

Abstract
Fungal natural products are a rich resource for bioactive molecules. To fully exploit this potential it is necessary to link genes to metabolites. Genetic information for numerous putative biosynthetic pathways has become available in recent years through genome sequencing. However, the lack of solid methodology for genetic manipulation of most species severely hampers pathway characterization. Here we present a simple PCR based approach for heterologous reconstitution of intact gene clusters. Specifically, the putative gene cluster responsible for geodin production from Aspergillus terreus was transferred in a two step procedure to an expression platform in A. nidulans. The individual cluster fragments were generated by PCR and assembled via efficient USER fusion prior to transformation and integration via re-iterative gene targeting. A total of 13 open reading frames contained in 25 kb of DNA were successfully transferred between the two species enabling geodin synthesis in A. nidulans. Subsequently, functions of three genes in the cluster were validated by genetic and chemical analyses. Specifically, ATEG_08451 (gedC) encodes a polyketide synthase, ATEG_08453 (gedR) encodes a transcription factor responsible for activation of the geodin gene cluster and ATEG_08460 (gedL) encodes a halogenase that catalyzes conversion of sulochrin to dihydrogeodin. We expect that our approach for transferring intact biosynthetic pathways to a fungus with a well developed genetic toolbox will be instrumental in characterizing the many exciting pathways for secondary metabolite production that are currently being uncovered by the fungal genome sequencing projects.
Introduction
Fungal natural products constitute a rich resource for bioactive secondary metabolites [1], [2]. To fully exploit this potential, it is essential to identify the genes required for the biosynthesis of these compounds. This process is becoming progressively easier due to the rapidly increasing number of fungal genomes that have been fully sequenced; and since the genes involved in the production of a given secondary metabolite often cluster together in the same region of a chromosome [1]–[3]. Importantly, the genome sequencing projects have revealed that the number of putative gene clusters for secondary metabolite production greatly exceeds the number of known natural products in a given fungus, hence, indicating that most fungal compounds are yet to be discovered.
The prerequisite for genetic exploration of the huge reservoir of undiscovered biosynthetic pathways is solid methodologies for cultivating, propagating and genetically manipulating the producing species. However, the vast majority of newly sequenced organisms fail to meet these requirements, hence, hampering pathway elucidation and exploitation. An attractive solution to this problem is to transfer pathways into another fungus where the methodology is well established. This approach has been used successfully to investigate several gene clusters [4]–[6]. All of these studies apply a strategy where individual genes in a cluster are PCR amplified, cloned, and integrated sequentially into either a random or defined locus. One advantage of this strategy is that it allows easy engineering of individual genes prior to integration in the host strain. In addition, it is possible to insert the foreign gene(s) into a well characterized locus that supports high expression levels [7]. Inserting the genes into a known locus also simplifies strain validation. In a recent example of this strategy, Itoh et al. [6] transferred five genes from Aspergillus fumigatus to A. oryzae allowing the authors to deduce the biosynthetic route for the meroterpenoid pyropyripene A. However, the strategy may be limited to reconstitution of simple pathways depending on only a small number of genes since assembly of multistep pathways will require several rounds of tedious iterative integration steps or enough genetic markers.
For multistep pathways it is therefore desirable to transfer entire gene clusters from the natural producer to the expression host in one or a few steps. This requires a host, which can efficiently express all genes in the cluster and correctly splice the resulting transcripts. Assembly of multiple genes and PCR fragments can efficiently be performed in Saccharomyces cerevisiae by recombination based methods [8]. However, the demand for splicing disfavors S. cerevisiae as expression host for this gene transfer strategy as only little splicing occurs in yeast. In contrast, correct and efficient splicing of heterologous transcripts by another filamentous fungus is likely. Activation of the cluster in the new host requires as a minimum that the chromatin structure at the insertion site is in an open configuration and that transcription factors exist that recognize the individual promoters in the cluster. The latter may be facilitated by the fact that many clusters appear to contain one or more genes that encode transcription factors. In a pioneering study, Bergmann et al. showed that expression of such a gene activated the entire aspyridone gene cluster in A. nidulans [9]. The potential of transferring entire clusters to an expression host has been demonstrated by Sakai et al. who managed to produce citrinin in A. oryzae [10]. To obtain this feat, they isolated and transformed a cosmid from a Monascus purpureus library containing all six genes required for production of citrinin into this production host. Here constitutive expression of the citrinin pathway regulator encoded by ctnA dramatically increased citrinin production in the heterologous host. However, construction and screening of cosmid libraries is not simple and a versatile PCR based method that facilitates the transfer of entire gene clusters from the natural producer to the expression host is desirable.
We have previously developed a versatile PCR based expression platform that can be used for heterologous expression in A. nidulans of one or a pair of genes from the defined locus IS1, which supports a high level of gene expression [4], [7]. Here, we demonstrate how this platform can be expanded to allow transfer of an entire gene cluster from another fungus into IS1.
Results and Discussion
2.1 Method for PCR based reconstruction of fungal gene clusters in a heterologous host
Our method for transfer of large DNA fragments relies on successive gene targeting events that introduce ∼15 kb fragments into a defined locus. In the example of the method presented here, we transfer a gene cluster into IS1 taking advantage of a vector set we have previously developed for this purpose [7]. In our method, fragments covering the entire gene cluster are PCR amplified, combined via USER fusion into ∼15 kb fragments, and inserted into the integration vector by USER cloning (Figure 1A) [11], [12]. The first fragment to be integrated is assembled in the vector and integrated into IS1 as we have described previously for integration of single genes [7]. The following fragments are integrated as an extension of the previous one by using one of two different markers, argB and pyrG, for selection. In principle, an indefinite number of integrations can be done, since the marker from the previous integration is excised as the new fragment with the other marker integrates (Figure 1B). This principle is referred to as re-iterative gene targeting [13]. Importantly, marker replacements allows for a simple selection scheme for identification of correctly targeted strains. If, as in our case, the pyrG marker is flanked by a direct repeat, we recommend to use the pyrG marker in the last integration step, as the pyrG marker subsequently can be removed by direct repeat recombination if desirable [14]. In this manner both markers are available in the finalized strain providing a marker repertoire for additional genetic engineering.
Figure 1. Schematic overview of the PCR based USER cloning strategy for transfer of entire gene clusters from one fungus to another.

