

Abstract
Correcting aberrant folds that develop during protein folding disease states is now an active research endeavor that is attracting increasing attention from both academic and industrial circles. One particular approach focuses on developing or identifying small molecule correctors or pharmacological chaperones that specifically stabilize the native fold. Unfortunately, the limited screening platforms available to rapidly identify or validate potential drug candidates are usually inadequate or slow because the folding disease proteins in question are often transiently folded and/or aggregation-prone, complicating and/or interfering with the assay outcomes. In this review, we outline and discuss the numerous platform options currently being employed to identify small molecule therapeutics for folding diseases. Finally, we describe a new stability screening approach that is broad based and is easily applicable toward a very large number of both common and rare protein folding diseases. The label free screening method described herein couples the promiscuity of the GroEL binding to transient aggregation-prone hydrophobic folds with surface plasmon resonance enabling one to rapidly identify potential small molecule pharmacological chaperones.
Keywords: Protein misfolding, missense mutations, pharmacological chaperones, GroEL chaperonin, Surface Plasmon Resonance
1. INTRODUCTION: IDENTIFYING THE CHALLENGE- STABILIZING MOVING TARGETS
The common text book presentation of proteins and protein structure often gives a novice the impression that proteins are stable rigid entities, where protein ligands interact with defined three dimensional binding sites with lock and key precision, and whose structures rarely deviate from their final folds. Also within these beginning texts, optimal protein folding reactions are usually depicted as thermodynamically favorable events. Proteins that are large enough and have a suitable number of hydrophobic residues collapse to form stabilized interior cores, folding into a final structure guided by local interactions. An optimal folding reaction is one where folding proceeds on a fixed path toward a final functional state with very little deviation or structure exploration from the final fold. While most proteins usually reach their most thermodynamically stable states, one has to bear in mind that it is also true that most proteins possess stabilization free energies within the range of 5–15 kcal/mole. Thus, any small increase in temperature will result in a significant rise in the transient appearance of partially folded proteins. In addition, the simple notion that protein conformations only populate very narrow highly defined, near singular energy states is essentially incorrect. In reality, proteins exist in multiple conformational states whose equilibrium fluctuations and functionally important motions combine to create a rough multidimensional folding energy landscape [1–3]. Proteins are no longer viewed simply as vestibules for simple lock and key binding reactions that only acquire highly limited stable conformations. Rather, proteins are dynamic metastable entities [4], where small environmental changes or missense mutations can readily shift populations toward misfolded forms, sometimes resulting in accumulations of aggregated or proteolytically susceptible species. Interestingly, a significant portion of the E. coli proteome exhibit stabilities in the 4 kcal/mole range. The realization that proteins, particularly disease proteins, are highly dynamic entities or moving targets must frame our attempts to develop broad based screening systems aimed at stabilizing the fold.
In this short review, we shall explore the current in vitro methods that are used to identify potential protein stabilizers which may be useful in ameliorating protein folding defects in vivo. The overall goal in correcting protein folding defects is to increase levels of properly folded proteins while simultaneously decreasing the amounts of aberrantly folded aggregation-prone protein populations. We shall examine in silico approaches where the structure of the target protein is used as a template to search for potential ligands that lead to a stabilization of the global fold. Historically (within at least the last decade), this has been a prevalent method of choice in characterizing and in some cases, developing small molecule “pharmacological chaperones” [5, 6]. The realization that this particular strategy may be fruitful comes from sound thermodynamic principles involving ligand stabilization of proteins [7]. The rationale behind concerted efforts to “correct the fold” with small molecule pharmacological chaperones is bolstered by observations that a large number mutant missense forms of proteins can be “rescued” from their aberrant folds if the these mutant proteins are either expressed at lower temperatures or if folding osmolytes are present at high enough concentrations (reviewed in [8]).
Demonstrating that simple addition of folding osmolytes or lowering the temperature of expression rescues the misfolded protein are the type of first pass experiments that have to be performed prior to initiating a search for small molecule protein stabilizers or pharmacological chaperones. It is now recognized that subtle folding mutations can sometimes manifest their deleterious effects even when organisms experience relatively moderate thermal or chemical stress conditions [9]. In developing specific pharmacological chaperones, we shall examine in detail both in silico and solution based screening systems, highlighting some recent successes. We shall examine the limits to the current in silico or in vitro screening approaches for identifying stabilizers that result from the absence of a defined protein structure (particularly of the partially folded aggregation prone form) or direct aggregation interferences respectively. This discussion leads to the realization that there is a real need to develop robust in vitro methods to test for compound efficacy in ameliorating misfolding reactions while stabilizing the native fold. Finally, in the last part of this review, we introduce a new chaperonin-based screening platform that shows promise in rapidly identifying small molecule protein stabilizers of the native fold even in instances where a large population of the target protein initially exists as partially folded or transiently folded species. This approach should enable researchers to expand the set of proteins that can be screened for folding rescue to include those misfolded or aggregation prone disease proteins that may be inherently difficult to stabilize.
2. PROTEIN FOLDING DISEASES
The actual number of diseases that are linked to changes in protein folding states may be very large. According to estimates, diseases that involve protein folding and misfolding reactions could comprise up to at least 30–50% of the total human disease states at any particular time. These disease states involve not only common missense mutants, but also include activated proteins whose normal lifespan within cells must be tightly controlled to prevent aberrant uncontrolled cell growth (e.g. some cancers). The common folding diseases from missense mutations result in defective protein folding reactions, defects in trafficking and aggregations (Table 1: An abbreviated list of protein misfolding diseases). Within this folding disease set of proteins, we can also include viral or bacterial toxin proteins whose transitions during infections involve small to large scale unfolding and refolding (e.g. toxins transitioning from soluble to membrane insertable states) reactions.
Table 1.
An Abbreviated List of Protein Misfolding Diseases
| Folding Disease | Protein Involved | Reference |
|---|---|---|
| α-synuclein | Parkinson’s disease | [10] |
| Amyloid β | Alzheimer’s disease | [11] |
| Tau | Alzheimer’s disease | [12] |
| Superoxide dismutase I | Amyotrophic lateral sclerosis | [13] |
| Huntingtin | Huntington’s disease | [14] |
| Cystic fibrosis transmembrane regulator | Cystic fibrosis | [15] |
| β-glucosidase | Gaucher disease | [16] |
| α-Galactosidase A | Fabry disease | [17] |
| Aquaporin-2 | Nephrogenic diabetes insipidus | [18] |
| β2-Microglobulin | Heamodialysis associated amyloidosis | [19] |
| Transthyretin | Familial amyloid polyneuropathy | [20] |
Missense mutations by themselves lead to a wide variety of these diseases accounting for 48% of all reported human disease-causing alleles [21]. A single protein may have a wide variety of missense mutations, each of which might lead to various misfolding reactions. This is common not only in cancers [22–25], but also in other genetic diseases like Cystic fibrosis, Phenylketonuria [21], and Friedreich’s ataxia [26] to name a few. Cystic fibrosis itself is caused by 1910 different mutations where missense mutations are responsible for about 40% of the total disease set (www.genet.sickkids.on.ca/cftr). Advances in genome sequencing have and will make it more facile to search for mutations and correlate these mutations with disease states. More importantly, the wide varieties of mutations certainly complicates the search for global therapeutics to treat individual protein folding diseases [27].
3. STRATEGIES FOR TREATMENT OF MISFOLDING DISEASES
3.1. The Promise of Gene Therapies
Gene-based therapy efforts have depended on the premise that correcting the gene in question will effectively allow one to deliver a correctly folded therapeutic protein to particular sites within the body or targeted organs. The specific folding diseases in question usually cannot be rescued by simply supplying the properly folded protein as a replacement. Gene therapy methods are very simple in theory, but this therapeutic option has not been realized to date. Among the reasons for its failure is the realization that many human diseases are actually multi-gene diseases with complex interactions. Thus, the best candidates for gene therapy are single gene disorders that are not subjected to complex regulation [28]. In some particular instances it is noted that one only needs to restore function by producing a minimal concentration of correctly folded protein, sometimes leading to the clinical rescue of the disease in question. In this latter case, directly correcting single defective gene targets should yield a favorable outcome but unfortunately, this general approach is fraught with problems that have yet to be resolved [29–33]. Given that a general disease phenotype may be the result of many different genotypes, a single therapeutic approach may also fail to provide a complete treatment for a particular disease state. One may therefore be required to formulate specific targeted therapies for each disease variant or class of variants.
A novel offshoot of gene therapy is the RNA interference approach. RNA interference uses short RNA sequences that target the protein mRNA, which enhances its degradation, leading to general decrease in protein concentration [34]. This approach may be useful in diseases that result in a “gain of toxic function” due to increased protein expression and/or protein aggregation. Reducing the aberrant protein levels should potentially lead to decreased aggregation and toxicity. One such study was shown to be successful in decreasing the deleterious effects that results in aberrant accumulation and misfolding of mutant alpha anti-trypsin (AAT) protein levels within mouse model systems [35]. Functionally, AAT is involved in maintaining the elasticity of connective tissue in the bronchial tubes. AAT mutants that can no longer inhibit resident elastases fail to prevent the protease dependent deterioration of lung elasticity, ultimately leading to emphysema. In addition to emphysema, these AAT missense mutants aggregate, inhibiting or compromising the general cellular secretion pathways in the liver, resulting in a toxic “gain-of-function” phenotype that sometimes leads to liver cirrhosis. In the work by Li and colleagues [35], it was observed that specific shRNA treatments decreased the levels of the misfolded protein within mutant mouse models, preventing large-scale aggregation and associated liver cirrhosis. Unfortunately, this approach does not prevent emphysema per se because the mutant protein levels within the serum remain below normal. Overall, gene therapy approaches still need further development to address important issues regarding gene insertion safety and efficacy.
3.2. Towards Direct Correction of the Aberrant Fold
The idea that one can use novel therapeutic approaches to correct the actual misfolding events in diseases such as the cystic fibrosis was initially proposed in a seminal hypothesis paper by Thomas and Pederson in 1992 [36]. For this particular disease, a prominent temperature sensitive defect in the folding and trafficking of the cystic fibrosis transmembrane regulator (CFTR) leads defects in Cl− ion transport, which affect the epithelial cells in the intestine, gall bladder, pancrease and the respiratory system. After synthesis, the mutant CFTR protein fails to reach a mature state but instead remains in a complex with the molecular chaperones calnexin in the ER lumen [37] and Hsp70 in the cytosol [38]. For the wild type CFTR, the protein folds properly, resulting in decreased chaperone interactions and is subsequently transported to the plasma membrane. The missense folding mutant protein remains in a complex with the chaperones in the ER, trapping the mutant protein within the ER, subsequently leading to failed export and trafficking to the plasma membrane. The levels of these trapped CFTR-chaperone complexes are dramatically increased for the various missense misfolded mutant proteins [39]. An unusual feature of CFTR biogenesis is that even with the wild type protein, some reports suggest that ~ 75% of synthesized protein is degraded via ER assisted degradation [40]. Initial landmark studies by Thomas, Pederson and Welch found that the proper processing, folding and trafficking of particular CFTR missense mutants to the plasma membrane is actually enhanced at lower temperatures or when folding osmolytes such as tri-methylamine N-Oxide (TMAO), glycerol or Myo-inositol are added to cells containing the mutant protein [41]. When one shifts the temperature back to 37°C, Cl− transport persists transiently until the mutant transmembrane protein is subsequently degraded.
