

Abstract
Objectives
Randomised controlled trials report group-level treatment effects. However, an individual patient confronting a treatment decision needs to know whether that person's expected treatment benefit will exceed the expected treatment harm. We describe a flexible model for individualising a treatment decision. It individualises group-level results from randomised trials using clinical prediction guides.
Methods
We constructed models that estimate the size of individualised absolute risk reduction (ARR) for the target outcome that is required to offset individualised absolute risk increase (ARI) for the treatment harm. Inputs to the model include estimates for the individualised predicted absolute treatment benefit and harm, and the relative value assigned by the patient to harm/benefit. A decision rule recommends treatment when the predicted benefit exceeds the predicted harm, value-adjusted. We also derived expressions for the maximum treatment harm, or the maximum relative value for harm/benefit, above which treatment would not be recommended.
Results
For the simpler model, including one kind of benefit and one kind of harm, the individualised ARR required to justify treatment was expressed as required ARRtarget(i)=ARIharm(i) × RVharm/target(i). A complex model was also developed, applicable to treatments causing multiple kinds of benefits and/or harms. We demonstrated the applicability of the models to treatments tested in superiority trials (either placebo or active control, either fixed harm or variable harm) and non-inferiority trials.
Conclusions
Individualised treatment recommendations can be derived using a model that applies clinical prediction guides to the results of randomised trials in order to identify which individual patients are likely to derive a clinically important benefit from the treatment. The resulting individualised prediction-based recommendations require validation by comparison with strategies of treat all or treat none.
Keywords: STATISTICS & RESEARCH METHODS
Article summary.
Article focus
Randomised controlled trials provide relative group-level estimates of the beneficial and harmful effects of a treatment. However, the absolute size of those effects may vary across individuals according to their baseline risk.
Models have been described previously to individualise results of superiority placebo-control trials in the case of a variable benefit/fixed harm scenario.
Key messages
We provide a generalised model to individualise treatment recommendations. We start from the definition of the Clinically Important Difference: the size of treatment benefit that offsets the treatment harm, after adjusting for the patient's values.
The model applies to a variable benefit and a fixed or variable harm, and to superiority (placebo and active control) and non-inferiority trials. It can accommodate more than one kind of benefit and/or harm.
Clinical Prediction Guides are used to individualise the predicted risk of the target event and of the harm at trial entry.
Strengths and limitations of this study
Strengths: the model adopts an individual perspective and is flexible and timely. It allows the calculation of an individual's maximum predicted absolute risk increase for the treatment harm, or the maximum relative value for harm/target, that would overturn the treatment decision.
Limitations: economic costs are not modelled; uncertainty will exist for some quantities entering the model; the model awaits empirical validation.
Introduction
For questions of treatment and prevention, randomised controlled trials (RCTs) provide the most valid evidence concerning the benefits and, often, the harms of the intervention. However, RCTs typically report only group-level results, whereas treatment effects may depend importantly on characteristics of individual patients.
A clinical prediction guide (CPG)1–3 uses patient-specific risk data to predict the level of risk for a clinical outcome of interest for an individual patient. CPGs applied to participants in clinical trials can predict the individual patient's level of risk at trial entry (baseline risk (BLR)) for the target outcome at which the treatment is directed, and also for harm caused by the treatment. If the relative risk reduction for the target outcome (or relative risk increase for the harm) is constant across the range of BLR, then the absolute treatment effects can be predicted in individual patients: absolute risk reduction (ARR) for the target outcome (the treatment benefit) and absolute risk increase (ARI) for the treatment harm.
But what size of ARR for the target benefit is sufficiently large to justify acceptance of a treatment that carries with it the potential for benefit and harm? That depends on the frequency of the harm caused by treatment, and the relative importance of the harm compared to the benefit. The size of treatment benefit that is large enough to offset the treatment harm is the patient's clinically important difference (CID).
The concept of CID has been incorporated in several prior formulations: the threshold ARR (inverted, the threshold number needed to treat4), the threshold for agreeing with treatment,5 the decision threshold (inverted, the number willing to treat (NWT)).6 Each of these constructs embodies the concept that for treatment to be justified, the predicted treatment benefit must exceed the predicted harm for that individual.
Absolute treatment benefits vary directly with BLR for the target benefit: for an effective treatment, the higher the BLR, the greater the predicted absolute benefit. When modelling absolute treatment effects across individuals, the assumed model usually has incorporated a variable benefit, but a fixed harm.4–7 However, the absolute size of treatment harms may also vary across individuals, in which case a variable benefit/variable harm model would apply. The two models are illustrated in figure 1.
Figure 1.

Models for individualising treatment. Variable benefit/fixed harm (A) and variable benefit/variable harm (B) models are shown. In each model, treatment benefit, modelled as absolute risk reduction for the target event, varies directly with baseline risk for the target event. Treatment harm is modelled as the absolute risk increase for the harm of treatment. Harm is then value-adjusted based on a relative value (RV) assigned to the treatment harm as compared with the target event prevented. With a fixed harm (A), the absolute risk increase for the harm of treatment is constant. With a variable harm (B), the absolute risk increase for the harm of treatment varies with the baseline risk for the harm. As indicated by the arrow in each panel, the point at which the value-adjusted treatment harm intersects the treatment benefit defines the clinically important difference (CID) for the treatment benefit.
The objectives of this report were: (1) to derive an expression for CID that is flexible and applicable to either fixed or variable treatment harm and (2) to describe a generalised model for deriving a treatment recommendation based on CID, using group-level estimates of treatment effects provided by RCTs and CPGs for prediction of individualised absolute treatment benefit and harm.
Methods
We define CID as the size of benefit from the treatment that offsets the harm of the treatment. We define a benefit as the reduction of the occurrence of the target outcome, expressed as the negative outcome, for example, death, rather than the positive outcome, for example, survival. When the benefit is defined categorically, CID is the required ARR for the target outcome (ARRtarget) obtained with the treatment compared with the control. The control can be no treatment (or placebo) or an active control. The model contains parameters for the predicted individualised treatment benefit, the predicted individualised treatment harm and the patient's values. The model accommodates more than one kind of benefit and more than one kind of harm. No economic cost, either direct or indirect, is included in the model.