In the illustrated case, the geodin gene cluster in A. terreus is PCR amplified, cloned, and integrated into the IS1 locus in A. nidulans. A) ORFs GedA-GedL are depicted as arrows. The yellow and green arrows represent the ORFs encoding the transcription factor and the PKS, respectively. Remaining ORFs are represented by red arrows. Arrow size is proportional to ORF length and arrow direction indicates genomic orientation. Numbers above the gene cluster specify sequence in base pairs. Genomic DNA fragments and cloning vectors are amplified as PCR products using primers extended with uracil-containing tails. The tails contain matching sequences (indicated by identical colors) allowing for PCR product assembly in a single USER Fusion reaction. For the geodin cluster, all putative ORFs are fused into two fragments, which are individually inserted into a vector prepared for gene targeting. Blue boxes labeled up (upstream) and dw (downstream) represent targeting sequences for homologous recombination into IS1 in the first gene-targeting event. The targeting sequences in the second integration event are represented in gray and blue and consist of the overlapping region between Fragment 1 and 2 and the downstream part of IS1, respectively. Genetic markers used for selection are depicted in orange (argB) and purple (AFpyrG). The sizes of uracil-containing tails, vector elements and PgpdA fragment are not drawn to scale. B) The first gene-targeting event introduces the first fragment into IS1 by homologous recombination between IS1 up and down-sequences as indicated. The second gene-targeting event introduces the second fragment using the overlapping region of the Fragment 1 and 2 (gray) and the downstream section of IS1 as targeting sequences. Note that additional DNA can be inserted in subsequent gene-targeting events. For example, a third fragment can be inserted by using the downstream end of fragment 2 and the downstream region of IS1 as targeting sequences. See text for details concerning use and recycling of markers.
In the present study we demonstrate the potential of our method by transferring the geodin gene cluster from A. terreus to A. nidulans. This cluster was chosen, firstly, because it contains a gene encoding a putative transcription factor, which potentially could facilitate activation of the other genes in the cluster. Secondly, the biosynthetic pathway for geodin production is partially characterized (Figure 2) [15]–[21], which simplifies the delineation of the cluster size. Thirdly, the geodin pathway shares several steps with the monodictyphenone pathway including production of several common intermediates/products e.g. emodin [22]. We therefore envisioned, that the chance of producing geodin in A. nidulans would be increased, as the corresponding endogenous enzymes could complement geodin enzymes that might not be functional. Moreover, shared intermediates would likely be non-toxic to the host.
Figure 2. Proposed pathway for geodin production.
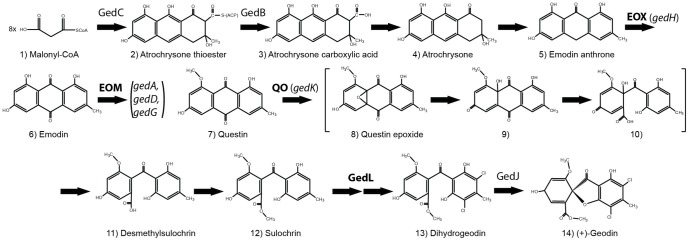
The PKS (ACTS, [20]), thioesterase (ACTE, [20]) and dihydrogeodin oxidase previously linked to genes as well as the sulochrin halogenase identified in this study (highlighted in bold) are denoted by their ged-annotation. Enzymatic reactions for which the enzyme has been characterized but the gene not identified are marked in bold as EOX = emodin anthrone oxygenase, EOM = emodin-O-methyltransferase and QO = questin oxygenase. Reactions involving compounds 8-10 shown in brackets are inferred reactions proposed by Henry and Townsend based on a similar intra-molecular rearrangement in aflatoxin biosynthesis [26], [27].
2.2 Delineation of the putative geodin producing gene cluster in A. terreus
Three enzymes involved in geodin production have previously been linked to genes. Specifically, dihydrogeodin oxidase, a polyketide synthase, and a thioesterase are encoded by ATEG_08458 [18], ATEG_08451 [19]–[21], and ATEG_08450 [20], respectively, see Aspergillus Comparative Sequencing Project database (Broad Institute of Harvard and MIT, http://www.broadinstitute.org/). As indicated by the gene numbers these genes localize in close proximity to each other strongly suggesting that a gene cluster responsible for production of geodin exists. Three additional enzymes required for geodin biosynthesis have been characterized biochemically in vitro: emodin anthrone oxygenase [15], emodin O-methyltransferase [16] and questin oxygenase [17]. Moreover, the occurrence of chlorine atoms in geodin suggests the involvement of a halogenase.
To explore the possibility that genes encoding these four enzymatic activities were also present in this region, we examined all annotated open reading frames (ORFs) positioned between ATEG_08458 and ATEG_08450, as well as 20 kb upstream of ATEG_08458 and downstream of ATEG_08450. Among the ORFs in this region, none had a functional annotation corresponding to these enzymatic activities. We therefore subjected all annotated ORFs in this regions to a functional prediction using the BLAST algorithm [23] from NCBI and the HHpred software [24]. This analysis uncovered three genes that could encode putative methyltransferases (ATEG_08449, ATEG_08452 and ATEG_08456), one ORF that may encode an oxygenase carrying out a Baeyer-Villiger oxidation (ATEG_08459), and a putative halogenase (ATEG _08460), see Table 1.
Table 1. Summary of characterized and putative ORFs in the A. terreus geodin gene cluster.
| BROAD annotation | Proposed annotation | Enzyme class | Validation | Reference |
| ATEG_08449 | GedA | Putative O-methyl transferase* | BLAST, Hhpred | This study |
| ATEG_08450 | GedB | β-lactamase type thioesterase | Heterologous expression, in vitro assays | [20] |
| ATEG_08451 | GedC | Polyketide synthase | Deletion mutants, heterologous expression, in vitro assays | [19]–[21] |
| ATEG_08452 | GedD | Putative O-methyl transferase* | BLAST, Hhpred | This study |
| ATEG_08453 | GedR | Putative transcription factor | Deletion mutant, quantitative RT-PCR | This study |
| ATEG_08454 | GedE | Putative gluthathione-S-transferase | Annotation from BROAD, BLAST, Hhpred | This study |
| ATEG_08455 | GedF | Putative oxidoreductase | BLAST, Hhpred | This study |
| ATEG_08456 | GedG | Putative SAM-dependent-methyltransferase* | BLAST, Hhpred | This study |
| ATEG_08457-2 | GedH | Putative emodin anthrone oxidase, similar to HypC | BLAST, 34% amino acid identity | This study |
| ATEG_08457-1 | GedI | Putative mdpH homolog | BLAST, 46% amino acid identity | This study |
| ATEG_08458 | GedJ | Dihydrogeodin oxidase | Enzymatic assays, protein sequencing | [18] |
| ATEG_08459 | GedK | Putative Bayer Villiger-type oxidase | Enzymatic assays, Identity inferred in this study | [17] |
| ATEG_08460 | GedL | Sulochrin halogenase | Deletion mutant, functional complementation | This study |
All similarity percentages indicate identities at the amino acid level.
One of these three putative ORFs is likely to encode the emodin O-methyltransferase described by Chen et al [16].
