used a stable isotope labeled cell line as an internal standard to spike into tissues and quantitate protein expression changes.136 Liao et al showed the reverse also works, where stable isotope labeled brain tissue can be spiked into primary neuronal cells to quantitate protein expression.137 By using a ratio of ratio approach to quantitate, the internal standard is spiked into the experimental system as well as the control and this controls for systematic errors and does not require the internal standard to be exactly the same as either system.138 Stable isotope labeling of animals allows the use of tissues and organs for the study of diseases. Of additional interest is that tissues and organs are collections of many different cell types and thus are systems of systems which in the end will require studies to understand how these communities of cells function.
Figure 4.
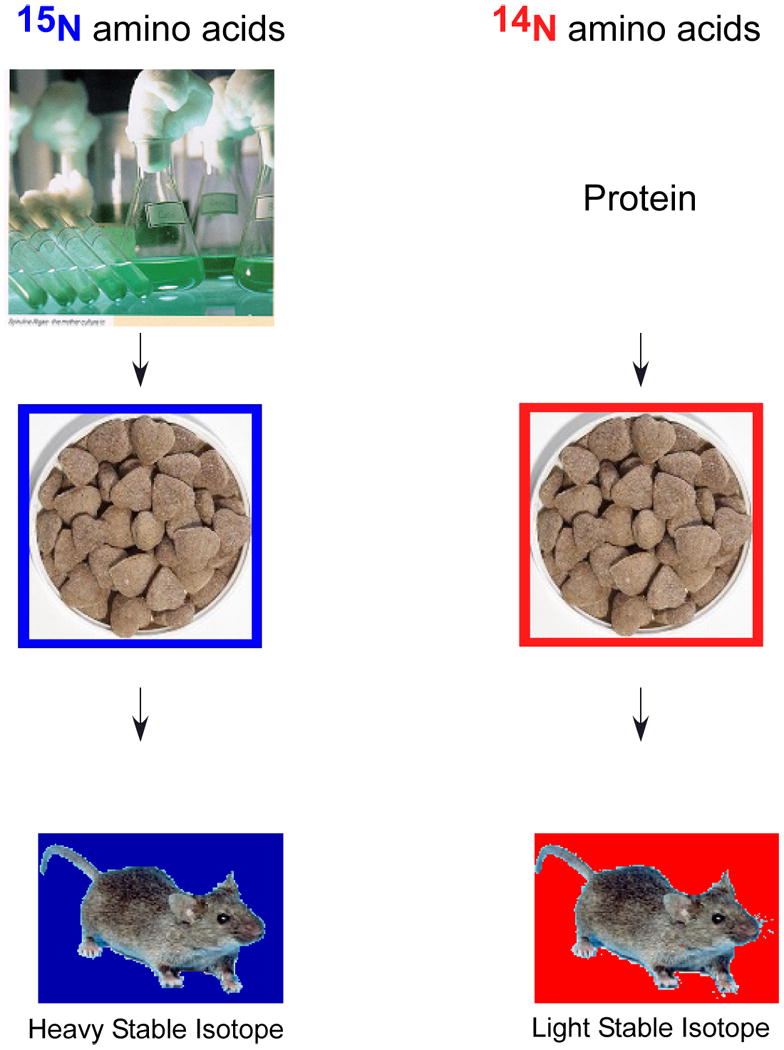
Mice and rats can be labeled with heavy stable isotopes by controlling the protein source in their food. By adding protein labeled with heavy and light stable isotopes to protein free chow, the source of protein in the diet of the animals is restricted to the heavy or light isotopes. These amino acids are incorporated into the new proteins metabolically synthesized by the cells of the animal.
Quantitation/Identification Paradox
Large-scale methods to identify proteins have naturally led to strategies that also try to simultaneously measure the amount of protein present using the methods described above. Being able to both identify and quantify proteins allows the determination of changes in biological systems with perturbations or stimulations. In shotgun proteomics, this creates a paradox. To identify proteins in complex systems in a comprehensive manner requires fast scanning instruments and highly efficient chromatography to maximize peak capacity for MSMS. The instrument should rapidly collect data for a peptide and then move on to a new peptide. Peptide quantitation requires the collection of sufficient data points to make an accurate measurement of the differences between two states. The competing demands, brevity in measurement versus persistence in measurement, leads to trade-offs in the quality of data used for quantitation since limits of detection for peptide identification most often exceeds the limit of quantitation. A solution to this problem is to optimize measurements for identification and quantify well enough to observe the trends in changes which can then be measured more accurately and precisely with more focused mass spectrometry methods. This strategy was used by Dong et al to measure genetically perturbed changes in the insulin signaling pathway in C. elegans.74 Another issue is the measurement of “presence or absence” in quantitation experiments. Most software tools require both the heavy and light isotopically labeled peptides be present for a measurement to be calculated. When the ratio of peptides exceeds 10 to 1, quantitation efficiency begins to drop-off and large changes can be missed.139 Some label-free methods such as spectral counting are better able to determine large changes, but these methods tend to be less accurate than labeling methods.140,141 Better methods are needed to observe large changes in abundance which can be very important in biological systems.