Other proteins also show temperature-sensitive processing and folding. A recent addition to an expanding list of temperature sensitive folding proteins is the epidermal growth factor filulin like extracellular matrix protein -1 (EFEMP1), where misfolding of a single site mutant, R345W, results in early onset age-related macular degeneration. This single site amino acid substitution caused significant protein secretion defects. This secretion defect can be relieved in part by decreasing the growth temperature, leading to an increase in correct disulfide formation and folding simply by reducing translation loads by adding cycloheximide [42].
Welch and colleagues demonstrated early on that global “chemical chaperones” could be utilized in a manner similar to temperature to not only increase CFTR expression but also improve chloride transport [43]. Chemical chaperones or osmolytes are small organic molecules that can thermodynamically stabilize and protect proteins from large scale denaturation. In vivo, the cells frequently accumulate such osmolytes at high concentrations during long-term stress to globally stabilize native folds. Commonly, increases in these folding osmolyte conditions normally do not affect other cellular catalytic processes such as Km values or catalytic rates of enzymes [44]. The one drawback with accumulating chemical chaperones is that they are not specific and will affect the physical properties of a broad class of proteins that are present within the cell. The general thermodynamic mechanism that defines this stabilization effect in most proteins is that the osmolytes specifically destabilize the unfolded state due to exposed peptide backbones, resulting in the favorable burial and hence folding of the native fold [45]. Curiously, the addition of folding osmolytes to transiently-folded missense mutants found in many protein folding diseases results in a rescue of the native fold, solubilization and in some cases, the regain of native function
Membrane proteins can also be rescued using this indirect strategy. For example, the osmolyte glycerol rescues the misfolding of the water selective channel protein aquaporin (AQP2) found in the loop of Henle. These proteins are essential for water transport in the kidney and deletion or missense mutations in the AQP2 protein leads to Nephrogenic Diabetes Insipidus, a disease in which the kidney fails to concentrate urine. These mutations lead to improper localization and trafficking of the AQP2 protein, which has been attributed to impaired ER processing and ERAD [53]. Some mutants are also unable to form oligomers and are mistransported to the basolateral membrane instead of the epithelial membrane [54]. Aquaporin mutants are similar to CFTR mutants in that both are membrane proteins where specific missense mutations prevent efficient transport and processing. It was observed that the elevation in concentrations of myo-inositol (an osmolyte that occurs naturally at high concentration in the kidneys to counteract the effect of urea [55]) also increases the expression of the aquaporin mutants. In summary, utilizing folding osmolytes as a general therapeutic strategy is not generally applicable for specific folding diseases because of global stabilization effects, lack of specificity, and the high concentration requirement (~ Molar concentrations) for pharmacological efficacy.
3.3. The New Paradigm Part 1: Targeting the Proteostasis Network
The proteostastic network within the cell comprises all of the machinery that controls the protein steady state levels where the processes of protein synthesis and folding is balanced against degradation leading to a healthy functional proteome [30, 56]. Specifically, this network controls the concentration, conformation and the binding interactions of proteins, ultimately insuring that the functional form of the protein is present at physiologically relevant steady state levels. The steady state concentration can be affected by changes in several cellular environments including fluctuations in temperature, the appearance of naturally occurring mutations, as well as the intrinsic kinetic competition between folding, misfolding and concentration dependent aggregation. These dynamic steady state conditions are maintained and are controlled by a multitude of cellular proteostatic networks. These networks comprise roughly 17 different pathways including in total about 1000 different assistant proteins such as folding enzymes, chaperones and trafficking components. These proteostatic network proteins act through transcriptional and post-translational mechanisms to balance protein folding and function capacity by reducing protein synthesis, by enhancing folding and repair processes, and/or by mediating degradation [57]. These proteostatic processes preserves the synergy and integrity of the proteome within an organism during developmental changes, environmental stress, infectious disease and aging. As discussed previously, some proteins are dynamic metastable entities that exist in rapid equilibria between their folded and partially unfolded forms [31]. This inherent metastability is very important for certain protein functions, such as their interaction with other biomolecules, for their catalytic reactions or in instances where global unfolding and refolding processes are necessary for specific trafficking and proper localization.
Recently, the idea of developing novel therapeutics to control the target-rich proteostasis system has gained considerable interest within the pharmaceutical industry. The thought behind this strategy rests on the notion that control of the proteostatic network and its broad biological components or processes should influence the lifespan, folding integrity, and steady state levels of proteins within cellular environments. Proteostatic targets include all of the genetic regulatory elements that controls the protein levels within the proteostatic network, the protein production machinery, membrane transport systems, molecular chaperone networks (e.g. Hsp90 and cofactors as targets), stress sensing systems (unfolded protein response), or control of the protein degradation machinery. All of these networks are integrated to control protein levels within this massive protein homeostatic system. Most importantly, it is apparent that these systems are in fact finely tuned to respond to the aspects of protein stress brought on by normal developmental processes as well as frequent environmental stress perturbations (reviewed in [58]).
Viable methods to control the proteostasis network, which controls the protein steady state levels are being developed on a broad front (reviewed in [57, 58]). Corrective control may entail deliberately increasing protein levels of a target protein through enhanced folding and rescue by boosting the action of the chaperone protein networks. A classical example where the successful control of proteostatic networks is efficacious relates to concerted efforts of combinational therapies being used to increase the proper fold and correct trafficking of cystic fibrosis membrane regulator protein missense mutants [57].
Alternatively, one may also specifically develop therapeutics to decrease steady state levels of certain proteins. This latter approach could be applied in instances where increases in toxic “gain-of-function” mutants lead to increased aggregation, and or mistrafficking. The aberrant accumulation of these disease proteins lead to a decline in steady state levels by aggregation through mass action effects, sometimes leading to an actual decrease in functional protein. The particular strategies commonly used to regulate proteostatic networks are outlined in a number of excellent monographs by Powers et al., and Morimoto [57, 58].
As stated above, proteostatic strategies can enhance the folding of missense mutant populations by initially increasing the amount of soluble protein either by decreasing protein degradation or increasing the level of chaperone proteins. Curiously, a further improvement in folding can be accomplished by subsequently stabilizing the fold through the binding of small molecule pharmacological chaperones. For example, this combinatorial “polypharmacological” approach of introducing both proteostatic regulators and pharmacological chaperones has been observed to boost properly folded protein levels of a specific glucocerebrosidase mutant (L444P) when a small molecule proteostatic regulator MG-132, (celastrol) was supplied in tandem with a glucocerebrosidase pharmacologic chaperone called N-(n-Nonyl)-1-deoxynojirimycin (NN-DNJ). In this particular instance, application of this combinatorial drug approach is synergistic because one can simultaneously increase the expression and folded protein levels of the L444P mutant 4 to 6 fold over the implementation of the individual strategies alone [6].
3.4. The New Paradigm part 2: Directly Correcting the Fold with Pharmacological Chaperones
It is a well-known thermodynamic observation that small molecule ligands that bind to the native state of a protein generally lead to a further stabilization that protein, where the bound ligand state shifts the intrinsic dynamic equilibrium to favor the native fold population. This paradigm holds for mutant proteins as well. As an example, vasopressin receptor antagonists stabilize the protein receptor, decreasing proteosomal degradation, leading to a 15 fold increased expression of that receptor on the cell surface [59]. Similarly, ER permeable indoles, quinolones and erythromycin macrolide agonists and antagonists of the gonadotrophin-releasing hormone receptor were utilized to increase the proper trafficking and expression of the protein within the proper membrane system [60]. These observations indicated that small molecule ligands, which bind specifically to the mutant protein, could be used as a therapeutic option. Such small molecules are often called “pharmacological chaperones”. In contrast to “chemical chaperones” (e.g. osmolytes), this class of small molecule chaperones specifically acts on single targets and can be effective at very low concentrations. These pharmacological chaperones tend to bind to the native fold of the protein, leading to a stabilization of the proper fold, kinetically decreasing the population of partially folded states that may be subject to wholesale degradation and/or aggregation.
4. IDENTIFYING PHARMACOLOGICAL CHAPERONES
4.1. In Silico Screening Approaches
In silico screening methods that identify candidate pharmacological chaperones involve computational techniques that examine the interactions of model compounds at “drug-gable” sites for proteins where a representative three dimensional structure exists. A major factor associated with favoring this mode of screening is the observation that in vitro> 100,000 compound screens can be extremely costly per run including assay development. In silico screening simulations can potentially reduce the search space for effective potential small molecule, making validation screening more efficient provided one has a druggable site [61]. In-silico methods are utilized at various stages of the lead discovery process. They can be used to exclude false positives from a primary screen and can speed up docking studies to simulate protein-ligand binding as well as to screen for scaffold similarities. An advantage with in silico screening is that it is not restricted to synthesized compounds on hand. An ideal pharmacophore for a particular protein as well as a virtual library of related compounds can be easily generated using computational methods.
As one example, Lansbury and colleagues applied in silico methods to identify potential ligand candidates that could prevent a particular superoxide dismutase dimer missense mutant (A4V) from dissociating into aggregation prone monomers, presumably leading to Amyotrophic Lateral Sclerosis for these patients [62]. A recent successful application of this approach came from an in silico approach where a derived small molecule stabilizer of the tetramer of transthyretin (TTR) polyneuropathy mutants called tafamidis meglumine was tested via in silico type screens before it was mass produced and tested in Phase III trials. This particular small molecule pharmacological chaperone effectively prevents dissociation of the TTR tetramer, thereby preventing deleterious amyloid formation in this particular disease [63]. Transthyretin is a plasma protein that acts as a transporter for thyroxine and is a retinol binding protein. It usually exists as a homo-tetramer but in case of mutations, the monomer can lead to the formation of aggregates and amyloids. Prior to the development of this potential pharmacological chaperone, the only treatments that existed for Transthyretin amyloidosis used gene therapy and liver transplantation methods, both of which fraught with many complications. It is important to note that the time span for developing a successful pharmacological chaperone for this particular disease was approximately ~ 10 years.
Another class of protein folding diseases where promising pharmacological chaperones are being developed specifically target lysosomal enzymes wherein, misfolding of acid-alpha-galactosidase (alpha-Gal A) and acid-beta-glucosidase (GCase) results in defects in lipid metabolism leading to Fabry and Gaucher diseases, respectively. The alpha-Gal A and GCase enzymes hydrolyze the sphingolipids globotriaosyl- and glucosylceramide, respectively. Missense mutants of these proteins are improperly folded and fail to effectively exit the endoplasmic reticulum. Pharmacological chaperones such as isofagomine (IFG) and 1-deoxygalactonijirimycin (DGJ), specifically bind to the active sites of GCase and alpha-Gal A, stabilizing these proteins and allowing them to efficiently exit the ER [64]. Structural and thermodynamic analysis of these interactions indicates that this strategy of specifically targeting the stabilization of the active site is a sound option as long as the pharmacological chaperone dissociates from that active site once the enzyme reaches its destination of function [65, 66]. In this instance, these compounds are classified as reversible inhibitors [7].
