

Abstract
A high-fat diet is a major risk factor for atherosclerosis. We conducted a longitudinal investigation to determine whether vascular endothelial senescence is involved in the mechanism by which a high-fat diet promotes atherogenesis. We challenged 10 baboons (Papio sp.) with a high-cholesterol high-fat (HCHF) diet for 7 weeks. In addition to multiple changes in plasma lipid profiles, inflammatory status, and endothelial functions in each individual, we found that levels of total serum cholesterol (TSC) and monocyte chemotactic protein-1 (MCP-1) were negatively and significantly correlated with endothelial nitric oxide synthase (eNOS) levels in endothelial cells while the levels of tumor necrosis factor α (TNF-α) and interleukin 6 (IL-6) were significantly correlated with nitric oxide levels in plasma within this time window. Most important, we observed that senescence-associated β-galactosidase (SA-β-gal) activities in endothelial cells harvested at 7 weeks after initiation of HCHF diet were significantly elevated by comparison with cells isolated from the same animals prior to dietary challenge. The SA-β-gal activities correlated significantly with the elevations of TSC, LDL-cholesterol, HDL-cholesterol, and IL-8 after 7 weeks of HCHF diet and with the changes of TSC and TNF-α levels after 3 weeks of HCHF diet. Our data indicate that the HCHF diet caused hyperlipidemia and prominent inflammation, which subsequently will cause endothelial dysfunction and promote senescence. The present study is the first to demonstrate the sequential and interactive changes as a consequence of an HCHF dietary challenge and establish a potential mechanism underlying the etiology of diet-induced atherogenesis in a nonhuman primate.
Keywords: High-cholesterol high-fat diet, lipidemic profiling, inflammatory response, vascular endothelial senescence, atherosclerosis, in vivo animal model
Introduction
Atherosclerosis is the major cause of cardiovascular diseases in western countries [1,2]. Although many determinants of atherosclerosis are recognized, thorough and elaborate research on its etiology continues to be a high priority, because these determinants do not by themselves account for the reasons why only a fraction of individuals exposed to dietary and other environmental risk factors develop the conditions [3,4]. Epidemiological and clinical intervention studies have demonstrated that a high-fat diet damages the endothelium, which lines the inside surface of blood vessels [3,5,6]. Therefore, understanding how high-fat diet interacts with endothelium is of paramount importance.
Ingesting a high-fat, high cholesterol diet (HCHF) raises plasma cholesterol levels and alters vascular endothelial integrity and barrier function, enabling an abnormal amount of LDL to infiltrate the subendothelial layer and elicit either necrotic changes [7-9] or apoptotic manifestations [10-13]. Within the past decade, vascular senescence has been found to play a more important role in atherogenesis than previously thought. Two types of cellular senescence: replicative senescence resulting from finite division, and stress-induced senescence resulting from constant DNA damage and persistent mitogenic and stress stimulation [14,15], are closely associated with vascular senescence [16]. Arterial samples from humans with atherosclerotic lesions contain endothelial cells that exhibit morphological features of senescence [17,18]. The vascular cells undergoing atherosclerosis stain positively for senescent-associated β-galactosidase (SA-β-gal), an enzymatic marker for endothelial senescence [19-22]. Progressive cell senescence has been observed in regions of arteries susceptible to atherosclerosis in humans and animals, but not in nonatherosclerotic areas [23,24].
Few investigations have addressed directly the interrelationships among an HCHF diet, cellular senescence, and atherogenesis elicited by diet, especially in human subjects. We previously demonstrated that endothelial cells isolated from baboon femoral arteries after a 7-week HCHF dietary challenge exhibited a dramatic increase of senescence characteristics by comparison with cells isolated from the same animals prior to the dietary challenge and that the senescence was independent of telomere shortening [25]. It is not clear whether this is a surrogate phenomenon or if the diet is a direct causative factor; the mechanisms that underlie these changes are not known. While many other investigators also have observed that a high-fat diet is highly associated with endothelial dysfunction and stress [26-28], it is important to determine whether endothelial dysfunction becomes a trigger that relates to senescence. This unsolved question is particularly important for depicting how HCHF dietary challenge causes atherosclerosis.
We hypothesize that a high-fat diet induces blood vessel senescence, which consequently contributes to endothelial damage. By using an established nonhuman primate model, we determined the changes in three compartments including lipid profile, inflammatory status, and endothelial dysfunction and senescence during the dietary challenge [25,29-31]. In the present study, we evaluated the interrelationship among these variables and identified two factors that were involved in endothelial senescence in baboons fed the HCHF diet: 1) total serum cholesterol (TSC) levels were found to be correlated with decreased endothelial nitric oxide synthase (eNOS) content and with elevated SA-β-gal activities in endothelial cells; and 2) increased levels of tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6) were present in the circulation at week 3 after dietary challenge, and the change in TNF-α apparently mediated prominent endothelial senescence observed at 7 weeks after feeding HCHF diet. Therefore, it is likely that high-fat diet causes endothelial senescence by directly creating dysfunction of endothelial cells and by indirectly provoking a systemic proinflammatory status in the circulation. Our results illuminate how an HCHF diet is involved in atherogenesis in a nonhuman primate model, helping to explain the path by which an individual’s susceptibility to atherosclerosis is determined.
Materials and methods
Study design for HCHF dietary challenge
This study reanalyzed the samples that had been collected previously [25], when we challenged the subjects with HCHF diet for 7 weeks. Blood samples were taken at 0, 3, and 7 weeks of the challenge to measure the lipid profiles, cytokines, and other circulating molecules related to cardiovascular disease (CVD). Endothelial cells were collected via femoral artery biopsy at 0 weeks (left side) and 7 weeks (right side) of dietary challenge. In contrast with previous investigations, we conducted a longitudinal comparison of both circulating factors and blood vessels under the influence of the atherogenic diet to robustly delineate how HCHF diet damages vascular endothelium.
Animals and sample preparations
Ten adult baboons (six males and four females, ages 7–15 years, mean=10.8 years) were enrolled in this study; they had been classified as high responders because their serum LDL-cholesterol concentrations increased substantially in response to an HCHF diet [32]. All baboons used in this study were maintained at the Southwest National Primate Research Center. They were housed in outdoor group caging and, except during the dietary challenge, were fed chow ad libitium (Harlan Teklad SWF Primate Diet, Madison, WI), supplemented with seeds and corn. The composition of the HCHF diet has been described in previous reports [25,29-31]. Arteries were biopsied by experienced veterinarians as described previously and endothelial cells were isolated from the subjects [31,33]. Blood samples for plasma were drawn aseptically into the tubes containing EDTA. All samples were centrifuged at 3000 rpm for 30 minutes. Plasma was aliquoted and stored at -80°C. The Texas Biomedical Research Institute Institutional Animal Care and Use Committee approved the study.