Future Prospects
George postulated that the unexpectedly low number of genes in humans is necessitated by the need to have a useful immune system.142 If the sequence space of humans were too large, pathogens could readily evade the immune system. Functional diversity must then result from small changes in proteins rather than from completely new sequences. Alternate splicing and covalent modification create this functional diversity. To fully understand human biology, we must begin to understand the functional roles of protein isoforms and modifications and thus we need technology to readily separate and measure protein isoforms and modifications in a functional context. As we've learned with histones and their complex sets of modifications, patterns of a modification or patterns of different types of modifications together may create a higher order of regulation.143
To fill this need, robust methods to measure molecular weight and determine sequence for intact proteins would be ideal. Technology for “top down” mass spectrometry is still developing and will require significant innovation to reduce the cost and complexity of mass spectrometers to democratize its use.144-147 In the intermediate term, MS analyzers to sequence and characterize longer polypeptides in the range of 5-10,000 Da have improved dramatically in the last few years, but proteases or chemical cleavage methods to cleave proteins in to 5-10K pieces are needed. Most proteases produce smaller peptides, although Wu et al recently reported a bacterial protease, OmpT, that is a rare cutter protease producing on average polypeptides 6.3 kDa or greater.147 Higher resolution mass spectrometers coupled with ETD should permit ready characterization of these medium sized polypeptides. 146
Protein complexes represent a higher order structure within cells (Figure 5). Determining how protein isoforms or modified forms (now referred to as “proteoforms”) affect the function or activity of complexes is a next step. When protein complexes are studied they are often enriched through a single “bait” protein that may exist in many different complexes. Consequently when analyzed all the components of the multiple complexes are identified without knowing to what specific complex they belong. By isolating the different complexes consisting of this bait and then identifying all the proteins and proteoforms present, the functions of the individual complexes can be better dissected. Furthermore, the composition of complexes is dynamic and thus higher throughput strategies for global analyses of complexes, as well as methods to dissect out the composition of individual complexes with cellular changes are needed.148,149 Determining proteoform information in the context of individual protein complexes will help sort out the functional roles of proteoforms and the individual protein complexes.
Figure 5.
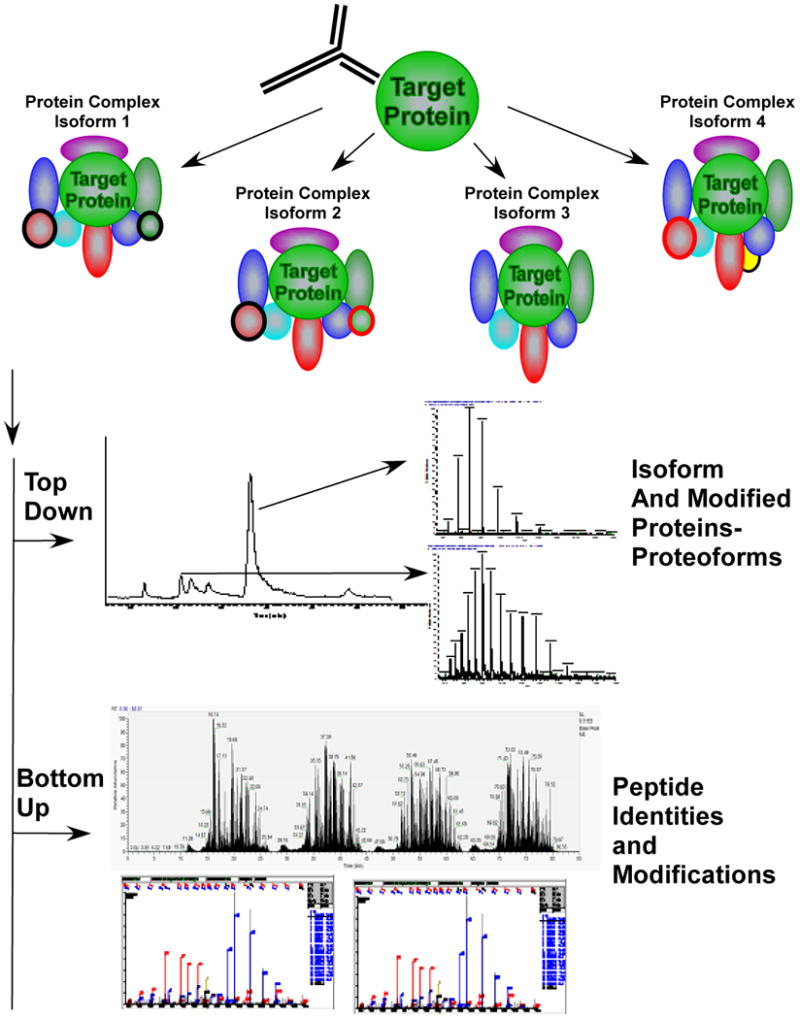
A future challenge to proteomics is deciphering the structures of protein complexes where different proteins, be they isoforms or modified forms may associate at different times or in different locations within a cell with a specific “core” protein. The core protein is depicted as the “target protein” of the enrichment process. To fully characterize the complexes associating with a specific target protein will require methods to separate or enrich the individual complexes, methods such as top down or native mass spectrometry of the complexes or components of the complexes. This data can identify modification status or protein isoforms (proteoforms) present in the complexes. Bottom-up mass spectrometry can accurately identify the proteins present in the complex and modification sites.