When applied to individualise a treatment recommendation, the model provides an individualised required ARRtarget. A decision rule recommends the treatment when the patient’s predicted ARRtarget is greater than her required ARRtarget.
Data requirements
Table 1 summarises the required quantities for entry into the model, distinguishing between group-level measures and individual-level predictions.
Table 1.
Quantities required for a generalised model for individualising treatment recommendations
| Element | Group-level measures |
Individualised predictions |
||
|---|---|---|---|---|
| Quantity | Measured as | Quantity | Predicted as | |
| Benefits | RRRtrial | 1−RRtrial or 1−HRtrial | ARRi | RRRtrial×BLRi for benefit* |
| Harms fixed variable |
ARItrial RRItrial |
Risktreated−control RRtrial−1 or HRtrial−1 |
ARIi |
ARItrial used as ARIi RRItrial×BLRi for harm† |
| Values | RV | Vharm/Vbenefit | RVi | Provide a range of RVs centred on typical group-level RV‡ |
*Estimate BLRi for benefit using CPG for individualised prediction of outcome comprising the benefit.
†Estimate BLRi for a variable harm using CPG for individualised prediction of outcome comprising the harm.
‡Estimate typical RV from formal utility-based analyses, patient groups or expert opinion.
ARItrial, absolute risk increase for a fixed harm; ARRi, ARIi, BLRi, RVi, individualised predicted estimates; BLR, baseline risk (risk in control group); CPG, clinical prediction guide; HR, hazard ratio; RR, relative risk; RRItrial, relative risk increase for a variable harm, from RCT(s) or best evidence; RRRtrial, relative risk reduction observed in RCT(s); RV, relative value.
Group-level quantities
Most of the shown group-level quantities are used to generate individualised estimates. For treatment benefits, the required group-level measure is the relative risk reduction. The data source can be a meta-analysis of large RCTs or a single large RCT. The required group-level quantities for the harms depend on the type of harm, fixed or variable. For fixed harms, we use a group-level absolute quantity, the ARI (ARIharm). For variable harms, we use a group-level relative quantity, the relative risk increase (RRIharm). Whether fixed or variable, the estimate of the treatment effect for harms comes from a meta-analysis of large RCTs, a single large RCT or best available observational evidence. Values are entered as the relative value (RV) of the harm compared with the target benefit. A group-level RVharm/target can be derived from formal utility-based analyses, patient groups or expert opinion.
Individual-level quantities
The individualised treatment benefit is expressed as ARR (ARRtarget(i)). The individualised treatment harm is expressed as ARI (ARIharm(i)). For BLR, we mean the risk in the reference group (the control arm in the trial), whether it is represented by patients on no treatment or placebo or by patients on an existing treatment. Table 2 summarises the role of CPGs in individualising predicted treatment benefits and harms.
Table 2.
Clinical Prediction Guides (CPG) for individualising treatment effects
| Type of trial | Type of control | CPG to predict control risk for target event: population | CPG to predict control risk for harm: population |
|---|---|---|---|
| Superiority trial | Placebo or no treatment | CPG developed on patients on placebo or no treatment | Fixed harm: CPG not needed |
| Variable harm: CPG developed on patients on placebo or no treatment* | |||
| Active control (EET) | CPG developed on patients on EET† | Fixed harm: CPG not needed | |
| Variable harm: CPG developed on patients on EET | |||
| Non-inferiority trial | Active control (EET) | CPG developed on patients on EET† | Fixed harm: CPG not needed |
| Variable harm: CPG developed on patients on EET |
*If a validated CPG developed on treated patients is used (see worked example on warfarin), the individualised risk for the harm off treatment can be obtained by dividing the risk on treatment by the group-level relative risk for the harm with the treatment compared with placebo or no treatment.
†If a validated CPG developed on patients on placebo or no treatment is used, the individualised risk for the target event while on EET can be obtained by multiplying the risk off treatment by the group-level relative risk for the target event on EET compared with placebo or no treatment.
EET, established effective therapy.
Benefits modelling: The model allows the predicted ARRtarget(i) to increase for increasing BLRs for the target outcome (BLRtarget(i)), according to the equation: predicted ARRtarget(i)=RRRtarget×BLRtarget(i). The group-level RRRtarget is assumed to be constant across different BLRs. The BLRtarget(i) for the target benefit is estimable using a validated CPG.
Harms modelling. In the case of a fixed harm, the group-level estimate (ARIharm(trial)) is used for the predicted ARIharm(i). No CPG is needed to predict an individualised harm. When the receipt of the treatment per se is modelled as a fixed harm (as with risk/discomfort), that harm is experienced by every treated patient, so the ARIharm(trial) for the harm is constantly equal to 1.0 (100%). In the case of a variable harm across patients, the predicted ARIharm(i) is calculated by multiplying the group-level RRIharm by the individualised BLR for that harm (BLRharm(i)). The group-level RRIharm is assumed to be constant across different BLRs. The BLRharm(i) can be estimated using a validated CPG.
Values modelling. An individual RV (RVharm/target(i)) assigned by the patient enters the model. We recognise that an RVharm/target(i) may not be ascertained reliably. Therefore, we modelled a range of RVs centred on a group-level RV.
When more than one benefit and/or more than one harm is included, for each benefit and harm the specific ARRi/ARIi/RVi is separately calculated or assigned as above.
Construction of models for individualising a treatment recommendation
We constructed two models: a simple model where there is one kind of treatment benefit and one kind of treatment harm; and a complex model where there is more than one kind of benefit and/or more than one kind of harm. In both cases, the model equation is solved for the required ARRtarget(i) to offset the treatment harm(s), given the predicted ARIharm(i) and RVharm/target(i). The same basic equation can then be used to calculate:
The maximum ARIharm(i) above which treatment would not be justified, given the predicted ARRtarget(i) and RVharm/target(i).
The maximum RVharm/target(i) above which treatment would not be justified, given the predicted ARRtarget(i) and ARIharm(i).