Unexpectedly, none of the annotated ORFs were found to encode the emodin anthrone oxygenase (Figure 2). To investigate this apparent dilemma, we searched the literature for other oxygenases catalyzing a similar reaction. Via this effort, we found an oxygenase that catalyzes conversion of norsolorinic acid anthrone to norsolorinic acid, a step towards aflatoxin production in A. flavus [25]. This recently identified enzyme is encoded by the gene hypC. Inspired by these findings, we used the sequence of HypC to conduct pair-wise alignments to putative proteins encoded by alternative ORFs in the proposed geodin gene cluster. One short putative ORF encodes a protein of 150 amino acid residues with an overall identity of 34% with the 210 residues of HypC. Moreover, the conserved amino acid residues were primarily positioned in catalytic regions or conserved domains (Figure S1, [25]). Interestingly, the putative ORF is oriented in the opposite direction of ATEG_08457. This strongly indicates that the region at ATEG_08457 is wrongly annotated and contains two separate ORFs that we now denote ATEG_08457-1 (the originally annotated ATEG_08457.1) and ATEG_08457-2 (the new putative HypC homolog). Specifically, we suggest that ATEG_08457-2 and ATEG_08457-1 are positioned on A. terreus supercontig 12, base pairs 1307175-1307627 and 1308053-1308540, respectively.
Finally, we inspected the remaining ORFs in the region for activities relevant for production of geodin. Among these, one (ATEG_08454) was functionally annotated as a gluthatione-S-transferase and two ORFs (ATEG_08455 and ATEG_08457-1) uncovered by the BLAST analysis displayed similarity to oxidoreductases and MdpH, respectively. The latter is a protein of unknown function required for emodin synthesis in A. nidulans [22]. To substantiate our predictions of the involvement of these putative genes, we conferred literature on similar biosynthetic pathways. The requirement for an oxidoreductase in geodin biosynthesis has previously been proposed by Henry and Townsend [26], [27], while Simpson suggested the involvement of both a glutathione-S-transferase and an oxidoreductase in the biosynthesis of xanthones in A. nidulans [28].
In addition to genes involved in the biosynthetic steps towards geodin, we noticed the presence of a gene, ATEG_08453, which encodes a putative transcription factor. The position of this gene within the putative geodin gene cluster suggests that it could regulate the activity of all genes in the cluster. In summary, our analysis suggests that the geodin gene cluster spans 25 kb and contains 13 putative ORFs (Figure 1A). Gene numbers, functional predictions and published data resulting from the entire analysis are presented in Table 1.
2.3 Strategy for transferring the geodin cluster from A. terreus into A. nidulans
Two vectors, containing 12 kb (Fragment 1) and 15 kb (Fragment 2) of the putative geodin gene cluster, respectively, were constructed by USER fusion by merging four individual PCR fragments in the first vector and seven PCR fragments in the second (Figure 1A). Assembling the geodin pathway from PCR fragments offers the possibility of introducing defined changes in the DNA sequence prior to integration via the many tools and methods for PCR based genetic engineering. In the present case, we inserted the strong constitutive promoter, PgpdA, of A. nidulans upstream of ATEG_08453, which encodes the putative transcription factor described above, with the intention that this modification would activate transcription of all genes in the geodin cluster after its integration into IS1. Importantly, a fragment of the geodin cluster (2 kb) is included in both constructs to serve as the upstream targeting sequence for the second gene-targeting event as illustrated in Figure 1B.
2.4 Production of geodin in A. nidulans
Using the two vectors constructed above, the putative geodin gene cluster (ged) was successfully transferred to an A. nidulans reference strain as well as to a strain where the entire monodictyphenone gene cluster (mdpA-LΔ) had been deleted. Geodin production in the mdpA-LΔ strains would indicate that all genes in the geodin cluster are functionally expressed, while geodin formation in the reference strain could be mediated via metabolites produced in the monodictyphenone pathway. The ability of the recombinant strains to produce geodin on minimal medium was analyzed by UHPLC-HRMS. The presence of geodin in fungal extracts was identified by comparison of retention time, accurate mass spectra, and isotope ratio to an authentic geodin standard. These analyses demonstrated that both ged+ and ged+ mdpA-LΔ produced geodin (Figure 3A). Consequently, we suggest that the putative transcription factor, ATEG_08453, is renamed gedR and that the enzymes in the cluster (ATEG_08449-08452 + ATEG_08454-08460) are renamed gedA-L. In different experiments, we observed that the amount of geodin is reproducibly higher in the ged+ strain (40–70 µg/plate) as compared to the amount in the ged+ mdpA-LΔ strain (2–4 µg/plate), which does not produce emodin via the mdp cluster. We therefore speculate that natively produced emodin, or other intermediates towards emodin production, could be converted into geodin in the ged+-strains.
Figure 3. Production of geodin in A. nidulans ged+ strains.

A) Left panels depict extracted ion chromatograms (ESI-) of geodin m/z 396.9876 ± 0.005 amu from fungal extracts of ged+, ged+ mdpA-LΔ and reference strains (Bruker maXis system). An authentic geodin standard is included for comparison. The mass spectra of the putative geodin peak in ged+ and the authentic geodin standard are depicted in panels to the right. B) and C) ESI− chromatograms of geodin m/z 396.9876 ± 0.005 amu (B)) and sulochrin m/z 331.0812± 0.005 amu (C)) extracted from ged+ mdpA-LΔ (grey), ged+ mdpA-LΔ gedLΔ, (blue), ged+ mdpA-LΔ gedCΔ (purple) and ged+ mdpA-LΔ gedRΔ (red).
2.5 Genetic characterization of the geodin cluster in A. nidulans
One reason for transferring a gene cluster into a host with a well-developed genetic toolbox is the possibility for further characterization of the cluster. To demonstrate this possibility, we decided to investigate the functionality of three key genes in the cluster, gedC, gedR and gedL encoding the PKS, the putative regulator, and the putative halogenase, respectively. We focused our efforts on the ged+ mdpA-LΔ strains, as they provide a genetic background with no risk of complementation by mdp enzymes. UHPLC-HRMS analysis of strains grown on minimal medium revealed that all three deletion strains were unable to synthesize geodin (Figure 3B), thereby confirming that geodin is indeed produced from the reconstituted cluster and that the corresponding proteins of all three genes were functional in A. nidulans and play a role in geodin biosynthesis. We note the presence of a co-eluting isobaric compound, seen as the broad peak (6.7–7.3 min) in Figure 3B. However, this compound is not geodin as it does not contain a chlorine isotopic pattern. In agreement with previous analyses [19]–[21], no intermediates of the proposed geodin pathway (Figure 2) accumulated in the ged+ gedCΔ mdpA-LΔ strain, which is expected, as the PKS responsible for geodin formation is absent.