It is expected that mass spectrometers will continue to evolve at a fast pace through technological advances and fierce commercial competition. Instruments will scan faster with better sensitivity to create better tools for proteomics and further increase capabilities for biological discovery. Mass spectrometry will continue to drive the discovery of new and important biology.
Acknowledgments
I would like to acknowledge funding support from the following NIH grants: P41 RR011823/GM103533, R01 MH067880-09, P01 AG031097-03, R01 HL079442-07. I would like to thank Claire M. Delahunty, PhD for a careful reading of the paper.
References
- 1.Malcolm AD. Nature. 1978;275:90. doi: 10.1038/275090a0. [DOI] [PubMed] [Google Scholar]
- 2.Couzin-Frankel J. Science. 2009;234:1504. doi: 10.1126/science.324_1504. [DOI] [PubMed] [Google Scholar]
- 3.Grayson MA. Measuring Mass, From Positive Rays to Proteins. Chemical Heritage Press; Philadelphia: 2002. [Google Scholar]
- 4.Biemann K, Gapp G, Seibl J. J Am Chem Soc. 1959;81:2274. [Google Scholar]
- 5.Biemann K, Lioret C, Asselineau J, Lederer E, Polonsky J. Biochim Biophys Acta. 1960;40:369. doi: 10.1016/0006-3002(60)91370-6. [DOI] [PubMed] [Google Scholar]
- 6.Biemann K, Cone C, Webster BR, Arsenault GP. J Am Chem Soc. 1966;88:5598. doi: 10.1021/ja00975a045. [DOI] [PubMed] [Google Scholar]
- 7.Gohlke RS. Analytical Chemistry. 1959;31:535. [Google Scholar]
- 8.Nau H, Kelley JA, Biemann K. J Am Chem Soc. 1973;95:7162. doi: 10.1021/ja00802a048. [DOI] [PubMed] [Google Scholar]
- 9.Lucas F, Barber M, Wolstenholme WA, Geddes AJ, Graham GN, Morris HR. Biochem J. 1969;114:695. doi: 10.1042/bj1140695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herlihy WC, Royal NJ, Biemann K, Putney SD, Schimmel PR. Proc Natl Acad Sci U S A. 1980;77:6531. doi: 10.1073/pnas.77.11.6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barber M, Bordoli RS, Sedgwick RD, Tyler AN. Nature. 1981;293:270. [Google Scholar]
- 12.Morris HR, Panico M, Barber M, Bordoli RS, Sedgwick RD, Tyler A. Biochem Biophys Res Commun. 1981;101:623. doi: 10.1016/0006-291x(81)91304-8. [DOI] [PubMed] [Google Scholar]
- 13.Morris HR, Panico M, Taylor GW. Biochemical and Biophysical Research Communications. 1983;117:299. doi: 10.1016/0006-291x(83)91575-9. [DOI] [PubMed] [Google Scholar]
- 14.Hunt DF, Bone WM, Shabanowitz J, Rhodes J, Ballard JM. Analytical Chemistry. 1981;53:1704. [Google Scholar]
- 15.Hunt DF, Yates JR, 3rd, Shabanowitz J, Winston S, Hauer CR. Proc Natl Acad Sci U S A. 1986;83:6233. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito Y, Takeuchi T, Ishii D, Goto M, Mizuno T. J Chromatogr. 1986;358:201. doi: 10.1016/s0021-9673(01)90330-9. [DOI] [PubMed] [Google Scholar]
- 17.Caprioli RM, Fan T, Cottrell JS. Anal Chem. 1986;58:2949. doi: 10.1021/ac00127a012. [DOI] [PubMed] [Google Scholar]
- 18.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Science. 1989;246:64. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 19.Smith RD, Udseth HR. Nature. 1988;331:639. doi: 10.1038/331639a0. [DOI] [PubMed] [Google Scholar]
- 20.Smith RD, Loo JA, Edmonds CG, Barinaga CJ, Udseth HR. Anal Chem. 1990;62:882. doi: 10.1021/ac00208a002. [DOI] [PubMed] [Google Scholar]
- 21.Novotny M, Zlatkis A. Chromatogr Rev. 1971;14:1. doi: 10.1016/0009-5907(71)80010-8. [DOI] [PubMed] [Google Scholar]
- 22.Kennedy RT, Jorgenson JW. Analytical Chemistry. 1989;61:1128. doi: 10.1021/ac00180a012. [DOI] [PubMed] [Google Scholar]
- 23.Hunt DF, Henderson RA, Shabanowitz J, Sakaguchi K, Michel H, Sevilir N, Cox AL, Appella E, Engelhard VH. Science. 1992;255:1261. doi: 10.1126/science.1546328. [DOI] [PubMed] [Google Scholar]
- 24.Davis MT, Stahl DC, Hefta SA, Lee TD. Analytical Chemistry. 1995;67:4549. doi: 10.1021/ac00120a019. [DOI] [PubMed] [Google Scholar]
- 25.Sokolow S, Steiner U, Lewis JR, editors. USPTO. Finnigan Corporation; USA: 4,947,315. System for controlling instrument using a levels data structure and concurrently running compiler task and operator task 1990
- 26.Yates JR, III, Eng JK, McCormack AL, Schieltz D. Analytical Chemistry. 1995;67:1426. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]
- 27.Davis MT, Stahl DC, Swiderek KM, Lee TD. Methods. 1994;6:304. [Google Scholar]
- 28.National Technical Information Service: Springfield, Virginia, 1990.