Results
Algebraic solution to the model
We derived the following equations to describe the model (see Appendix for algebraic details).
Simple model: one kind of treatment benefit, one kind of treatment harm
Required ARRtarget(i)
The required size of the ARRtarget that offsets the treatment harm, value-adjusted, for the patient i can be calculated as (Appendix section 1, equations (1) and (2))
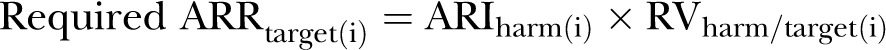 |
(m1) |
The equation includes the particular condition of a fixed harm when the ARItrial can substitute for the ARIi. When the treatment receipt is considered the harm, the ARItrial is 1.0 and so the ARR(target)i is directly predictable from the RVharm/target(i) as
| (m2) |
Decision rule: In case of fixed harm and variable harm, the treatment would be justified for the patient i when
| (d1) |
Maximum ARIharm(i) and maximum RVharm/target(i)
The maximum ARIharm(i) above which the treatment would not be justified for the patient i can be calculated as (Appendix equations (1) and (3))
| (m3) |
Decision rule: The treatment would not be justified for the patient i when
| (d2) |
where the predicted ARIharm(i) can be fixed (=ARItrial) or variable.
Similarly, the maximum RVharm/target(i) above which the treatment would not be justified for the patient i can be calculated as (Appendix equations (1) and (4))
| (m4) |
Decision rule: The treatment would not be justified for the patient i when
| (d3) |
Complex model: multiple treatment benefits, multiple treatment harms
The model can be generalised to incorporate additional treatment benefits other than the reduction of the target outcome, and multiple harms, whether fixed or variable (Appendix section 2). A harm may have a fixed as well as a variable component. In that case, the fixed and variable components would be entered as separate harms, along with their separate RVs. The size of the ARRtarget, which is required to offset the value-adjusted treatment harms and which accounts for other treatment benefits, is calculated for the patient i as (Appendix equations (5), (6) and (7))
 |
(m5) |
where k is the number of treatment harms, m is the total number of treatment benefits, and the benefit(2) to benefit(m) are the benefits other than the target benefit. Every RV(i) is expressed as the value assigned to each outcome, prevented or caused by the treatment, compared with the value of the target benefit.
Decision rule: Similar to the case of only one benefit and one harm, the treatment would be justified for the patient i when
| (d1) |
The complex model can be used to predict the individualised maximum allowed ARI for a target harm and the maximum RV for the target benefit compared with a target harm, above which the treatment is not justified.
Applicability of the model
Theoretically, the model is applicable to every situation tackling the choice between two treatment strategies. Three examples are proposed to show the applicability of the model to individualised treatment recommendations: a superiority trial with a variable benefit/fixed harm scenario; a superiority trial with a variable benefit/variable harm scenario and the case of non-inferiority trials.
Superiority trial: variable benefit, fixed harm. Rosuvastatin for primary prevention of cardiovascular events
The Justification for the Use of Statins in Prevention (JUPITER) trial8 evaluated the effect of rosuvastatin versus placebo for reduction of cardiovascular events in apparently healthy men and women with low-density lipoprotein cholesterol levels <3.4 mmol/L and elevated high-sensitivity C reactive protein. The primary outcome was a composite of myocardial infarction, stroke, arterial revascularisation, hospitalisation for unstable angina or cardiovascular death. The group-level result showed a substantial relative benefit of rosuvastatin (HR 0.56, 95% CI 0.46 to 0.69). This is equivalent to an RRRtarget of 0.44 (95% CI 0.31 to 0.54). Nevertheless, the individual's absolute benefit with rosuvastatin will vary according to her BLR (BLRi.). Validated CPGs exist to predict the BLR for cardiovascular events. The Framingham risk score,9 for example, predicts the risk of cardiovascular events at 10 years combining risk factors such as age, gender, smoking, total and high-density lipoprotein cholesterol levels, systolic blood pressure and hypertension. Dorresteijn et al6 used the group-level quantities provided by the JUPITER study and CPGs, either existing9 10 or newly developed,6 to individualise the predicted BLRi and absolute effect of rosuvastatin at 10 years (ARR(target)i) among JUPITER's participants. They found an approximate 20-fold variation in the predicted BLR(target)i. Thus, the predicted ARR(target)i varied from about 1–20% at 10 years, with a slightly different patient stratification depending on the CPG used. Dorresteijn and colleagues then evaluated the application of these individualised predictions to recommend the treatment. They defined the ‘Number Willing to Treat (NWT)’ as the number of patients one is willing to treat in exchange for the prevention of one target outcome event. Its inverse ratio (1/NWT) was defined as the ‘decision threshold’ and is equivalent to the required ARR(target)i defined for our model. They considered that the treatment receipt per se constituted the harm (fixed harm). Thus, the required ARR(target)i (ie, 1/NWT) equalled the RVharm/target(i) (m2). They examined how the treatment recommendations varied across a range of hypothetical values for NWT.
Superiority trial: variable benefit, variable harm. Warfarin to prevent cardioembolic events in patients with atrial fibrillation
Six RCTs compared warfarin versus placebo/no treatment in patients with non-valvular atrial fibrillation to reduce the occurrence of stroke and systemic cardioembolism. Hart et al11 meta-analysed those RCTs and found a pooled RRR for cardioembolic events (RRRstroke) of 0.64—or 64%—(95% CI 0.49 to 0.74). On the other hand, warfarin was associated with a pooled RRI for major extracranial bleeding (RRIbleed) of 1.3—or 130%—(95% CI 0.08 to 3.89; note: Hart et al11 included the intracranial haemorrhages among the strokes in the efficacy analyses).