Measurements of plasma lipid profiles and CVD-related biomarkers
The lipid profiles included TSC levels, which were measured with Roche-Diagnostics standards and kits (Roche, IN), and triglycerides, measured with Liquicolor (Stanbio, TX). Lipoproteins containing Apo B were precipitated using heparin manganese (Diasorin, MN); HDL-C was measured in the supernatant. LDL-C was calculated by subtracting HDL-C from TSC. High sensitivity C-reactive protein (CRP) concentrations were measured with a commercial kit (Kamiya Biomedical, WA) using a latex particle immunoturbiometric method. We determined oxidized LDL concentrations with an ELISA kit (ALPCO Diagnostics, NH).
Measurements of plasma cytokines by Luminex
The human cytokine Lincoplex kit (Linco Research Inc, MO) was used to measure plasma IL-6, IL-8, MCP-1 and TNF-α levels according to the manufacturer’s instructions. RANTES was determined using the Lincoplex kit (Linco Research Inc, MO). Bead signals were analyzed on a Luminex 100IS instrument. The between-assay coefficient of variation for the control products in these immunoassays ranged from 5.3% to 11.1% for IL-6, IL-8, MCP-1, and TNF-α and measured 1.6% for RANTES.
Measurements of eNOS and nitric oxide in cell lysates and plasma
We measured eNOS concentrations in cell lysates with an ELISA kit (R&D Systems, MN); their concentrations were normalized by protein contents in cell lysates with the use of Lowry reagents (Sigma, MO). We measured nitric oxide in plasma by a colorimetric assay using a kit (R&D Systems, MN).
Cytochemical staining and quantification for SA-β-gal activity
Endothelial cells were seeded in 8-well Lab-Tek culture slides. At the end of the experiment, cell staining was conducted using a kit (Cell Signaling, CA). Quantitative analysis of enzymatic activity, as defined by the positively stained area in histochemistry slides, was performed using the software Image-Pro 4.5.1 (MediaCybernetics, MD). Using a 40x objective lens, we randomly selected 150 cells per slide, captured the specific color that was identical to the products of SA-β-gal, and used the area measurement option provided by the software program to count area sizes and objective intensities. SA-β-gal activities before and after dietary challenge were compared statistically using the method described below.
Statistical analysis
Data were expressed as mean±SD and represented the results of at least three independent experiments with duplicate sets for each run. The statistical analysis of data between experimental groups was performed by Spearman coefficient (ρ values) and by paired Student’s test using statistical software JMP 5.1 (JMP, SAS, NC) [34]. Differences between groups were considered significant at p<0.05.
Results
Systemic and interactive changes in response to HCHF diet challenge
During 7 weeks of dietary challenge, we documented the changes in multiple factors in circulation and endothelial functionalities in cells obtained from femoral arteries. In peripheral blood, we found that HCHF diet caused lipid increases in all ten baboons by comparison to basal level (0 weeks). Levels of TSC (Figure 1A), LDL-C (Figure 1B), triglycerides (Figure 1C), apolipoprotein B (Apo B, Figure 1D), and HDL-C (Figure 1E) were elevated 3 weeks after initiation of HCHF diet and held steady over the remaining course of the 7-week challenge. Oxidized LDL was increased at 3 weeks, but not significantly (Figure 2A). Next, we analyzed inflammatory status after dietary challenge. Levels of proinflammatory cytokines TNF-α (Figure 1F) and IL-6 (Figure 1G) increased significantly after 3 weeks of HCHF diet but were negligible after 7 weeks of diet. Both TNF-α and IL-6 decreased significantly between 3 and 7 weeks after dietary challenge. Levels of chemokines including RANTES (Figure 1H), IL-8 (Figure 1I), and MCP-1 (Figure 1J) increased after 3 weeks of dietary challenge and remained high at 7 weeks; significant elevations were seen in IL-8 and MCP-1 only at 7 weeks. C-reactive protein (CRP) levels did not vary significantly during the dietary challenge but exhibited greater interindividual variation at baseline. This variation diminished with the progression of dietary challenge (Figure 2B). We saw dramatic endothelial dysfunction during the dietary challenge, as shown in eNOS content (Figure 1K) and nitric oxide in plasma (Figure 1L). We observed a sharp and significant decline in eNOS content at 0 weeks and at 7 weeks after HCHF diet (no samples were collected at week 3). We also observed a gradual decrease in nitric oxide concentration in plasma during the 7-week time frame, which corresponded to intracellular eNOS decline. It is noteworthy that interindividual variations of many determinants, as mentioned above, were elevated at 3 weeks after HCHF diet; the variances became gr-eater after 7 weeks, as shown in Table 1.
Figure 1.

Effect of HCHF diet on lipid profiles and endothelial dysfunctionalities. TSC showed significant increases at 3 and 7 weeks by comparison to 0 weeks. Significant increase of LDL-C level was seen at 7 weeks; Apo B was significantly increased only at 3 weeks. Significant increases of HDL-C were observed at 3 and 7 weeks while no statistically change was seen at 3 and 7 weeks. TNF-α and IL-6 were significantly elevated after 3 weeks of dietary challenge and declined to negligible levels at 7 weeks. RANTES, IL-8, and MCP-1 in plasma remained elevated; eNOS contents were determined in cell lysates at 0 and 7 weeks after dietary challenge; and nitric oxide was determined in plasma at 0, 3, and 7 weeks after dietary challenge. The eNOS expression was significantly down-regulated to a great extent by dietary challenge. Significant nitric oxide decrease was observed between 0 and 7 weeks after dietary challenge, while no change was seen between 0 and 3 weeks after dietary challenge. Paired Student’s t-test was used to compare means between 0 and either 3 or 7 weeks after dietary challenge. *p<0.05, **p<0.01, and ***p<0.001.
Figure 2.
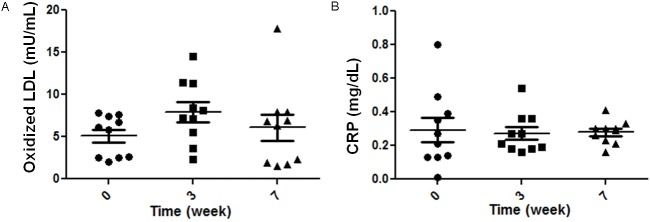
Oxidized LDL and CRP changes in plasma during dietary challenge. Oxidized LDL (Panel A) and CRP (Panel B) were shown during HCHF dietary challenge. No statistical significance was found in either oxidized LDL or CRP during 7 weeks. However, larger inter-individual variations were seen at 0 weeks by comparison with 7 weeks after dietary challenge. Data were shown in mean±SD as they are marked with black lines; significances were analyzed by paired Student’s t-test; n=10.
Table 1.