- 29.Council NR. Mapping and sequencing the human genome. 1988 [Google Scholar]
- 30.Eng J, McCormack A, Yates J. J Am Soc Mass Spectrom. 1994;5:976. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 31.Yates JR, III, Eng JK, McCormack AL. Anal Chem. 1995;67:3202. doi: 10.1021/ac00114a016. [DOI] [PubMed] [Google Scholar]
- 32.McCormack AL, Schieltz DM, Goode B, Yang S, Barnes G, Drubin D, Yates IJR. Analytical Chemistry. 1997;69:767. doi: 10.1021/ac960799q. [DOI] [PubMed] [Google Scholar]
- 33.Hunt DF, Giordani AB, Rhodes G, Herold DA. Clin Chem. 1982;28:2387. [PubMed] [Google Scholar]
- 34.McCormack AL, Eng JK, Yates IJR. Methods: A companion to Methods in Enzymology. 1994;6:274. [Google Scholar]
- 35.Washburn MP, Wolters D, Yates JR., 3rd Nature Biotechnology. 2001;19:242. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 36.Yates IR, III, McCormack AL, Link AJ, Schieltz DM, Eng JK, Hays L. Analyst. 1996;121:65R. doi: 10.1039/an996210065r. [DOI] [PubMed] [Google Scholar]
- 37.Link AJ, Carmack E, Yates IJR. International Journal of Mass Spectrometry and Ion Processes. 1997;160:303. [Google Scholar]
- 38.Yates IJR., III J Mass Spectrom. 1998;33:1. doi: 10.1002/(SICI)1096-9888(199801)33:1<1::AID-JMS624>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 39.Schirmer EC, Florens L, Guan T, Yates JR, 3rd, Gerace L. Science. 2003;301:1380. doi: 10.1126/science.1088176. [DOI] [PubMed] [Google Scholar]
- 40.Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. Nature. 2003;426:570. doi: 10.1038/nature02166. [DOI] [PubMed] [Google Scholar]
- 41.Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvik BM, Yates JR., 3rd Nat Biotechnol. 1999;17:676. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- 42.Blonder J, Conrads TP, Yu LR, Terunuma A, Janini GM, Issaq HJ, Vogel JC, Veenstra TD. Proteomics. 2004;4:31. doi: 10.1002/pmic.200300543. [DOI] [PubMed] [Google Scholar]
- 43.Wu CC, MacCoss MJ, Howell KE, Yates JR., 3rd Nat Biotechnol. 2003;21:532. doi: 10.1038/nbt819. [DOI] [PubMed] [Google Scholar]
- 44.Blackler AR, Speers AE, Ladinsky MS, Wu CC. J Proteome Res. 2008;7:3028. doi: 10.1021/pr700795f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manza LL, Stamer SL, Ham AJ, Codreanu SG, Liebler DC. Proteomics. 2005;5:1742. doi: 10.1002/pmic.200401063. [DOI] [PubMed] [Google Scholar]
- 46.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Nat Methods. 2009;6:359. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 47.Motoyama A, Yates JR., 3rd Anal Chem. 2008;80:7187. doi: 10.1021/ac8013669. [DOI] [PubMed] [Google Scholar]
- 48.Liu H, Lin D, Yates JR., 3rd Biotechniques. 2002;32:898. doi: 10.2144/02324pt01. [DOI] [PubMed] [Google Scholar]
- 49.Cristobal A, Hennrich ML, Giansanti P, Goerdayal SS, Heck AJ, Mohammed S. Analyst. 2012;137:3541. doi: 10.1039/c2an35445d. [DOI] [PubMed] [Google Scholar]
- 50.Shen Y, Zhao R, Berger SJ, Anderson GA, Rodriguez N, Smith RD. Anal Chem. 2002;74:4235. doi: 10.1021/ac0202280. [DOI] [PubMed] [Google Scholar]
- 51.Motoyama A, Venable JD, Ruse CI, Yates JR., 3rd Anal Chem. 2006;78:5109. doi: 10.1021/ac060354u. [DOI] [PubMed] [Google Scholar]
- 52.MacNair JE, Lewis KC, Jorgenson JW. Anal Chem. 1997;69:983. doi: 10.1021/ac961094r. [DOI] [PubMed] [Google Scholar]
- 53.Altelaar AF, Heck AJ. Curr Opin Chem Biol. 2012;16:206. doi: 10.1016/j.cbpa.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 54.Luo Q, Yue G, Valaskovic GA, Gu Y, Wu SL, Karger BL. Anal Chem. 2007;79:6174. doi: 10.1021/ac070583w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yue G, Luo Q, Zhang J, Wu SL, Karger BL. Anal Chem. 2007;79:938. doi: 10.1021/ac061411m. [DOI] [PubMed] [Google Scholar]
- 56.Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR, 3rd, Hays L, Morgan WF, Petrini JH. Cell. 1998;93:477. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- 57.Hazbun TR, Malmstrom L, Anderson S, Graczyk BJ, Fox B, Riffle M, Sundin BA, Aranda JD, McDonald WH, Chiu CH, Snydsman BE, Bradley P, Muller EG, Fields S, Baker D, Yates JR, 3rd, Davis TN. Mol Cell. 2003;12:1353. doi: 10.1016/s1097-2765(03)00476-3. [DOI] [PubMed] [Google Scholar]