Several CPGs to predict the risk of stroke and bleeding have been developed and validated in patients with atrial fibrillation. Using the individual predictions for the BLR for stroke (BLRstroke(i)) and for bleeding (BLRbleed(i)), the absolute beneficial effect and also the absolute adverse effect with warfarin can be individualised as ARRstroke(i)=RRRstroke×BLRstroke(i) and ARIbleed(i)=RRIbleed×BLRbleed(i), respectively. As an example, for the prediction of the BLRstroke(i), we adopted the CHADS2 score developed on patients off anticoagulation.12 For the prediction of the BLRbleed(i), we adopted the HEMORR2HAGES score.13 Since the HEMORR2HAGES score was developed on patients on warfarin,13 the corresponding BLRbleed(i) off warfarin was calculated by dividing the predicted risk on warfarin by 2.3, which is the reported relative risk for major bleeding for warfarin compared with placebo.11 The results are shown in table 3. The predicted ARRstroke(i) varied from 1.22% to 11.65%/year and the predicted ARIbleed(i) varied from 1.07% to 6.95%/year. Comparing the individualised predictions for the benefit and the harm, value-adjusted, we then obtained individualised treatment recommendations for warfarin. A range of plausible values of the RVbleed/stroke was examined.
Table 3.
Framework for application of prognostic risk scores for variable treatment benefit, variable treatment harm to particularise a treatment recommendation
| Risk for stroke (CHADS2) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Score | 0 | 1 | 2 | 3 | 4 | 5 | 6 | ||||
| Predicted BLRstroke (%/year) | 1.9 | 2.8 | 4.0 | 5.9 | 8.5 | 12.5 | 18.2 | ||||
| Risk for bleed (HEMORR2HAGES) |
Predicted ARRstroke (95% CI) %/year* | 1.22 (0.93 to 1.41) | 1.79 (1.37 to 2.07) | 2.56 (1.96 to 2.96) | 3.78 (2.89 to 4.37) | 5.44 (4.17 to 6.29) | 8.00 (6.13 to 9.25) | 11.65 (8.92 to 13.5) | |||
| Score | Predicted BLRbleed, %/year warfarin |
Predicted ARIbleed (95% CI) %/year† |
Required ARRstroke (95% CI) @ RVbleed/stroke 0.6 %/year |
Tentative treatment recommendation‡ | |||||||
| Yes | No | ||||||||||
| 0 | 1.9 | 0.83 | 1.07 (0.07 to 3.23) | 0.64 (0.04 to 1.94) | T | T | T | T | T | T | T |
| 1 | 2.5 | 1.09 | 1.41 (0.09 to 4.24) | 0.85 (0.05 to 2.54) | T | T | T | T | T | T | T |
| 2 | 5.3 | 2.30 | 3.00 (0.18 to 8.95) | 1.80 (0.11 to 5.37) | DT | CC | T | T | T | T | T |
| 3 | 8.4 | 3.65 | 4.75 (0.29 to 14.2) | 2.85 (0.17 to 8.52) | DT | DT | DT | T | T | T | T |
| 4 | 10.4 | 4.52 | 5.88 (0.36 to 17.6) | 3.53 (0.22 to 10.6) | DT | DT | DT | T | T | T | T |
| ≥ 5 | 12.3 | 5.35 | 6.95 (0.43 to 20.8) | 4.17 (0.26 to 12.5) | DT | DT | DT | DT | T | T | T |
Example: Warfarin versus placebo for stroke reduction in patients with atrial fibrillation.
*Predicted ARRstroke and 95% CI if RRRstroke is 0.64 (0.49 to 0.74) using warfarin.11
†Predicted ARIbleed and 95% CI if RRIbleed is 1.30 (0.08 to 3.89) using warfarin.11
‡Tentative treatment recommendations are based on predicted point estimates for ARRstroke and ARIbleed. Uncertainties in these estimates, indicated by 95% CIs above, must also be considered before actual treatment recommendations can be derived.
ARI, absolute risk increase; ARR, absolute risk reduction; BLR, baseline risk; CC, close all; DT, do not treat; RV, relative value; T, treat.
Required ARRstroke(i) to justify warfarin
To justify warfarin, the predicted ARRstroke(i) should be greater than the required ARRstroke(i) (d1), that is, greater than ARIbleed(i)×RVbleed/stroke(i) (m1). Table 3 summarises the results of the application of the model to individualise warfarin recommendation in a hypothetical patient population, according to the coclassification of patients based on the CHADS2 and HEMORR2HAGES scores. We arbitrarily chose a group-level RV for a bleed/stroke of 0.6, an RV calculated from a lost-utility analysis over a 10-year time frame.4 Table 3 shows the resulting treatment decisions for each of the 42 cells formed according to the CHADS2 and HEMORR2HAGES scores.
As a base case, the table was obtained using for RRRstroke the point estimate (0.64).11 Since a treatment is accepted as superior compared with placebo/no treatment only when the upper bound of the 95% CI for the relative risk for the target outcome is below 1, we repeated the example using for RRRstroke a value of 0.49 (corresponding to the upper bound for RRstroke 0.51). In that case, the predicted ARRstroke(i) is reduced and slightly fewer patients would be recommended for treatment. For example, a CHADS2 3 and HEMORR2HAGES 4 patient would now not be recommended for warfarin treatment (results not shown). We also repeated the example, using for RRIbleed a value of 3.89, corresponding to the upper bound of the 95% CI for RRIbleed. Now, considerably fewer patients would be recommended for treatment (data not shown). The major differences in who would be recommended for treatment arise primarily from the great uncertainty in the estimate for ARIbleed in this example. We caution that table 3 is presented only as a framework for presenting particularised treatment recommendations in a variable benefit/variable harm scenario. The recommendations shown there are based only on point estimates, and should not be accepted without taking into account the uncertainties in the estimates for ARRstroke and ARIbleed in deriving the treatment recommendations.
Maximum ARIbleed(i) above which warfarin would not be justified
Figure 2 shows how the maximum ARIbleed(i) (m3) varies according to the different CHADS2 scores and different values of RVbleed/stroke(i) centred on a group-level RVbleed/stroke of 0.6.
Figure 2.