In some instances, treatments using specific binding cofactors can rescue certain instances where misfolding is prevalent. Specifically, the misfolding of phenylalanine hydroxylase results in the metabolic disease phenylketonuria (PKU) because a cofactor is missing or is not synthesized in abundance. A subpopulation of patients could be effectively treated with large doses of tetrahydropterin [27]. This cofactor is necessary to catalyze the hydroxylation of phenylalanine yielding tyrosine. While this particular strategy did not arise from in silico screening efforts, ligand based knowledge from biochemical and structural efforts can also enable drug development efforts to proceed when one pays specific attention to the biochemistry of the target system.
In silico screening methods requires one to have adequate and existing knowledge of detailed structural information describing the protein (~ 2 Å resolution or better) before initiating an in silico screen. This is a difficult proposition for many of the proteins that are prone to misfolding, particularly in situations where the three dimensional structure of the mutant structure is unavailable. In the one instance noted above, Lansbury and colleagues were fortunate to acquire a crystal structure of the mutant A4V aggregation prone superoxide dismutase mutant enabling them to target a potential druggable binding site that only existed for this particular mutant [67]. Once promising lead compound candidates are identified after a massive in silico screening procedure, the proposed stabilizing interaction still has to be validated using in vitro classical thermodynamic stability assays such as isothermal calorimetry, denaturant titrations and differential thermal stability methods.
4.2. Putting Strategy to Practice: The CFTR Folding Consortium
As noted above, there are numerous misfolding mutations that result in cystic fibrosis (CF). One therapeutic strategy for CF includes increasing the amount of CFTR on the epithelial surface by stabilizing the protein fold with small molecules called correctors. Another strategy is to utilize potentiator/activator molecules that increase the chloride ion conductance from mutants of CFTR that are already present on the cell surface. Several studies have been carried out to identify novel correctors/potentiators. One of the first high throughput small molecule screens identified several classes of correctors from more than 150,000 compounds [68]. Another screening identified six correctors after screening more than 160,000 compounds [69]. Development of these lead compounds by Vertex pharmaceuticals have resulted in the potentiator VX-770, which has cleared phase III clinical trials and the corrector VX-809 that currently in Phase II clinical studies [68, 69]. Both these studies utilized cell based functional assays. For example, the positive readout for some cell based assays rely on immunohistochemical methods to quantitate the amount of CFTR that becomes successfully expressed on cell surface [70]. Since CFTR is a membrane protein that has been difficult to purify in large quantities, most of the screening assays used for identifying the potential correctors have been cell-based rather protein-based systems. The question still remains as to whether some of the candidate corrector compounds identified directly stabilize the CFTR protein or indirectly affect the proteostatic networks within the cell.
5. CURRENT METHODOLOGIES FOR IDENTIFYING PHARMACOLOGICAL CHAPERONES - HIGH-THROUGHPUT SCREENING APPROACHES
Using pharmacological chaperones to directly stabilize the native state of proteins and rescue misfolded proteins from degradation is an attractive proposition to treat protein misfolding diseases. In vitro screens for pharmacological chaperones take advantage of large combinatorial chemistry platforms. Screening for misfolding diseases is, however, a problematic proposition because the assay usually has to be specifically designed for each misfolding protein. In addition, it is highly conceivable that each missense mutation requires a specific individual pharmacological chaperone rather than one global corrector. As mentioned previously, the problem of assay design must recognize the fact that the potential binding targets for misfolding diseases are actually moving targets because transient partially unfolded states are in constant states of flux. It is highly conceivable that some candidate compounds may actually affect the protein folding reactions that occur as the polypeptide is being synthesized and emerges from the ribosome. In this instance it is entirely reasonable to use a fragment based substrate as a target protein. The challenge for developing a broad-based screen for folding diseases is that any test platform must be able to detect stabilizers that influence the kinetic behavior of the mutants in question whether they are a complete folded protein or a critical folding fragment of that protein. The next sections will describe the most popular in vitro direct protein targeted screening systems and detection methods that are available.
5.1. Aggregation Based Detection Methods
A large set of protein stability assays depend on the inherent aggregation propensities of the proteins when perturbing conditions are introduced (Fig. 2- Reaction 1). A multi-well format can be used to measure large scale aggregation by spectroscopic means (light scattering, turbidity). Alternatively, one can also measure the aggregates directly using ultracentrifugation or size exclusion chromatography. These latter assays are frequently used to evaluate long term stability of protein solutions used in formulation development. In the presence of protein stabilizers or specific ligands, the aggregation signals should decline. These aggregation assays are sometimes lengthy involving time frames that range from several hours to even days. The length of time involved in these assays makes it difficult to implement larger scale screens to identify stabilizers in a chemical library that contains thousands of compounds. However, this type of aggregation assay is still useful to validate potential ligand candidates from small sets of compounds or compounds that have been identified from in silico screens. Most notably, a general long-term aggregation assay was used as the base platform to validate the phase III drug Tafamadis as a potential amyloidosis inhibitor of transthyretin wild type and mutant aggregation [63].
Fig. 2.

Within the general reaction scheme, the changes in the amounts of three prominent reaction species are detected to determine if a stabilizing ligand/corrector of the fold is present. (1) The aggregation based screening system relies on the appearance or disappearance of downstream aggregates. (2) The fluorescence thermal scanning systems rely on the appearance of partially folded forms of the protein to determine when a potential ligand/corrector is present. (3) The chaperonin based capture system discussed in this review detects the appearance or decrease in the initial transient fold, giving this assay the advantage of identifying potential stabilizing ligands/correctors under near physiological solution conditions.
Aggregation assays in general typically only measure the appearance of large sized particles. There can be instances where smaller aggregates (e.g. dimers) form but are not easily detected, which sometimes leads to false positive readouts. It is also possible that a small molecule candidate may simply interfere with the general aggregation surface rather than stabilizing the protein directly, again resulting in a false positive read-out. With this in mind, this latter result requires one to further validate the compounds with secondary screens to rule out these false positive outcomes. As a general screening platform, aggregation based assays are particularly problematic for disease proteins that are already aggregation prone under normal physiological solution conditions. The difficulty here is that the test protein may be extremely difficult to purify in a reasonable folded form that is not aggregated at the start of the assay.
It is worth mentioning however, that some progress has been made in developing cell-based aggregation screening methods. These are particularly useful in identifying broad based small molecule therapeutics that could function either as targeted correctors (i.e. directly correct the fold) or as broad based potentiators (modulating proteostatic networks). In one instance, a novel anti-aggregation bacterial based systems were used to identify inhibitors of 1–42 Aβ chimera that undergoes export through the using a novel ssTorA-Aβ42-Bla construct that uses the two arginine translocation mechanism. Any inhibitors of the aggregation of this particular construct results in efficient translocation of monomeric construct. Once valid compounds were identified, further validation was confirmed using a thioflavin T based aggregation assay. [71] Another useful in vivo aggregation detection system developed by Ignatova and Gierasch used a specific FlAsH (4,3 bis(1,3,2-dithioarsolan-2yl) fluorescein label that was incorporated into aggregation-prone proteins to monitor the reversal of in vivo protein aggregation when known cellular osmolytes were effectively transported into the cells [72]. It is highly conceivable that these particular approaches using fluorescence based measurements could be developed into a high throughput cellular based assay.
5.2. Thermal Scanning Fluorescence Methodologies
Another method that has gained popularity as a moderate-throughput screening system is a fluorescence-based assay that detects folding intermediates as the protein unfolds during a thermal denaturation [73]. As a protein thermally denatures, a fluorescent dye (e.g. SYPRO-Orange) binds to transiently exposed hydrophobic regions of molten globule states, which increases the dye quantum yield, generating a kinetically dependent transition fluorescent profile (Fig. 2-reaction 2). For a well-behaved protein, the denaturation profile resembles a thermal denaturation curve comprised of a predenaturation region, a transition region and a post denaturation signal. Any ligand that presumably stabilizes the native protein form will shift the equilibrium toward the folded state and the entire fluorescence dependent kinetic transition profile shifts rightward signifying that a potential stabilizer is responsible this shift Fig. (2). These types of thermal scanning assays are very rapid, can be adapted to use simple PCR based thermal systems and can be designed to use very little protein [74, 75].
5.3. The Need for In Vitro Screening Platforms for Transiently Stable Disease Proteins
The most difficult set of proteins to evaluate and identify protein stabilizers for are the metastable misfolding disease proteins. A common property of these transiently stable proteins is that they readily aggregate under near physiological conditions even before the protein systems are stressed (e.g. temperature, pH, denaturant, etc.). Furthermore, at least for the thermofluor based systems, oligomeric proteins are also extremely difficult if not impossible to evaluate because they have a tendency to aggregate prior to complete unfolding. For both intrinsically metastable proteins and oligomers, the thermal melts usually do not follow “well-behaved” kinetic transition profiles. Membrane proteins are even more problematic since the fluorescent dyes will interact with the detergents, lipid components, and hydrophobic portions of the folded membrane protein before exposing these proteins to solution stress conditions. In addition to the non-ideal transition profiles, the dye binding itself may perturb the equilibrium between the folded and unfolded populations. Also problematic is the fact that ligand binding occurs under non-physiological conditions (high temperatures) and secondary titrations at near physiological temperatures are needed to confirm binding affinities. Lastly, large quantities of proteins are required (1 mg/96 well plate) and the precious mutant proteins are no longer salvageable once the assay has been performed.
An optimal assay platform is defined as one that would be broadly applicable for metastable proteins at near physiological solutions and would include the oligomer and membrane protein classes. As with most high-throughput protein stabilization assays, it would also be highly desirable if the efficacy of protein stabilizer candidates could be evaluated using small quantities of the protein. As an additional bonus, it would be extremely useful to be able to salvage the protein of interest after the assay is complete, which would be useful for hard-to-purify misfolding disease proteins. Most importantly, in our view, the best type of assay would evaluate the protein stability of the dynamic equilibrium states that naturally exist prior to larger scale unfolding and aggregation (Fig. 2- reaction 3).
Once potential lead compounds are found, one can utilize cell-screening assays to determine if the pharmacological chaperone is effective in rescuing the fold in vivo. Conn and coworkers used a strategy where candidate non antagonistic compounds were tested in a cellular environment to determine if proper folding and trafficking of misfolded membrane receptor mutants of GPCR and Vasopressin 2 receptor occurred [76]. While cell based aggregation or transport assays certainly can speed the detection of potential “corrector or potentiator” drug candidates, without further validation with the protein target itself, the exact nature of the stabilizing/rescue mechanism will remain unclear. For example, less specific proteostatic effectors may certainly increase the trafficking efficiency and hence folding by a variety of mechanisms by either increasing chaperone protein interactions or decreasing proteasome function or both but these same compounds may not have any direct influence on stabilizing the protein target itself. As stated previously, combinatorial approaches using both proteostatic and direct pharmacological chaperones seems to enhance protein product formation while preventing deleterious aggregation.