Means and variances of lipid profiles, CVD-related biomarkers, and cytokines in response to HCHF diet (n=10)
| 0 weeks | 3 weeks | 7 weeks | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Mean | Variance | Mean | Variance | Mean | Variance | |
| TSC (mg/mL) | 102 | 423 | 190 | 2987 | 209 | 4728 |
| LDL-C (mg/mL) | 43 | 241 | 96 | 3344 | 113 | 7175 |
| Triglycerides (mg/mL) | 47 | 156 | 57 | 389 | 49 | 619 |
| Apo B (mg/mL) | 31 | 153 | 54 | 689 | 50 | 1475 |
| HDL-C (mg/mL) | 59 | 160 | 94 | 529 | 97 | 657 |
| Oxidized LDL (mU/L, x104) | 5 | 6 | 8 | 16 | 6 | 24 |
| CRP (mg/mL) | 0.3 | 0.05 | 0.27 | 0.01 | 0.28 | 0 |
| RANTES (pg/mL) | 116 | 15269 | 235 | 8236 | 208 | 31126 |
| IL-6 (pg/mL) | 16 | 44 | 48 | 172 | 1 | 1 |
| IL-8 (pg/mL) | 131 | 12672 | 261 | 17531 | 287 | 56266 |
| MCP-1 (pg/mL) | 31 | 45 | 41 | 211 | 52 | 182 |
| TNF-α (pg/mL) | 8 | 5 | 16 | 79 | 1 | 1 |
Correlations of pathophysiological responses induced by HCHF diet challenge
To reveal the systemic and interactive events involved in the HCHF-diet-induced pathological process, we used pairwise and nonparametric analysis to study the relationships among multiple variables and identified several significant correlations, as listed in Table 2. Among these positive correlations, some were expected, such as levels of TSC and Apo B (p<0.001 at 3 weeks, p<0.01 at 7 weeks); TSC and HDL-C (p<0.001 at 3 weeks, p<0.01 at 7 weeks, data not shown); Apo B and oxidized LDL (p<0.001 at 3 weeks, p<0.001 at 7 weeks); TNF-α and IL-6 (p<0.001 at both 3 weeks and 7 weeks); and multiple significant correlations among cytokines and chemokines. When we investigated correlations of inflammatory reactions with lipid-related molecules, we found that TSC levels did not significantly correlate with TNF-α at either 3 weeks or 7 weeks, but a significant correlation existed at 3 weeks for IL-6 (p<0.05). However, TSC levels were significantly correlated with three chemokines determined at both 3 and 7 weeks (RANTES, IL-8, and MCP-1). Apo B levels after 3 weeks were significantly correlated with both TNF-α and IL-6; their correlations disappeared at 7 weeks. Other correlations, such as levels of Apo B and oxidized LDL and oxidized LDL, TNF-α, and IL-6 were also high. The decline of eNOS content in endothelial cells was negatively and significantly correlated with TSC levels (Table 3, Spearman ρ=-0.6549, p<0.01, n=8). MCP-1 was also highly correlated with eNOS content (Table 3, Spearman ρ=-0.7549, p<0.001, n=8). Reduced nitric oxide levels at 7 weeks were associated with increased levels of proinflammatory cytokines TNF-α and IL-6. These data imply that ingesting a high-fat diet results in an interactive pathological alteration in circulation, which is capable of causing endothelial cells to be dysfunctional.
Table 2.
Multivariate analysis of correlations between lipid profiles and cytokines during HCHF dietary challenge
| 3 weeks | 7 weeks | ||||
|---|---|---|---|---|---|
|
|
|||||
| Variable | By variable | Spearman ρ | p values | Spearman ρ | p values |
| TSC | Apo B | 0.7703 | <0.0001 | 0.5575 | 0.0014 |
| TSC | Oxidized LDL | 0.5370 | 0.0148 | 0.3115 | 0.0938 |
| TSC | RANTES | 0.5862 | 0.0067 | 0.4738 | 0.0082 |
| TSC | IL-8 | 0.6363 | 0.0026 | 0.4479 | 0.0131 |
| TSC | MCP-1 | 0.5235 | 0.0178 | 0.5236 | 0.0030 |
| TSC | IL-6 | 0.4934 | 0.0271 | -0.0571 | 0.7645 |
| TSC | TNF-α | 0.3449 | 0.1364 | -0.0969 | 0.6105 |
| Apo B | Oxidized LDL | 0.7068 | 0.0005 | 0.8016 | <0.0001 |
| Apo B | RANTES | 0.5412 | 0.0137 | 0.6166 | 0.0003 |
| Apo B | IL-8 | 0.6202 | 0.0035 | 0.5843 | 0.0007 |
| Apo B | MCP-1 | 0.4712 | 0.0360 | 0.1969 | 0.2970 |
| Apo B | IL-6 | 0.5415 | 0.0137 | 0.2569 | 0.1706 |
| Apo B | TNF-α | 0.4731 | 0.0351 | 0.2203 | 0.2420 |
| Oxidized LDL | RANTES | 0.4421 | 0.0510 | 0.5162 | 0.0035 |
| Oxidized LDL | IL-8 | 0.4135 | 0.0699 | 0.4296 | 0.0178 |
| Oxidized LDL | MCP-1 | 0.5008 | 0.0245 | 0.1604 | 0.3972 |
| Oxidized LDL | IL-6 | 0.7212 | 0.0003 | 0.4877 | 0.0063 |
| Oxidized LDL | TNF-α | 0.5424 | 0.0135 | 0.4136 | 0.0231 |
| IL-6 | RANTES | 0.4606 | 0.0410 | 0.2202 | 0.2422 |
| IL-6 | IL-8 | 0.3909 | 0.0883 | 0.1253 | 0.5094 |
| IL-6 | MCP-1 | 0.7909 | <0.0001 | 0.0663 | 0.7276 |
| IL-6 | TNF-α | 0.7871 | <0.0001 | 0.8699 | <0.0001 |
| TNF-α | RANTES | 0.5805 | 0.0073 | 0.3279 | 0.0769 |
| TNF-α | IL-8 | 0.1465 | 0.5378 | -0.0167 | 0.9303 |
| TNF-α | MCP-1 | 0.8788 | <0.0001 | 0.1319 | 0.4872 |
| IL-8 | RANTES | 0.4045 | 0.0769 | 0.3280 | 0.0768 |
| MCP-1 | RANTES | 0.6511 | 0.0019 | 0.3518 | 0.0566 |
| MCP-1 | IL-8 | 0.2496 | 0.2885 | 0.0661 | 0.7287 |
Table 3.
