

Abstract
Dysfunctions of dopaminergic homeostasis leading to either low or high dopamine (DA) levels are causally linked to Parkinson's disease, schizophrenia, and addiction. Major sites of DA synthesis are the mesencephalic neurons originating in the substantia nigra and ventral tegmental area; these structures send major projections to the dorsal striatum (DSt) and nucleus accumbens (NAcc), respectively. DA finely tunes its own synthesis and release by activating DA D2 receptors (D2R). To date, this critical D2R-dependent function was thought to be solely due to activation of D2Rs on dopaminergic neurons (D2 autoreceptors); instead, using site-specific D2R knock-out mice, we uncover that D2 heteroreceptors located on non-DAergic medium spiny neurons participate in the control of DA levels. This D2 heteroreceptor-mediated mechanism is more efficient in the DSt than in NAcc, indicating that D2R signaling differentially regulates mesolimbic- versus nigrostriatal-mediated functions. This study reveals previously unappreciated control of DA signaling, shedding new light on region-specific regulation of DA-mediated effects.
Introduction
Dopamine (DA) is a key modulator of motor, emotional, hormonal, and cognitive functions. Midbrain DA neurons form four dopaminergic pathways identified on the topographical and functional properties of their projections arising from specific DA nuclei [e.g., ventral tegmental area (VTA), substantia nigra (SN) compacta (SNc)], which innervate regions involved in the control of specialized brain functions (reward, movement, etc.) (Björklund and Dunnett, 2007). Recent evidence is at odds with classical views on the homogeneity of DA neurons, showing different characteristics and specialization even of DA neurons located within the same nucleus (i.e., VTA) (Lammel et al., 2011). These findings suggest a high degree of functional diversity, which is very likely determined by intrinsic and extrinsic events that converge on DA neurons to modulate their functions.
DA D2 receptor (D2R)-mediated signaling has a relevant role in this respect (De Mei et al., 2009); indeed, DA acting in an autocrine manner stimulates D2Rs expressed by DAergic neurons (here referred to as autoreceptors), which inhibit DA synthesis and release, as well as the firing of DA neurons. DA moreover activates D2Rs on neurons receiving DA afferents (here referred to as heteroreceptors), which control the release of heterologous neurotransmitters such as glutamate (Bamford et al., 2004a,b), GABA (Drew et al., 1990; Centonze et al., 2002), and acetylcholine (Cragg, 2005; T. Zhang et al., 2009), which in return stimulate/inhibit DA release.
Present knowledge, however, posits that the D2R-mediated control of DA synthesis and release is essentially cell-autonomous; in agreement, knock-out of D2Rs completely abolishes D2 autoreceptor functions (L'Hirondel et al., 1998; Benoit-Marand et al., 2001; Rouge-Pont et al., 2002; Schmitz et al., 2002). Nevertheless, in D2R-null mice, both D2 autoreceptors and heteroreceptors were simultaneously ablated; lack of selective pharmacological, chemical, or genetic approaches to isolate D2R-mediated responses at these two sites induced us to generate mice with specific deletions of D2Rs either in DA neurons (D2 autoreceptor KO) or in striatal medium spiny neurons (MSNs; D2 heteroreceptor KO), using the CRE-loxP system (Branda and Dymecki, 2004). Mice carrying D2R-floxed alleles were mated with transgenic lines expressing the CRE under specific promoters. We achieved the selective ablation of D2 autoreceptors in SNc and VTA using the engrailed-1 (En1) CRE line (Kimmel et al., 2000), generating D2Rfloxflox/En1Cre/+ mice; and in MSNs, the primary neurons of the dorsal striatum (DSt) and nucleus accumbens (NAcc), using the D1Cre line (Lemberger et al., 2007), generating D2Rfloxflox/D1Cre/+ mice. The analysis of these mouse models allowed us to determine the site-specific role of D2R signaling in DA-mediated events.
We find that the selective loss of D2 autoreceptors in SNc and VTA unmasks prominent feedback mechanisms regulating DA release (Paladini et al., 2003). Importantly, we identified that these inhibitory feedback loops are D2R-mediated and, interestingly, they have a relatively larger impact in the DSt than in the NAcc, in agreement with differential regulation of DA release in these areas (L. Zhang et al., 2009). These results further our knowledge on the DAergic system and might provide insights on the physiopathological mechanisms underlying human pathologies and addiction.
Materials and Methods
Animals.
Mice were group housed under standard conditions (12 h light/dark cycles) with food and water ad libitum. All experiments were performed in 8- to 12-week-old mice and in accordance to the institutional animal care and use committees of the University of California Irvine, National Institute of Health, and institutional guidelines.
Drugs.
(-)Quinpirole hydrochloride, haloperidol, and cocaine hydrochloride were from Sigma; baclofen was from Tocris Bioscience. Drugs were dissolved directly in 0.9% NaCl with the exception of haloperidol, which was dissolved as previously described (Usiello et al., 2000).
Generation of mice.
To generate D2R-floxed alleles, a LoxP site was first inserted 5′ of the 900 bp NcoI genomic fragment of the D2R gene containing exon 2; to allow selection of the recombinant ES cells, a neomycin cassette (pGKneo), flanked by LoxP sites, was inserted at the 3′ end of the NcoI genomic fragment (Fig. 1A). This generated the LoxP-Exon2-LoxP-pGKneo-LoxP construct, which contained an additional HindIII site allowing discrimination between the WT and floxed alleles (Fig. 1A). The endogenous NcoI fragment of the D2R gene was then replaced with this construct in the KpnI-SalI genomic fragment of 6.5 Kb (Fig. 1A). The engineered KpnI-SalI fragment was then used to electroporate ES cells (129/S6 H1). Chimeras obtained from the injection of positive clones successfully transmitted the mutation to the progeny and homozygous D2Rfloxflox mice were obtained. D2Rfloxflox mice were then mated with En1Cre mice to generate D2Rfloxflox/En1Cre/+ animals and D1Cre mice to generate D2Rfloxflox/D1Cre/+ mice. Thereby, D2Rfloxflox/En1Cre/+ and D2Rfloxflox/D1Cre/+ mice are two independent lines, which have been analyzed independently from each other. Mutant and control mice for each line used in this study are in the same genetic background (87.5% C57BL6 × 12.5% 129 SV). During the establishment of the two mutants lines, we selected by Southern analyses only mice containing the excision of the neomycine cassette.
Figure 1.
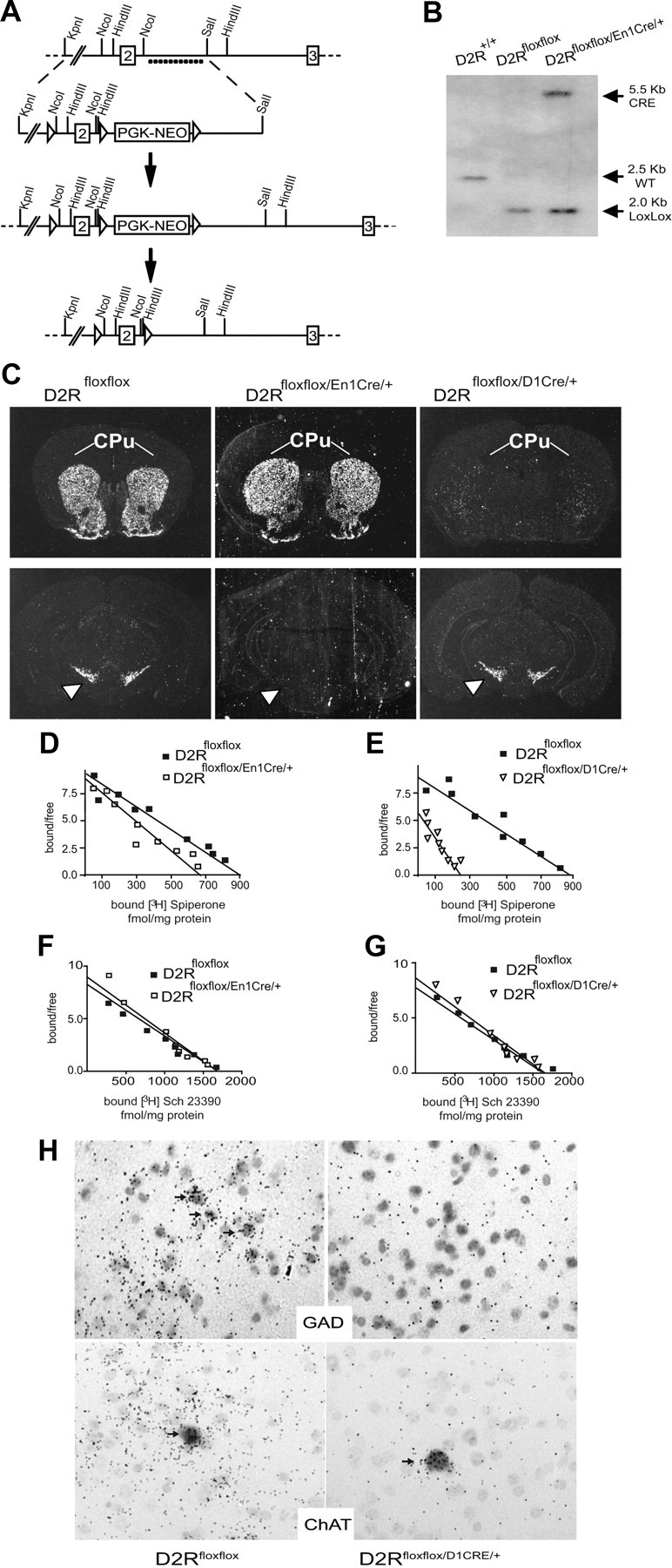
Generation and characterization of D2Rfloxflox/En1Cre/+ and D2Rfloxflox/D1Cre/+ mice. A, Scheme representing the strategy to generate D2Rfloxflox mice. ◁, Lox P site, dots indicate the probe used for genotyping. B, Representative Southern blot of genomic DNA from D2R+/+, D2Rfloxflox, and D2Rfloxflox/En1Cre/+ digested with HindIII and hybridized with 32P-labeled D2R and CRE probes; the genotype and size of the obtained fragments are as indicated. C, In situ hybridization using the D2R Exon2 probe showing selective ablation of D2R in SN and VTA (white arrowheads) for the D2Rfloxflox/En1Cre/+ and in the CPu (white lines) and NAcc for the D2Rfloxflox/D1Cre/+ mice. D, Scatchard analysis of the saturation isotherm for binding of the D2R antagonist [3H]-spiperone to striatal membranes from D2Rfloxflox (■; Bmax = 884 ± 25 fmol/mg protein; Kd = 171 ± 49 pm) and D2Rfloxflox/En1Cre/+ (□; Bmax = 690 ± 37 fmol/mg protein; Kd = 132 ± 21 pm) (n = 3). E, Scatchard analysis of the saturation isotherm for binding of the D2R antagonist [3H]-spiperone to striatal membranes from D2Rfloxflox (■; Bmax = 875 ± 28 fmol/mg protein; Kd = 184 ± 52 pm) and D2Rfloxflox/D1Cre/+ (▿; Bmax = 259 ± 35 fmol/mg protein; Kd = 198 ± 58 pm). Values are given ± SEM (n = 3). F, Scatchard analysis of the saturation isotherm for binding of the D1 DA receptor antagonist [3H]-SCH23390 in striatal extracts from D2Rfloxflox (■; Bmax = 1703 ± 204 fmol/mg protein; Kd = 297 ± 93 pm) and D2Rfloxflox/En1Cre/+ (□; Bmax = 1682 ± 168 fmol/mg protein; Kd = 354 ± 115 pm) mice (n = 3). G, Scatchard analysis of the saturation isotherm for binding of the D1 DA receptor antagonist [3H]-SCH23390 in striatal extracts from D2Rfloxflox (■; Bmax = 1695 ± 269 fmol/mg protein; Kd = 278 ± 85 pm) and D2Rfloxflox/D1Cre/+ (▿; Bmax = 1686 ± 198 fmol/mg protein; Kd = 354 ± 104 pm). H, Double in situ hybridizations of sections from D2Rfloxflox and D2Rfloxflox/D1Cre/+ mice using a 35S-labeled D2R-specific exon 2 probe together with the DIG-labeled Gad1 or ChAT probes, as indicated. Magnification 40×.