- 58.Conaway JW, Florens L, Sato S, Tomomori-Sato C, Parmely TJ, Yao T, Swanson SK, Banks CA, Washburn MP, Conaway RC. FEBS Lett. 2005;579:904. doi: 10.1016/j.febslet.2004.11.031. [DOI] [PubMed] [Google Scholar]
- 59.Sato S, Tomomori-Sato C, Parmely TJ, Florens L, Zybailov B, Swanson SK, Banks CA, Jin J, Cai Y, Washburn MP, Conaway JW, Conaway RC. Mol Cell. 2004;14:685. doi: 10.1016/j.molcel.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 60.Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, Bauch A, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A, Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, Seraphin B, Kuster B, Neubauer G, Superti-Furga G. Nature. 2002;415:141. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 61.Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dumpelfeld B, Edelmann A, Heurtier MA, Hoffman V, Hoefert C, Klein K, Hudak M, Michon AM, Schelder M, Schirle M, Remor M, Rudi T, Hooper S, Bauer A, Bouwmeester T, Casari G, Drewes G, Neubauer G, Rick JM, Kuster B, Bork P, Russell RB, Superti-Furga G. Nature. 2006;440:631. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- 62.Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, Yang L, Wolting C, Donaldson I, Schandorff S, Shewnarane J, Vo M, Taggart J, Goudreault M, Muskat B, Alfarano C, Dewar D, Lin Z, Michalickova K, Willems AR, Sassi H, Nielsen PA, Rasmussen KJ, Andersen JR, Johansen LE, Hansen LH, Jespersen H, Podtelejnikov A, Nielsen E, Crawford J, Poulsen V, Sorensen BD, Matthiesen J, Hendrickson RC, Gleeson F, Pawson T, Moran MF, Durocher D, Mann M, Hogue CW, Figeys D, Tyers M. Nature. 2002;415:180. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 63.Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, Punna T, Peregrin-Alvarez JM, Shales M, Zhang X, Davey M, Robinson MD, Paccanaro A, Bray JE, Sheung A, Beattie B, Richards DP, Canadien V, Lalev A, Mena F, Wong P, Starostine A, Canete MM, Vlasblom J, Wu S, Orsi C, Collins SR, Chandran S, Haw R, Rilstone JJ, Gandi K, Thompson NJ, Musso G, St Onge P, Ghanny S, Lam MH, Butland G, Altaf-Ul AM, Kanaya S, Shilatifard A, O'Shea E, Weissman JS, Ingles CJ, Hughes TR, Parkinson J, Gerstein M, Wodak SJ, Emili A, Greenblatt JF. Nature. 2006;440:637. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- 64.Malovannaya A, Lanz RB, Jung SY, Bulynko Y, Le NT, Chan DW, Ding C, Shi Y, Yucer N, Krenciute G, Kim BJ, Li C, Chen R, Li W, Wang Y, O'Malley BW, Qin J. Cell. 2011;145:787. doi: 10.1016/j.cell.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guruharsha KG, Rual JF, Zhai B, Mintseris J, Vaidya P, Vaidya N, Beekman C, Wong C, Rhee DY, Cenaj O, McKillip E, Shah S, Stapleton M, Wan KH, Yu C, Parsa B, Carlson JW, Chen X, Kapadia B, Vijayraghavan K, Gygi SP, Celniker SE, Obar RA, Artavanis-Tsakonas S. Cell. 2011;147:690. doi: 10.1016/j.cell.2011.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sauerwald A, Zhu W, Major TA, Roy H, Palioura S, Jahn D, Whitman WB, Yates JR, 3rd, Ibba M, Soll D. Science. 2005;307:1969. doi: 10.1126/science.1108329. [DOI] [PubMed] [Google Scholar]
- 67.Liu Y, Patricelli MP, Cravatt BF. Proc Natl Acad Sci U S A. 1999;96:14694. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kislinger T, Cox B, Kannan A, Chung C, Hu P, Ignatchenko A, Scott MS, Gramolini AO, Morris Q, Hallett MT, Rossant J, Hughes TR, Frey B, Emili A. Cell. 2006;125:173. doi: 10.1016/j.cell.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 69.Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, Bolouri MS, Ray HN, Sihag S, Kamal M, Patterson N, Lander ES, Mann M. Cell. 2003;115:629. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- 70.Carlton JM, Angiuoli SV, Suh BB, Kooij TW, Pertea M, Silva JC, Ermolaeva MD, Allen JE, Selengut JD, Koo HL, Peterson JD, Pop M, Kosack DS, Shumway MF, Bidwell SL, Shallom SJ, van Aken SE, Riedmuller SB, Feldblyum TV, Cho JK, Quackenbush J, Sedegah M, Shoaibi A, Cummings LM, Florens L, Yates JR, Raine JD, Sinden RE, Harris MA, Cunningham DA, Preiser PR, Bergman LW, Vaidya AB, van Lin LH, Janse CJ, Waters AP, Smith HO, White OR, Salzberg SL, Venter JC, Fraser CM, Hoffman SL, Gardner MJ, Carucci DJ. Nature. 2002;419:512. doi: 10.1038/nature01099. [DOI] [PubMed] [Google Scholar]