According to the proposed biosynthetic route for geodin production, the halogenase accepts sulochrin as substrate and adds two chlorine atoms to form dihydrogeodin (Figure 2). Consistent with the hypothesis that gedL encodes the sulochrin halogenase, sulochrin accumulated significantly in the ged+ gedLΔ mdpA-LΔ strain (1.2 – 1.8 µg/plate), but was undetectable in the gedR or gedC deletion strains (Figure 3C). To confirm that this lack of halogenase activity was due to the gedL deletion we reintroduced the gedL ORF at another ectopic site, IS3, which is a site located on a chromosome different from the one harboring IS1, see Figure S2. Surprisingly, no production of geodin was observed in this strain (Figure S3A). This prompted us to perform a BLAST search of the GenBank database [23] using the amino acid sequence of the current ATEG_08460.1 gene model as query. Strikingly, the majority of the best hits were enzymes that contain additional 49 amino acid residues in their N-terminus, including a conserved MSIP/MSVP motif at the very N-terminal end, see Figure S4A. Interestingly, intron prediction based on Augustus [29] predicts an intron just upstream of the AUG proposed by the current gene model (ATEG_08460.1). Taking this into account and by using an ATG further upstream in the gedL gene, a very similar extension can be generated for GedL, see Figure S4B and C. We therefore inserted a larger fragment of the gedL locus that includes this new ATG as well as its native UTR sequence into IS2 [4] in the ged+ gedLΔ mdpA-LΔ strain. In this strain, geodin was produced in ample amounts (4.0 – 6.8 µg/plate) strongly suggesting that gedL indeed encodes the sulochrin halogenase. Interestingly, in this strain, targeted analysis of the UHPLC-HRMS data and comparison to an in-house metabolite database [30], revealed 0.04 – 0.06 µg/plate of sulochrin and trace amounts of monochlor-sulochrin indicating that chlorine is added in two discrete catalytic steps, see Figure S3B.
To investigate whether GedR regulates the genes of the geodin cluster in A. nidulans, we performed a gene specific mRNA transcript analysis by quantitative RT-PCR in the ged+ mdpA-LΔ and ged+ gedRΔ mdpA-LΔ strains for all genes in the geodin gene cluster where a putative homolog is present in the monodictyphenone cluster (Table 1). This analysis demonstrated that transcription of all seven selected genes was down regulated in the absence of GedR. Most prominently transcription from four of the genes (gedF, G, H, and K) was reduced to less than 10% of the level obtained in the ged+ mdpA-LΔ strain (Figure 4). We note that the de novo annotated candidate gene for the emodin anthrone oxidase, gedH (ATEG_08457-2), is transcribed in both the ged+ mdpA-LΔ and ged+ mdp+ strains. In addition, its expression levels in the two strains were different from those obtained for gedI (ATEG_08457-1). Together, these observations strongly indicate that gedR encodes a transcription factor, which activates the expression of the genes that are involved in geodin synthesis and that gedH is a genuine ORF.
Figure 4. The A. terreus transcription factor GedR is important for gene expression in the geodin gene cluster in A. nidulans.
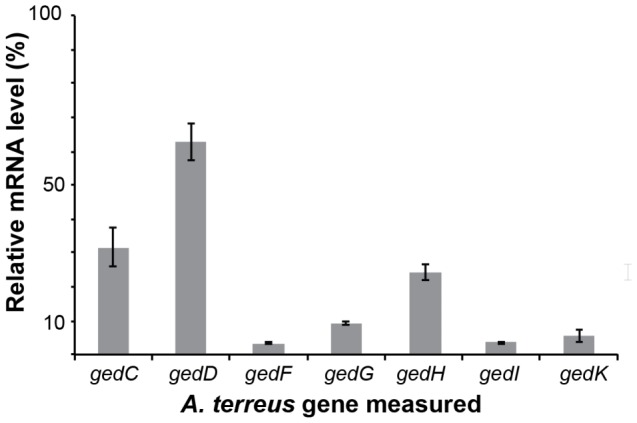
Transcription levels of selected ged-genes in ged + mdpA-LΔ, gedRΔ strains relative to the corresponding levels in ged + mdpA-LΔ strains.
Inspired by these results, we next tested whether GedR would activate the gedR promoter. To this end we inserted a lacZ reporter gene under the control of the native gedR promoter into IS3, in ged+ mdpA-LΔ and ged+ gedRΔ mdpA-LΔ strains. On MM medium containing 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal), colonies formed by the PgpdA-lacZ positive control strain were strongly blue, see Figure S5. The center of the colonies formed by ged+ mdpA-LΔ PgedR-lacZ strain exhibited slightly blue color. However, this level of blue represents background as it did not differ from the amount and location of blue color produced by the negative control strain ged+ mdpA-LΔ, see Figure S5. In agreement with this, a quantitative RT-PCR analysis showed that the lacZ mRNA level was only modestly increased (1.5 fold) in the ged + mdpA-LΔ PgedR-lacZ strain as compared to a ged + gedRΔ mdpA-LΔ PgedR-lacZ strain, but this difference was not statistically significant (p = 0.08). Thus, GedR is not sufficient to induce expression from gedR in A. nidulans.
The fact that geodin production was significantly higher in the ged+ than in the ged+ mdpA-LΔ strains prompted us to investigate whether GedR could also activate transcription of the mdp cluster. Specifically, we compared transcription from mdpG, encoding the monodictyphenone PKS, in the ged+ and the reference strains. In agreement with our hypothesis the mdpG transcript was easily detectable in the ged+ strain, but undetectable in the reference, see Figure S6.
2.6 Conservation of gene clusters resembling the geodin cluster in other fungal species
Finally, we speculated whether gene clusters of a similar organization could be found in other sequenced fungal species as emodin is well-known to serve as precursor to a wide range of natural products [31]–[34]. Comparison of the geodin gene cluster to all Aspergillus genomes available at the Aspergillus Comparative Sequencing Project database (Broad Institute of Harvard and MIT, http://www.broadinstitute.org/) revealed the presence of putative gene clusters in A. fumigatus and A. fischerianus containing putative homologs of 12 of the 13 annotated ORFs in the geodin cluster (the halogenase, gedL, is absent). The internal organization of the putative clusters in A. fumigatus (Afu4g14450-14580) and A. fischerianus (101790-101920) were identical to the geodin cluster with the exception of an inversion affecting the five ORFs gedG- gedK. Moreover, the amino acid identities between biosynthetic enzymes in A. terreus and A. fumigatus/A. fischerianus were in average 58% and 60%, respectively. The conservancy across these three species further substantiates our delineation of the geodin cluster and hints that the putative clusters in A. fumigatus and A. fischerianus may encode the biosynthesis for a similar compound. Both species are known to produce trypacidin [35], which differs from geodin only by the absence of chlorines and the presence of an additional methyl group [36]. In agreement with the structural differences between geodin and trypacidin, the putative A. fumigatus and A. fischerianus gene clusters contain one additional putative methyltransferase, but lack the putative halogenase. Thus, the two putative clusters are candidates for trypacidin gene clusters.