Multivariate analysis of some CVD-related factors correlated to endothelial dysfunctional alterations during HCHF dietary challenge
| 3 weeks | 7 weeks | ||||
|---|---|---|---|---|---|
|
|
|||||
| Variable | By variable | Spearman ρ | p values | Spearman ρ | p values |
| TSC | Nitric Oxide | 0.0150 | 0.9500 | -0.2919 | 0.1244 |
| eNOS | NA | NA | -0.6549 | 0.0059 | |
| Apo B | Nitric Oxide | 0.3555 | 0.1240 | 0.1923 | 0.3175 |
| eNOS | NA | NA | -0.1520 | 0.5741 | |
| TNF-α | Nitric Oxide | 0.0652 | 0.7848 | 0.4347 | 0.0185 |
| eNOS | NA | NA | 0.2959 | 0.2659 | |
| IL-6 | Nitric Oxide | 0.0056 | 0.9814 | 0.4519 | 0.0138 |
| eNOS | NA | NA | 0.4752 | 0.0629 | |
| IL-8 | Nitric Oxide | -0.1845 | 0.4363 | -0.1701 | 0.3775 |
| eNOS | NA | NA | 0.3723 | 0.1556 | |
| MCP-1 | Nitric Oxide | -0.0552 | 0.8173 | -0.3029 | 0.1102 |
| eNOS | NA | NA | -0.7594 | 0.0006 | |
| eNOS | Nitric Oxide | NA | NA | 0.5800 | 0.0234 |
NA, not available; no endothelial cultures were harvested at this time point.
Endothelial senescence induced by HCHF diet and potential factors contributing to the changes
To test whether HCHF diet could induce premature senescence of vascular endothelium, we compared cell growth and senescent marker cells obtained from the same subjects prior to HCHF diet. We observed their growth status, an alternative parameter of cellular proliferative potential (Table 4). We noticed that the endothelial cells obtained from animal ID#11017 after it had been fed HCHF diet could not be passaged because they did not attach firmly to culture dishes and were lost during medium change. Some cells from animal ID#8272 were attached to the culture dish but did not survive after passage. Endothelial cells from two other animals (ID#13676 and ID#10041) did not grow well in vitro after dietary challenge. Because these anomalies do not occur often with cultures derived from baboons fed a basal diet, these data imply that HCHF diet caused serious damage to the endothelium. Then, we quantified cellular activities of SA-β-gal, a comprehensive marker for cell senescence, before and after HCHF diet (Figure 3). Significantly increased SA-β-gal activity was seen in cells after HCHF dietary challenge, indicating that a high-fat dietary challenge resulted in endothelial senescence. In order to determine which factors are relevant to endothelial senescence as a result of HCHF diet, we investigated the correlations between circulating determinants and endothelial changes throughout the treatment. We found a significant association of plasma TSC, LDL-C, and HDL-C levels 7 weeks after dietary challenge with SA-β-gal activities in endothelial cells, but the association with the levels of Apo B was not statistically significant (Table 5). TSC levels were negatively and significantly associated with eNOS contents in endothelial cells harvested after dietary challenge, indicating that elevated TSC levels may have impacted intracellular eNOS content and led to dysfunctional endothelial performance. When we assessed correlations of plasma TNF-α and IL-6 levels after HCHF dietary challenge with SA-β-gal activities in endothelial cells, we noted that a time-dependent pathological process was involved in diet-induced responses. TNF-α and IL-6 levels after 7 weeks on HCHF diet did not relate significantly to endothelial senescence, possibly because the concentrations of these substances were too low to be accurately measured. However, when we correlated TNF-α and IL-6 levels after 3 weeks on HCHF diet to SA-β-gal activities after 7 weeks of dietary challenge, we found that TNF-α, but not IL-6, had a positive association with SA-β-gal activities in endothelial cells harvested at 7 weeks (Table 5).
Table 4.
Endothelial growth rate at baseline and 7 weeks after HCHF diet
| Animal ID | 0 Weeks | 7 Weeks |
|---|---|---|
| 11017 | ++++ | – |
| 13676 | ++++ | + |
| 10041 | ++++ | + |
| 12345 | ++++ | ++++ |
| 11850 | ++++ | ++++ |
| 14811 | ++++ | ++++ |
| 14705 | ++ | ++ |
| 8272 | ++++ | ± |
| 14704 | ++ | ++ |
| 14818 | ++++ | ++++ |
Endothelial growth rate was estimated based on the number of passage times in vitro under standard culture conditions. ++++, indicates that ECs had highly proliferative ability and were able to be passaged from 6-9 times; ++, indicates moderate proliferative ability and ability to be passaged from 3-5 times; +, indicates poorly proliferative ability with capacity of one or two passages; ±, indicated that no cells survived after first passage; –, indicated no cells were harvested at the end of culture.
Figure 3.
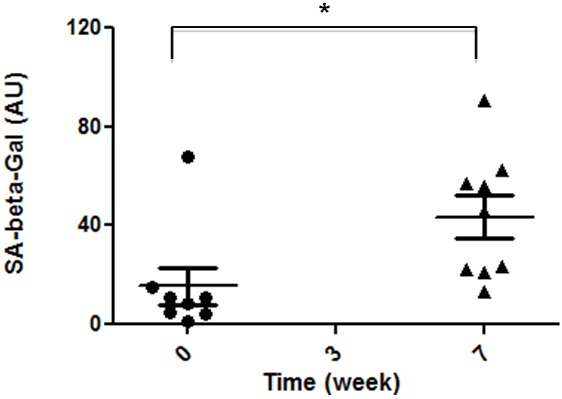
Effects of HCHF dietary challenge on SA-β-gal activities in endothelial cultures. Endothelial cells were harvested before and after dietary challenge and cultured on chamber slides. Cell staining and quantitative analysis of enzymatic activities are described in the section Materials and Methods. Results of SA-β-gal activities at 0 and 7 weeks after dietary challenge were compared; statistical significance existed between them (p<0.05, n=8).
Table 5.
Correlation of SA-β-gal activities after 3 and 7 weeks of HCHF dietary challenge with CVD-related factors measured 3 weeks of dietary challenge
| Variables | Between 0 and 3 weeks | Between 0 and 7 weeks | ||
|---|---|---|---|---|
|
| ||||
| Spearman ρ | p values | Spearman ρ | p values | |
| TSC | 0.754 | 0.005 | 0.615 | 0.011 |
| LDL-C | -0.108 | 0.799 | 0.668 | 0.005 |
| HDL-C | -0.619 | 0.102 | 0.509 | 0.044 |
| Apo B | 0.402 | 0.195 | 0.402 | 0.065 |
| Oxidized LDL | -0.667 | 0.071 | 0.193 | 0.474 |
| CRP | -0.371 | 0.227 | -0.371 | 0.227 |
| TNF-α | 0.669 | 0.002 | -0.337 | 0.280 |
| IL-6 | 0.505 | 0.093 | -0.418 | 0.170 |
| MCP-1 | 0.535 | 0.073 | 0.246 | 0.359 |
| RANTES | 0.334 | 0.288 | 0.229 | 0.393 |
| IL-8 | 0.024 | 0.977 | 0.641 | 0.007 |
| eNOS | NA | NA | -0.127 | 0.732 |
NA, not available; no endothelial cultures were harvested at this time point.