Genotyping.
Southern blot analyses were performed on genomic DNA extracted from tail biopsies of D2Rfloxflox, D2Rfloxflox/En1Cre/+, and D2Rfloxflox/D1Cre/+ mice, digested with HindIII, separated on 1% agarose gel and blotted to Hybond membranes (Millipore) (Baik et al., 1995). Immobilized genomic DNA was hybridized to the mouse 32P-labeled NcoI-SalI D2R genomic fragment (Fig. 1B) and to a BamHI 32P-labeled CRE probe containing the CRE coding region.
In situ hybridization.
Brains were rapidly dissected, snap frozen in isopentane-dry ice, and stored at −80°C. Coronal cryostat sections (10 μm) were hybridized with 35S-CTP-labeled D2R exon 2-specific probe, as previously described (Baik et al., 1995; Welter et al., 2007). Double in situ hybridizations were performed on sections from D2Rfloxflox and D2Rfloxflox/D1Cre/+ mice using a 35S-labeled D2R-specific exon 2 probe together with the digoxigenin (DIG)-labeled choline acetyl transferase (ChAT) or glutamic acid decarboxylase (Gad)1 probes. Radioactive and DIG-labeled probes were prepared as previously described (Baik et al., 1995; Clark et al., 2001). Pictures were taken with an SP5 microscope (DMRE; Leica).
Ligand-binding assays.
D2R ligand-binding assays were performed on striatal membranes using [3H]-Spiperone (specific activity, 91 Ci mmol−1; GE Healthcare). Binding assays were performed as previously described (Baik et al., 1995; Usiello et al., 2000), performed in triplicates, and repeated three times. D1R ligand-binding assays were carried on striatal membranes (15 μg) using [3H]-SCH23390 (specific activity, 85 Ci mmol−1; GE Healthcare), with triplicates of each point; experiments were repeated three times. Binding assays were performed as previously described (Baik et al., 1995). Results were analyzed with Prism 4.0 (GraphPad).
Behavioral analyses.
Behavioral experiments were performed using male mice (8- to 12-weeks-old); in each experiment and for each line, knock-out and control (D2Rfloxflox) mice were issued from the same mating. Four days before experiments, mice were moved to soundproof behavioral rooms. The open field was a white square box (30 × 30 cm; 70 lux); the home cage was a transparent plastic box (20 × 30 cm). Activity was followed by a video-tracking system (Viewpoint) for 1 h. An observer blind to mouse genotype scored rearing activity during the first 5 min of each test. Quinpirole (i.p.) effect was observed in animals not habituated to the testing cage (20 × 30 cm) and analyzed for 30 min; see Figure 9 for quinpirole's effect observed in animals exposed to the open field for 30 min. Cocaine (i.p.) was administered after habituation to a new home cage (1 h) and activity recorded for 30 min. Catalepsy by haloperidol (i.p.) was evaluated by the bar test; the time spent in a cataleptic position was scored 3 h after treatment, cutoff was given at 120 s, as previously described (Usiello et al., 2000). The rotarod test was performed by placing mice on the rotating rod and measuring their fall latencies. Rotations were increased from 4 to 40 rpm over a period of 5 min. Mice were given 3 trials/d on 4 consecutive days; the best performance from each day was used for analysis. Rotarod data were analyzed by ANOVA with repeated measures.
Figure 9.
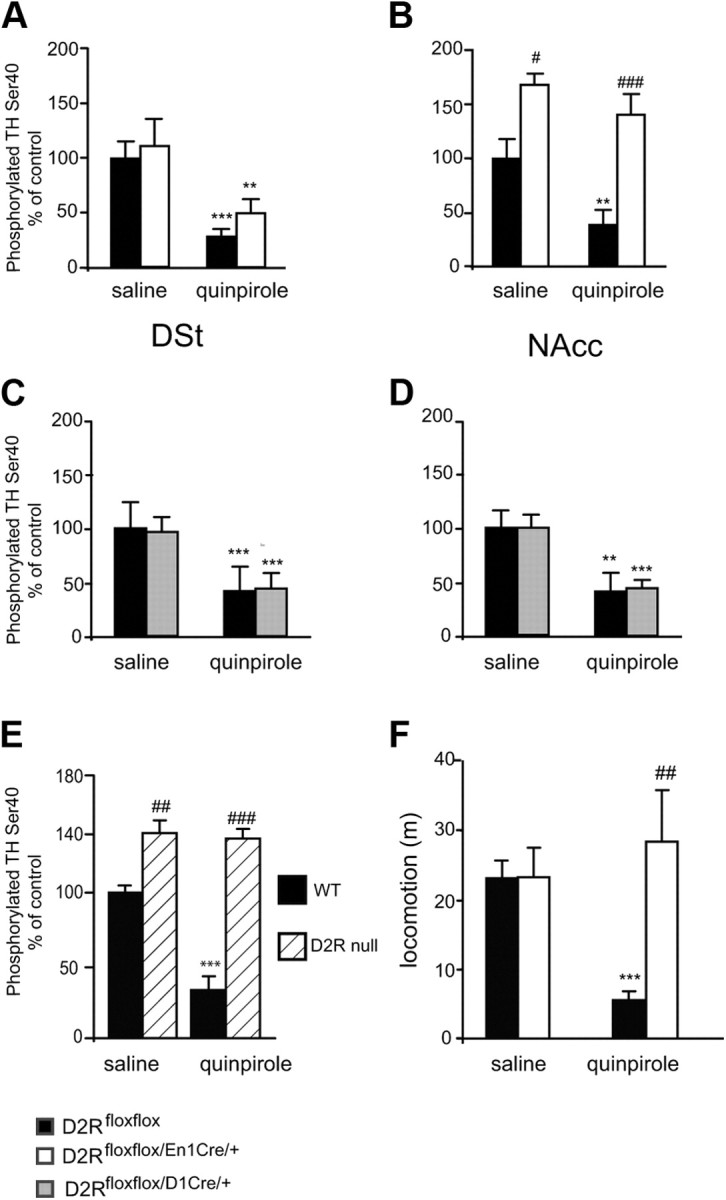
Effect of loss of D2 autoreceptors or heteroreceptors on TH phosphorylation. Western blot analyses of pTH-Ser40 levels in DSt and NAcc extracts (30 μg/sample) of D2Rfloxflox/En1Cre/+, D2Rfloxflox/D1Cre/+, and D2Rfloxflox mice treated either with saline or with quinpirole (0.2 mg/kg), as indicated. Values represent the ratio of pTH-Ser40/total TH; the values of the ratio from saline-treated D2Rfloxflox mice were arbitrarily set at 100%. A, Ratios obtained from analyses of DSt extracts from D2Rfloxflox (black bars) and D2Rfloxflox/En1Cre/+ (white bars). Two-way ANOVA shows no genotype effect (F(1,17) = 1.848, p > 0.05). B, Same as in A for NAcc extracts, D2Rfloxflox (black bars) and D2Rfloxflox/En1Cre/+ (white bars). Two-way ANOVA shows Genotype effect: F(1,17) = 33.126 p < 0.001. C, Same analysis as in A, but using DSt extracts from D2Rfloxflox (black bars) and D2Rfloxflox/D1Cre/+ (gray bars). Two-way ANOVA no Genotype effect: F(1,18) = 1.984 p > 0.05. D, Same analysis as in B but using NAcc extracts from D2Rfloxflox (black bars) and D2Rfloxflox/D1Cre/+ (gray bars). Two-way ANOVA no Genotype effect: F(1,18) = 1.976 p > 0.05. Values are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 versus saline control. D2Rfloxflox versus D2Rfloxflox/En1Cre/+, #p < 0.05, ##p < 0.01. E, Ratio obtained from analyses of DSt extracts (30 μg/sample) from D2R-null mice treated either with saline or quinpirole (0.2 mg/kg), genotypes are as indicated. Values represent the ratio of pTH-Ser40/total TH; the values of the ratio from saline-treated D2R mice were arbitrarily set at 100%. A significant increase of pTH-Ser40 was observed in D2R-null mice, which was not affected by quinpirole treatment compared with WT extracts. Treatment × Genotype: F(1,17) = 34.142, p < 0.01.***p < 0.001 versus saline; WT versus D2R−/−: ##p < 0.01; ###p < 0.001. F, D2Rfloxflox (black bars) and D2Rfloxflox/En1Cre/+ (white bars) treated either with saline or quinpirole (0.2 mg/kg), as indicated, were exposed to the open field for 30 min. While quinpirole induced motor sedation in D2Rfloxflox mice, no effect of this drug was observed in D2Rfloxflox/En1Cre/+ mice under these conditions. Treatment × Genotype: F(1,33) = 10.94. Saline versus quinpirole: ***p < 0.001; D2Rfloxflox treated versus D2Rfloxflox/En1Cre/+ treated: ##p < 0.01.