Materials and Methods
Strains and media
Escherichia coli strain DH5α was used to propagate all plasmids. Genomic DNA from the geodin producing A. terreus IBT15722 strain was used as template for PCR amplification of the geodin cluster. A. nidulans strains are shown in Table S1. A. nidulans strains were grown on solid glucose minimal medium (MM) prepared as described by Cove [37], but with 1 % glucose, 10 mM NaNO3 and 2 % agar. MM was supplemented with 10 mM uridine (Uri), 10 mM uracil (Ura), and/or 4 mM L-arginine (Arg) when required. Solid plates containing 5-fluoroorotic acid (5-FOA) were made as MM+Uri+Ura medium supplemented with filter sterilized 5-FOA (Sigma-Aldrich) to a final concentration of 1.3 mg/ml.
Vector construction
All vectors were made by USER cloning and USER fusion [7], [11]. All PCR products were amplified in 35 cycles using proof-reading PfuX7 polymerase [38]. Next USER fusions of vector and inserts were performed as previously described [12]. Reactions were incubated for 20 min at 37 °C, followed by 20 min at 25 °C before transformation into E. coli.
The pU2111-3 vector was constructed by USER fusion of 5 PCR amplified fragments: 1) vector backbone for propagation in E. coli (amplified with primers DH110/DH111), 2) US (upstream) targeting sequence for insertion in IS3 (DH112/DH113), 3) PgpdA-S::UEC::TtrpC (DH114/DH115), 4) A. fumigatus pyrG (marker) (DH116/DH117) and 5) DS (downstream) targeting sequence for insertion in IS3 (DH118-DH119). UEC: uracil excision cassette. Template for fragments 1 and 3: pU1111, for fragments 2 and 5: A. nidulans genomic DNA, and for fragment 4: pDEL2 [13]. p2110-3-lacZ was constructed by combing an AsiSI and Nb.BtsI pU2111-3 vector fragment with a PCR product containing the E. coli lacZ gene (amplified from pU2110-1-lacZ using motni136/motni137 [7] as primers) by USER cloning. The plasmid p2010-3-PgedR-lacZ was constructed by USER fusion of 5 PCR amplified fragments: 1) gedR promoter sequence (DH120/DH121, template: NID677 genomic DNA with), 2) lacZ::TtrpC::AFpyrG (motni136/DH122, template: p2111-3), 3) vector backbone for propagation in E. coli (DH110/DH111 template: pU1111), 4) DS targeting sequence for insertion in IS3 (DH123/DH119 template: pU2111-3) and 5) US targeting sequence for insertion in IS3 (DH112/DH113). The vectors pU2110-3-ATEG_08460.1 and pU2110-2-gedL were constructed by combining the PCR fragments generated with the primers JBN K35/K36 and JBN W77/W78 with pU2111-3 and pU2111-2 [4] vector fragments, respectively, by USER fusion. Prior to USER fusion both plasmids were digested with AsiSI and Nb.BtsI to create the vector fragments.
The two integration vectors containing the geodin gene cluster, pU1110-1-ged1 (containing Fragment 1) and pU2000-ged2 (containing Fragment 2), were made as follows. Primers for generating all PCR fragments for Fragment 1 and 2 assemblies are shown in Table S2. pU1110-1-ged1 was constructed by combining all relevant fragments for Fragment 1 assembly into an AsiSI and Nb.BtsI pU1111-1 vector fragment by USER fusion. The vector fragment of pU2000-ged2 is based on two PCR fragments generated by using primers 77/422 and 421/70 as well as pJ204 [7] and pU2111-1, respectively, as templates. These two PCR products and all relevant fragments for Fragment 2 assembly were then combined by USER fusion. The inserts in the two integration vectors were fully sequenced (StarSEQ, Germany).
Construction of A. nidulans strains
Protoplastation and gene-targeting procedures were performed as described by Nielsen et al [14] using either argB or AFpyrG as marker. All strains were verified by PCR analysis using spores as the source of DNA. Prior to PCR, the samples were incubated for 25 min at 98°C to liberate genomic DNA. This treatment was followed by touch-down PCR programs with annealing temperatures ranging from 64–56°C. Reactions were carried out in 35 cycles using 40 µL volume with less than 1000 spore (one light stab in the colony with a pipette tip).
The ged+ (NID677) and ged+ mdpA-LΔ (NID695) strains were obtained by transformation of the relevant gene targeting substrates into NID74 [39] or NID356, respectively. The gene targeting substrates containing Fragment 1 and Fragment 2 were liberated from pU1111 (NotI) and pU2052 (SwaI), respectively, and gel purified (GFXTM, GE Healthcare) prior to transformation. The resulting strains, NID677 and NID695, were subjected to counter selection on 5-FOA, generating NID802 and NID823, in order to recycle the AFpyrG marker, hence, allowing the use of this marker for subsequent gene deletions of gedC, gedL and gedR. These deletions were made as described in [14] using the primers listed in Table S2.
The strains NID1291 and NID1297, expressing lacZ under the control of ATEG_08453 promoter was made by transforming a gene targeting substrate liberated from pU2010-3-PgedR-lacZ by digestion with SwaI into NID823 and NID925, respectively. A control strain expressing lacZ under the constitutive promoter PgpdA ¸ NID1278, was constructed by integrating the gene-targeting substrate liberated from pU2110-3-lacZ by SwaI. Both gene-targeting substrates integrate into IS3 located between genes AN4770 and AN4769 on chromosome III, at position 1047840-1051735, by homologous recombination (See Figure S2).
The halogenase complementation strains, ATEG_08460.1 and gedL, were made in the NID1279 background, a pop-out recombinant strain from NID843. Digestion of vector pU2110-3-ATEG_08460.1 and pU2110-2-gedL liberated gene-targeting constructs for transformation and integration in IS3 and IS2, respectively.
Chemical characterization of A. nidulans strains
All strains were grown as three point stab inoculations for 7 days at 37°C in the dark on solid MM-media. Extraction and analysis of metabolites were performed by 2 methods: i) The agar plug extraction method described by Smedsgaard [40], using a total of 1 cm2 of a colony, followed by analysis using reversed phase separation UHPLC- UV/VIS -HRMS on a maXis G3 quadrupole time of flight (qTOF) mass spectrometer (Bruker Daltonics, Bremen, Germany) connected to an Ultimate 3000 UHPLC system (Dionex, Sunnyvale, CA), and equipped with a 10 cm Kinetex C18 column (Phenomenex Torrance, Ca, USA) running a 10–100% acetonitrile gradient system in 10 min at 40°C; ii) more concentrated samples were made by extracting metabolites from a total of 15 cm2 of a colony using 12 ml solvent (ethyl acetate-dichloromethane-methanol-formic acid 60∶30∶15∶1 v/v/v) in a 16-ml vial. The extract was evaporated to dryness with N2 flow and redissolved in 0.5 ml methanol and analyzed by reversed phase separation an Agilent 1290 UHPLC coupled to an Agilent 6550 qTOF (Santa Clara, CA, USA) equipped with a electrospray source, and equipped with a 25 cm Agilent Poroshell phenyl hexyl running a 10–100% acetonitrile gradient system in 15 min 60°C. Both MS instruments were mainly operated in ESI− as geodin and related compounds ionizes best here in this mode [30]. Identification and quantification of geodin and sulochrin (BioAustralis, Smithfield, NSW, Australia) were based on comparison of peak area, retention time, accurate mass (±1.5 ppm), isotope pattern and adduct pattern to quantitative authentic standards. Non quantitative standards representing 5-O-methylsulochrin; sulochrin-2'-methylether; isosulochrin; 3-O-demethylsulochrin; trypacidin; and emodin were also included in the analyses. Other intermediates were identified by comparison to an internal reference standard database (∼1500 compounds) [30]. For the identification of geodin in NID695, high resolution MS (50 000 FWHM) and mass accuracy (< 1.5 ppm) of the maXis G3 was needed to exclude a non chlorine containing co-eluting isobaric compound, seen as the broad peak in Figure 3 (6.7–7.3 min) that impaired the identification of geodin. In the following strains geodin was further verified by better chromatographic separation on a 25 cm phenyl-hexyl column on the Agilent UHPLC-qTOF.