Discussion
Elevated plasma cholesterol induced by a high-fat diet is a critical factor for atherogenesis; it not only results in the deposition of lipids in the atheromatous lesions but also contributes to the primary injury initiating atherogenesis [5-8]. Traditionally, it has been recognized that high levels of lipids in the circulation result in endothelial damage either by apoptosis or necrosis, which are probably the predominant cellular mechanisms underlying the pathogenesis of atherosclerosis [9,10,35]. During the past decade, accumulating evidence has implied that vascular senescence also contributes to atherosclerosis; that aging vasculature persists as the most significant risk factor [16]; and that multiple cardiovascular risk factors promote premature vascular senescence [36-39]. These observations have led to the emerging concept that “vascular age” rather than chronological age is the best predictor of cardiovascular disease [24,40]. Previously, we confirmed that HCHF diet diminished endothelial functions [31] and resulted in higher levels of SA-β-gal activities [25]. In this study, we investigated factors that were associated with endothelial senescence resulting from a short-term HCHF dietary challenge. The results suggest that a high-fat diet plays a major role in inducing endothelial injury that initiates atherogenesis.
The use of a large nonhuman primate model enables us to reveal how high fat diet causes vascular endothelial damage [25,31,33,41-43] because baboons and humans share similar characteristics of physiology and aging [44,45]. In addition, this model gives us experimental access to observe spatial- and temporal-dependent changes in three compartments that reflect the integrative nature of this pathological process. To our knowledge, this is the first experiment that details the variables involved in diet-induced vascular endothelial alteration and demonstrates their interactions. We found that an HCHF diet not only elevated levels of plasma lipid (Figure 1, panels A-E) but also resulted in gross systemic inflammatory responses (Figure 1, panels F-J). For many years, conflicting results have been presented in regard to inflammatory markers with risk of atherosclerosis [46-49]. In our longitudinal study, we defined the dynamics of molecules involved in systemic inflammation. The data illustrate the different roles of inflammatory cytokines in response to dietary challenge and provide an experimental basis for the up- and down-regulation of the inflammatory cytokine network of this disease. The findings have provided helpful insights into understanding the pathological processes in human subjects.
To determine which factors cause endothelial senescence as a result of HCHF diet, we investigated the correlations between circulating determinants and endothelial changes throughout the dietary challenge. First, we found a significant association of plasma TSC, LDL-C, and HDL-C levels 7 weeks after dietary challenge with SA-β-gal activities in endothelial cells, but the association with the levels of Apo B was not statistically significant (Table 5). That observation suggested that plasma cholesterol, either in LDL or in HDL, may be an important factor in the initiation of vascular endothelial senescence. Although the importance of LDL cholesterol in the development of atherosclerosis has long been recognized, the role of HDL cholesterol has not been considered to as great an extent. Our study indicated that both LDL-C and HDL-C levels were significantly associated with endothelial senescence resulting from HCHF diet. This finding may explain why many people remain at high risk of atherosclerosis even though therapeutic intervention has progressively reduced their LDL-C level.
To explore the mechanism underlying this scenario, we further observed that TSC levels were negatively and significantly associated with eNOS contents in endothelial cells harvested after dietary challenge, indicating that elevated TSC levels had impacted intracellular eNOS content and led to dysfunctional endothelial performance (Table 3). Therefore, we speculate that elevated plasma cholesterol mediates endothelial senescence via nitrosative stress, a condition resulting from low nitric oxide (NO) concentrations and thus that reduction of NO levels may be the first step in pathogenesis of atherosclerosis. Our results are consistent with previous in vitro studies that concluded that reduced bioavailability of endothelium-synthesized NO disrupts the regulation of vascular tone, structure, and function, which is the key mechanism mediating reduced endothelium-dependent dilation [36,50,51]. Dietary alterations regulate eNOS activity and NO synthesis in endothelial cells in rodents [52-56]. Here, we provide in vivo evidence supporting the concept that plasma cholesterol could directly promote endothelial dysfunction in a nonhuman primate. Our results also suggest that proinflammatory cytokines may induce endothelial senescence as a consequence of a high-fat diet. When we assessed correlations of plasma TNF-α and IL-6 levels after HCHF dietary challenge with SA-β-gal activities in endothelial cells, we noted a time-dependent pathological process involved in diet-induced responses. TNF-α and IL-6 levels after 7 weeks on HCHF diet did not relate significantly to endothelial senescence, possibly because the concentrations of these substances were too low to be accurately measured. However, when we correlated TNF-α and IL-6 levels after 3 weeks on HCHF diet to SA-β-gal activities after 7 weeks of dietary challenge, we found that TNF-α, but not IL-6, had a positive association with SA-β-gal activities in endothelial cells harvested at 7 weeks (Table 3). This result indicates that earlier proinflammatory stimuli resulting from HCHF diet might have consequences for endothelial damage, and TNF-α might well be responsible. This finding supports not only the concept that proinflammatory TNF-α is probably a significant factor driving endothelial senescence [15,24,40], at least under our experimental conditions but also suggests that TNF-α mediates diet-induced vascular injury. IL-6 is also a potent molecule that drives vascular senescence; elevated IL-6 in circulation is currently considered to be a prominent feature of aging [15,37]. However, the correlation in this small sample size was not significant (p=0.093).
The manner in which inflammatory networks cause cellular senescence has been debated for years [15,16,57]. It has not been determined whether inflammatory factors are causes or consequences of cellular senescence because inflammation is systemic and diffuse in nature. Because ours was a longitudinal study, we were able to investigate the impact of high-fat diet on inflammatory profiling at different time points and to identify time-dependent events that are potentially important in the development of cellular pathology. We demonstrated that proinflammatory cytokines IL-6 and TNF-α behaved differently from chemokines (MCP-1, RANTES, and IL-8) and that there are two phases in the appearance of inflammatory status. IL-6 and TNF-α were dramatically elevated in the early phase, from 0 to 3 weeks after diet, and then dropped to undetectable levels after 7 weeks of high-fat diet (Figure 1F and 1G). However, RANTES, IL-8, and MCP-1 remained at high levels beyond the first 3 weeks of dietary challenge (Figure 1H-J). Based on these observations, we propose here that consumption of a high-fat diet could initiate a proinflammatory status, as evidenced by IL-6 and TNF-α increases, which then triggers premature senescence of endothelial cells. The declines in circulating IL-6 and TNF-α in the second phase of the dietary challenge might be a result of senescence of immune cells that released these cytokines after an increased burst of free radicals upon high-fat dietary challenge [58]. The three chemokines that we studied were constantly released into circulation. The continuous increase of chemokines might be a result of release from activated inflammatory cells, and might also be caused in part by release from senescent cells, since numerous reports have indicated that secretion of these factors is a typical feature, termed senescence-associated secretory phenotype [15,57,59]. We found that only IL-8 levels at 7 weeks correlated significantly with SA-β-gal activities (p=0.007, n=8), but not at 3 weeks, of HCHF diet. Therefore, we conclude that proinflammatory responses resulting from dietary insult are the triggers of premature senescence and that the senescent cells originate chemokine production. Song and Goldstein reported that senescing induces a proinflammatory phenotype; senescent cells are also considered a source of chronic inflammation that is associated with many age-related pathophysiological processes and diseases [15,60].