Western blots.
Animals treated with saline or quinpirole (0.02 or 0.2 mg/kg, respectively; i.p.) were killed 30 min after the administration of the compound. Heads were immediately immersed in liquid nitrogen for 6 s. Brains were frozen, punched in the region of interest, and processed as previously described (Svenningsson et al., 2000) to preserve phosphorylations. Protein determination was made by BCA. Equal amounts of proteins (30 μg) were loaded onto 10% SDS/PAGE and transferred to PVDF membranes (Bio-Rad). Western analyses were performed using antibodies directed against phosphor-Ser40-TH (1:1500; Millipore), total TH (1:3000; Millipore), and α-tubulin (1:5000; Sigma); goat anti-rabbit secondary antibodies (1:5000; Millipore) were used and blots revealed with ECL Plus (GE Healthcare). Quantifications were performed using NIH ImageJ (version 1.42q) software.
Analysis of DA and its metabolites by HPLC
Tissue punches from DSt, NAcc, and PFC were homogenized by sonication in 30 volumes (w/v) of ice-cold 100 mm HClO4. The homogenates were centrifuged for 20 min at 15,000 g, 4°C. The supernatants were recovered and passed through 0.2 μm filters and monoamines and metabolites quantified in the filtrates by HPLC-EC. The HPLC system consisted of a BASi LC-4C detector coupled to a BASi LCEC radial flow cell. Flow was provided by a Shimadzu LC-20AD solvent delivery module, preceded by an online degasser series 1100 from Agilent. The chromatograms were analyzed using PowerChrom software (eDAQ). Monoamines in 10 μl of tissue filtrate were separated on a 1 × 100 mm UniJet microbore 5 μm C-8 column (BASi). The mobile phase consisted of 24 mm Na2HPO4, 0.3 mm sodium octyl sulfate, 27.4 mm citric acid, 107 μm EDTA and 11% (v/v) MeOH, pH adjusted to 4.5 with NaOH. The flow was set at 100 μl/min and the potential was set at +750 mV relative to an Ag/Cl reference electrode. Elution times were as follows (in min): 3,4-dihydroxyphenylacetic acid (DOPAC), 2.6; dopamine, 5.1; homovanillic acid (HVA), 6.4.
Amperometry and cyclic voltammetry.
Adult male D2Rfloxflox and D2Rfloxflox/En1Cre/+ or D2Rfloxflox/D1Cre/+ were analyzed the same day. Slices were cut at 250 μm and allowed to recover at room temperature for 1.5 h. Recording temperature was ∼30°C, and carbon fiber electrodes were placed close to corpus callosum (laterodorsal to dorsal section). For amperometry (AMP), a constant voltage of +500 mV was used and traces were digitally filtered using a Gaussian filter (250 Hz cutoff frequency). For cyclic voltammetry (CV), electrodes were calibrated before and after each experiment to estimate DA concentration. Each protocol was performed independently, except for the paired-pulse experiments performed in CV. Eight prepulses were applied to achieve constant DA release. Single-pulse DA levels and decay parameters were measured using the eighth prepulse. Paired-pulse experiments were performed using AMP (0.5 and 1 s) and CV (5–60 s). Trains of five or 10 pulses at 20 Hz were analyzed by CV. Quinpirole was applied for 10 min, then removed and slices were washed for additional 10 min.
Cell-attached electrophysiological recordings from adult mouse brain slices.
Horizontal midbrain mouse slices (200 μm) preparation, recovery, and external solutions were made as described previously (Kapfhamer et al., 2010). Cells were visualized with an upright microscope using infrared differential interference contrast illumination and cell-attached voltage-clamp recordings were made with a Multiclamp 700A amplifier (Molecular Devices). The external solution for electrophysiology studies was carbogen (95% O2/5% CO2)-bubbled artificial CSF containing 126 mm NaCl, 2.5 mm KCl, 1.2 mm NaH2PO4, 1.2 mm MgCl2, 2.4 mm CaCl2, 18 mm NaHCO3, 11 mm glucose, pH 7.2–7.4, and 301–305 mOsm. Electrodes (2–4 MΩ) contained the following (in mm): 117 cesium methansulfonic acid, 20 HEPES, 0.4 EGTA, 2.8 NaCl, 5 TEA-Cl, 2.5 MgATP, 0.25 MgGTP (pH 7.2–7.4), 275–285 mOsm. Baclofen (2 μm) and quinpirole (2 μm) were applied by bath superfusion.
Statistical analysis.
Statistical analyses were performed independently for each line by comparing each knock-out to its own WT (D2Rfloxflox) littermates. We opted for this type of analysis because, despite both lines being generated from the common D2Rfloxflox mice, these mice were subsequently mated with two independent lines of CRE mice. Therefore, the resulting D2Rfloxflox/En1Cre/+ and D2Rfloxflox/D1Cre/+ and the respective WT littermates are two independent lines, which were obtained and analyzed at different times. All values are given as mean ± SEM. Student's t test was used for direct comparisons between D2Rfloxflox and knock-outs of each line; ANOVA was used to determine group effects and interactions followed by the appropriate post hoc comparison (Bonferroni; p < 0.05 was considered statistically significant).
Results
Generation of site-specific D2 knock-out mice
To generate site-specific D2R mutants, exon2 of the D2R gene was flanked by loxP sites, thus generating D2Rfloxflox mice (Fig. 1A). D2Rfloxflox mice are undistinguishable from their WT controls as assessed by molecular, biochemical, and behavioral analyses (data not shown); these mice breed and reproduce normally. Two independent lines of site-specific D2R knock-out mice were generated using the D2Rfloxflox mice. To abolish D2Rs expression from DAergic neurons, D2Rfloxflox mice were mated with mice carrying the CRE recombinase under the control of the En1 gene promoter (Kimmel et al., 2000), thus generating D2Rfloxflox/En1Cre/+ mice (here referred as D2 autoreceptor KO). Deletion of D2Rs in MSNs neurons was achieved by mating of D2Rfloxflox mice with the D1CRE transgenic line (Lemberger et al., 2007) in which the DA D1R gene promoter drives the CRE expression, generating D2Rfloxflox/D1Cre/+ mice (here referred as D2 heteroreceptor KO). Southern blot analyses confirmed the presence of the D2R floxed alleles and CRE gene in both lines (Fig. 1B, D2Rfloxflox/En1Cre/+). En1-CRE expression is restricted to the mesencephalon (Simon et al., 2001), and it did not affect the number of DAergic neurons in D2Rfloxflox/En1Cre/+ mice as established by stereological counts of midbrain sections labeled with an anti-TH antibody (WT = 7807 ± 920 TH+ cells; D2Rfloxflox/En1Cre/+ = 7723 ± 640; p = 0.137; n = 3/genotype). In situ hybridization analyses, using the D2R exon 2-specific probe, showed the selective ablation of D2R transcripts in the SNc and VTA in D2Rfloxflox/En1Cre/+ but not in the caudate–putamen (CPu; Fig. 1C); conversely, in D2Rfloxflox/D1Cre/+, D2R mRNAs were selectively ablated from MSNs neurons, leaving the SNc and VTA expression unaltered (Fig. 1C) compared with D2Rfloxflox (here referred to as control or WT) brain sections. The ablation of D2R at the protein level was supported by ligand binding analyses on striatal membranes using a D2-specific ligand (3H-spiperone); we observed a 20% reduction of D2R binding sites in D2Rfloxflox/En1Cre/+ (Fig. 1D) and 70% reduction in D2Rfloxflox/D1Cre/+ (Fig. 1E) compared with WT littermates of each line. Importantly, in both lines of site-specific D2R mutants, D1R mRNA and protein levels were not altered, as established by qRT-PCR (data not shown) and ligand binding experiments (Fig. 1F,G).
The specificity of the D1Cre line in selectively ablating D2R in the MSNs of D2Rfloxflox/D1Cre/+ mice (Fig. 1C) but not in striatal cholinergic interneurons, where the D1R is not expressed (Rivera et al., 2002), was further confirmed by double immunostaining/in situ analyses (Fig. 1H). Indeed, using the D2R exon 2 radioactive probe and antibodies directed either against GAD as marker of MSNs (gabaergic neurons; Fig. 1H, top) or ChAT as marker of cholinergic interneurons (Fig. 1H, bottom), we observed the absence of D2R in GAD+ neurons but not in ChAT+ interneurons. Other types of striatal interneurons (calbindin+, calretin+, NPY+, parvalbumin+) were not labeled by the D2R probe in either WT or D2Rfloxflox/D1Cre/+ mice (data not shown). In addition, in support of D2R ablation in MSNs, as previously observed in D2R-null mice (Baik et al., 1995), a 61% (p < 0.01) increase of enkephalin mRNA expression was found by qRT-PCR on mRNAs from D2Rfloxflox/D1Cre/+ (n = 5) compared with D2Rfloxflox (n = 5) mice.