6. USING NATURE’S SENTRIES TO DETECT AND CAPTURE TRANSIENT MISFOLDS AS THEY FORM
To maintain cellular and protein homeostasis, cells use a large variety of protein molecular chaperones to insure proper folding, trafficking, protein transport and protein degradation. In this section we will briefly summarize and evaluate the utility of the most prominent chaperone classes to detect and capture transient dynamic folds. There are many classes of common chaperone proteins that can be potential candidates for interacting with partially folded proteins as they arise. These chaperone proteins including the Hsp60s (chaperonins), Hsp70, Hsp90, Hsp100 and small Hsps (17–25 kDa) classes. With the exception of the small Hsps, the other listed chaperones actively cycle between ATP and ADP bound states, changing their binding affinities for partially folded proteins, effectively inhibiting deleterious aggregation while favoring folding, unfolding, and refolding of most cellular proteins. In the absence of external stress, these cycling systems constantly maintain steady state populations of folded or transportable proteins. In vivo, during stress, these chaperone proteins initially bind to increasing unfolded protein populations and are titrated away from suppressing the folding or oligomerization of regulatory proteins (e.g. HSF). It is these oligomerized states that then bind to DNA regulatory elements up-regulating the stress response (i.e. increased chaperone production). The appearance of a protein that has a tendency to kinetically partition toward misfolded populations imparts an added stress on the normal protein homeostatic system. Before chaperones are developed as detection platforms, one has to evaluate the how efficient these chaperones are in capturing and tightly binding these transient kinetic folds or misfolds. In most instances, the nucleotide-driven fluctuation in the binding affinities of chaperone proteins insures that the chaperones maintain short interaction times with their substrates. Indeed, it is detrimental for cells to possess very tight binding chaperones since these high affinity states could effectively sequester the inherently metastable proteins away from their soluble states within the cellular milieu. However, in devising a useful protein stability screening platform, we propose that it is actually desirable to identify a chaperone system or systems that can capture and sequestering transient folds, thus serving as an effective kinetic trap of the initially transient states as they appear Fig. (2- reaction 3, Fig. 3).
Fig. 3.

Proteins, such as folding disease proteins that naturally equilibrate with partially folded forms readily bind to the high affinity chaperonin. The decision that a potential corrector or stabilizer of the native fold is present simply depends on the observable increases or decreases in chaperonin binding. In this new reaction diagram, downstream aggregation reactions are initially prevented by the chaperonin. Negative result - If a lead compound candidate does not stabilize the target protein in question, the chaperonin will bind to this protein in a time dependent manner. Positive result - If a stabilizing ligand is present that binds to the native fold, the chaperonin binding reaction will be diminished. This chaperonin sink assay is dependent on a simple yes/no decision point.
7. IDENTIFYING THE BEST CHAPERONE CAPTURE PLATFORM
7.1. Hsp70/90 Chaperone Proteins
Both the Hsp70 and Hsp90 proteins are very dynamic during their ATP/ADP cycling reactions resulting in highly variable substrate capture and binding states. These chaperones are selected to interact transiently with exposed linear regions or dynamic folds, sometimes maintaining proteins their metastable active states. For instance, the Hsp70 class of chaperones exist in a number of different nucleotide bound states, exhibiting a wide range of binding affinities and interaction states. In addition to the multiple substrate interaction states, both of the Hsp90 and Hsp70 chaperones interact with multiple cofactors. In the case of the Hsp70, Hsp40 binds to and transfers potential protein substrates to the Hsp70 and stimulates Hsp70 dependent ATP hydrolysis [77, 78]. Another cochaperone (e.g. GrpE in E.coli) facilitates nucleotide exchange. Both of these stimulatory and exchange functions diminishes interaction times of the substrate with the Hsp70 system. The substrate free states of both of these chaperones do not possess large protein binding sites, which kinetically diminishes their capture efficiency potentials. As for Hsp90, this protein appears to exist in at least two different substrate-binding states (open and closed) in solution even in the absence of protein substrates [79]. Furthermore, this chaperone protein has particularly wide array of co-chaperone proteins whose general role appears to impart specificity for Hsp90 binding to particular client proteins.
Small molecules that modulate Hsp70 activity could be a potential therapeutic strategy for misfolding diseases like Alzheimer’s disease. Hsp70 over expression has also been shown to affect aggregation of huntingtin [80] and alpha-synuclein [81]. Hsp70 has also been thought to be involved in the stability of Tau protein and its association with micro-tubules [82], thus potentially preserving the toxic species. Hsp70 activators maintain the Tau concentration while the inhibitors were found to decrease Tau levels. The inhibitors include benzothiazines dyes such as Methylene blue and Azure C and flavones like myricetin [83]. These inhibitor compounds affect the rate of ATP conversion by modulating the alpha helical lid closure. Decreasing ATP hydrolysis using inhibitors led to decreased Hsp70 affinity for tau, leading to an increase in tau ubiquitinylation [83]. These potential small molecule activators or inhibitors appear to be proteostatic effectors rather than pharmacological chaperones. This approach decreases some of the complications associated with Hsp90 drugs such as induction of heat shock response. The wide variation in binding affinities, the specificity requirements of co-chaperones with particular protein substrates and the smaller protein binding sites diminishes the effectiveness for using the Hsp90 and 70 classes of chaperones to capture multiple dynamic protein folding states. This is not to say that these particular chaperone proteins will not be useful in capturing some specific proteins but again, the small size of the binding sites, and the reality that higher binding affinities may higher binding affinities may coincide with multi-protein functional states complicates their use as a broad based screening system.
7.2. Hsp100 Class –Aggregate Binding Proteins
The Hsp100 classes of chaperones are usually called disaggregases due to their ability to disrupt and dissociate protein aggregates. They are mostly expressed during stress conditions in bacterial, plant and fungal cells. The Hsp100 class or proteins are generally ring shaped molecules that belongs to the AAA+ family and utilize ATP hydrolysis to disassemble misfolded molecular complexes that exist in smaller protein aggregates. The N-terminal domain of the E.coli Hsp100 protein, ClpB, is thought to be directly involved in binding to aggregates [84]. It carries out the disaggregation reaction by physically translocating or threading the target protein substrate through its central channel [85, 86]. During this process the substrate gradually unfolds perhaps due to a specific ratcheting movements caused by two unstructured loops within the central channel [87, 88]. The unfolded protein may either refold or reaggregate if other chaperones are unavailable to capture newly unfolded protein. These Hsp100 proteins have also been thought to be involved in prion propagation [89] and they require ATP to carry out the remodeling of protein structures such as stable beta sheets [90]. The bacterial ClpB has also been implicated in the invasion and survival of several bacterial pathogens such as Mycobacterium tuberculosis [91], Plasmodium falciparum [92] and Leischmania donovani [93].
From a protein substrate capture standpoint, the binding sites of the ring chaperone proteins are presumably large enough to make them attractive kinetic sinks. The Hsp100 class is particularly useful because can bind large exposed hydrophobic regions of proteins and even small aggregates. However, one of the most serious drawbacks in using Hsp100 oligomers as general capture platforms for both aggregates and aggregation prone protein substrates is that they exist as highly dynamic rather than stable oligomers. It turns out that their oligomeric states undergo assembly and disassembly that is governed by the concentration of the Hsp100 protein, nucleotide levels and ionic strength conditions [94–98].
7.3. GroEL: The Optimal Detection Platform to Trap Partially Folded Intermediates
It has been known for some time that increases in GroEL levels enables the E. coli cell to suppress mutations in several genes encoding diverse proteins in vivo by serving as an active cycling protein buffer that prevents protein misfolding [99]. Gatenby and colleagues first demonstrated that temperature sensitive mutants containing destabilizing point mutations in the his operon could survive heat shock when GroE chaperonins levels within the cell were elevated. Recently, Tawfik and colleagues demonstrated that elevated levels of the GroE chaperonin system can also serve as a buffer or evolutionary capacitor to temporarily maintain proper folds for missense mutants until distant secondary site suppressor mutants could accumulate, thus reestablishing protein stability and most interestingly, allowing for the selection of new evolved functions [100]. The range of proteins that GroEL is able to bind and sequester appears to be extremely broad. Not only does GroEL interact with and fold literally hundreds of non-E. coli proteins, Horwich and colleagues demonstrated that this broad substrate utilization extends inside its E. coli host. Specifically, these researchers used a temperature sensitive polypeptide trap GroEL binding mutant [101], E461K, which is no longer able to release polypeptides even when ATP and the cochaperonin GroES are present. Upon a slight temperature upshift (20 to 30°C), cells containing this mutation showed that a massive amount of intrinsic E. coli proteins from all functional classes precipitated into inclusion bodies. Furthermore, electron micrograph visualization of some of the affected cells clearly showed that GroEL formed large continuous tube-like head to tail like aggregates within the E. coli cytosol, most likely linked via trapped polypeptides that expose two broad hydrophobic binding surfaces that now bind adjacent GroEL oligomers [101].
Early seminal observations by Lorimer’s group lead to the realization that the high- affinity nucleotide-free form of GroEL can bind to partially folded proteins that are kinetically linked to the native fold via a dynamic equilibrium [102]. Furthermore, they also determined that GroEL binding diminished almost completely when saturating concentrations of a stabilizing ligand toward the folded target substrate protein was present. This was demonstrated with dihydrofolate reductase (DHFR) and its ligand NADPH. Our laboratory also demonstrated that an SH oxidized form of rhodanese could partition onto GroEL even though a disulfide reduction system was present [103]. This latter partitioning reaction was prevented when oxygen was rigorously removed, which prevents cysteine oxidation. The abilities of the nucleotide-free high-affinity form of GroEL to easily trap folding intermediates lead us to propose that we can utilize this form of the GroEL chaperonin to act as a general thermodynamic and kinetic sink, allowing us to capture kinetically transient misfolded forms that were hydrophobic. This efficient GroEL capture/partitioning reaction should enable us to use the intrinsic dynamic equilibrium between folded and misfolded forms to screen for stabilizers for a wide variety of protein misfolding diseases [102, 103]. As described by Edward Eisenstein, the GroEL chaperonin can be thought of as a “Promiscuous Antibody” that can bind to non-native folding intermediates as hydrophobic surfaces become transiently exposed to the aqueous environment. This makes it an excellent tool to kinetically trap and thus detect the transient partially folded intermediates that lead to misfolding diseases. The advantage of directly measuring transient forms in this capacity is that it enables one to rapidly monitor the initial unfolding reaction rather than detecting later occurring massive unfolding reactions such as dye binding to molten globule populations or large scale aggregation Fig. (2-reactions 2,3, Fig. 3). This chaperonin dependent kinetic partitioning reaction also allows one to accelerate the protein stabilizer assay for aggregation prone proteins (e.g. transthyretin mutants), leading to a dramatic decrease in assay time [104]. Since GroEL is always in excess compared with the transient concentration of partially folded proteins, the partitioning kinetics usually follows a pseudo first order reaction allowing one to speed up the detection/partitioning reaction by increasing the GroEL concentration [103]. Additionally, GroEL is a stable heat shock protein that can easily tolerate elevated temperatures, allowing one to control the kinetic rates of unfolding. Since the nucleotide free GroEL used has a very stringent binding surface, potentially mimicking typical in vivo chaperone surfaces, pharmacological chaperones that stabilize the misfolding protein in presence of GroEL may prevent partitioning onto intracellular chaperones, leading to an improvement in protein stabilization and perhaps decreasing specific protein degradation.