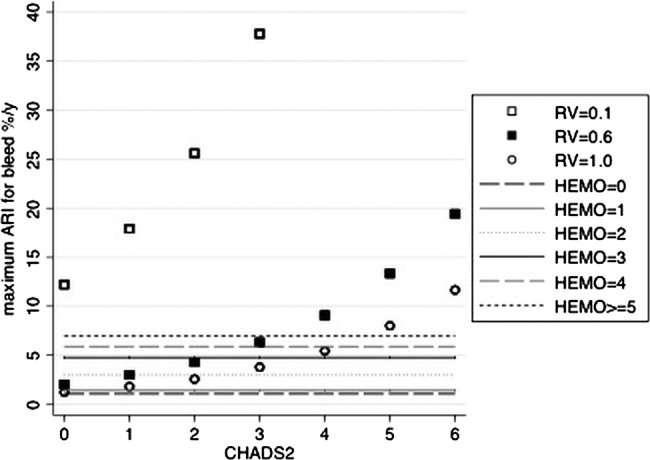
Maximum ARIbleed for treatment to be justified, by CHADS2 score and relative valuestroke/bleed. The scatter plot shows the maximum ARIbleed (%/year) above which warfarin would not be justified, according to the CHADS2 score and different RVbleed/stroke. The horizontal lines depict the predicted ARIbleed with warfarin for each HEMORR2HAGES score. As examples: at RVbleed/stroke 0.6, we would treat CHADS2 score 0 patients only if their predicted ARIbleed given warfarin were less than 2%/year. Accordingly, we would treat HEMORR2HAGES score 0–1 patients because their predicted ARIbleed (1.1, 1.4%/year (table 3)) is less than 2%/year. We would not treat HEMORR2HAGES score ≥2 patients because their predicted ARIbleed (3–7%/year (table 3)) is greater than 2%/year. Again at RVbleed/stroke 0.6, we would treat CHADS2 score 2 patients only if their predicted ARIbleed were less than 4.3%/year. Thus, we would treat HEMORR2HAGES score 0–2 patients because their predicted ARIbleed (1.1–3%/year (table 3)) is less than 4.3%/year. We would not treat HEMORR2HAGES ≥3 patients because their predicted ARIbleed (4.8–7%/year (table 3)) is greater than 4.3%/year. At the RVbleed/stroke set higher or lower than 0.6, fewer patients or more patients, respectively, would be recommended for treatment according to the model. ARI, absolute risk increase; RV, relative value.
Maximum RVbleed/stroke(i) above which warfarin would not be justified
Similarly, given the CHADS2 and the HEMORR2HAGES scores of the patient, the model can calculate which is the maximum RVbleed/stroke(i) (m4) such that if the patient assigns an RVbleed/stroke higher than this maximum, warfarin would not be justified. The variation of the maximum RVbleed/stroke(i) according to the different CHADS2 and HEMORR2HAGES scores is depicted in figure 3.
Figure 3.
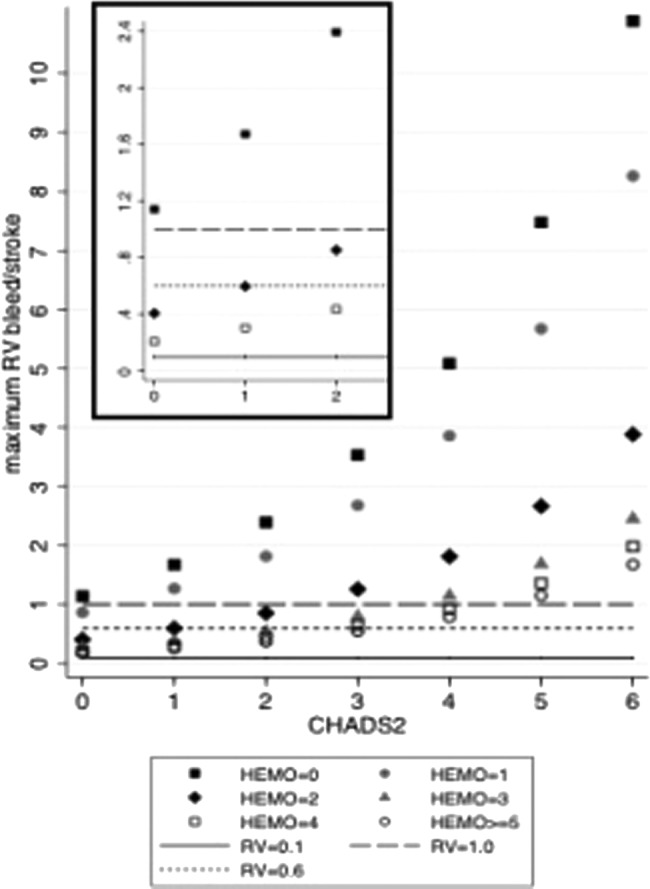
Maximum RVbleed/stroke for treatment to be justified, by CHADS2 score and HEMORR2HAGES score. The scatter plot shows the variation of the maximum RVbleed/stroke according to CHADS2 and HEMORR2HAGES (abbreviated as HEMO) scores. The horizontal lines depict three illustrative maximum relative values. The model predicts the maximum RVbleed/stroke to vary over a range between 0.1 (ie, a value assigned to a stroke 10 times higher than that assigned to a major bleeding) and about 10 (ie, a value assigned to a major bleeding 10 times higher than that assigned to a stroke). As examples, the insert zooms in the results for patients with a CHADS2 score of 0–2 and HEMO scores of 0, 2 and 4. Among patients with a CHADS2 score of 0, warfarin would be recommended for HEMO 0 patients if their RVbleed/stroke were <1.1; for HEMO 2 patients, if their RVbleed/stroke were <0.4; for HEMO 4 patients if their RVbleed/stroke were <0.2. For patients with a CHADS2 score of 2, warfarin would be recommended for HEMO 0 patients if their RVbleed/stroke were <2.3; for HEMO 2 patients if their RVbleed/stroke were <0.8; for HEMO 4 patients if their RVbleed/stroke were <0.4. RV, relative value.