D2R-mediated control of motor activity
One prominent function of D2Rs is the control of motor activity (Baik et al., 1995). Interestingly, loss of autoreceptors in D2Rfloxflox/En1Cre/+ mice does not affect horizontal motor activity in the home cage compared with WT siblings (Fig. 2A). On the contrary, deletion of D2 heteroreceptors in the MSNs in D2Rfloxflox/D1Cre/+ mice resulted in profound impairments of motor activity with respect to their WT littermates (Fig. 2B). In addition, coordination of movements of D2Rfloxflox/En1Cre/+ mice was not different from that of WT littermates, as assessed by the rotarod test (Fig. 2C); in contrast, this function was greatly affected in D2Rfloxflox/D1Cre/+ mice compared with their WT siblings (Fig. 2D). Next, we compared the motor activity of each mutant with the respective WT littermates in the open field, an unfamiliar environment with an anxiogenic component. Interestingly, D2Rfloxflox/En1Cre/+ mice were hyperresponsive to this environment in both the horizontal and vertical movements compared with their WT littermates (Fig. 2E), while D2Rfloxflox/D1Cre/+ mice showed motor impairment consistent with their behavior in the home cage compared with their WT siblings (Fig. 2F). Furthermore, D2Rfloxflox/En1Cre/+ mice presented an augmented motor response to acute administration of cocaine, which was well above that of control siblings (Fig. 2G). Conversely, cocaine-treated D2Rfloxflox/D1Cre/+ mice showed a blunted response to the motor-activating effect of the drug compared with WT littermates (Fig. 2H). These results indicate that the loss of D2R in DAergic neurons or in MSN target neurons differently affect motor behavior and the acute response to psychostimulants. Hyperactivity in the open field and to cocaine, but not in the home cage in D2 autoreceptor mutants, suggests increased DA activation of the mesolimbic pathway (Hooks and Kalivas, 1995). Conversely, the blunted motor response of D2Rfloxflox/D1Cre/+ mutants in each of the conditions tested shows the key role of D2R in MSNs in the regulation of motor behavior.
Figure 2.

Loss of D2 autoreceptors or heteroreceptors differently affects motor behavior. A, Locomotion and rearing activity of D2Rfloxflox (black bars) and D2Rfloxflox/En1Cre/+ (white bars) mice in the home cage (n = 9–10). No difference was observed between genotypes. Student's t test, p > 0.05. B, Locomotion and rearing activity of D2Rfloxflox (black bars) and D2Rfloxflox/D1Cre/+ (gray bars) mice in the home cage (n = 9–10). Student's t test, ***p < 0.001. C, Motor coordination as assessed using the rotarod test. Values represent the average ± SEM of fall latency (s) across 4 d, D2Rfloxflox (■) and D2Rfloxflox/En1Cre/+ (□) mice. Both genotypes showed similar latencies to fall (F(3,36) = 1.178, p > 0.05) and improved their performance with time (F(3,36) = 14.426, p < 0.001). D, Motor coordination as assessed using the rotarod test. Values represent the average ± SEM of fall latency (s) across 4 d, D2Rfloxflox (■) and D2Rfloxflox/D1Cre/+ (▩) mice. D2Rfloxflox/D1Cre/+ showed significantly shorter latencies to fall (F(3,34) = 6.239 p < 0.05). E, Locomotion and rearing activity in the open field of D2Rfloxflox (black bars) and D2Rfloxflox/En1Cre/+ (white bars) (n = 9–10). Student's t test, *p < 0.05 and rearing **p < 0.01. F, Locomotion and rearing activity in the open field of D2Rfloxflox (black bars) and D2Rfloxflox/D1Cre/+ (gray bars) (n = 9–10). Student's t test, **p < 0.01, ***p < 0.001. G, Motor activity after acute cocaine injection (15 mg/kg) recorded for 30 min of D2Rfloxflox (black bars) and D2Rfloxflox/En1Cre/+ (white bars). Two-way ANOVA Treatment × Genotype: F(1,34) = 5.249, p < 0.05. Saline versus treated: *p < 0.05, **p < 0.01. D2Rfloxflox/En1Cre/+ versus D2Rfloxflox: #p < 0.05. H, Motor activity after acute cocaine injection (15 mg/kg) recorded for 30 min for D2Rfloxflox (black bars) and D2Rfloxflox/D1Cre/+ (gray bars) mice. Two-way ANOVA Treatment × Genotype: F(1,49) = 5.759, p < 0.05. Saline versus treated: *p < 0.05. D2Rfloxflox/D1Cre/+ versus D2Rfloxflox: #p < 0.05, ###p < 0.001.
Behavioral responses of auto- and hetero-D2R mutants to pharmacological challenges
To further characterize the respective contribution of auto- and hetero-D2Rs in motor control, we analyzed the behavioral response of D2Rfloxflox/En1Cre/+ mice to pharmacological challenge using dopaminergic D2-like specific ligands. Haloperidol, a D2-like antagonist, induces catalepsy through blockade of striatal D2Rs (Boulay et al., 2000). Accordingly, haloperidol induced catalepsy in the bar test in both D2Rfloxflox/En1Cre/+ mutants and WT littermates (Fig. 3A), while in D2Rfloxflox/D1Cre/+ mice, haloperidol exhibited a minimal effect that was detected only at the highest dose tested (4 mg/kg; Fig. 3B) compared with WT siblings.
Figure 3.

Behavioral effects of D2-like agonists and antagonists in D2Rfloxflox/En1Cre/+ and D2Rfloxflox/D1Cre/+ mice. A, Haloperidol-induced catalepsy in D2Rfloxflox (black bars) and D2Rfloxflox/En1Cre/+ (white bars) mice. Two-way ANOVA Treatment effect (F(3,81) = 135.637, p < 0.001) and a trend toward genotype effect (F(1,81) = 3.906, p = 0.052). B, Haloperidol-induced catalepsy in D2Rfloxflox (black bars) versus D2Rfloxflox/D1Cre/+ (gray bars) mice. Two-way ANOVA Genotype × Treatment effect (F(2,57) = 14.635, p < 0.001). C, Differential quinpirole induced motor sedation in D2Rfloxflox (black bars) versus D2Rfloxflox/En1Cre/+ (white bars) mice. Two-way ANOVA significant Genotype (F(1,48) = 18.967, p < 0.001) and Treatment effects (F(2,48) = 17.878, p < 0.001). D, Quinpirole induced sedation in D2Rfloxflox (black bars) versus D2Rfloxflox/D1Cre/+ (gray bars) mice. Two-way ANOVA: Genotype × Treatment (F(2,37) = 8.284 p < 0.01). Saline versus treated *p < 0.05; **p < 0.01; ***p < 0.001; D2Rfloxflox/En1Cre/+ treated versus D2Rfloxflox treated #p < 0.05; ##p < 0.01. D2Rfloxflox/D1Cre/+ treated versus D2Rfloxflox treated, +++p < 0.001.
Interestingly, haloperidol at the lowest concentration used (0.04 mg/kg) was more effective in inducing catalepsy in D2Rfloxflox/En1Cre/+ than in WT littermates (Fig. 3A). These results indicate that haloperidol-induced catalepsy is mostly induced by blockade of D2 heteroreceptors on striatal neurons since it is present in D2Rfloxflox/En1Cre/+ mutants but almost completely absent in D2Rfloxflox/D1Cre/+ mice (Fig. 3B).
Quinpirole, a D2-specific agonist, elicits motor sedation (Usiello et al., 2000) at low doses; this effect is thought to depend on activation of D2 autoreceptors and the consequent reduction of DA release. D2Rfloxflox/En1Cre/+ and D2Rfloxflox/D1Cre/+ mice, together with the WT littermates of each line, were thus administered 0.02 or 0.2 mg/kg quinpirole and their motor behavior recorded in the home cage. Quinpirole inhibited motor behavior in WT mice at both doses tested (F(2,25) = 18.048, p < 0.0001). In D2Rfloxflox/En1Cre/+ mice, in contrast, quinpirole did not inhibit motor behavior at a dose of 0.02 mg/kg, while it induced a significant reduction at 0.2 mg/kg (Fig. 3C), albeit less strong than in WT littermates. In D2Rfloxflox/D1Cre/+ mice, the motor suppressant effect of quinpirole was significant with respect to saline-treated littermates only at 0.2 mg/kg (Fig. 3D), possibly due to the already highly reduced motor skills of these animals; at 0.02 mg/kg the treatment did not reach statistical significance. These data suggest that the motor sedative effect of low doses of quinpirole is due to activation of both D2 autoreceptors and heteroreceptors, although the activation of D2 autoreceptors has a more prominent impact on this parameter.
These results were puzzling as conventional opinion is that motor sedation is solely a function of autoreceptors (Bello et al., 2011) and thereby should be lost in D2Rfloxflox/En1Cre/+ mice; thus, we performed ex vivo brain slice electrophysiology to compare the regulation of firing of SNc and VTA neurons by quinpirole in these animals and their WT littermates.
Electrophysiological characterization of DA neurons in D2Rfloxflox/En1Cre/+ mice
Recordings were performed in cell-attached mode to preserve the natural intracellular milieu of the neurons. SNc neurons from animals of both genotypes possessed an action potential waveform of ∼1.5–1.8 ms under the cell-attached configuration (Ungless et al., 2004), and were Ih (hyperpolarization-activated current) positive when examined after breaking into whole-cell mode at the end of the experiment; these features are consistent with dopaminergic neurons, which represent the majority of SNc and VTA neurons (Margolis et al., 2006; Lammel et al., 2008).
D2Rs negatively regulate firing in midbrain dopamine neurons by activating hyperpolarizing G-protein coupled inward rectifier K+ (GIRK) channels (Mercuri et al., 1997; Beckstead et al., 2004). Interestingly, while SNc and VTA neurons from control mice showed strong suppression of firing by quinpirole (2 μm) in vitro, neurons from D2Rfloxflox/En1Cre/+ did not (Fig. 4A,C), indicating a loss of functional D2 autoreceptors from dopaminergic neurons in these mice. In addition, the GABAB receptor agonist baclofen (2 μm) was able to strongly inhibit firing in both control and D2Rfloxflox/En1Cre/+ neurons, suggesting that activation of GIRK channels by a receptor other than D2R remained intact in D2Rfloxflox/En1Cre/+ mice (Fig. 4B,D) (Mercuri et al., 1997; Bartlett et al., 2005). Thus, electrophysiological testing clearly established that the autoreceptor functions regulating the firing of DA neurons are absent in D2Rfloxflox/En1Cre/+ mice.