8. INITIAL CONSTRUCTION OF GROEL-TRAP: IMMOBILIZED GROEL-SEPHAROSE BEAD PLATFORMS
Our initial assay setups took advantage of the large size of GroEL. We were able to nonspecifically couple this protein onto sepharose beads to construct a surface that could bind partially folded proteins as they transiently formed in solution. By immobilizing GroEL onto large sepharose beads, one can easily separate a kinetically partitioned protein that binds to GroEL from the remaining soluble population using either simple ultra filtration methods or by directly examining the time dependent loss of soluble protein in the presence of the immobilized GroEL capture platforms [105]. The partitioning rates are observed to be higher with free GroEL rather than the immobilized GroEL but experience has demonstrated that the separation of bound GroEL-target substrate complex from the solution cannot be done rapidly. Some progress was observed when the separation of immobilized GroEL-protein substrate complexes from the free protein substrate populations was accomplished using large pore ultra filtration devices. This method of separation also lends itself to be more amenable to scale-up and automation of the screening system. Another advantage of using GroEL bead approach is that, even on the bead surface, the GroEL itself can be easily regenerated and reused by treating the GroEL-beads with a mixture of ATP and a folding osmolyte such as glycerol or sucrose [105] resulting in a dissociation of the bound target protein substrate. We have also found that increases in the rate of formation and concentration of the transient-folded species using mild denaturing conditions (e.g. low urea (~ 1 M) and slight temperature elevation(~45°C)) increases the overall GroEL capture rate [102, 104, 105]. This approach is particularly useful for proteins that do not populate significant amounts of kinetically transient forms of partially folded species under less harsh conditions. The ability to subtly control kinetic partitioning rates of normal folding reactions onto the high affinity form of GroEL expands the utility of this capture method, thus allowing one to identify stabilizers or stabilizing conditions of natively folded proteins as well as folding disease variants.
Unfortunately, despite our demonstration that we can effectively separate the GroEL-protein substrate from the soluble “natively folded” protein substrate, the bead-based nature of the assay still proved to be a bit cumbersome as a reliable moderate-throughput screening platform. From the standpoint of evaluating transient aggregation prone metastable disease states, the most severe drawback of using an immobilized chaperonin bead system is that any intrinsic large-scale aggregation of the test protein in solution inhibits the facile kinetic capture of the target protein by the chaperonin, leading to a false positive result. To overcome this difficulty it was reasoned that a viable alternative and superior solution that avoids this premature aggregation problem prior to GroEL binding is to immobilize the protein substrate itself and probe the folding integrity of the target protein itself using surface plasmon resonance (SPR).
9. DETECTION OF SMALL MOLECULE STABILIZERS OF TRANSIENT FOLDS USING GROEL-SPR BASED SYSTEMS
Much of the past work done with surface plasmon resonance (SPR) and the GroE chaperonins focused on mechanism studies of chaperonin function (e.g. ATP dependent complex formation of the GroE complex and substrate release). There were however, a couple of instances where direct GroEL binding to a non-native polypeptide that was immobilized on the SPR chip surfaces was examined [107, 108]. These particular studies demonstrated that one can successfully attach a protein substrate onto a chip surface that is either partially unfolded or is in rapid equilibrium with a partially folded form and monitor the extent of binding of the high affinity (nucleotide free) GroEL protein onto these immobilized target proteins. Due to its large molecular mass (802kDa), any GroEL binding to the immobilized protein will result in a massive signal deflection in resonance signal and this signal is proportional to the amount of GroEL that binds and by default reveals the unfolded/partially folded character of the target proteins. Another added advantage of using the chaperonin as a detection for partially folded proteins is that this binding can be readily reversed by adding ATP to the GroEL-protein complex and the process can be repeated numerous times [107, 108. Murai et al. [108], showed that GroEL will bind to a partially folded from of a protein (reduced α-lactalbumin) coupled onto an SPR chip but most importantly from the standpoint of constructing our chaperonin dependent protein stabilizer detection system, they also noted that GroEL was unable to bind to the native folded state of this particular protein. The GroEL binding experiments with reduced and oxidized α-lactalbumin were verified using a BIACore 3000 SPR system and the results are shown in Fig. (4).
Fig. 4.

Using a similar setup reported by Murai et al., Surface Plasmon resonance technologies was used to determine if the immobilized form of protein alpha lactalbumin has acquired it’s proper fold compared to its partially folded form (disulfide reduced form) by comparing the extent of chaperonin binding to each species. When the protein has acquired its proper fold, there is a dramatic signal decline in SPR response units as chaperoninbinding decreases.
Typical binding constants of partially folded proteins binding to the high affinity nucleotide free form of GroEL approach antibody-antigen interactions in some instances [109]. Through simple mass action effects, dynamic proteins will bind and become sequestered as a partially folded state within the binding site of GroEL [102, 103]. When this dynamic equilibrium is perturbed in such a way as to shift protein populations towards larger steady-state concentrations of the non-native form by, for instance, introducing non-denaturing urea (1 M) or moderate heat (~ 45°C) the observed partitioning reaction accelerates dramatically [103, 109]. In all cases, GroEL dissociation is easily achieved by adding ATP and GroES or in our case, ATP and osmolytes [111]. This combination of activators facilitate refolding largely due to a decrease in GroEL binding affinity for partially folded hydrophobic proteins and effectively regenerates the SPR chip surface.
The binding of the chaperonin to the kinetically transient partially unfolded species in solution prevents refolding (and rebinding with oligomers) and results in a substantial decrease in protein misfolding and overall prevention of large-scale aggregation [104, 109]. Instead of the relying on hour time scales to measure downstream aggregates Fig. (2- Reaction 3), the high-affinity nucleotide-free GroEL chaperonin based capture approach traps and holds transient folds as they appear and prevents them from refolding. Most interestingly, oligomers that dissociate into monomers can be trapped by the high affinity chaperonin, preventing reassociation of those oligomers, resulting in an apparent acceleration in the disappearance of the oligomer. The ability of this form of the chaperonin to directly interrogate the partially unfolded species also allowed us to distinguish between the kinetic properties of different mutant proteins (See TTR examples [104]).
Attaching the target protein of interest to an immobilized support enables one to examine the stability/detection of the transient fold by determining how potential stabilizers affect the substrate partitioning onto the chaperonin in real time. To evaluate the effectiveness of a test compound at stabilizing mutant or native folds, the signal output differences between a chaperonin bound state and the decrease in this signal due to protein stabilization defines a readily observable signal range to evaluate large compound sets Fig. (7). Furthermore, one only has to follow the association kinetic profile differences rather than the complete association/dissociation kinetic profiles to score the potential effectiveness of the test compound ability to stabilize the fold of the protein of interest Fig. (5 and 7). False positive results (i.e. interference with chaperonin binding directly) can be easily ruled out by employing secondary screens with other known chaperonin (unrelated) protein substrates (e.g. using DHFR or reduced partially folded 3lactalbumin). Once a positive result is verified, a full evaluation of the binding equilibrium can be easily confirmed using standard biophysical binding measurements (e.g. ITC, fluorescence etc). This novel approach of evaluating protein stabilizers using a chaperonin based SPR detection system is supported by the observation that the real-time ligand stabilization of two of proteins DHFR (+NADPH) and CFTR-NBD1(+GTP), shows virtually no GroEL binding when these stabilizing ligands are present Fig. (6). As an aside, it is important to note that GTP does not interact with GroEL [104, 109]. Furthermore, the process of immobilizing the target substrate on the SPR chip, at least for the three systems evaluated here, do not appear to interfere with the ligand-protein stabilization itself. It is apparent from these examples that immobilizing the target protein onto the chip and probing the folding stability of the target protein substrate with the chaperonin appears to be highly efficacious. From the standpoint of screening larger combinatorial chemistry arrays for potential stabilizers of transiently stable missense mutants, protein target immobilization procedures used here avoids premature aggregation reactions, which normally impedes solution based protein stability measurements.
Fig. 7.

Base Assay readout for moderate/high-throughput protein stabilizer screens using Chaperonin-based SPR detection technology. Positive hits of potential stabilizers would result in a decrease in the Chaperonin based signal in the Association phase. Secondary screens with known Chaperonin substrates will rule out false positive outcomes (e.g. inhibition of chaperonin binding site).
Fig. 5.

In this schematic diagram, the basis of the SPR-chaperonin based stabilizing ligand detection system is presented. A) If the compound of interest does not bind to the immobilized protein, the chaperonin will readily bind to the immobilized protein, resulting in a large SPR signal. B) If the potential corrector/stabilizer (Ligand L now bound to the protein) binds to the protein of interest, than the chaperonin binding will diminish, resulting in a decrease in the SPR signal.
Fig. 6.

In Panel A, one can rapidly identify naturally occurring protein binding ligands that result in the stabilization of immobilized Dihydrofolate reductase (DHFR). As demonstrated by Viitanen et al., (1991), the presence of a stabilizing ligand such as NADPH results in a marked decline in DHFR binding to the GroEL chaperonin. This outcome results in a decrease in the SPR signal due to the decrease in GroEL chaperonin binding to the immobilized DHFR. In Panel B, the SPR signal from GroEL chaperonin binding to the destabilized, immobilized CFTR-NBD1 protein was significantly diminished when a known stabilizing ligand such as GTP was present in solution. Importantly, the GroEL chaperonin exclusively binds ATP and whereas GTP fails to bind to the chaperonin.
10. OUTLOOK
The experiments presented here indicate that the real time measurements of chaperonin binding to transiently stable proteins immobilized on SPR chips provides a viable platform system to rapidly screen and identify potential protein stabilizers that exist within large chemical compound arrays. In addition, this assay is not limited to evaluating well-behaved single domain proteins. The CFTR-NBD1 protein used in the above experiments readily aggregated at physiological pH values and moderate temperatures in the absence of its nucleotide stabilizer. It is also highly probable that this SPR attachment method will be useful in evaluating the stabilities of oligomeric proteins. In this latter situation, one may need to develop more specific immobilization procedures to create homogenous target protein populations with defined orientations. The use of specific attachment platforms involving His-tagged proteins with Ni-NTA chips, biotin-strepavidin or GST-glutathione SPR chips, for example, are just some of the few variations that are available to achieve specific orientations of oligomers.