Individualising recommendations for a non-inferior treatment
Application of model to non-inferiority trials
The objective of a non-inferiority trial is to show that the effect of a new treatment on a target outcome is not worse, compared with an established effective treatment (EET), by more than a prespecified margin. This ‘non-inferiority margin’ is the maximum loss of efficacy that is considered acceptable in exchange for a hypothesised reduction in harm, value-adjusted. At the design phase, the non-inferiority margin is expressed as either an absolute or relative increase in the target event rate. A group-level RVharm/benefit is at least implicit when setting the specified margin. When interpreting the results of a non-inferiority trial at the group level, the CI for the observed treatment effect on the target outcome is compared with the non-inferiority margin. If the bound of the CI that reflects the maximal estimate for inferiority is less than the margin (does not ‘cover’ the margin), then it is concluded that the new treatment is non-inferior to EET.
In non-inferiority trials, the CID for a patient can be expressed as the required reduction of the harm which exactly compensates for the allowed increase of the target outcome, value-adjusted. Thus, for application to non-inferiority trials, the equation m1 can be rewritten as:
Individualisation of the results of a trial demonstrating group-level non-inferiority
We individualise group-level results by using CPGs, as applicable (table 2), to predict BLR(i) and thereby absolute treatment effects on the target outcome (ARItarget(i)) and the treatment harm (ARRharm(i)). ARItarget(i) is derived as BLRtarget(i) × RRItrial. ARRharm(i) is derived as BLRharm(i) × RRRtrial. We value-adjust the treatment harm for the RVharm/target(i). We then compare the individualised predictions of treatment effects on the target outcome and on the harm to derive individualised treatment recommendations. A recommendation to treat with the non-inferior therapy would result when the predicted reduction in harm, value-adjusted, exceeds the predicted loss of efficacy, that is, when ARRharm(i) × RVharm/target(i) > ARItarget(i) (or, holding the same terminology as for superiority trials, when predicted ARRharm(i) > required ARRharm(i)).
To examine the worst case, we then repeat the comparison of reduction in harm and loss of efficacy by calculating ARItarget(i) using not the point estimate for RRItrial but the bound of its CI that reflects the maximal inferiority of the new treatment.
Discussion
We presented an extension of the previously described models to individualise treatment recommendations, based on the use of CPGs to predict individual-level treatment effects, adjusted for the relative importance assigned by the patient to different outcomes.
Strengths
The adoption of an individual-level perspective represents the fundamental feature of the model. The individualising process requires the conversion of group-level into individual-level treatment effects and the use of the patient's values.14 The model presented here is more flexible than models for individualising treatment recommendations described previously.4 5 Either a fixed or a variable harm is accommodated in our model. LaHaye et al15 developed a decision aid specifically designed to individualise antithrombotic therapy in patients with atrial fibrillation that included a variable benefit/variable harm scenario and also the patient's RVbleed/stroke. However, they did not explicitly conceptualise and generalise the underlying model. We showed the adaptability of our model to treatments causing multiple kinds of benefits and harms, as well as to non-inferiority trials. The concepts of the maximum ARIharm and maximum RVharm/benefit that would overturn the clinical decision had not been developed previously. The model is timely, given the increasing number of very large RCTs providing precise group-level estimates of treatment harms as well as treatment benefits, and the recent rapid rise in validated CPGs, catalogued and searchable in EvidenceUpdates,3 which makes the individualisation of those group-level quantities more feasible.
Limitations
In our model, we did not include economic costs, either direct or indirect. Like clinical benefits and harms, economic costs can be fixed or variable across patients. This raises the question of whether a group-level cost-effectiveness analysis of a treatment can be individualised.16 A step in that direction is to apply prognostic models to particularise group-level information on cost-effectiveness according to the predicted risk and patient subgroup.17 Our model provides a method for individualising the consequences of treatment. However, analyses of incremental cost-effectiveness or cost-utility at the individual level are constrained at present by the lack of reliable individualised data on the incremental direct and indirect costs of treatment.
Use and appropriateness of CPGs for individualising recommendations
We generically explained why, how and when model building requires the use of CPGs. CPGs are developed for different purposes. A particular application of a CPG is to individualise risk predictions in the control group of an RCT. There are some desirable features of a CPG for this specific application. In box 1, we provide an aid to guide the user in the search for and the evaluation of an appropriate CPG for individualising the group-level results of the RCT of interest.
Box 1. How to use a Clinical Prediction Guide (CPG) on risk prediction to individualise the results of an RCT.
Relevance
Will the CPG help me in making individualised risk predictions for patients in the control group of the randomized controlled trial (RCT) of interest?
Were the patients on whom the CPG was developed or validated similar to the RCT's control group in regard to their clinical characteristics?
Does the treatment status of the patients on whom the CPG was developed match that of the RCT's control group, that is, each on no treatment or placebo; each on established effective therapy?
Does the CPG provide the absolute risk (or is it at least derivable) for the outcome of interest (target event or harm), in a specified period of time, according to risk factors/risk score?
Validity
Are the predictions made by the CPG valid?
-
How was the CPG developed?
Was the CPG developed on a well defined and representative sample of patients prospectively followed up?
-
How well did the CPG perform in the population of derivation?
Was the CPG's calibration tested? How accurate were the predictions of the absolute risk, that is, how good was the agreement between predictions and observed outcome?
Were the CPG's discrimination (c-statistic) and reclassification tested? How good were they?
Did the CPG undergo internal validation to quantify and eventually adjust for overfitting/optimism?
- Did the CPG undergo external validation?
- Was the CPG's performance tested in patients different from those on whom it was developed? How good was it?
Precision
How precise were the predictions of the absolute risk, that is, how wide was the uncertainty around the provided estimates?
In the case of a variable benefit/variable harm, we look for two different CPGs to classify the patients according to the ‘baseline’ risks for the target event and for the harm. In this case, the predictions resulting from this coclassification might be constrained by a possible within-patient correlation between the two variable risks, since the target event and the harm may share some risk factors or may not be independent outcomes.