Figure 4.
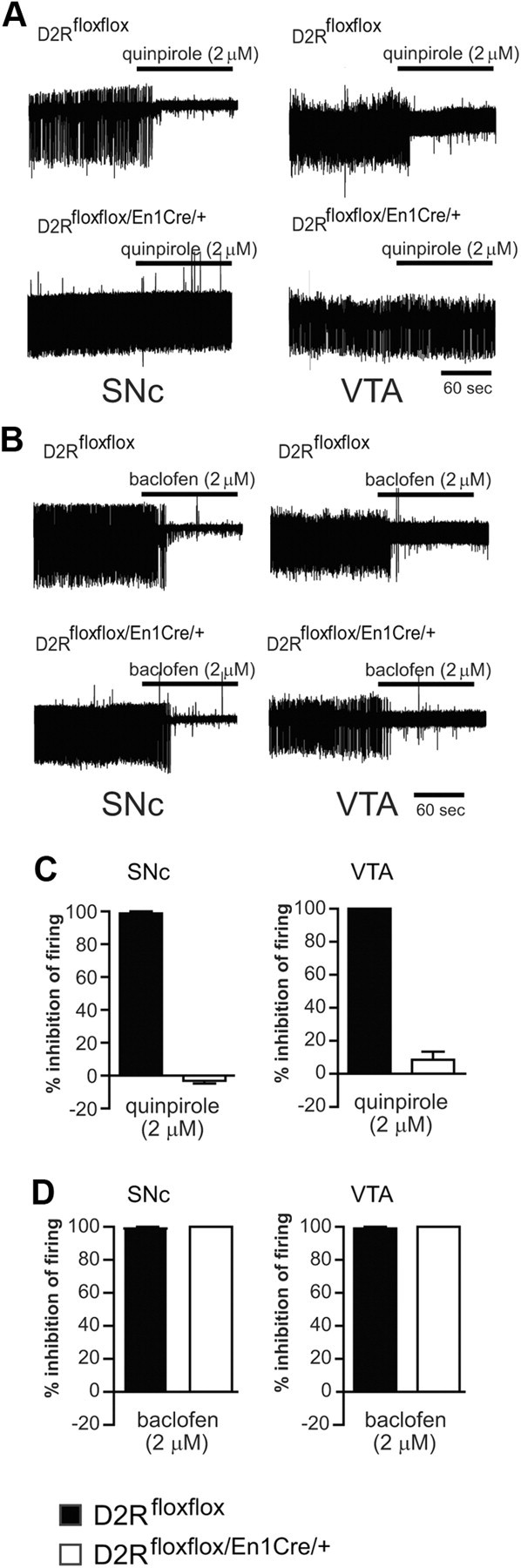
Electrophysiological characterization of SNc and VTA neurons of D2Rfloxflox/En1Cre/+ mice. A, Loss of presynaptic D2 autoreceptor-mediated inhibition of firing in SNc and VTA of D2Rfloxflox/En1Cre/+ mice compared with control mice. B, Normal GABAB-mediated inhibition of firing in both genotypes (representative examples). C, Percentage of firing inhibition by quinpirole (2 μm) in SNc (left) and VTA (right) in D2Rfloxflox (black bars) and D2Rfloxflox/En1Cre/+ (white bars) (n = 4 for each condition; for both regions: p < 0.05 D2Rfloxflox vs D2Rfloxflox/En1Cre/+, ranked sum test). D, Percentage of firing inhibition by baclofen (2 μm) in SNc (left; n = 4 D2Rfloxflox/En1Cre/+, n = 3 D2Rfloxflox) and VTA (right; n = 4 for both genotypes) [not significant D2Rfloxflox (black bars) vs D2Rfloxflox/En1Cre/+ (white bars) in either region].
Both D2 autoreceptors and heteroreceptors inhibit evoked DA overflow
While the recordings indicate a lack of quinpirole effect on DA cells firing in D2Rfloxflox/En1Cre/+ mice, the effect of the drug on behavior suggests that D2 heteroreceptors might also regulate DA release. To examine this hypothesis, we first measured DA and its metabolites DOPAC and HVA by HPLC in tissue punches homogenates from the NAcc, DSt, and cortex of D2Rfloxflox/En1Cre/+ and D2Rfloxflox/D1Cre/+ mice and the WT littermates for each line. Interestingly, while no difference from WT was found in the DSt (Fig. 5A), we measured a significant increase of DA in the NAcc and cortex of D2Rfloxflox/En1Cre/+ mice compared with WT littermates (Fig. 5B,C). In structures from D2Rfloxflox/D1Cre/+ mice, levels of DA and its metabolites were unaltered with respect to WT littermates (Fig. 5D–F).
Figure 5.

Determination of DA and its metabolites in D2Rfloxflox, D2Rfloxflox/En1Cre/+, and D2Rfloxflox/D1Cre/+ mice. Values indicate the percentage of DA, DOPAC, and HVA with respect to D2Rfloxflox levels, which were arbitrarily set at 100%. A–C, Determination were made by HPLC on extracts from tissue punches of DSt (A), NAcc (B), and PFC (C) from D2Rfloxflox (black bars) and D2Rfloxflox/En1Cre/+ (white bars). D–F, DSt (D), NAcc (E), and PFC (F) from D2Rfloxflox (black bars) and D2Rfloxflox/D1Cre/+ (gray bars). Statistical analyses were performed by the Student's t test: *p < 0.05; **p < 0.01 (n = 7–8).
We then compared the effects D2 autoreceptors and heteroreceptors deletion on evoked DA overflow by performing CV in acute brain slices of DSt (Heien and Wightman, 2006). In agreement with previous results obtained in the D2R-null mice (Schmitz et al., 2002, 2003), DA overflow evoked by a single stimulus in the DSt of D2Rfloxflox/En1Cre/+ mice was decreased with respect to control littermates (Fig. 6A,B).
Figure 6.

Characterization of DA overflow by CV in D2Rfloxflox/En1Cre/+ and D2Rfloxflox/D1Cre/+ mice. Ablation of either D2 autoreceptors or D2 heteroreceptors in MSNs affects DA overflow. Analysis of DA overflow by CV. A, Representative traces for cyclic voltammetry experiments in the DSt of D2Rfloxflox and D2Rfloxflox/En1Cre/+, as indicated. Scale bars: vertical, 0.15 μm; horizontal, 2.0 s. B, Single-pulse stimulation in D2Rfloxflox/En1Cre/+ mice (white bar) (n = 19–20). C, Quinpirole (Quin)-mediated inhibition of DA overflow in: D2Rfloxflox/En1Cre/+ mice (○, 50 nm; □, 500 nm) and D2Rfloxflox (●, 50 nm; ■, 500 nm) (50 nm: n = 7, F(1,261) = 8.46, p = 0.0039; 500 nm: n = 7–8, F(1,204) = 23.52, p < 0.001). D, Representative traces for cyclic voltammetry experiments in the DSt of D2Rfloxflox and D2Rfloxflox/D1Cre/+, as indicated. Scale bars: vertical, 0.15 μm; horizontal, 2.0 s. E, Single-pulse stimulation in D2Rfloxflox/D1Cre/+ mice (n = 35–37). F, Quinpirole-mediated inhibition of DA overflow in D2Rfloxflox/D1Cre/+ (▩) and D2Rfloxflox (■) mice (500 nm: n = 9–10, F(1,274) = 33.23, p < 0.0001). Two-tailed unpaired t test was used in B and E. In C and F, significance was determined by F test, which compares the individual bottom values obtained for each genotype from a nonlinear regression with an average bottom value obtained from a global regression using both genotypes simultaneously. *p < 0.05, ***p < 0.001.
Quinpirole's effect on DA overflow is absent in D2R-null mice (Schmitz et al., 2002, 2003). Surprisingly, however, D2Rfloxflox/En1Cre/+ mice still exhibited significant dose-dependent inhibition of evoked DA release by quinpirole (Fig. 6C). These results strongly suggest that the D2 autoreceptor is not the only D2R that participates in the inhibition of evoked DA release.
To determine whether D2 heteroreceptors located on MSNs can inhibit evoked DA overflow, we examined D2Rfloxflox/D1Cre/+ mice and their WT littermates. Importantly, D2Rfloxflox/D1Cre/+ mice also displayed 20% lower evoked DA overflow than WT siblings (Fig. 6D,E), consistent with a role for D2 heteroreceptors located on MSNs in the presynaptic regulation of DA release. Consistently, quinpirole's inhibition of DA overflow was attenuated in D2Rfloxflox/D1Cre/+ mice (Fig. 6F), supporting the hypothesis that D2 receptors located on MSNs contribute to the inhibition of evoked DA release.
D2 heteroreceptors and autoreceptors control evoked DA release, but only autoreceptors control reuptake
Because evoked DA overflow measured by CV depends on both DA release and reuptake by the DA transporter (DAT) (Schmitz et al., 2001), we also recorded evoked DA release by AMP, a procedure in which the event amplitudes are less affected by DAT activity, to confirm differences in DA overflow. Consistent with CV results, both D2Rfloxflox/En1Cre/+ and D2Rfloxflox/D1Cre/+ mice exhibited smaller relative peak amplitudes than their respective control mice, confirming that there is indeed lower evoked release in both mutant lines, as shown by representative traces in Figure 7, A (D2Rfloxflox: 74.2 ± 16.6 pA, n = 10; D2Rfloxflox/En1Cre/+: 32.7 ± 3.85 pA, n = 10, p < 0.05) and B (D2Rfloxflox: 417 ± 47.7 pA, n = 20; D2Rfloxflox/D1Cre/+: 275 ± 36.9 pA, n = 21, p < 0.05).
Figure 7.