Since chaperonin binding generates such a rather large SPR signal, one can use relatively low concentrations of target protein for assay development. This is particularly useful in situations where one has to work with limiting concentrations of missense disease proteins since these proteins often express low levels of soluble populations during over expression and production, making them difficult to purify in large quantities. Also, since the chaperonin binds to general hydrophobic residues as well as hydrophobic patches, the folding state of the target protein can vary from partially folded to near completely unfolded forms. Curiously, this promiscous hydrophobic binding property of GroEL for hydrophobic protein fragments and short peptides initially complicated early purification procedures (e.g see [106]). Here however, this promiscuous binding property is extremely useful for the general detection of exposed hydrophobic surfaces and for capturing transient folding intermediates. Furthermore, it is entirely possible that prior to screening, one is not limited to obtain a completely folded protein. For instance, one can specifically couple partially folded proteins or important folded fragments directly onto SPR surfaces. A similar strategy was previously employed to effectively fold difficult to fold partially folded protein substrates. In this particular instance, we used a folding osmolyte/chaperonin-assisted folding system to fold an orientation specific affinity-tagged chimeric protein directly on an immobilized bead surface [105].
The broad-based binding affinity of a chaperonin platform system should allow us to rapidly identify potential-pharmacological chaperones that display various modes of stabilization. It is highly conceivable for example, that one does not have to be limited with identifying potential small molecule pharmacological chaperones that only bind to protein active sites. The chaperonin detection system can potentially identify small molecule stabilizers that bind transiently at distant sites (resembling allosteric modulators) that facilitate specific folding nucleation events or stabilize dimer interfaces preventing dissociation (e.g. SOD mutants in Ray et al. 2005). Alternatively, it is also conceivable that this chaperonin-SPR assay setup could be used to screen for potential small molecule protein destabilizers rather than stabilizers. This latter approach could be useful for targeting and destabilizing activated oncogenic proteins.
With respect to stabilizing transient folds of misfolding disease proteins, it is worth noting that the chaperoning binding detection system described herein provides an advantage over simpler SPR based small-molecule binding to immobilized proteins alone for the following reasons. 1) The chaperonin based detection of protein stability may be more relevant to assess pharmacological chaperone stabilization of target proteins within the intracellular environment. In this instance, a small molecule binding and stabilization interaction that diminishes or removes stringent chaperonin binding or partitioning onto a target protein could certainly increase the steady state concentration of the target protein within the cellular environment. 2) The chaperonin based SPR platform not only provides a large signal that is easy to detect, it also directly monitors the loss or diminished exposure of the aggregation prone hydrophobic interaction surfaces on the target protein even in an immobilized state. 3) The chaperonin captures kinetically transient populations that may be hard to observe under normal physiological conditions. This is particularly true for disease proteins that slowly dissociate and aggregate. From a biotechnology screening standpoint, the assay costs for these system platforms will also decline since the chaperonin binding can be completely reversed when ATP and osmolytes are added. This procedure removes the once bound chaperonin Fig. (4, [106]), resulting in a regeneration of the target protein surface. Also, SPR setups can be easily coupled with robotic compound delivery systems to generate genuine automated protein stabilizer screening systems for transiently stable proteins. In summary, the most important advantage that this chaperonin-based SPR detection system offers to the protein folding disease field is that it is extremely broad-based in design, enabling researchers to rapidly identify viable protein stabilizer drug candidates for a wide variety of both common as well as rare protein folding disease systems.
Fig. 1.
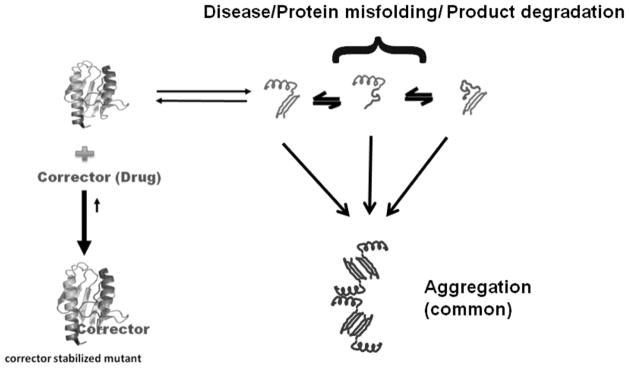
Rapid identification of potential stabilizers or therapeutics for folding diseases is difficult because disease proteins are often transiently stable. In this simplified reaction scheme, the folded protein of interest is in rapid equilibrium with a set of transient folding intermediates, which also have a tendency to aggregate. In the presence of a suitable corrector (stabilizing ligand), the dynamic equilibrium shifts towarda stable fold (lower left hand corner).
Table 2.
Mutants That Demonstrate Chemical Osmolyte Rescue
Acknowledgments
This work was supported Grant funds from the Institute for Advancing Medical Innovation (MTF), National Institutes of Health, National Institute for Allergy and Infectious Disease (NIAID) RO1 Al090085 (MTF), and from the National Center for Research Resources (5P20RR017708-10) and the National Institute of General Medical Sciences (8 P20 GM103420-10) from the National Institutes of Health (NZ, PG).
Footnotes
This article is published as part of a themed issue on Protein Misfolding in Conformational Disorders, Guest Edited by Cláudio M. Gomes (ITQB/UNL).
Send Orders of Print-Reprints and e-Reprints at reprints@benthamscience.net
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflicts of interest.
References
- 1.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4(1):10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 2.Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254(5038):1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 3.Onuchic JN, Luthey-Schulten Z, Wolynes PG. Theory of protein folding: the energy landscape perspective. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh K, Dill K. Cellular proteomes have broad distributions of protein stability. Biophys J. 2010;99(12):3996–4002. doi: 10.1016/j.bpj.2010.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morello JP, Petaja-Repo UE, Bichet DG, Bouvier M. Pharmacological chaperones: a new twist on receptor folding. Trends Pharmacol Sci. 2000;21(12):466–469. doi: 10.1016/s0165-6147(00)01575-3. [DOI] [PubMed] [Google Scholar]
- 6.Mu TW, Ong DS, Wang YJ, Balch WE, Yates JR, 3rd, Segatori L, Kelly JW. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell. 2008;134(5):769–781. doi: 10.1016/j.cell.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ringe D, Petsko GA. What are pharmacological chaperones and why are they interesting? J Biol. 2009;8(9):80. doi: 10.1186/jbiol186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajan RS, Tsumoto K, Tokunaga M, Tokunaga H, Kita Y, Arakawa T. Chemical and pharmacological chaperones: appli cation for recombinant protein production and protein folding diseases. Curr Med Chem. 2011;18(1):1–15. doi: 10.2174/092986711793979698. [DOI] [PubMed] [Google Scholar]
- 9.Henriques BJ, Fisher MT, Bross P, Gomes CM. A polymorphic position in electron transfer flavoprotein modulates kinetic stability as evidenced by thermal stress. FEBS Lett. 2011;585(3):505–510. doi: 10.1016/j.febslet.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 10.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 11.Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature. 1992;360(6405):672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 12.Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A. 1986;83(11):4044–4048. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung WY, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 14.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90(3):549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 15.Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O’Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63(4):827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- 16.Ron I, Horowitz M. ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum Mol Genet. 2005;14(16):2387–2398. doi: 10.1093/hmg/ddi240. [DOI] [PubMed] [Google Scholar]
- 17.Eng CM, Desnick RJ. Molecular basis of Fabry disease: mutations and polymorphisms in the human alpha-galactosidase A gene. Hum Mutat. 1994;3(2):103–111. doi: 10.1002/humu.1380030204. [DOI] [PubMed] [Google Scholar]
- 18.Mulders SM, Bichet DG, Rijss JP, Kamsteeg EJ, Arthus MF, Lonergan M, Fujiwara M, Morgan K, Leijendekker R, van der Sluijs P, van Os CH, Deen PM. An aquaporin-2 water channel mutant which causes autosomal dominant nephrogenic diabetes insipidus is retained in the Golgi complex. J Clin Invest. 1998;102(1):57–66. doi: 10.1172/JCI2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gejyo F, Homma N, Suzuki Y, Arakawa M. Serum levels of beta 2-microglobulin as a new form of amyloid protein in patients undergoing long-term hemodialysis. N Engl J Med. 1986;314(9):585–586. doi: 10.1056/NEJM198602273140920. [DOI] [PubMed] [Google Scholar]
- 20.Saraiva MJ, Birken S, Costa PP, Goodman DS. Amyloid fibril protein in familial amyloidotic polyneuropathy, Portuguese type. Definition of molecular abnormality in transthyretin (prealbumin) J Clin Invest. 1984;74(1):104–119. doi: 10.1172/JCI111390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waters PJ, Parniak MA, Akerman BR, Scriver CR. Characterization of phenylketonuria missense substitutions, distant from the phenylalanine hydroxylase active site, illustrates a paradigm for mechanism and potential modulation of phenotype. Mol Genet Metab. 2000;69(2):101–110. doi: 10.1006/mgme.2000.2965. [DOI] [PubMed] [Google Scholar]
- 22.Cai Y, Wang J, Ren C, Ittmann M. Frequent heterogeneous missense mutations of GGAP2 in prostate cancer: implications for tumor biology, clonality and mutation analysis. PLoS One. 2012;7(2):e32708. doi: 10.1371/journal.pone.0032708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stehr H, Jang SH, Duarte JM, Wierling C, Lehrach H, Lappe M, Lange BM. The structural impact of cancer-associated missense mutations in oncogenes and tumor suppressors. Mol Cancer. 2011;10:54. doi: 10.1186/1476-4598-10-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tomasson MH, Xiang Z, Walgren R, Zhao Y, Kasai Y, Miner T, Ries RE, Lubman O, Fremont DH, McLellan MD, Payton JE, Westervelt P, DiPersio JF, Link DC, Walter MJ, Graubert TA, Watson M, Baty J, Heath S, Shannon WD, Nagarajan R, Bloomfield CD, Mardis ER, Wilson RK, Ley TJ. Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood. 2008;111(9):4797–4808. doi: 10.1182/blood-2007-09-113027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minde DP, Anvarian Z, Rudiger SG, Maurice MM. Messing up disorder: how do missense mutations in the tumor suppressor protein APC lead to cancer? Mol Cancer. 2011;10:101. doi: 10.1186/1476-4598-10-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koutnikova H, Campuzano V, Koenig M. Maturation of wild-type and mutated frataxin by the mitochondrial processing peptidase. Hum Mol Genet. 1998;7(9):1485–1489. doi: 10.1093/hmg/7.9.1485. [DOI] [PubMed] [Google Scholar]