Uncertainty in group-level estimates and patient values
The results of an RCT are usually provided as point estimates accompanied by a measure of variability (CI). Often, as shown in the example in table 3, the within-trial estimates for the harm have been characterised by high imprecision. However, this situation may be improving with the increasing reports of very large active-control RCTs.18
Probably the major source of uncertainty is the patient's RVharm/benefit and its elicitation. The scenario presented to the patient should uniformly include the major clinical outcomes of the treatment decision, including death if relevant, and the time frame of the consequences of the decision. Decision aids, which are tools specifically designed to prepare the patient to participate in the decision process, have been shown to improve patient knowledge and involvement, especially when they target explicit values clarification.19
One may embed in the calculation of the individual quantities a measure of the variance (eg, SE) of the group-level measures entering the model.20 Additionally, one may estimate how much that uncertainty can affect the individual predictions in the most pessimistic direction, that is, using the CI bounds for the group-level estimate of the treatment effect on target corresponding to the worst scenario. We proposed an alternative approach to deal with the uncertainty around the quantities entering the model. We provided formulas for estimating the individualised maximum ARIharm and RVharm/benefit above which the decision to treat would be overturned.
Future research objectives
Resolution of uncertainty. In applying our model, methods are needed to resolve uncertainty arising from imprecision in the estimates of treatment benefit and treatment harm derived from group-level results from RCTs. In this paper, we addressed uncertainty by resorting to sensitivity analyses utilising bounds of CIs on treatment effects. However, in the field of cost-effectiveness analysis, investigators progressed to approaches dealing simultaneously with the stochastic uncertainty of all the quantities entering the model. These approaches include non-parametric bootstrapping, Fieller's theorem and Bayesian methods.21 We suggest as a future goal that such methods be explored for their applicability to resolution of uncertainty in clinical harm/clinical benefit analyses.
Net benefit and model validation. Vickers et al5 conceived a method to empirically test whether individualised recommendations based on CPG-based predictions of absolute treatment effects, value-adjusted, would actually result in a greater net benefit in real life compared with a policy of treating all patients or treating none. The method utilises the distribution of predicted individualised treatment effects in the randomly allocated treatment and control groups of a large RCT. One combines the patients whose predicted ARRtarget(i) exceeded the required ARRtarget(i) who were randomised to the treatment group, and the patients whose predicted ARRtarget(i) did not exceed their required ARRtarget(i) who were randomised to the control group. Those are the respective patients who would or would not be recommended for treatment and who used prediction-based treatment in real life. One then compares the observed outcomes in the trial of that combined group with the outcomes for the treatment arm of the RCT. The superiority of the prediction-based policy is validated if its net benefit is greater than the net benefit of treat all, or treat none. The empirical result, in the examples of Vickers et al5 and later Dorresteijn et al,6 was that a prediction-based policy was superior, but only within a limited range of the required ARRtarget(i). If the required ARRtarget(i) was extreme in either the low or high direction, a policy of treat all or treat none, respectively, was preferred.
Vickers et al5 and Dorresteijn et al6 used this approach to validate individualised recommendations in a fixed harm scenario, where the harm was receipt of the treatment per se. Nevertheless, the same approach can be used to validate individualised recommendations in variable harm scenarios, and for treatments tested in non-inferiority as well as superiority trials. As with Vickers’ method in general, individual-patient trial data must be available to identify the patients whose predicted ARRtarget(i) did or did not exceed their required ARRtarget(i).
Supplementary Material
Acknowledgments
We thank Dr Brian Haynes, Nancy Wilczynski and Dr Alfonso Iorio at the Health Information Research Unit, McMaster University Health Sciences, for stimulating discussions and support.
Appendix: Algebraic derivation of the models
Legend:
Target = target outcome that the treatment can prevent
Harm = any increase of an adverse outcome due to the treatment
CID = clinically important difference
ARR = absolute risk reduction
ARI = absolute risk increase
V = value
RV = relative value
1. Derivation of the simple model (one benefit, one harm)
The CID corresponds to the ARR for the target benefit sufficiently large to exactly offset the treatment harm. Allowing for a different value assigned to the target outcome prevented by the treatment and to the harm caused by the treatment (Vtarget and Vharm, respectively), the condition at the CID can be expressed algebraically as:
| (1) |
1.1. Algebraic solution for the required ARRbenefit to offset the treatment harm
Dividing each side of the equation (1) by Vtarget
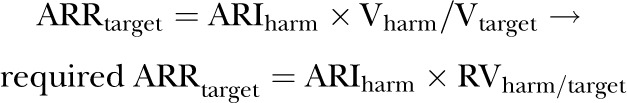 |
(2) |
1.2. Algebraic solution for the maximum ARIharm above which treatment would not be justified
Dividing each side of the equation (1) by Vharm
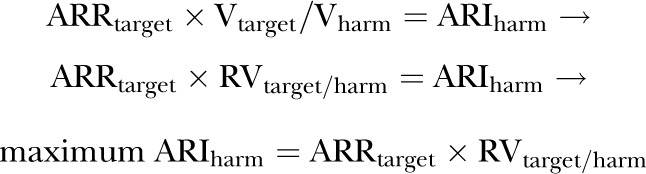 |
(3) |
or, expressed in terms of RVharm/target
1.3. Algebraic solution for the maximum RVharm/target above which treatment would not be justified
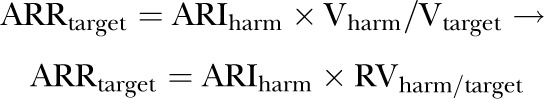 |
dividing each side of equation by ARIharm
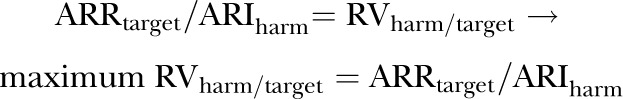 |
(4) |
2. Derivation of the complex model (multiple benefits, multiple harms)
Legend:
Benefit = any reduction of an adverse outcome additional to the target outcome.