Based on our results, we propose a model that describes how high-fat diet induces vascular endothelial senescence in baboons, as shown in Figure 4. Elevated total cholesterol depletes NO availability, and Apo B and/or oxidized LDL elicits a high proinflammatory status; both propel the senescence process. We propose that individual variation in these factors and interactions among them in response to high-fat dietary challenge are responsible for some of the inter-individual differences in susceptibility to atherogenic pathogenesis.
Figure 4.
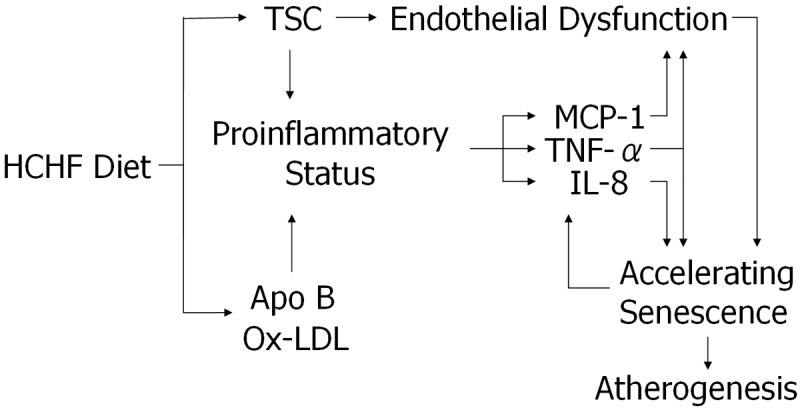
Model of factors that regulate endothelial senescence by HCHF. Based on the associations, we suggest that endothelial senescence is a dominant endothelial damage mechanism in response to HCHF diet. Our data suggest that an HCHF diet initiates the sequential and interactive changes, which eventually cause endothelial senescence. We hypothesize that elevated TSC levels at both 3 and 7 weeks of dietary challenge cause endothelial senescence. TNF-α levels at 3 weeks of dietary challenge also are closely related to vascular endothelial senescence. TSC and Apo B may exert differently in inducing endothelial senescence under our hypothesis, elevated TSC levels devastate endothelial ability to synthesize NO and result in endothelial dysfunction; while increased Apo B levels, accompanying by increased oxidized LDL, seem to provoke an inflammatory scenario that initiates and accelerates endothelial senescence. Therefore, we conclude that high-fat diet causes endothelial senescence via multiple pathways. Vascular endothelial senescence may be the underlying mechanism of endothelial damage that triggers the pathogenesis of diet-induced atherosclerosis.
The scope of this study imposes several limitations. The power to examine associations with variables characterized during the HCHF dietary challenge needs improvement; expanding the sample size will almost certainly reveal additional significant relationships that were not detected in this study as a result of larger interindividual variations. Future research with a larger sample size would be expected to confirm our findings in a robust manner and explain some of the conflicting findings in this field [15,40]. Interestingly, some well-known CVD-related risk factors, such as Apo B, oxidized LDL, and CRP, did not exert significant roles in diet-induced vascular damage in our model, although we found oxidized LDL to be strongly correlated to MCP-1, IL-6, and TNF-α (Table 2), indicating that they might have an indirect role or inappropriate time points of sampling. Two additional notable limitations of this study are 1) that we were not able to measure other senescent markers besides SA-β-gal activities in this study and 2) that the activities were measured in isolated endothelial cells. The growth status of endothelial cells was observational. We plan future investigations in which we will use blood vessels in situ and examine both p16INK-pRb-β-gal and p53-pRb-β-gal signaling pathway activations to detail the molecular nature of this process. Finally, although we concluded that the high-fat dietary challenge stressed endothelial cells and caused premature senescence, whether these senescent cells become focal loci that develop into atherosclerotic lesion remains to be determined.
Despite these remaining questions, our results clearly established that 1) even a short-term exposure (7 weeks) to a high-fat diet can have devastating pathological effects on vascular endothelial cells, and 2) physiological responses during the first 3 weeks of high-fat diet may have lasting effects that are manifested in endothelial cells at a later time. These conclusions significantly impact the traditional perception that a high-fat diet is detrimental only if exposure to it is of long duration. They also have important implications for the design of experiments aimed at understanding the progression of events that lead to diet-induced atherogenesis. Assessment of physiological effects of diet after 3 weeks of exposure may be essential to understanding mechanisms of atherogenesis as well as individual variation in extent and severity of atherosclerosis years after the initiation of the dietary challenge.
Acknowledgements
This study was supported by the National Institutes of Health (grants P01 HL028972; NIH grants P51 RR013986, C06 RR016228, and C06 RR017332 provided infrastructure support for the research). Qiang Shi designed the study, collected endothelial cells, conducted functional analyses, analyzed data, and wrote the first draft of the manuscript; Peter J. Hornsby contributed to the development of hypotheses and interpretation of data; Qinghe Meng contributed to statistical analysis; Jane F. VandeBerg conducted lipid profiling and CVD-related biomarker determinations; and John L. VandeBerg contributed to the experimental design, interpretation of data, and manuscript writing. The authors thank Michelle M. Leland, Catherine Jett, and Linda Talley for the blood collections, biopsies, and cytokine measurements.
Disclosure of conflict of interest
None.