Analysis of DA reuptake in D2Rfloxflox/En1Cre/+ and D2Rfloxflox/D1Cre/+ mice. Only D2Rfloxflox/En1Cre/+ mice present changes in DAT function, whereas D2Rfloxflox/D1Cre/+ do not, as assessed by the analysis of the decay parameters and the nomifensine-mediated increase of DA overflow. Black bars, D2Rfloxflox; white bars, D2Rfloxflox/En1Cre/+; gray bars, D2Rfloxflox/D1Cre/+. A, B, Representative traces for amperometric experiments in the DSt; the genotype corresponding to each trace is indicated. Stimulation artifacts were digitally removed to improve visualization. Scale bars: A: vertical, 20 pA; horizontal, 0.3 s. B: vertical, 240 pA; horizontal, 0.3 s. C, Decay parameters were calculated from the DA overflow spikes obtained from CV experiments showing that D2Rfloxflox/En1Cre/+ mice (white bars) exhibit a significant change in spike width (t1/2) compared with D2Rfloxflox mice (black bars). The decay time from the 75% to 25% of the spike (fall time) and the decay constant (τ) presented no changes (n = 16–17). Decay parameters calculated by AMP show a minimal but not significant decrease in the fall time and decay constant (p = 0.08 and 0.07, respectively; n = 10) in D2Rfloxflox/En1Cre/+ mice. D, Decay parameters calculated from the DA overflow spikes in D2Rfloxflox/D1Cre/+ mice (gray bars) showed no significant changes in spike width, decay time, or decay constant (CV: n = 13–14; AMP: n = 10) compared with D2Rfloxflox mice (black bars). E, DA overflow measured in D2Rfloxflox/En1Cre/+ mice (□) during nomifensine-mediated blockade of DAT does not reach the same levels of WT mice (■) (nomifensine: 5 μm; n = 4; increase in [DA], D2Rfloxflox: 2.36 μm; D2Rfloxflox/En1Cre/+: 0.92). F, Nomifensine-mediated blockade of DAT induces a significant increase in DA overflow in D2Rfloxflox/En1Cre/+ mice (□) when compared with the baseline-normalized response with respect to WT mice (■) (nomifensine: 5 μm; n = 4; increase in [DA], D2Rfloxflox: ∼2-fold; D2Rfloxflox/En1Cre/+: ∼2.7-fold; F(1,89) = 8.61, p = 0.004). Maximal DA-overflow concentration and normalized response were calculated using a nonlinear regression that takes in consideration the plateau and the rising independently. In F, significance was determined by an F test, which compares the individual top values obtained for each genotype from the nonlinear regression with an average top value obtained from a global regression using both genotypes simultaneously. Two-tailed unpaired t test with Welch's correction was used in C and D.
DA reuptake can be estimated with CV from the kinetic parameters of falling phase of the signal: of these, the duration at half-height is a particularly reliable indicator of reuptake (Mosharov and Sulzer, 2005). We noted a small but significant increase in DA reuptake in D2Rfloxflox/En1Cre/+ mice compared with WT siblings, whereas reuptake by D2Rfloxflox/D1Cre/+ mice was identical to WT littermates (Fig. 7C,D).
In addition, we observed that while the DAT inhibitor nomifensine (5 μm) did not increase the amplitude of evoked DA overflow of D2Rfloxflox/En1Cre/+ mice to levels of the WT siblings (0.92 vs 2.36 μm, respectively; Fig. 7E), the normalized increase of peak amplitude was higher in the mutant (2.8- vs 2.0-fold increase, respectively; Fig. 7F). This enhanced response to nomifensine is consistent with a greater relative contribution of DAT activity to peak amplitude in the mutant, and is consistent with D2 autoreceptor inhibition of reuptake. Together, these experiments indicate that D2 receptors at two distinct locations, on MSN and on dopaminergic neurons, each act to regulate basal release, and that D2 agonists acting on both receptors decrease evoked release. Only the D2 autoreceptor, however, appears to inhibit reuptake by DAT.
D2 autoreceptors and heteroreceptors regulate the kinetics of synaptic recovery
Recovery of evoked DA release in slices requires a long rest, >60 s. It has been presumed that a portion of the delayed rate of recovery is due to a presynaptic inhibition by the DA released during the first pulse onto D2 autoreceptors, but this assumption has not been clearly tested. We first measured the rate of recovery using paired stimuli. We did not observe differences from WT littermates for either mutant line at interpulse intervals of >1 s (Fig. 8A,B). As expected, the paired pulse ratio was significantly higher at 1 and 2 Hz in D2Rfloxflox/En1Cre/+ mice (Fig. 8A), consistent with evoked DA release in the first pulse inhibiting the autoreceptor in WT mice.
Figure 8.
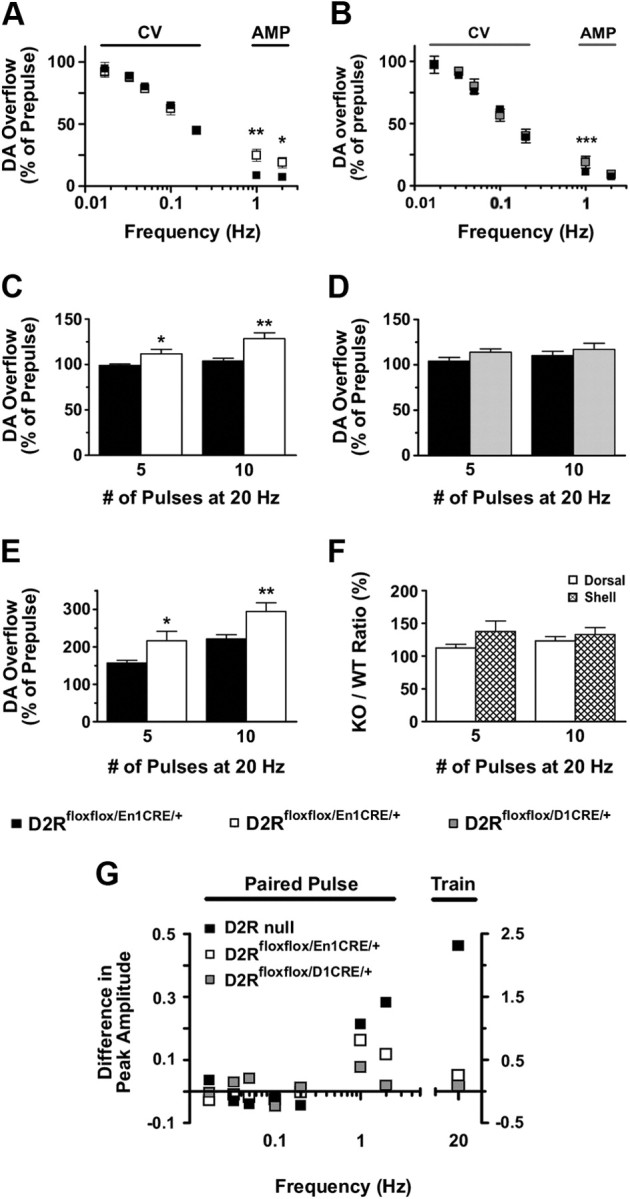
DA release probability in D2Rfloxflox/En1Cre/+ and D2Rfloxflox/D1Cre/+ compared with WT mice. Ablation of D2 autoreceptors increases release probability at both low and high frequencies, whereas ablation of D2 heteroreceptors in MSNs increases release probability only at low frequencies in the DSt. A, Paired-pulse analysis (CV: 0.017–0.2 Hz, n = 4–5; AMP: 1 and 2 Hz, n = 6–7) in D2Rfloxflox/En1Cre/+ mice (□) displayed enhanced recovery at 1 and 2 Hz compared with WT littermates (■) [AMP (Genotype): F(1,22) = 28.46, p < 0.0001]. B, Paired-pulse analysis (CV: n = 6; AMP: n = 12–13) in D2Rfloxflox/D1Cre/+ mice (▩) displayed enhanced recovery only at 1 Hz compared with WT littermates (■) [AMP (Interaction): F(1,37) = 12.81, p = 0.001]. C, Train of pulses at 20 Hz displayed enhanced DA overflow in D2Rfloxflox/En1Cre/+ mice (white bars) with respect to control littermates (black bars) (CV; n = 6–12; genotype: F(1,32) = 18.98, p = 0.0001). D, Train of pulses at 20 Hz displayed no changes in DA overflow in D2Rfloxflox/D1Cre/+ mice (gray bars) compared with controls (black bars) (CV; n = 8–12; genotype: F(1,36) = 2.76, p = 0.11). E, Train of pulses at 20 Hz displayed enhanced DA overflow in the shell of the NAcc of D2Rfloxflox/En1Cre/+ (white bars) versus control littermates (black bars) (n = 6–9; F(1,29) = 21.37, p < 0.0001). F, Comparisons of the ratios D2Rfloxflox/En1Cre/+/D2Rfloxflox of values of DA-overflow produced by a train of pulses between the DSt and the shell of NAcc for five and 10 pulses (□, DSt;  , shell NAcc; F(1,27) = 4.34, p = 0.046). G, Comparison of the DA release probability in D2R-null (■), D2Rfloxflox/En1Cre/+ (□), and D2Rfloxflox/D1Cre/+ (▩) mice. Summary diagram of the difference between the D2R-null, D2Rfloxflox/En1Cre/+, and D2Rfloxflox/D1Cre/+ mice (as indicated) compared with their respective WT controls in the paired-pulse and train of pulses experiments. The frequencies represent interpulse intervals of 60, 30, 20, 10, 5, 1, 0.5, and 0.05 s. Two-way ANOVA with Bonferroni's post hoc test for genotype was used in all panels. *p < 0.05, **p < 0.01, ***p < 0.001.