At the CID, the sum of treatment benefits offsets the sum of treatment harms. Allowing for different values for every outcome prevented or caused by treatment, this can be expressed algebraically as:
 |
(5) |
where m is the total number of treatment benefits, the benefit(2) to benefit(m) are the benefits other than the target one, and k is the number of treatment harms. Or, likewise:
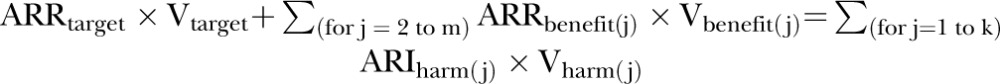 |
(6) |
Subtracting 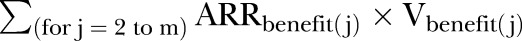 from both sides and dividing both sides for Vtarget, we can obtain the required ARRtarget such that the total treatment benefits offset the total treatment harms:
from both sides and dividing both sides for Vtarget, we can obtain the required ARRtarget such that the total treatment benefits offset the total treatment harms:
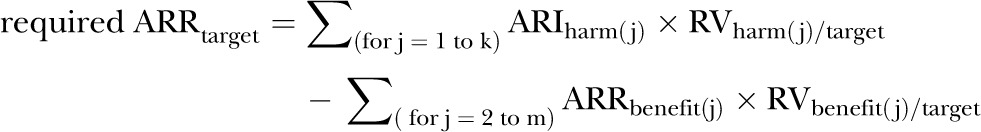 |
(7) |
where every RV is expressed as the value of that outcome, prevented or caused by the treatment, compared with the value assigned to the target outcome.
Footnotes
Contributors: MM designed and carried out the data analyses, interpreted the results, and drafted and revised the manuscript. JCS designed and carried out the data analyses, interpreted the results, and drafted and revised the manuscript. Each author had full access to all the data (including statistical reports and tables) in the study and can take responsibility for the integrity of the data and the accuracy of the data analysis. All authors have read and approved the final manuscript.
Funding: This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: MM and JCS do not have support from and have no relationships with any company that might have an interest in the submitted work in the previous 3 years; MM and JCS have no non-financial interests that may be relevant to the submitted work.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: No additional data are available.
References
- 1.Wong SS, Wilczynski NL, Morgan D, et al. Developing optimal search strategies for detecting sound clinical prediction studies in MEDLINE. AMIA Annu Symp Proc 2003:728–32 [PMC free article] [PubMed] [Google Scholar]
- 2.Holland JL, Wilczynski NL, Haynes RB; Hedges Team Optimal search strategies for identifying sound clinical prediction studies in EMBASE. BMC Med Inform Decis Mak 2005;5:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Website for accessing an up-to-date register of critically-appraised clinical prediction guides, searchable by topic. http://plus.mcmaster.ca/evidenceupdates
- 4.Sinclair JC, Cook RJ, Guyatt GH, et al. When should an effective treatment be used? Derivation of the threshold number needed to treat and the minimum event rate for treatment. J Clin Epidemiol 2001;54:253–62 [DOI] [PubMed] [Google Scholar]
- 5.Vickers AJ, Kattan MW, Sargent D. Method for evaluating prediction models that apply the results of randomized trials to individual patients. Trials 2007;8:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dorresteijn JA, Visseren FL, Ridker PM, et al. Estimating treatment effects for individual patients based on the results of randomized trials. BMJ 2011;343:d5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glasziou PP, Irwig LM. An evidence-based approach to individualizing treatment. BMJ 1995;311:1356–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008;359:2195–207 [DOI] [PubMed] [Google Scholar]
- 9.National Cholesterol Education Program Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III). JAMA 2001;285:2486–97 [DOI] [PubMed] [Google Scholar]
- 10.Ridker PM, Buring JE, Rifai N, et al. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: the Reynolds risk score. JAMA 2007;297:611–19 [DOI] [PubMed] [Google Scholar]
- 11.Hart RG, Pearce LA, Aguilar MI. Meta-analysis: antithrombotic therapy to prevent stroke in patients who have nonvalvular atrial fibrillation. Ann Intern Med 2007;146:857–67 [DOI] [PubMed] [Google Scholar]
- 12.Gage BF, Waterman AD, Shannon W, et al. Validation of clinical classification schemes for predicting stroke. JAMA 2001;285:2864–70 [DOI] [PubMed] [Google Scholar]
- 13.Gage BF, Yan Y, Milligan PE, et al. Clinical classification schemes for predicting hemorrhage: results from the National Registry of Atrial fibrillation (NRAF). Am Heart J 2006;151:713–19 [DOI] [PubMed] [Google Scholar]
- 14.McAlister FA, Straus SE, Guyatt GH, et al. Users’ guides to the medical literature: XX. Integrating research evidence with the care of the individual patient. Evidence-Based Medicine Working Group. JAMA 2000;283:2829–36 [DOI] [PubMed] [Google Scholar]
- 15.LaHaye SA, Gibbens SL, Ball DG, et al. A clinical decision aid for the selection of antithrombotic therapy for the prevention of stroke due to atrial fibrillation. Eur Heart J 2012;33:2163–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ioannidis JP, Garber AM. Individualized cost-effectiveness analysis. PLoS Med 2011;8:e1001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mark DB, Hlarky MA, Califf RM, et al. Cost-effectiveness of thrombolytic therapy with tissue plasminogen activator as compared with streptokinase for acute myocardial infarction. N Engl J Med 1995;332:1418–24 [DOI] [PubMed] [Google Scholar]
- 18.Miller CS, Grandi SM, Shimony A, et al. Meta-analysis of efficacy and safety of new oral anticoagulants (dabigatran, rivaroxaban, apixaban) versus warfarin in patients with atrial fibrillation. Am J Cardiol 2012;110:453–60 [DOI] [PubMed] [Google Scholar]
- 19.Stacey D, Bennett CL, Barry MJ, et al. Decision aids for people facing health treatment or screening decisions. Cochrane Database Syst Rev 2011;10:CD001431. [DOI] [PubMed] [Google Scholar]
- 20.Walter SD, Sinclair JC. Uncertainty in the minimum event risk to justify treatment was evaluated. J Clin Epidemiol 2009;62: 816–24 [DOI] [PubMed] [Google Scholar]
- 21.Glick HA, Briggs AH, Polsky D. Quantifying stochastic uncertainty and presenting results of cost-effectiveness analyses. Expert Rev Pharmacoecon Outcomes Res 2001;1:25–36 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.