References
- 1.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 2.Binder CJ, Chang MK, Shaw PX, Miller YI, Hartvigsen K, Dewan A, Witztum JL. Innate and acquired immunity in atherogenesis. Nat Med. 2002;8:1218–1226. doi: 10.1038/nm1102-1218. [DOI] [PubMed] [Google Scholar]
- 3.Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–2051. doi: 10.1161/ATVBAHA.108.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weiss JN, Karma A, MacLellan WR, Deng M, Rau CD, Rees CM, Wang J, Wisniewski N, Eskin E, Horvath S, Qu Z, Wang Y, Lusis AJ. “Good enough solutions” and the genetics of complex diseases. Circ Res. 2012;111:493–504. doi: 10.1161/CIRCRESAHA.112.269084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ross R, Harker L. Hyperlipidemia and atherosclerosis. Science. 1976;193:1094–1100. doi: 10.1126/science.822515. [DOI] [PubMed] [Google Scholar]
- 6.Ross R. Cell biology of atherosclerosis. Annu Rev Physiol. 1995;57:791–804. doi: 10.1146/annurev.ph.57.030195.004043. [DOI] [PubMed] [Google Scholar]
- 7.Bocan TM, Mueller SB, Uhlendorf PD, Ferguson E, Newton RS. Dietary and mechanically induced rabbit iliac-femoral atherosclerotic lesions: a chemical and morphologic evaluation. Exp Mol Pathol. 1991;54:201–277. doi: 10.1016/0014-4800(91)90031-r. [DOI] [PubMed] [Google Scholar]
- 8.Davenport WD Jr, Ball CR. Diet-induced atrial endothelial damage--a scanning electron-microscopic study. Atherosclerosis. 1981;40:145–152. doi: 10.1016/0021-9150(81)90032-0. [DOI] [PubMed] [Google Scholar]
- 9.Mitchinson MJ, Hardwick SJ, Bennett MR. Cell death in atherosclerotic plaques. Curr Opin Lipidol. 1996;7:324–329. doi: 10.1097/00041433-199610000-00011. [DOI] [PubMed] [Google Scholar]
- 10.Bennett MR, Evan GI, Schwartz SM. Apoptosis of human vascular smooth muscle cells derived from normal vessels and coronary atherosclerotic plaques. J Clin Invest. 1995;95:2266–2274. doi: 10.1172/JCI117917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Patschan S, Goligorsky MS. Stress-induced premature senescence of endothelial cells. J Nephrol. 2008;21:337–344. [PubMed] [Google Scholar]
- 12.Davis N, Katz S, Wylie-Rosett J. The effect of diet on endothelial function. Cardiol Rev. 2007;15:62–66. doi: 10.1097/01.crd.0000218824.79018.cd. [DOI] [PubMed] [Google Scholar]
- 13.Mallat Z, Tedgui A. Apoptosis in the vasculature: mechanisms and functional importance. Br J Pharmacol. 2000;130:947–962. doi: 10.1038/sj.bjp.0703407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Bio. 2005;37:961–976. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 15.Freund A, Orjalo AV, Desprez PY, Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med. 2010;16:238–246. doi: 10.1016/j.molmed.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. 2012;111:245–259. doi: 10.1161/CIRCRESAHA.111.261388. [DOI] [PubMed] [Google Scholar]
- 17.Ross R. The pathogenesis of atherosclerosis--an update. N Engl J Med. 1986;314:488–500. doi: 10.1056/NEJM198602203140806. [DOI] [PubMed] [Google Scholar]
- 18.Burrig KF. The endothelium of advanced arteriosclerotic plaques in humans. Arterioscler Thromb. 1991;11:1678–1689. [PubMed] [Google Scholar]
- 19.Kumazaki T, Kobayashi M, Mitsui Y. Enhanced expression of fibronectin during in vivo cellular aging of human vascular endothelial cells and skin fibroblasts. Exp Cell Res. 1993;205:396–402. doi: 10.1006/excr.1993.1103. [DOI] [PubMed] [Google Scholar]
- 20.Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000;113:3613–3622. doi: 10.1242/jcs.113.20.3613. [DOI] [PubMed] [Google Scholar]
- 21.Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res. 2007;100:15–26. doi: 10.1161/01.RES.0000256837.40544.4a. [DOI] [PubMed] [Google Scholar]
- 22.Erusalimsky JD, Kurz DJ. Endothelial cell senescence. Handb Exp Pharmacol. 2006:213–248. doi: 10.1007/3-540-36028-x_7. [DOI] [PubMed] [Google Scholar]
- 23.Erusalimsky JD, Kurz DJ. Cellular senescence in vivo: its relevance in ageing and cardiovascular disease. Exp Gerontol. 2005;40:634–642. doi: 10.1016/j.exger.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 24.Erusalimsky JD. Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol. 2009;106:326–332. doi: 10.1152/japplphysiol.91353.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi Q, Hubbard GB, Kushwaha RS, Rainwater D, Thomas CA 3rd, Leland MM, Vandeberg JL, Wang XL. Endothelial senescence after high-cholesterol, high-fat diet challenge in baboons. Am J Physiol Heart Circ Physiol. 2007;292:H2913–H2920. doi: 10.1152/ajpheart.01405.2006. [DOI] [PubMed] [Google Scholar]
- 26.Hennig B, Toborek M, McClain CJ, Diana JN. Nutritional implications in vascular endothelial cell metabolism. J Am Coll Nutr. 1996;15:345–358. doi: 10.1080/07315724.1996.10718609. [DOI] [PubMed] [Google Scholar]
- 27.Hennig B, Toborek M, McClain CJ. High-energy diets, fatty acids and endothelial cell function: implications for atherosclerosis. J Am Coll Nutr. 2001;20:97–105. doi: 10.1080/07315724.2001.10719021. [DOI] [PubMed] [Google Scholar]
- 28.Jebelovszki E, Kiraly C, Erdei N, Feher A, Pasztor ET, Rutkai I, Forster T, Edes I, Koller A, Bagi Z. High-fat diet-induced obesity leads to increased NO sensitivity of rat coronary arterioles: role of soluble guanylate cyclase activation. Am J Physiol Heart Circ Physiol. 2008;294:H2558–H2564. doi: 10.1152/ajpheart.01198.2007. [DOI] [PubMed] [Google Scholar]
- 29.Singh AT, Rainwater DL, Kammerer CM, Sharp RM, Poushesh M, Shelledy WR, VandeBerg JL. Dietary and genetic effects on LDL size measures in baboons. Arterioscler Thromb Vasc Biol. 1996;16:1448–1453. doi: 10.1161/01.atv.16.12.1448. [DOI] [PubMed] [Google Scholar]
- 30.Rainwater DL, Kammerer CM, Carey KD, Dyke B, VandeBerg JF, Shelledy WR, Moore PH Jr, Mahaney MC, McGill HC Jr, VandeBerg JL. Genetic determination of HDL variation and response to diet in baboons. Atherosclerosis. 2002;161:335–343. doi: 10.1016/s0021-9150(01)00658-x. [DOI] [PubMed] [Google Scholar]
- 31.Shi Q, VandeBerg JF, Jett C, Rice K, Leland MM, Talley L, Kushwaha RS, Rainwater DL, Vandeberg JL, Wang XL. Arterial endothelial dysfunction in baboons fed a high-cholesterol, high-fat diet. Am J Clin Nutr. 2005;82:751–759. doi: 10.1093/ajcn/82.4.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rainwater DL, Kammerer CM, Mahaney MC, Rogers J, Cox LA, Schneider JL, VandeBerg JL. Localization of genes that control LDL size fractions in baboons. Atherosclerosis. 2003;168:15–22. doi: 10.1016/s0021-9150(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 33.Shi Q, Wang J, Wang XL, VandeBerg JL. Comparative analysis of vascular endothelial cell activation by TNF-alpha and LPS in humans and baboons. Cell Biochem Biophys. 2004;40:289–303. doi: 10.1385/CBB:40:3:289. [DOI] [PubMed] [Google Scholar]