RNA isolation and quantitative RT-PCR
RNA isolation from the A. nidulans strains and subsequent quantitative RT-PCR reactions were done as previously described in [7] except that biomass for RNA isolation was prepared with a Tissue-Lyser LT (Qiagen) by treating samples for 1 min at 45 MHz. The A. nidulans histone 3 encoding gene, hhtA (AN0733) was used as an internal standard for normalization of expression levels. All primers used for quantitative RT-PCR are shown in Table S2.
Bioinformatic analysis of gedL (ATEG_08460.1)
Alignment illustrations and sequence were made in CLC Main Workbench 6.8.4.; Alignment parameters Gap open costs = 10, Gap extension cost = 1. All sequences in the alignments can be retrieved from Genbank, by the following references for the putative halogenases: Aspergillus oryzae ref|XP_001818590.1|, Aspergillus terreus ref|XP_001217599.1| and the A. terreus genome sequence resource at the Broad Institute, Bipolaris sorokiniana gb|EMD66881.1|, Chaetomium chiversii (RadH) gb|ACM42402.1|, Chaetomium globosum ref|XP_001227515.1|, Pochonia chlamydosporia (Rdc2) gb|ADM86580.1| [39] and Talaromyces stipitatus ref|XP_002486044.1|. Intron prediction of the A. terreus ATEG_08460.1 locus was done based on the Augustus gene prediction resource [29].
Concluding Remarks
We have described the complete and targeted transfer of all 13 genes of the geodin gene cluster from A. terreus to A. nidulans through a sequential integration approach enabling A. nidulans to synthesize geodin. In principle, this strategy can be used to reconstitute gene clusters of any size as the sequential integrations are based on marker recycling. In addition, defined promoters can easily be introduced in front of relevant genes in the cluster of interest. Importantly, we demonstrate that the cluster can be genetically dissected for clarification of its biochemical potential. We therefore envision that our method will significantly speed up the uncovering of biochemical pathways in fungi where the genome has been sequenced.
Supporting Information
Identification of putative HypC homolog encoded by gedH ( ATEG_08457-2 ) in the A. terreus geodin gene cluster. Pairwise alignment of putative emodin anthrone oxidase, GedH (ATEG_08457-2), from A. terreus and norsolinic anthrone oxidase, HypC, from A. flavus. The conserved DUF-1772 domain and putative catalytic regions proposed by Ehrlich et al [25] are highlighted in green and blue, respectively.
(TIF)
Schematic overview of the integration of a gene-expression cassette into the integration site, IS3, by homologous recombination. IS3 is located between genes AN4770 and AN4769 on chromosome III. The cassette consists of six parts: upstream targeting sequence (US), promoter (P, in this case 0.5 kb PgpdA), your favorite gene (YFG), terminator (T, TtrpC), marker (in this case AFpyrG flanked by direct), and the downstream targeting sequence (DS). The orientations of the genes AN4770 and AN4769 are indicated by green arrows. The sizes of US, DS and the intergenic region are 1984 bp, 1911 bp, and 3007 bp, respectively.
(TIF)
Complementation of halogenase deficiency. A) Left panel: detection of sulochrin (-ESI, EIC(m/z 331.0812)); right panel: detection of geodin (-ESI, EIC(m/z 396.9876)). Strains for halogenase analysis, from top to bottom: NID843 (gedLΔ); NID1280 (gedLΔ, IS3::PgpdA-ATEG_08460.1); and NID1306 (gedLΔ, IS2::gedL). B) Ratio of geodin, monochlor-sulochrin, and sulochrin in the NID1306 strain, including ESI−-MS spectrum of monochlor-sulochrin showing the isotopic pattern and the mass deviations relative to the theoretical masses. Reference standards of geodin and sulochrin were included in all runs (data not shown).
(TIF)
Identification of the likely start codon of gedL . A) Alignment of the top hits in a BLAST search for ATEG_08460.1 homologs shows that they contain a very conserved 48 amino acid residue addition in the N-terminus. Amongst the homologs, Rdc2, has been characterized as a halogenase by [39] MLAS is the predicted N-terminus of ATEG_08460.1. Drawing is not to scale. B) The position of putative exons and intron in the 5’end of gedL as predicted by the Augustus software [29]. The predicted protein sequence encoded by exon 1 and by the first section of exon 2 is indicated. C) Full alignment of the halogenase homologs and GedL based on the GedL sequence derived from the new start codon.
(TIF)
Expression of lacZ under the control of the gedR promoter ( PgedR ). Left panel: the positions of the strains on the plate are shown in the right panel. NID823 (ged+ mdpA-LΔ) is the reference strain without the lacZ gene. NID1278 is a control strain containing the PgpdA-lacZ construct in IS3. The NID1291 (ged+ mdpA-LΔ PgedR-lacZ) strain carries PgedR-lacZ in IS3. The strains were stabbed on MM containing X-gal and incubated three days at 37 °C in the dark before photography.
(TIF)
Constitutive expression of gedR induces transcription of the A. nidulans gene mdpG . mdpG mRNA levels in reference (NID1) and in the ged+ strain (NID677) were evaluated by quantitative RT-PCR. For each strain, RNA was extracted as described in Materials and Method and the RNA samples analyzed in triplicate by quantitative RT-PCR. The samples were loaded and analyzed by 1% agarose gel-electrophoresis as indicated in the figure.
(TIF)
Primers list. The sequence of PCR products # 1-3 corresponds to supercontig 12: 1302695-1315163 (ATEG_08454-08460) in the genome sequenced isolate NIH2624 (IBT28053). Similarly, PCR products # 4-6 and 8 corresponds to supercontig 12: 1289727-1304484 (ATEG_08449-08454) in NIH2624 (IBT28053). * = For reference see Hansen et al ([7]).