, shell NAcc; F(1,27) = 4.34, p = 0.046). G, Comparison of the DA release probability in D2R-null (■), D2Rfloxflox/En1Cre/+ (□), and D2Rfloxflox/D1Cre/+ (▩) mice. Summary diagram of the difference between the D2R-null, D2Rfloxflox/En1Cre/+, and D2Rfloxflox/D1Cre/+ mice (as indicated) compared with their respective WT controls in the paired-pulse and train of pulses experiments. The frequencies represent interpulse intervals of 60, 30, 20, 10, 5, 1, 0.5, and 0.05 s. Two-way ANOVA with Bonferroni's post hoc test for genotype was used in all panels. *p < 0.05, **p < 0.01, ***p < 0.001.
To examine the kinetics of recovery at shorter intervals, we used trains of pulses. In this case, the first stimuli released by far the most DA, and subsequent pulses released smaller stable amounts that contributed to the peak amplitude. In D2Rfloxflox/En1Cre/+ mice, trains of five or 10 pulses at 20 Hz produced a small but significant increase in DA overflow that was greater than in WT littermates (∼1.1- and 1.24-fold, respectively; Fig. 8C), but was far less robust than the relative increase observed in the D2R-null mice (Fig. 8G). The D2Rfloxflox/D1Cre/+ mice showed no differences from their WT controls (Fig. 8D).
As mechanisms controlling DA release are differentially regulated in the dorsal and ventral striatum (L. Zhang et al., 2009), and hyperactivity to a novel environment as well as to psychostimulants (as observed in the D2Rfloxflox/En1Cre/+ mice; Fig. 2E,G) has been associated with activation of the mesolimbic system (Hooks and Kalivas, 1995), we explored the possibility that DA overflow might be differently regulated in the NAcc versus the DSt of D2Rfloxflox/En1Cre/+ mice compared with WT.
Evoked DA overflow in control slices was threefold smaller in the NAcc shell than in the DSt (L. Zhang et al., 2009; T. Zhang et al., 2009), limiting the use of some protocols due to a lower signal-to-background ratio. We were, however, able to successfully examine single-pulse stimulation and trains of pulses in the NAcc shell. Single-pulse evoked DA overflow was decreased by ∼41% in D2Rfloxflox/En1Cre/+ mice compared with WT littermates (WT: 0.21 ± 0.02, n = 18; D2Rfloxflox/En1Cre/+: 0.12 ± 0.01, n = 15). Interestingly, trains of pulses at five and 10 pulses displayed ∼1.3- and 1.4-fold increase in DA overflow, respectively (Fig. 8E,F), compared with WT littermates. These results suggest that the D2 heteroreceptor-mediated mechanisms governing DA release are less efficient on VTA neurons than in neurons originating in the SNc and projecting to the DSt (DA overflow NAcc: 59% vs DSt: 37%, of control, respectively). These results might also explain why motor hyperactivity is only observed when D2Rfloxflox/En1Cre/+ mice are exposed to novelty (Hooks and Kalivas, 1995) or administered with psychostimulants (Aragona et al., 2008), both conditions principally stimulating the mesolimbic DAergic pathway.
Autoreceptor-mediated control of DA synthesis: inhibition of TH phosphorylation
D2 autoreceptors also play an important role in regulating DA synthesis by inhibiting the activity of TH, the rate-limiting enzyme in DA synthesis. TH activity is regulated by phosphorylation at multiple sites (Haycock and Haycock, 1991; Lindgren et al., 2000); TH phosphorylation at Ser40 (pTH-Ser40) is mostly cAMP and PKA-dependent and inhibited by D2R-mediated signaling (Lindgren et al., 2001).
In light of the effect of quinpirole in CV results showing a D2 heteroreceptor-dependent mechanism on DA release, we analyzed its effect on DA synthesis. pTH-Ser40 levels were analyzed by Western blot in DSt and NAcc extracts from D2Rfloxflox/En1Cre/+, D2Rfloxflox/D1Cre/+, and respective WT littermates using pTH-Ser40-specific antibodies and reported as pTH-Ser40/total TH ratio (Fig. 9). These analyses showed that total TH expression is unaltered in mutant mice compared with controls. D2 autoreceptor removal in D2Rfloxflox/En1Cre/+ mice does not affect levels of pTH-Ser40 in the DSt compared with WT littermates (Fig. 9A); conversely, pTH-Ser40 increases in the NAcc (Fig. 9B), in agreement with HPLC measurements of DA content in this structure and the smaller reduction of DA release by a single pulse and the higher ratio with train of pulses in the NAcc with respect to the DSt of D2Rfloxflox/En1Cre/+ mice.
No difference in pTH-Ser40 levels in DSt and NAcc were observed between D2Rfloxflox/D1Cre/+ and control littermates with either saline or quinpirole treatment (Fig. 9C,D).
Thus, in line with CV results, quinpirole at 0.2 mg/kg decreased pTH-Ser40 in the DSt of D2Rfloxflox/En1Cre/+ as in WT siblings (Fig. 9A). This event does not appear to be mediated by D3Rs, since in similar experiments using D2R−/− mice (Baik et al., 1995), we could not detect an inhibition by quinpirole of pTH-Ser40 (Fig. 9E). In addition, electrophysiological testing of dopaminergic neurons of both the SN and VTA of D2Rfloxflox/En1Cre/+ mice showed absence of the hyperpolarizing effect of quinpirole, further excluding the intervention of D3Rs, also expressed by these neurons, in the observed effects. We might speculate that, in the absence of D2 autoreceptors, lack of activation of GIRKs (Martel et al., 2011) would cause a higher calcium influx that might activate calcium/calmodulin-dependent mechanisms, leading to the activation of phosphodiesterases and consequent lowering of cAMP-dependent signaling (Chéramy et al., 1994).
Importantly, in the same treatment conditions (0.2 mg/kg), the pTH-Ser40 level in extracts from the NAcc was not affected (Fig. 9B). Since quinpirole does not affect pTH-Ser40 in extracts from D2R-null mice (Fig. 9E), these results clearly indicate the presence of a D2R-activated circuit that controls TH activity in the DSt, which is unmasked in the absence of D2 autoreceptors.
The differential effect of quinpirole in extracts from the NAcc versus the DSt shows a more prominent role of D2 autoreceptors in the NAcc in the control of TH phosphorylation, and thereby of DA synthesis. This might underlie the higher DA content found by HPLC in the NAcc and cortex of D2Rfloxflox/En1Cre/+ mice (Fig. 5B,C) compared with the same structures in control littermates. These results are interesting since they show a differential effect of loss of D2 autoreceptors in neurons belonging to the VTA (projecting to the NAcc) or to the SN (projecting to the DSt). In the DSt, in the absence of D2 autoreceptors, D2 heteroreceptor-mediated inhibitory feedbacks are unmasked; these effects are likely minor or absent in the VTA. The differential results between NAcc and DSt on pTH-Ser40 levels also suggest that the hyperactivity of D2Rfloxflox/En1Cre/+ mice might be generated by conditions that mostly involve the activation of the VTA neurons of the mesolimbic pathway.
Thus, we hypothesized that quinpirole might no longer affect motor activity in D2Rfloxflox/En1Cre/+ mice if administered in a novel environment, as opposed to what was observed the home cage (Fig. 3C). To test this hypothesis, we exposed D2Rfloxflox/En1Cre/+ and WT littermates to the open field after quinpirole treatment. Interestingly, and in support of our hypothesis, under these conditions, quinpirole did not reduce locomotion in D2Rfloxflox/En1Cre/+ mice (Fig. 9F) while it did in control WT littermates.
Discussion
Dopamine dysfunctions are implicated in a broad range of neurological (Grace, 2008; Jenner, 2008), neuropsychiatric (Dagher and Robbins, 2009; van Os and Kapur, 2009), and endocrine (Melmed, 2008) disorders. Analyses of D2R-null mice (Baik et al., 1995; Kelly et al., 1998; Jung et al., 1999) have been fundamental in illustrating the great variety of physiological impairments caused by the loss of these receptors in vivo (Tirotta et al., 2010). However, whether these impairments were caused by loss of presynaptic D2 autoreceptors, leading to altered DA levels (L'Hirondel et al., 1998; Benoit-Marand et al., 2001; Rouge-Pont et al., 2002; Schmitz et al., 2002; Lindgren et al., 2003; Håkansson et al., 2004) or conversely by the absence of postsynaptic D2 heteroreceptors was not established.
To address these questions, we present two novel models, D2Rfloxflox/En1Cre/+ and D2Rfloxflox/D1Cre/+ mice, in which D2Rs have been selectively removed either from DAergic neurons of the SNc and VTA, or from MSNs in the striatum. Importantly, both lines have a normal life span and, in contrast to D2R-null mice, maintain normal reproductive capacity, due to the normal D2R expression at the pituitary level in both mutants. Analyses of these animals provided important information on the role of D2R in the control of DA release and locomotion.
We found that loss of presynaptic D2 autoreceptors appears to have a minimal impact on motor activity of mice under normal conditions, while D2R deletion on MSNs strongly affects motor activity and coordination. Thus, deletion of D2R in the MSNs mirrors the motor impairments observed by the total loss of D2Rs in D2R-null mice (Baik et al., 1995; Jung et al., 1999) and support the critical role of these receptors in modulating the activity of D2R-expressing MSNs of the indirect pathway (Gittis et al., 2011).
D2Rfloxflox/En1Cre/+ mice, however, are hyperactive when exposed to a novel environment, in particular during the early phase of exploration, and hyperresponsive to DAT blockade by cocaine. Conversely, similar to D2R-null mice (Welter et al., 2007), D2Rfloxflox/D1Cre/+ mice have a blunted motor response to acute cocaine challenge. These results underline the key role of D2 autoreceptor in the regulation of DA release in response to environmental and drug-induced motor challenges and the absolute requirement of D2R in MSNs to translate DA signaling into motor activity.
Importantly, while analyses of D2Rfloxflox/En1Cre/+ mice show that presynaptic D2 autoreceptors are responsible for the inhibition of firing of DAergic neurons, they also reveal that the regulation of DA synthesis and release is not solely regulated by presynaptic D2 autoreceptors on DA neurons, but that postsynaptic D2 heteroreceptors participate in these functions, very likely through regulation of striatal circuitries. Indeed, in CV experiments, both D2 autoreceptor and heteroreceptor mutants have decreased DA overflow with single pulses, showing a role for both D2 components; there is an inhibition of subsequent DA release at intervals of ∼1 s due to both; and finally, quinpirole exerts inhibition on DA overflow in both mutants.