- 34.Kroenke CH, Pletcher MJ, Lin J, Blackburn E, Adler N, Matthews K, Epel E. Telomerase, telomere length, and coronary artery calcium in black and white men in the CARDIA study. Atherosclerosis. 2012;220:506–512. doi: 10.1016/j.atherosclerosis.2011.10.041. [DOI] [PubMed] [Google Scholar]
- 35.Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, Goddard M, Bennett M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res. 2006;99:156–164. doi: 10.1161/01.RES.0000233315.38086.bc. [DOI] [PubMed] [Google Scholar]
- 36.Seals DR, Jablonski KL, Donato AJ. Aging and vascular endothelial function in humans. Clin Sci (Lond) 2011;120:357–375. doi: 10.1042/CS20100476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song Y, Shen H, Schenten D, Shan P, Lee PJ, Goldstein DR. Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2012;32:103–109. doi: 10.1161/ATVBAHA.111.236349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Asia Pacific Cohort Studies Collaboration. The impact of cardiovascular risk factors on the age-related excess risk of coronary heart disease. Int J Epidemiol. 2006;35:1025–1033. doi: 10.1093/ije/dyl058. [DOI] [PubMed] [Google Scholar]
- 39.Fenton M, Barker S, Kurz DJ, Erusalimsky JD. Cellular senescence after single and repeated balloon catheter denudations of rabbit carotid arteries. Arterioscler Thromb Vasc Biol. 2001;21:220–226. doi: 10.1161/01.atv.21.2.220. [DOI] [PubMed] [Google Scholar]
- 40.Ben-Porath I, Weinberg RA. When cells get stressed: an integrative view of cellular senescence. J Clin Invest. 2004;113:8–13. doi: 10.1172/JCI200420663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang XL, Wang J, Shi Q, Carey KD, VandeBerg JL. Arterial wall-determined risk factors to vascular diseases: a nonhuman primate model. Cell Biochem Biophys. 2004;40:371–388. doi: 10.1385/CBB:40:3:371. [DOI] [PubMed] [Google Scholar]
- 42.Shi Q, Cox LA, Glenn J, Tejero ME, Hondara V, Vandeberg JL, Wang XL. Molecular pathways mediating differential responses to lipopolysaccharide between human and baboon arterial endothelial cells. Clin Exp Pharmacol Physiol. 2010;37:178–184. doi: 10.1111/j.1440-1681.2009.05260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao J, Hondara V, Wang XL, Vandeberg JL, Shi Q. Profound influence of LDL oxidative status and monocyte co-cultures on baboon endothelial activation. Am J Cardiovasc Dis. 2012;2:1–11. [PMC free article] [PubMed] [Google Scholar]
- 44.Jeyapalan JC, Sedivy JM. Cellular senescence and organismal aging. Mech Ageing Dev. 2008;129:467–474. doi: 10.1016/j.mad.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311:1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- 46.Esposito K, Ciotola M, Sasso FC, Cozzolino D, Saccomanno F, Assaloni R, Ceriello A, Giugliano D. Effect of a single high-fat meal on endothelial function in patients with the metabolic syndrome: role of tumor necrosis factor-alpha. Nutr Metab Cardiovasc Dis. 2007;17:274–279. doi: 10.1016/j.numecd.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 47.Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 48.Turk JR, Carroll JA, Laughlin MH, Thomas TR, Casati J, Bowles DK, Sturek M. C-reactive protein correlates with macrophage accumulation in coronary arteries of hypercholesterolemic pigs. J Appl Physiol. 2003;95:1301–1304. doi: 10.1152/japplphysiol.00342.2003. [DOI] [PubMed] [Google Scholar]
- 49.Giannattasio C, Zoppo A, Gentile G, Failla M, Capra A, Maggi FM, Catapano A, Mancia G. Acute effect of high-fat meal on endothelial function in moderately dyslipidemic subjects. Arterioscler Thromb Vasc Biol. 2005;25:406–410. doi: 10.1161/01.ATV.0000152231.93590.17. [DOI] [PubMed] [Google Scholar]
- 50.Kroncke KD. Nitrosative stress and transcription. Biol Chem. 2003;384:1365–1377. doi: 10.1515/BC.2003.153. [DOI] [PubMed] [Google Scholar]
- 51.Rojas A, Romay S, Gonzalez D, Herrera B, Delgado R, Otero K. Regulation of endothelial nitric oxide synthase expression by albumin-derived advanced glycosylation end products. Circ Res. 2000;86:E50–4. doi: 10.1161/01.res.86.3.e50. [DOI] [PubMed] [Google Scholar]
- 52.Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, DeRicco J, Kasuno K, Irani K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.d’Uscio LV, Baker TA, Mantilla CB, Smith L, Weiler D, Sieck GC, Katusic ZS. Mechanism of endothelial dysfunction in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:1017–1022. doi: 10.1161/01.atv.21.6.1017. [DOI] [PubMed] [Google Scholar]
- 54.Mujynya-Ludunge K, Viswambharan H, Driscoll R, Ming XF, von Segesser LK, Kappenberger L, Yang Z, Vassalli G. Endothelial nitric oxide synthase gene transfer restores endothelium-dependent relaxations and attenuates lesion formation in carotid arteries in apolipoprotein E-deficient mice. Basic Res Cardiol. 2005;100:102–111. doi: 10.1007/s00395-004-0500-9. [DOI] [PubMed] [Google Scholar]
- 55.Raman KG, Gandley RE, Rohland J, Zenati MS, Tzeng E. Early hypercholesterolemia contributes to vasomotor dysfunction and injury associated atherogenesis that can be inhibited by nitric oxide. J Vasc Surg. 2011;53:754–763. doi: 10.1016/j.jvs.2010.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miller JD, Chu Y, Castaneda LE, Serrano KM, Brooks RM, Heistad DD. Vascular function during prolonged progression and regression of atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2013;33:459–465. doi: 10.1161/ATVBAHA.112.252700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McElhaney JE, Effros RB. Immunosenescence: what does it mean to health outcomes in older adults? Curr Opin Immunol. 2009;21:418–424. doi: 10.1016/j.coi.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Narita M, Young AR, Narita M. Autophagy facilitates oncogene-induced senescence. Autophagy. 2009;5:1046–1047. doi: 10.4161/auto.5.7.9444. [DOI] [PubMed] [Google Scholar]
- 60.Le J, Vilcek J. Tumor necrosis factor and interleukin 1: cytokines with multiple overlapping biological activities. Lab Invest. 1987;56:234–248. [PubMed] [Google Scholar]