(XLSX)
Acknowledgments
We would like to acknowledge Bjarne Gram Hansen for fruitful discussions throughout the project and Martin Engelhard Kogle for technical assistance.
Funding Statement
This work was supported by the Danish Research Agency for Technology and Production, grant 09-064967. The PhD studies of which this work was part of was funded by the Research School for Biotechnology at the faculty of Life Sciences, University of Copenhagen. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Wilson RA, Talbot NJ (2009) Fungal physiology - a future perspective. Microbiology-Sgm 155: 3810–3815. [DOI] [PubMed] [Google Scholar]
- 2. Hoffmeister D, Keller NP (2007) Natural products of filamentous fungi: enzymes, genes, and their regulation. Natural Product Reports 24: 393–416. [DOI] [PubMed] [Google Scholar]
- 3. Keller NP, Hohn TM (1997) Metabolic pathway gene clusters in filamentous fungi. Fungal Genetics and Biology 21: 17–29. [PubMed] [Google Scholar]
- 4. Hansen BG, Mnich E, Nielsen KF, Nielsen JB, Nielsen MT, et al. (2012) Involvement of a natural fusion of a cytochrome P450 and a hydrolase in mycophenolic acid biosynthesis. Applied and Environmental Microbiology 78: 4908–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heneghan MN, Yakasai AA, Halo LM, Song ZS, Bailey AM, et al. (2010) First heterologous reconstruction of a complete functional fungal biosynthetic multigene cluster. Chembiochem 11: 1508–1512. [DOI] [PubMed] [Google Scholar]
- 6. Itoh T, Tokunaga K, Matsuda Y, Fujii I, Abe I, et al. (2010) Reconstitution of a fungal meroterpenoid biosynthesis reveals the involvement of a novel family of terpene cyclases. Nature Chemistry 2: 858–864. [DOI] [PubMed] [Google Scholar]
- 7. Hansen BG, Salomonsen B, Nielsen MT, Nielsen JB, Hansen NB, et al. (2011) Versatile enzyme expression and characterization system for Aspergillus nidulans, with the Penicillium brevicompactum polyketide synthase gene from the mycophenolic acid gene cluster as a test case. Applied and Environmental Microbiology 77: 3044–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wingler LM, Cornish VW (2011) Reiterative recombination for the in vivo assembly of libraries of multigene pathways. Proceedings of the National Academy of Sciences of the United States of America 108: 15135–15140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bergmann S, Schumann J, Scherlach K, Lange C, Brakhage AA, et al. (2007) Genomics-driven discovery of PKS-NRPS hybrid metabolites from Aspergillus nidulans . Nature Chemical Biology 3: 213–217. [DOI] [PubMed] [Google Scholar]
- 10. Sakai K, Kinoshita H, Shimizu T, Nihira T (2008) Construction of a citrinin gene cluster expression system in heterologous Aspergillus oryzae . Journal of Bioscience and Bioengineering 106: 466–472. [DOI] [PubMed] [Google Scholar]
- 11. Nour-Eldin HH, Hansen BG, Norholm MH, Jensen JK, Halkier BA (2006) Advancing uracil-excision based cloning towards an ideal technique for cloning PCR fragments. Nucleic Acids Research 34: e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Geu-Flores F, Nour-Eldin HH, Nielsen MT, Halkier BA (2007) USER fusion: a rapid and efficient method for simultaneous fusion and cloning of multiple PCR products. Nucleic Acids Research 35: e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nielsen JB, Nielsen ML, Mortensen UH (2008) Transient disruption of non-homologous end-joining facilitates targeted genome manipulations in the filamentous fungus Aspergillus nidulans . Fungal Genetics and Biology 45: 165–170. [DOI] [PubMed] [Google Scholar]
- 14. Nielsen ML, Albertsen L, Lettier G, Nielsen JB, Mortensen UH (2006) Efficient PCR-based gene targeting with a recyclable marker for Aspergillus nidulans . Fungal Genetics and Biology 43: 54–64. [DOI] [PubMed] [Google Scholar]
- 15. Chen ZG, Fujii I, Ebizuka Y, Sankawa U (1995) Purification and characterization of emodinanthrone oxygenase from Aspergillus terreus . Phytochemistry 38: 299–305. [Google Scholar]
- 16. Chen ZG, Fujii I, Ebizuka Y, Sankawa U (1992) Emodin O-methyltransferase from Aspergillus terreus . Archives of Microbiology 158: 29–34. [DOI] [PubMed] [Google Scholar]
- 17. Fujii I, Ebizuka Y, Sankawa U (1988) A novel anthraquinone ring cleavage enzyme from Aspergillus terreus . Journal of Biochemistry 103: 878–883. [DOI] [PubMed] [Google Scholar]
- 18. Huang KX, Fujii I, Ebizuka Y, Gomi K, Sankawa U (1995) Molecular cloning and heterologous expression of the gene encoding dihydrogeodin oxidase, a multicopper blue enzyme from Aspergillus terreus . Journal of Biological Chemistry 270: 21495–21502. [DOI] [PubMed] [Google Scholar]
- 19. Askenazi M, Driggers EM, Holtzman DA, Norman TC, Iverson S, et al. (2003) Integrating transcriptional and metabolite profiles to direct the engineering of lovastatin-producing fungal strains. Nature Biotechnology 21: 150–156. [DOI] [PubMed] [Google Scholar]
- 20. Awakawa T, Yokota K, Funa N, Doi F, Mori N, et al. (2009) Physically discrete beta-lactamase-type thioesterase catalyzes product release in atrochrysone synthesis by iterative type I polyketide synthase. Chemistry & Biology 16: 613–623. [DOI] [PubMed] [Google Scholar]
- 21. Couch RD, Gaucher GM (2004) Rational elimination of Aspergillus terreus sulochrin production. Journal of Biotechnology 108: 171–178. [DOI] [PubMed] [Google Scholar]
- 22. Chiang YM, Szewczyk E, Davidson AD, Entwistle R, Keller NP, et al. (2010) Characterization of the Aspergillus nidulans monodictyphenone gene cluster. Applied and Environmental Microbiology 76: 2067–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic Local Alignment Search Tool. Journal of Molecular Biology 215: 403–410. [DOI] [PubMed] [Google Scholar]
- 24. Soding J, Biegert A, Lupas AN (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Research 33: W244–W248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ehrlich KC, Li P, Scharfenstein L, Chang PK (2010) HypC, the anthrone oxidase involved in aflatoxin biosynthesis. Applied and Environmental Microbiology 76: 3374–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Henry KM, Townsend CA (2005) Synthesis and fate of o-carboxybenzophenones in the biosynthesis of aflatoxin. Journal of the American Chemical Society 127: 3300–3309. [DOI] [PubMed] [Google Scholar]
- 27. Henry KM, Townsend CA (2005) Ordering the reductive and cytochrome P450 oxidative steps in demethylsterigmatocystin formation yields general insights into the biosynthesis of aflatoxin and related fungal metabolites. Journal of the American Chemical Society 127: 3724–3733. [DOI] [PubMed] [Google Scholar]
- 28. Simpson TJ (2012) Genetic and biosynthetic studies of the fungal prenylated xanthone shamixanthone and related metabolites in Aspergillus spp. revisited. Chembiochem 13: 1680–1688 10.1002/cbic.201200014. [DOI] [PubMed] [Google Scholar]
- 29. Stanke M, Morgenstern B (2005) AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Research 33: W465–W467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nielsen KF, Mansson M, Rank C, Frisvad JC, Larsen TO (2011) Dereplication of microbial natural products by LC-DAD-TOFMS. Journal of Natural Products 74: 2338–2348. [DOI] [PubMed] [Google Scholar]
- 31. Gatenbeck S, Malmström L (1969) On the biosynthesis of sulochrin. Acta Chemica Scandinavica 23: 3493–3497. [Google Scholar]
- 32.Steglich W, Arnold R, Losel W, Reininger W (1972) Biosynthesis of anthraquinone pigments in Dermocybe. J Chem Soc, Chem Commun 102–103.