- 71.Florens L, Washburn MP, Raine JD, Anthony RM, Grainger M, Haynes JD, Moch JK, Muster N, Sacci JB, Tabb DL, Witney AA, Wolters D, Wu Y, Gardner MJ, Holder AA, Sinden RE, Yates JR, Carucci DJ. Nature. 2002;419:520. doi: 10.1038/nature01107. [DOI] [PubMed] [Google Scholar]
- 72.Lasonder E, Ishihama Y, Andersen JS, Vermunt AM, Pain A, Sauerwein RW, Eling WM, Hall N, Waters AP, Stunnenberg HG, Mann M. Nature. 2002;419:537. doi: 10.1038/nature01111. [DOI] [PubMed] [Google Scholar]
- 73.Le Roch KG, Johnson JR, Florens L, Zhou Y, Santrosyan A, Grainger M, Yan SF, Williamson KC, Holder AA, Carucci DJ, Yates JR, 3rd, Winzeler EA. Genome Res. 2004;14:2308. doi: 10.1101/gr.2523904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dong MQ, Venable JD, Au N, Xu T, Park SK, Cociorva D, Johnson JR, Dillin A, Yates JR., 3rd Science. 2007;317:660. doi: 10.1126/science.1139952. [DOI] [PubMed] [Google Scholar]
- 75.MacCoss MJ, McDonald WH, Saraf A, Sadygov R, Clark JM, Tasto JJ, Gould KL, Wolters D, Washburn M, Weiss A, Clark JI, Yates JR., 3rd Proc Natl Acad Sci U S A. 2002;99:7900. doi: 10.1073/pnas.122231399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gatlin CL, Eng JK, Cross ST, Detter JC, Yates IJR. Analytical Chemistry. 2000;72:757. doi: 10.1021/ac991025n. [DOI] [PubMed] [Google Scholar]
- 77.Swaney DL, Wenger CD, Coon JJ. J Proteome Res. 2010;9:1323. doi: 10.1021/pr900863u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Proc Natl Acad Sci U S A. 2004;101:9528. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cech NB, Enke CG. Mass Spectrom Rev. 2001;20:362. doi: 10.1002/mas.10008. [DOI] [PubMed] [Google Scholar]
- 80.Anderson L, Hunter CL. Mol Cell Proteomics. 2006;5:573. doi: 10.1074/mcp.M500331-MCP200. [DOI] [PubMed] [Google Scholar]
- 81.Krisp C, McKay MJ, Wolters DA, Molloy MP. Anal Chem. 2012;84:1592. doi: 10.1021/ac2028485. [DOI] [PubMed] [Google Scholar]
- 82.Fonslow BR, Carvalho PC, Academia K, Freeby S, Xu T, Nakorchevsky A, Paulus A, Yates JR., 3rd J Proteome Res. 2011;10:3690. doi: 10.1021/pr200304u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fonslow BR, Stein BD, Webb KJ, Xu T, Choi J, Park SK, Yates JR. Nature Methods. 2012 doi: 10.1038/nmeth.2250. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Masselon C, Anderson GA, Harkewicz R, Bruce JE, Pasa-Tolic L, Smith RD. Anal Chem. 2000;72:1918. doi: 10.1021/ac991133+. [DOI] [PubMed] [Google Scholar]
- 85.Venable JD, Dong MQ, Wohlschlegel J, Dillin A, Yates JR. Nat Methods. 2004;1:39. doi: 10.1038/nmeth705. [DOI] [PubMed] [Google Scholar]
- 86.Silva JC, Gorenstein MV, Li GZ, Vissers JP, Geromanos SJ. Mol Cell Proteomics. 2006;5:144. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 87.Silva JC, Denny R, Dorschel C, Gorenstein MV, Li GZ, Richardson K, Wall D, Geromanos SJ. Mol Cell Proteomics. 2006;5:589. doi: 10.1074/mcp.M500321-MCP200. [DOI] [PubMed] [Google Scholar]
- 88.Purvine S, Eppel JT, Yi EC, Goodlett DR. Proteomics. 2003;3:847. doi: 10.1002/pmic.200300362. [DOI] [PubMed] [Google Scholar]
- 89.Gerber GE, Anderegg RJ, Herlihy WC, Gray CP, Biemann K, Khorana HG. Proc Natl Acad Sci U S A. 1979;76:227. doi: 10.1073/pnas.76.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Carr SA, Hauschka PV, Biemann K. J Biol Chem. 1981;256:9944. [PubMed] [Google Scholar]
- 91.Gibson BW, Poulter L, Williams DH. J Nat Prod. 1986;49:26. doi: 10.1021/np50043a002. [DOI] [PubMed] [Google Scholar]
- 92.Poulter L, Ang SG, Gibson BW, Williams DH, Holmes CF, Caudwell FB, Pitcher J, Cohen P. Eur J Biochem. 1988;175:497. doi: 10.1111/j.1432-1033.1988.tb14222.x. [DOI] [PubMed] [Google Scholar]
- 93.Gibson BW, Cohen P. Methods Enzymol. 1990;193:480. doi: 10.1016/0076-6879(90)93434-m. [DOI] [PubMed] [Google Scholar]
- 94.Michel H, Hunt DF, Shabanowitz J, Bennett J. J Biol Chem. 1988;263:1123. [PubMed] [Google Scholar]
- 95.Andersson L, Porath J. Analytical Biochemistry. 1986;154:250. doi: 10.1016/0003-2697(86)90523-3. [DOI] [PubMed] [Google Scholar]