- 27.Pey AL, Ying M, Cremades N, Velazquez-Campoy A, Scherer T, Thony B, Sancho J, Martinez A. Identification of pharmacological chaperones as potential therapeutic agents to treat phenylketonuria. J Clin Invest. 2008;118(8):2858–2867. doi: 10.1172/JCI34355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sands MS, Davidson BL. Gene therapy for lysosomal storage diseases. Mol Ther. 2006;13(5):839–849. doi: 10.1016/j.ymthe.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Alt M, Caselmann WH. Liver-directed gene therapy: molecular tools and current preclinical and clinical studies. J Hepatol. 1995;23(6):746–758. doi: 10.1016/0168-8278(95)80044-1. [DOI] [PubMed] [Google Scholar]
- 30.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319(5865):916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 31.Bernado P, Blackledge M. Structural biology: Proteins in dynamic equilibrium. Nature. 2010;468(7327):1046–1048. doi: 10.1038/4681046a. [DOI] [PubMed] [Google Scholar]
- 32.Flotte TR, Zeitlin PL, Reynolds TC, Heald AE, Pedersen P, Beck S, Conrad CK, Brass-Ernst L, Humphries M, Sullivan K, Wetzel R, Taylor G, Carter BJ, Guggino WB. Phase I trial of intranasal and endobronchial administration of a recombinant adeno-associated virus serotype 2 (rAAV2)-CFTR vector in adult cystic fibrosis patients: a two-part clinical study. Hum Gene Ther. 2003;14(11):1079–1088. doi: 10.1089/104303403322124792. [DOI] [PubMed] [Google Scholar]
- 33.Moss RB, Milla C, Colombo J, Accurso F, Zeitlin PL, Clancy JP, Spencer LT, Pilewski J, Waltz DA, Dorkin HL, Ferkol T, Pian M, Ramsey B, Carter BJ, Martin DB, Heald AE. Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: a randomized placebo-controlled phase 2B trial. Hum Gene Ther. 2007;18(8):726–732. doi: 10.1089/hum.2007.022. [DOI] [PubMed] [Google Scholar]
- 34.Hannon GJ. RNA interference. Nature. 2002;418(6894):244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 35.Li C, Xiao P, Gray SJ, Weinberg MS, Samulski RJ. Combination therapy utilizing shRNA knockdown and an optimized resistant transgene for rescue of diseases caused by misfolded proteins. Proc Natl Acad Sci U S A. 2011;108(34):14258–14263. doi: 10.1073/pnas.1109522108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas PJ, Ko YH, Pedersen PL. Altered protein folding may be the molecular basis of most cases of cystic fibrosis. FEBS Lett. 1992;312(1):7–9. doi: 10.1016/0014-5793(92)81399-7. [DOI] [PubMed] [Google Scholar]
- 37.Pind S, Riordan JR, Williams DB. Participation of the endoplasmic reticulum chaperone calnexin (p88, IP90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1994;269(17):12784–12788. [PubMed] [Google Scholar]
- 38.Yang Y, Janich S, Cohn JA, Wilson JM. The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi nonlysosomal compartment. Proc Natl Acad Sci U S A. 1993;90(20):9480–9484. doi: 10.1073/pnas.90.20.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meacham GC, Lu Z, King S, Sorscher E, Tousson A, Cyr DM. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999;18(6):1492–1505. doi: 10.1093/emboj/18.6.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ward CL, Kopito RR. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem. 1994;269(41):25710–25718. [PubMed] [Google Scholar]
- 41.Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358(6389):761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- 42.Hulleman JD, Kaushal S, Balch WE, Kelly JW. Compromised mutant EFEMP1 secretion associated with macular dystrophy remedied by proteostasis network alteration. Mol Biol Cell. 2011;22(24):4765–4775. doi: 10.1091/mbc.E11-08-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown CR, Hong-Brown LQ, Biwersi J, Verkman AS, Welch WJ. Chemical chaperones correct the mutant phenotype of the delta F508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones. 1996;1(2):117–125. doi: 10.1379/1466-1268(1996)001<0117:ccctmp>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yancey PH, Clark ME, Hand SC, Bowlus RD, Somero GN. Living with water stress: evolution of osmolyte systems. Science. 1982;217(4566):1214–1222. doi: 10.1126/science.7112124. [DOI] [PubMed] [Google Scholar]
- 45.Bolen DW, Rose GD. Structure and energetics of the hydrogen-bonded backbone in protein folding. Annu Rev Biochem. 2008;77:339–362. doi: 10.1146/annurev.biochem.77.061306.131357. [DOI] [PubMed] [Google Scholar]
- 46.Jia LY, Gong B, Pang CP, Huang Y, Lam DS, Wang N, Yam GH. Correction of the disease phenotype of myocilin-causing glaucoma by a natural osmolyte. Invest Ophthalmol Vis Sci. 2009;50(8):3743–3749. doi: 10.1167/iovs.08-3151. [DOI] [PubMed] [Google Scholar]
- 47.Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: A potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc Natl Acad Sci U S A. 2000;97(4):1796–1801. doi: 10.1073/pnas.97.4.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tamarappoo BK, Yang B, Verkman AS. Misfolding of mutant aquaporin-2 water channels in nephrogenic diabetes insipidus. J Biol Chem. 1999;274(49):34825–34831. doi: 10.1074/jbc.274.49.34825. [DOI] [PubMed] [Google Scholar]
- 49.Song JL, Chuang DT. Natural osmolyte trimethylamine N-oxide corrects assembly defects of mutant branched-chain alpha-ketoacid decarboxylase in maple syrup urine disease. J Biol Chem. 2001;276(43):40241–40246. doi: 10.1074/jbc.M107242200. [DOI] [PubMed] [Google Scholar]
- 50.Vassilev PM, Guo L, Chen XZ, Segal Y, Peng JB, Basora N, Babakhanlou H, Cruger G, Kanazirska M, Ye C, Brown EM, Hediger MA, Zhou J. Polycystin-2 is a novel cation channel implicated in defective intracellular Ca(2+) homeostasis in polycystic kidney disease. Biochem Biophys Res Commun. 2001;282(1):341–350. doi: 10.1006/bbrc.2001.4554. [DOI] [PubMed] [Google Scholar]
- 51.Ohnishi T, Ohnishi K, Wang X, Takahashi A, Okaichi K. Restoration of mutant TP53 to normal TP53 function by glycerol as a chemical chaperone. Radiat Res. 1999;151(4):498–500. [PubMed] [Google Scholar]
- 52.Leandro P, Lechner MC, Tavares de Almeida I, Konecki D. Glycerol increases the yield and activity of human phenylalanine hydroxylase mutant enzymes produced in a prokaryotic expression system. Mol Genet Metab. 2001;73(2):173–178. doi: 10.1006/mgme.2001.3172. [DOI] [PubMed] [Google Scholar]
- 53.Deen PM, Croes H, van Aubel RA, Ginsel LA, van Os CH. Water channels encoded by mutant aquaporin-2 genes in nephrogenic diabetes insipidus are impaired in their cellular routing. J Clin Invest. 1995;95(5):2291–2296. doi: 10.1172/JCI117920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marr N, Bichet DG, Lonergan M, Arthus MF, Jeck N, Seyberth HW, Rosenthal W, van Os CH, Oksche A, Deen PM. Heteroligomerization of an Aquaporin-2 mutant with wild-type Aquaporin-2 and their misrouting to late endosomes/ lysosomes explains dominant nephrogenic diabetes insipidus. Hum Mol Genet. 2002;11(7):779–789. doi: 10.1093/hmg/11.7.779. [DOI] [PubMed] [Google Scholar]
- 55.Novak JE, Turner RS, Agranoff BW, Fisher SK. Differentiated human NT2-N neurons possess a high intracellular content of myoinositol. J Neurochem. 1999;72(4):1431–1440. doi: 10.1046/j.1471-4159.1999.721431.x. [DOI] [PubMed] [Google Scholar]
- 56.Gidalevitz T, Kikis EA, Morimoto RI. A cellular perspective on conformational disease: the role of genetic background and proteostasis networks. Curr Opin Struct Biol. 2010;20(1):23–32. doi: 10.1016/j.sbi.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 58.Morimoto RI. The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb Symp Quant Biol. 2011;76:91–99. doi: 10.1101/sqb.2012.76.010637. [DOI] [PubMed] [Google Scholar]
- 59.Morello JP, Salahpour A, Laperriere A, Bernier V, Arthus MF, Lonergan M, Petaja-Repo U, Angers S, Morin D, Bichet DG, Bouvier M. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J Clin Invest. 2000;105(7):887–895. doi: 10.1172/JCI8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Janovick JA, Goulet M, Bush E, Greer J, Wettlaufer DG, Conn PM. Structure-activity relations of successful pharma cologic chaperones for rescue of naturally occurring and manu factured mutants of the gonadotropin-releasing hormone receptor. J Pharmacol Exp Ther. 2003;305(2):608–614. doi: 10.1124/jpet.102.048454. [DOI] [PubMed] [Google Scholar]
- 61.Davies JW, Glick M, Jenkins JL. Streamlining lead discovery by aligning in silico and high-throughput screening. Curr Opin Chem Biol. 2006;10(4):343–351. doi: 10.1016/j.cbpa.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 62.Nowak RJ, Cuny GD, Choi S, Lansbury PT, Ray SS. Improving binding specificity of pharmacological chaperones that target mutant superoxide dismutase-1 linked to familial amyo trophic lateral sclerosis using computational methods. J Med Chem. 2010;53(7):2709–2718. doi: 10.1021/jm901062p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA, Packman J, Powers ET, Wiseman RL, Foss TR, Wilson IA, Kelly JW, Labaudiniere R. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109(24):9629–9634. doi: 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cohen FE, Kelly JW. Therapeutic approaches to protein-misfolding diseases. Nature. 2003;426(6968):905–909. doi: 10.1038/nature02265. [DOI] [PubMed] [Google Scholar]
- 65.Lieberman RL, D’Aquino JA, Ringe D, Petsko GA. Effects of pH and iminosugar pharmacological chaperones on lysosomal glycosidase structure and stability. Biochemistry (Mosc) 2009;48(22):4816–4827. doi: 10.1021/bi9002265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lieberman RL, Wustman BA, Huertas P, Powe AC, Jr, Pine CW, Khanna R, Schlossmacher MG, Ringe D, Petsko GA. Structure of acid beta-glucosidase with pharmacological chaperone provides insight into Gaucher disease. Nat Chem Biol. 2007;3(2):101–107. doi: 10.1038/nchembio850. [DOI] [PubMed] [Google Scholar]