- 33. Franck B, Huper F (1966) A new principle of biosynthesis involving secoanthraquinones. Angew Chem Int Ed Engl 5: 728–729 10.1002/anie.196607282.4962479 [Google Scholar]
- 34. Sankawa U, Ebizuka Y, Shibata S (1973) Biosynthetic incorporation of emodin and emodinanthrone into the anthraquinonoids of Penicillium brunneum and P. islandicum . Tetrahedron Letters 14: 2125–2128. [Google Scholar]
- 35. Larsen TO, Smedsgaard J, Nielsen KF, Hansen MAE, Samson RA, et al. (2007) Production of mycotoxins by Aspergillus lentulus and other medically important and closely related species in section Fumigati. Med Mycol 45: 225–232. [DOI] [PubMed] [Google Scholar]
- 36.Afzal M, Davies JS, Hassall CH (1969) The biosynthesis of phenols. Part XIX. Synthesis of the polyhydroxybenzophenone derivative, sulochrin, of the gris-2',5'-diene-3,4'-dione, trypacidin, and of related compounds. J Chem Soc C 1721–1727.
- 37. Cove DJ (1966) Induction and Repression of Nitrate Reductase in Fungus Aspergillus Nidulans. Biochimica et Biophysica Acta 113: 51–&. [DOI] [PubMed] [Google Scholar]
- 38. Norholm MHH (2010) A mutant Pfu DNA polymerase designed for advanced uracil-excision DNA engineering. Bmc Biotechnology 10: 10.1186/1472–6750-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nielsen MT, Klejnstrup ML, Rohlfs M, Anyaogu DC, Nielsen JB, et al. (2013) Aspergillus nidulans synthesize insect juvenile hormones upon expression of a heterologous regulatory protein and in response to grazing by Drosophila melanogaster larvae. Plos One 10: 1317–journal.pone.0073369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smedsgaard J (1997) Micro-scale extraction procedure for standardized screening of fungal metabolite production in cultures. Journal of Chromatography A 760: 264–270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Identification of putative HypC homolog encoded by gedH ( ATEG_08457-2 ) in the A. terreus geodin gene cluster. Pairwise alignment of putative emodin anthrone oxidase, GedH (ATEG_08457-2), from A. terreus and norsolinic anthrone oxidase, HypC, from A. flavus. The conserved DUF-1772 domain and putative catalytic regions proposed by Ehrlich et al [25] are highlighted in green and blue, respectively.
(TIF)
Schematic overview of the integration of a gene-expression cassette into the integration site, IS3, by homologous recombination. IS3 is located between genes AN4770 and AN4769 on chromosome III. The cassette consists of six parts: upstream targeting sequence (US), promoter (P, in this case 0.5 kb PgpdA), your favorite gene (YFG), terminator (T, TtrpC), marker (in this case AFpyrG flanked by direct), and the downstream targeting sequence (DS). The orientations of the genes AN4770 and AN4769 are indicated by green arrows. The sizes of US, DS and the intergenic region are 1984 bp, 1911 bp, and 3007 bp, respectively.
(TIF)
Complementation of halogenase deficiency. A) Left panel: detection of sulochrin (-ESI, EIC(m/z 331.0812)); right panel: detection of geodin (-ESI, EIC(m/z 396.9876)). Strains for halogenase analysis, from top to bottom: NID843 (gedLΔ); NID1280 (gedLΔ, IS3::PgpdA-ATEG_08460.1); and NID1306 (gedLΔ, IS2::gedL). B) Ratio of geodin, monochlor-sulochrin, and sulochrin in the NID1306 strain, including ESI−-MS spectrum of monochlor-sulochrin showing the isotopic pattern and the mass deviations relative to the theoretical masses. Reference standards of geodin and sulochrin were included in all runs (data not shown).
(TIF)
Identification of the likely start codon of gedL . A) Alignment of the top hits in a BLAST search for ATEG_08460.1 homologs shows that they contain a very conserved 48 amino acid residue addition in the N-terminus. Amongst the homologs, Rdc2, has been characterized as a halogenase by [39] MLAS is the predicted N-terminus of ATEG_08460.1. Drawing is not to scale. B) The position of putative exons and intron in the 5’end of gedL as predicted by the Augustus software [29]. The predicted protein sequence encoded by exon 1 and by the first section of exon 2 is indicated. C) Full alignment of the halogenase homologs and GedL based on the GedL sequence derived from the new start codon.
(TIF)
Expression of lacZ under the control of the gedR promoter ( PgedR ). Left panel: the positions of the strains on the plate are shown in the right panel. NID823 (ged+ mdpA-LΔ) is the reference strain without the lacZ gene. NID1278 is a control strain containing the PgpdA-lacZ construct in IS3. The NID1291 (ged+ mdpA-LΔ PgedR-lacZ) strain carries PgedR-lacZ in IS3. The strains were stabbed on MM containing X-gal and incubated three days at 37 °C in the dark before photography.
(TIF)
Constitutive expression of gedR induces transcription of the A. nidulans gene mdpG . mdpG mRNA levels in reference (NID1) and in the ged+ strain (NID677) were evaluated by quantitative RT-PCR. For each strain, RNA was extracted as described in Materials and Method and the RNA samples analyzed in triplicate by quantitative RT-PCR. The samples were loaded and analyzed by 1% agarose gel-electrophoresis as indicated in the figure.
(TIF)
Primers list. The sequence of PCR products # 1-3 corresponds to supercontig 12: 1302695-1315163 (ATEG_08454-08460) in the genome sequenced isolate NIH2624 (IBT28053). Similarly, PCR products # 4-6 and 8 corresponds to supercontig 12: 1289727-1304484 (ATEG_08449-08454) in NIH2624 (IBT28053). * = For reference see Hansen et al ([7]).
(XLSX)