- 96.Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. Nat Biotechnol. 2006;24:1285. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 97.Tsur D, Tanner S, Zandi E, Bafna V, Pevzner PA. Nat Biotechnol. 2005;23:1562. doi: 10.1038/nbt1168. [DOI] [PubMed] [Google Scholar]
- 98.Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, White FM. Nat Biotechnol. 2002;20:301. doi: 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- 99.Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villen J, Li J, Cohn MA, Cantley LC, Gygi SP. Proc Natl Acad Sci U S A. 2004;101:12130. doi: 10.1073/pnas.0404720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Phanstiel DH, Brumbaugh J, Wenger CD, Tian S, Probasco MD, Bailey DJ, Swaney DL, Tervo MA, Bolin JM, Ruotti V, Stewart R, Thomson JA, Coon JJ. Nat Methods. 2011;8:821. doi: 10.1038/nmeth.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Swaney DL, Wenger CD, Thomson JA, Coon JJ. Proc Natl Acad Sci U S A. 2009;106:995. doi: 10.1073/pnas.0811964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP. Cell. 2010;143:1174. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kunz RC, McAllister FE, Rush J, Gygi SP. Anal Chem. 2012;84:6233. doi: 10.1021/ac301116z. [DOI] [PubMed] [Google Scholar]
- 104.Kubota K, Anjum R, Yu Y, Kunz RC, Andersen JN, Kraus M, Keilhack H, Nagashima K, Krauss S, Paweletz C, Hendrickson RC, Feldman AS, Wu CL, Rush J, Villen J, Gygi SP. Nat Biotechnol. 2009;27:933. doi: 10.1038/nbt.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Larsen MR, Thingholm TE, Jensen ON, Roepstorff P, Jorgensen TJ. Mol Cell Proteomics. 2005;4:873. doi: 10.1074/mcp.T500007-MCP200. [DOI] [PubMed] [Google Scholar]
- 106.Sano A, Nakamura H. Anal Sci. 2004;20:565. doi: 10.2116/analsci.20.565. [DOI] [PubMed] [Google Scholar]
- 107.Fonslow BR, Niessen SM, Singh M, Wong CC, Xu T, Carvalho PC, Choi J, Park SK, Yates JR., 3rd J Proteome Res. 2012;11:2697. doi: 10.1021/pr300200x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Aston FW. (Series 6).Philosophical Magazine. 1920;39:449. [Google Scholar]
- 109.Nier A, Gulbransen EA. Journal of the American Chemical Society. 1939;61:697. [Google Scholar]
- 110.Langen H, Fountoulakis M, Evers S, Wipf B, Berndt P. 3rd Siena 2D Electrophoresis Meeting: From Genome to Proteome. Siena, Italy: 1998. [Google Scholar]
- 111.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Nat Biotechnol. 1999;17:994. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 112.Oda Y, Huang K, Cross FR, Cowburn D, Chait BT. Proc Natl Acad Sci U S A. 1999;96:6591. doi: 10.1073/pnas.96.12.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Paša-Tolic L, Jensen PK, Anderson GA, Lipton MS, Peden KK, Martinovic S, Tolic N, Bruce JE, Smith RD. J Am Chem Soc. 1999;121:7950. [Google Scholar]
- 114.Chait BT, Cowburn D, Oda Y USPTO. Rockefeller University: Method for the comparative quantitative analysis of proteins and other biological material by isotopic labeling and mass spectroscopy. USA patent 6,391,649. 2002
- 115.Franza J, Robert B, Rochon YP. USA patent 6,653,076. Stable isotope metabolic labeling for analysis of biopolymers. 2003
- 116.Gerber SA, Scott CR, Turecek F, Gelb MH. J Am Chem Soc. 1999;121:1102. [Google Scholar]
- 117.Munchbach M, Quadroni M, Miotto G, James P. Anal Chem. 2000;72:4047. doi: 10.1021/ac000265w. [DOI] [PubMed] [Google Scholar]
- 118.Conrads TP, Alving K, Veenstra TD, Belov ME, Anderson GA, Anderson DJ, Lipton MS, Pasa-Tolic L, Udseth HR, Chrisler WB, Thrall BD, Smith RD. Anal Chem. 2001;73:2132. doi: 10.1021/ac001487x. [DOI] [PubMed] [Google Scholar]
- 119.Zhang X, Jin QK, Carr SA, Annan RS. Rapid Commun Mass Spectrom. 2002;16:2325. doi: 10.1002/rcm.864. [DOI] [PubMed] [Google Scholar]
- 120.Washburn MP, Ulaszek R, Deciu C, Schieltz DM, Yates JR., 3rd Anal Chem. 2002;74:1650. doi: 10.1021/ac015704l. [DOI] [PubMed] [Google Scholar]
- 121.Zhu H, Pan S, Gu S, Bradbury EM, Chen X. Rapid Commun Mass Spectrom. 2002;16:2115. doi: 10.1002/rcm.831. [DOI] [PubMed] [Google Scholar]