- 67.Ray SS, Nowak RJ, Brown RH, Jr, Lansbury PT., Jr Small-molecule-mediated stabilization of familial amyotrophic lateral sclerosis-linked superoxide dismutase mutants against unfolding aggregation. Proc Natl Acad Sci U S A. 2005;102(10):3639–3644. doi: 10.1073/pnas.0408277102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pedemonte N, Lukacs GL, Du K, Caci E, Zegarra-Moran O, Galietta LJ, Verkman AS. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest. 2005;115(9):2564–2571. doi: 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Van Goor F, Straley KS, Cao D, Gonzalez J, Hadida S, Hazlewood A, Joubran J, Knapp T, Makings LR, Miller M, Neuberger T, Olson E, Panchenko V, Rader J, Singh A, Stack JH, Tung R, Grootenhuis PD, Negulescu P. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol. 2006;290(6):L1117–1130. doi: 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- 70.Carlile GW, Robert R, Zhang D, Teske KA, Luo Y, Hanrahan JW, Thomas DY. Correctors of protein trafficking defects identified by a novel high-throughput screening assay. Chembiochem. 2007;8(9):1012–1020. doi: 10.1002/cbic.200700027. [DOI] [PubMed] [Google Scholar]
- 71.Lee LL, Ha H, Chang YT, DeLisa MP. Discovery of amyloid-beta aggregation inhibitors using an engineered assay for intracellular protein folding solubility. Protein Sci. 2009;18(2):277–286. doi: 10.1002/pro.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ignatova Z, Gierasch LM. Inhibition of protein aggregation in vitro and in vivo by a natural osmoprotectant. Proc Natl Acad Sci U S A. 2006;103(36):13357–13361. doi: 10.1073/pnas.0603772103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, Salemme FR. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen. 2001;6(6):429–440. doi: 10.1177/108705710100600609. [DOI] [PubMed] [Google Scholar]
- 74.Cummings MD, Farnum MA, Nelen MI. Universal screening methods and applications of ThermoFluor. J Biomol Screen. 2006;11(7):854–863. doi: 10.1177/1087057106292746. [DOI] [PubMed] [Google Scholar]
- 75.Lavinder JJ, Hari SB, Sullivan BJ, Magliery TJ. High-throughput thermal scanning: a general, rapid dye-binding thermal shift screen for protein engineering. J Am Chem Soc. 2009;131(11):3794–3795. doi: 10.1021/ja8049063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Janovick JA, Park BS, Conn PM. Therapeutic rescue of misfolded mutants: validation of primary high throughput screens for identification of pharmacoperone drugs. PLoS ONE. 2011;6(7):e22784. doi: 10.1371/journal.pone.0022784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mayer MP, Schroder H, Rudiger S, Paal K, Laufen T, Bukau B. Multistep mechanism of substrate binding determines chaperone activity of Hsp70. Nat Struct Biol. 2000;7(7):586–593. doi: 10.1038/76819. [DOI] [PubMed] [Google Scholar]
- 78.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11(8):579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Krukenberg KA, Street TO, Lavery LA, Agard DA. Conformational dynamics of the molecular chaperone Hsp90. Q Rev Biophys. 2011;44(2):229–255. doi: 10.1017/S0033583510000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sittler A, Lurz R, Lueder G, Priller J, Lehrach H, Hayer-Hartl MK, Hartl FU, Wanker EE. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum Mol Genet. 2001;10(12):1307–1315. doi: 10.1093/hmg/10.12.1307. [DOI] [PubMed] [Google Scholar]
- 81.Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science. 2002;295(5556):865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- 82.Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U S A. 2003;100(2):721–726. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jinwal UK, Miyata Y, Koren J, 3rd, Jones JR, Trotter JH, Chang L, O’Leary J, Morgan D, Lee DC, Shults CL, Rousaki A, Weeber EJ, Zuiderweg ER, Gestwicki JE, Dickey CA. Chemical manipulation of hsp70 ATPase activity regulates tau stability. J Neurosci. 2009;29(39):12079–12088. doi: 10.1523/JNEUROSCI.3345-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barnett ME, Nagy M, Kedzierska S, Zolkiewski M. The aminoterminal domain of ClpB supports binding to strongly aggregated proteins. J Biol Chem. 2005;280(41):34940–34945. doi: 10.1074/jbc.M505653200. [DOI] [PubMed] [Google Scholar]
- 85.Weibezahn J, Tessarz P, Schlieker C, Zahn R, Maglica Z, Lee S, Zentgraf H, Weber-Ban EU, Dougan DA, Tsai FT, Mogk A, Bukau B. Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell. 2004;119(5):653–665. doi: 10.1016/j.cell.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 86.Schlieker C, Tews I, Bukau B, Mogk A. Solubilization of aggregated proteins by ClpB/DnaK relies on the continuous extraction of unfolded polypeptides. FEBS Lett. 2004;578(3):351–356. doi: 10.1016/j.febslet.2004.11.051. [DOI] [PubMed] [Google Scholar]
- 87.Lum R, Tkach JM, Vierling E, Glover JR. Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J Biol Chem. 2004;279(28):29139–29146. doi: 10.1074/jbc.M403777200. [DOI] [PubMed] [Google Scholar]
- 88.Schlieker C, Weibezahn J, Patzelt H, Tessarz P, Strub C, Zeth K, Erbse A, Schneider-Mergener J, Chin JW, Schultz PG, Bukau B, Mogk A. Substrate recognition by the AAA+ chaperone ClpB. Nat Struct Mol Biol. 2004;11(7):607–615. doi: 10.1038/nsmb787. [DOI] [PubMed] [Google Scholar]
- 89.Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+] Science. 1995;268(5212):880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- 90.Doyle SM, Shorter J, Zolkiewski M, Hoskins JR, Lindquist S, Wickner S. Asymmetric deceleration of ClpB or Hsp104 ATPase activity unleashes protein-remodeling activity. Nat Struct Mol Biol. 2007;14(2):114–122. doi: 10.1038/nsmb1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Estorninho M, Smith H, Thole J, Harders-Westerveen J, Kierzek A, Butler RE, Neyrolles O, Stewart GR. ClgR regulation of chaperone and protease systems is essential for Mycobacterium tuberculosis parasitism of the macrophage. Microbiology. 2010;156(Pt 11):3445–3455. doi: 10.1099/mic.0.042275-0. [DOI] [PubMed] [Google Scholar]
- 92.de Koning-Ward TF, Gilson PR, Boddey JA, Rug M, Smith BJ, Papenfuss AT, Sanders PR, Lundie RJ, Maier AG, Cowman AF, Crabb BS. A newly discovered protein export machine in malaria parasites. Nature. 2009;459(7249):945–949. doi: 10.1038/nature08104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Krobitsch S, Clos J. A novel role for 100 kD heat shock proteins in the parasite Leishmania donovani. Cell Stress Chaperones. 1999;4(3):191–198. doi: 10.1379/1466-1268(1999)004<0191:anrfkh>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim KI, Cheong GW, Park SC, Ha JS, Woo KM, Choi SJ, Chung CH. Heptameric ring structure of the heat-shock protein ClpB, a protein-activated ATPase in Escherichia coli. J Mol Biol. 2000;303(5):655–666. doi: 10.1006/jmbi.2000.4165. [DOI] [PubMed] [Google Scholar]
- 95.Parsell DA, Kowal AS, Lindquist S. Saccharomyces cerevisiae Hsp104 protein. Purification and characterization of ATP-induced structural changes. J Biol Chem. 1994;269(6):4480–4487. [PubMed] [Google Scholar]
- 96.Schlee S, Groemping Y, Herde P, Seidel R, Reinstein J. The chaperone function of ClpB from Thermus thermophilus depends on allosteric interactions of its two ATP-binding sites. J Mol Biol. 2001;306(4):889–899. doi: 10.1006/jmbi.2001.4455. [DOI] [PubMed] [Google Scholar]
- 97.Watanabe YH, Motohashi K, Yoshida M. Roles of the two ATP binding sites of ClpB from Thermus thermophilus. J Biol Chem. 2002;277(8):5804–5809. doi: 10.1074/jbc.M109349200. [DOI] [PubMed] [Google Scholar]
- 98.Zolkiewski M. ClpB cooperates with DnaK, DnaJ, and GrpE in suppressing protein aggregation. A novel multi-chaperone system from Escherichia coli. J Biol Chem. 1999;274(40):28083–28086. doi: 10.1074/jbc.274.40.28083. [DOI] [PubMed] [Google Scholar]
- 99.Van Dyk TK, Gatenby AA, LaRossa RA. Demonstration by genetic suppression of interaction of GroE products with many proteins. Nature. 1989;342(6248):451–453. doi: 10.1038/342451a0. [DOI] [PubMed] [Google Scholar]
- 100.Tokuriki N, Tawfik DS. Chaperonin overexpression promotes genetic variation and enzyme evolution. Nature. 2009;459(7247):668–673. doi: 10.1038/nature08009. [DOI] [PubMed] [Google Scholar]
- 101.Chapman E, Farr GW, Usaite R, Furtak K, Fenton WA, Chaudhuri TK, Hondorp ER, Matthews RG, Wolf SG, Yates JR, Pypaert M, Horwich AL. Global aggregation of newly translated proteins in an Escherichia coli strain deficient of the chaperonin GroEL. Proc Natl Acad Sci U S A. 2006;103(43):15800–15805. doi: 10.1073/pnas.0607534103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Viitanen PV, Donaldson GK, Lorimer GH, Lubben TH, Gatenby AA. Complex interactions between the chaperonin 60 molecular chaperone and dihydrofolate reductase. Biochemistry (Mosc) 1991;30(40):9716–9723. doi: 10.1021/bi00104a021. [DOI] [PubMed] [Google Scholar]
- 103.Smith KE, Voziyan PA, Fisher MT. Partitioning of rhodanese onto GroEL. Chaperonin binds a reversibly oxidized form derived from the native protein. J Biol Chem. 1998;273(44):28677–28681. doi: 10.1074/jbc.273.44.28677. [DOI] [PubMed] [Google Scholar]
- 104.Naik S, Haque I, Degner N, Kornilayev B, Bomhoff G, Hodges J, Khorassani AA, Katayama H, Morris J, Kelly J, Seed J, Fisher MT. Identifying protein stabilizing ligands using GroEL. Biopolymers. 2010;93(3):237–251. doi: 10.1002/bip.21319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Katayama H, McGill M, Kearns A, Brzozowski M, Degner N, Harnett B, Kornilayev B, Matkovic-Calogovic D, Holyoak T, Calvet JP, Gogol EP, Seed J, Fisher MT. Strategies for folding of affinity tagged proteins using GroEL and osmolytes. J Struct Funct Genomics. 2009;10(1):57–66. doi: 10.1007/s10969-008-9053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Voziyan PA, Jadhav L, Fisher MT. Refolding a glutamine synthetase truncation mutant in vitro: identifying superior condi tions using a combination of chaperonins and osmolytes. J Pharm Sci. 2000;89(8):1036–1045. doi: 10.1002/1520-6017(200008)89:8<1036::aid-jps8>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 107.Lin Z, Eisenstein E. Nucleotide binding-promoted confor mational changes release a nonnative polypeptide from the Escherichia coli chaperonin GroEL. Proc Natl Acad Sci U S A. 1996;93(5):1977–1981. doi: 10.1073/pnas.93.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Murai N, Taguchi H, Yoshida M. Kinetic analysis of interactions between GroEL and reduced alpha-lactalbumin. Effect of GroES and nucleotides. J Biol Chem. 1995;270(34):19957–19963. doi: 10.1074/jbc.270.34.19957. [DOI] [PubMed] [Google Scholar]
- 109.Fenton WA, Horwich AL. GroEL-mediated protein folding. Protein Sci. 1997;6(4):743–760. doi: 10.1002/pro.5560060401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Martin J, Horwich AL, Hartl FU. Prevention of protein denaturation under heat stress by the chaperonin Hsp60. Science. 1992;258(5084):995–998. doi: 10.1126/science.1359644. [DOI] [PubMed] [Google Scholar]
- 111.Voziyan PA, Fisher MT. Chaperonin-assisted folding of glutamine synthetase under nonpermissive conditions: off-pathway aggregation propensity does not determine the co-chaperonin requirement. Protein Sci. 2000;9(12):2405–2412. doi: 10.1110/ps.9.12.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]