- 122.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Mol Cell Proteomics. 2002;1:376. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 123.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Mol Cell Proteomics. 2004;3:1154. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 124.Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C. Anal Chem. 2003;75:1895. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 125.Dephoure N, Gygi SP. Sci Signal. 2012;5:rs2. doi: 10.1126/scisignal.2002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bier DM. Eur J Pediatr. 1997;156:S2. doi: 10.1007/pl00014265. [DOI] [PubMed] [Google Scholar]
- 127.Krijgsveld J, Ketting RF, Mahmoudi T, Johansen J, Artal-Sanz M, Verrijzer CP, Plasterk RH, Heck AJ. Nat Biotechnol. 2003;21:927. doi: 10.1038/nbt848. [DOI] [PubMed] [Google Scholar]
- 128.Wu CC, MacCoss MJ, Howell KE, Matthews DE, Yates JR., 3rd Anal Chem. 2004;76:4951. doi: 10.1021/ac049208j. [DOI] [PubMed] [Google Scholar]
- 129.McClatchy DB, Dong MQ, Wu CC, Venable JD, Yates JR., 3rd J Proteome Res. 2007;6:2005. doi: 10.1021/pr060599n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.McClatchy DB, Liao L, Park SK, Venable JD, Yates JR. Genome Res. 2007;17:1378. doi: 10.1101/gr.6375007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.McClatchy D, Liao L, Park SK, Lee JH, Yates JR. J Proteome Res. 2012 doi: 10.1021/pr201176v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kruger M, Moser M, Ussar S, Thievessen I, Luber CA, Forner F, Schmidt S, Zanivan S, Fassler R, Mann M. Cell. 2008;134:353. doi: 10.1016/j.cell.2008.05.033. [DOI] [PubMed] [Google Scholar]
- 133.Savas JN, Toyama BH, Xu T, Yates JR, 3rd, Hetzer MW. Science. 2012;335:942. doi: 10.1126/science.1217421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.McClatchy DB, Liao L, Lee JH, Park SK, Yates JR., 3rd J Proteome Res. 2012;11:2467. doi: 10.1021/pr201176v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.McClatchy DB, Liao L, Park SK, Xu T, Lu B, Yates JR., Iii PLoS One. 2011;6:e16039. doi: 10.1371/journal.pone.0016039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ishihama Y, Sato T, Tabata T, Miyamoto N, Sagane K, Nagasu T, Oda Y. Nat Biotechnol. 2005;23:617. doi: 10.1038/nbt1086. [DOI] [PubMed] [Google Scholar]
- 137.Liao L, Sando RC, Farnum JB, Vanderklish PW, Maximov A, Yates JR. J Proteome Res. 2012;11:1341. doi: 10.1021/pr200987h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.MacCoss MJ, Wu CC, Liu H, Sadygov R, Yates JR., 3rd Anal Chem. 2003;75:6912. doi: 10.1021/ac034790h. [DOI] [PubMed] [Google Scholar]
- 139.Venable JD, Wohlschlegel J, McClatchy DB, Park SK, Yates JR., 3rd Anal Chem. 2007;79:3056. doi: 10.1021/ac062054i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Liu H, Sadygov RG, Yates JR., 3rd Anal Chem. 2004;76:4193. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 141.Zybailov B, Coleman MK, Florens L, Washburn MP. Anal Chem. 2005;77:6218. doi: 10.1021/ac050846r. [DOI] [PubMed] [Google Scholar]
- 142.George AJT. Trends in Immunology. 2002;23:351. doi: 10.1016/s1471-4906(02)02241-x. [DOI] [PubMed] [Google Scholar]
- 143.Jenuwein T, Allis CD. Science. 2001;293:1074. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 144.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL. Nature. 2011;480:254. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kellie JF, Catherman AD, Durbin KR, Tran JC, Tipton JD, Norris JL, Witkowski CE, 2nd, Thomas PM, Kelleher NL. Anal Chem. 2012;84:209. doi: 10.1021/ac202384v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ahlf DR, Compton PD, Tran JC, Early BP, Thomas PM, Kelleher NL. J Proteome Res. 2012;11:4308. doi: 10.1021/pr3004216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wu C, Tran JC, Zamdborg L, Durbin KR, Li M, Ahlf DR, Early BP, Thomas PM, Sweedler JV, Kelleher NL. Nat Methods. 2012;9:822. doi: 10.1038/nmeth.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Klockenbusch C, O'Hara JE, Kast J. Anal Bioanal Chem. 2012;404:1057. doi: 10.1007/s00216-012-6065-9. [DOI] [PubMed] [Google Scholar]
- 149.Chan JN, Vuckovic D, Sleno L, Olsen JB, Pogoutse O, Havugimana P, Hewel JA, Bajaj N, Wang Y, Musteata MF, Nislow C, Emili A. Mol Cell Proteomics. 2012;11:M111 016642. doi: 10.1074/mcp.M111.016642. [DOI] [PMC free article] [PubMed] [Google Scholar]