In support of a D2 heteroreceptor-mediated control of DA, we also found that activation of these receptors by quinpirole reduces motor activity and inhibits DA synthesis in the DSt of D2Rfloxflox/En1Cre/+ mice. These effects are D2R-specific, and could not be attributed to D3Rs, as they are: (1) absent in D2R-null mice (Boulay et al., 1999; Lindgren et al., 2003); (2) not present when dopaminergic neurons, which express D3Rs, are directly tested in electrophysiological analyses (Fig. 4); and (3) in conditions in which the dopaminergic mesolimbic pathway is activated (Fig. 9F). Thus, we propose that quinpirole-mediated effects are very likely dependent on D2R-mediated heterosynaptic control of neurotransmitters/circuits (Chéramy et al., 1994) acting on DA neurons.
Our results contrast with those previously reported (Bello et al., 2011) in a different presynaptic D2 autoreceptor mutant. Although several possibilities might account for this discrepancy, the most likely one is that presynaptic D2 autoreceptor deletion has been achieved through the use of a CRE line in which the DAT promoter drives the CRE expression (DAT-ires-CRE mice). DAT is poorly expressed in DA projections to the cortex, which might result into the persistence of D2R in cortical afferents. In addition, heterozygous DAT-ires-CRE mice have a reduced expression of DAT (Bäckman et al., 2006), which, together with the absence of D2 autoreceptors, might alter measures of the DA overflow obtained by electrical stimulations.
Altogether, our results suggest that the control of DA release is mostly provided by D2 autoreceptors, although D2Rs located on MSNs play a significant role, as seen by the loss of 20% of quinpirole's effect in D2Rfloxflox/D1Cre/+ mice. However, when D2 autoreceptors are absent, DA stimulation of D2 heteroreceptor-mediated feedback mechanisms become dominant in the control of DA neurons. Future studies will aim at identifying the neurotransmitter(s) involved in such regulation, including glutamate, GABA, acetylcholine, and endocannabinoids, each of which are likely candidates since they have been previously implicated in the control of DA release.
Of great relevance to recent reports on the differential activity of dopaminergic neurons (Lammel et al., 2008, 2011) of the SNc and VTA, we show a dichotomous D2R-dependent control of DA release in these structures. Thus, we conclude that D2Rs located in areas receiving dopaminergic afferents exert a stronger control over DA release from DAergic neurons projecting to the DSt than on those projecting to the NAcc.
These results suggests that the properties and identity of dopaminergic neurons of the SNc and VTA are in part dictated by afferents to these neurons, which are different in these two striatal compartments, and also by a D2R-mediated heteroreceptor control of neurotransmitters released from these terminals. Indeed, this regulation is completely absent in the D2R-null mouse model.
Thus, selective removal of D2Rs from dopaminergic or striatal neurons discloses important and unexpected findings, revealing an epistatic role of D2 heteroreceptors in maintaining appropriate DA levels. Interestingly, this modulatory influence allows D2Rfloxflox/En1Cre/+ mice to maintain normal motor behavior, which would have otherwise led to constant hyperactivity in any of the behavioral test performed. Nevertheless, when D2Rfloxflox/En1Cre/+ mice are exposed to a novel environment or to cocaine, the activation of DAergic neurons of the mesolimbic pathway and the subsequent higher DA release observed under conditions that simulate phasic responses may induce a hyperresponse. Future analyses at the behavioral and cellular level will help elucidate the influence of site-specific D2R deletion in complex dopamine-mediated behaviors.
Footnotes
This work was supported by INSERM-44790, NIH Grant DA024689, and European Community Grant EC LSHM-CT-2004-005166 (to E.B.). We are grateful to Drs. G. Schutz and W. Wurst for the generous gift of the D1CRE mice and En1Cre mice, respectively. We thank Andree Dierich and members of the IGBMC mouse facility for generating D2Rfloxflox mice; N. Tognazzi, A. Usiello, E. Erbs, R. Lutzing, C. Paparo for the initial interest in this work; E. Chen and A. Nasamran for technical assistance.
References
- Aragona BJ, Cleaveland NA, Stuber GD, Day JJ, Carelli RM, Wightman RM. Preferential enhancement of dopamine transmission within the nucleus accumbens shell by cocaine is attributable to a direct increase in phasic dopamine release events. J Neurosci. 2008;28:8821–8831. doi: 10.1523/JNEUROSCI.2225-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckman CM, Malik N, Zhang Y, Shan L, Grinberg A, Hoffer BJ, Westphal H, Tomac AC. Characterization of a mouse strain expressing Cre recombinase from the 3′ untranslated region of the dopamine transporter locus. Genesis. 2006;44:383–390. doi: 10.1002/dvg.20228. [DOI] [PubMed] [Google Scholar]
- Baik JH, Picetti R, Saiardi A, Thiriet G, Dierich A, Depaulis A, Le Meur M, Borrelli E. Parkinsonian-like locomotor impairment in mice lacking dopamine D2 receptors. Nature. 1995;377:424–428. doi: 10.1038/377424a0. [DOI] [PubMed] [Google Scholar]
- Bamford NS, Zhang H, Schmitz Y, Wu NP, Cepeda C, Levine MS, Schmauss C, Zakharenko SS, Zablow L, Sulzer D. Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron. 2004a;42:653–663. doi: 10.1016/s0896-6273(04)00265-x. [DOI] [PubMed] [Google Scholar]
- Bamford NS, Robinson S, Palmiter RD, Joyce JA, Moore C, Meshul CK. Dopamine modulates release from corticostriatal terminals. J Neurosci. 2004b;24:9541–9552. doi: 10.1523/JNEUROSCI.2891-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett SE, Enquist J, Hopf FW, Lee JH, Gladher F, Kharazia V, Waldhoer M, Mailliard WS, Armstrong R, Bonci A, Whistler JL. Dopamine responsiveness is regulated by targeted sorting of D2 receptors. Proc Natl Acad Sci U S A. 2005;102:11521–11526. doi: 10.1073/pnas.0502418102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckstead MJ, Grandy DK, Wickman K, Williams JT. Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron. 2004;42:939–946. doi: 10.1016/j.neuron.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Bello EP, Mateo Y, Gelman DM, Noaín D, Shin JH, Low MJ, Alvarez VA, Lovinger DM, Rubinstein M. Cocaine supersensitivity and enhanced motivation for reward in mice lacking dopamine D2 autoreceptors. Nat Neurosci. 2011;14:1033–1038. doi: 10.1038/nn.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit-Marand M, Borrelli E, Gonon F. Inhibition of dopamine release via presynaptic D2 receptors: time course and functional characteristics in vivo. J Neurosci. 2001;21:9134–9141. doi: 10.1523/JNEUROSCI.21-23-09134.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björklund A, Dunnett SB. Dopamine neuron systems in the brain: an update. Trends Neurosci. 2007;30:194–202. doi: 10.1016/j.tins.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Boulay D, Depoortere R, Perrault G, Borrelli E, Sanger DJ. Dopamine D2 receptor knock-out mice are insensitive to the hypolocomotor and hypothermic effects of dopamine D2/D3 receptor agonists. Neuropharmacology. 1999;38:1389–1396. doi: 10.1016/s0028-3908(99)00064-7. [DOI] [PubMed] [Google Scholar]
- Boulay D, Depoortere R, Oblin A, Sanger DJ, Schoemaker H, Perrault G. Haloperidol-induced catalepsy is absent in dopamine D(2), but maintained in dopamine D(3) receptor knock-out mice. Eur J Pharmacol. 2000;391:63–73. doi: 10.1016/s0014-2999(99)00916-4. [DOI] [PubMed] [Google Scholar]
- Branda CS, Dymecki SM. Talking about a revolution: the impact of site-specific recombinases on genetic analyses in mice. Dev Cell. 2004;6:7–28. doi: 10.1016/s1534-5807(03)00399-x. [DOI] [PubMed] [Google Scholar]
- Centonze D, Picconi B, Baunez C, Borrelli E, Pisani A, Bernardi G, Calabresi P. Cocaine and amphetamine depress striatal GABAergic synaptic transmission through D2 dopamine receptors. Neuropsychopharmacology. 2002;26:164–175. doi: 10.1016/S0893-133X(01)00299-8. [DOI] [PubMed] [Google Scholar]
- Chéramy A, Desce JM, Godeheu G, Glowinski J. Presynaptic control of dopamine synthesis and release by excitatory amino acids in rat striatal synaptosomes. Neurochem Int. 1994;25:145–154. doi: 10.1016/0197-0186(94)90033-7. [DOI] [PubMed] [Google Scholar]
- Clark SD, Nothacker HP, Wang Z, Saito Y, Leslie FM, Civelli O. The urotensin II receptor is expressed in the cholinergic mesopontine tegmentum of the rat. Brain Res. 2001;923:120–127. doi: 10.1016/s0006-8993(01)03208-5. [DOI] [PubMed] [Google Scholar]
- Cragg SJ. Singing to the tune of dopamine: focus on “Properties of dopamine release and uptake in the songbird basal ganglia.”. J Neurophysiol. 2005;93:1827–1828. doi: 10.1152/jn.01210.2004. [DOI] [PubMed] [Google Scholar]
- Dagher A, Robbins TW. Personality, addiction, dopamine: insights from Parkinson's disease. Neuron. 2009;61:502–510. doi: 10.1016/j.neuron.2009.01.031. [DOI] [PubMed] [Google Scholar]
- De Mei C, Ramos M, Iitaka C, Borrelli E. Getting specialized: presynaptic and postsynaptic dopamine D2 receptors. Curr Opin Pharmacol. 2009;9:53–58. doi: 10.1016/j.coph.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew KL, O'Connor WT, Kehr J, Ungerstedt U. Regional specific effects of clozapine and haloperidol on GABA and dopamine release in rat basal ganglia. Eur J Pharmacol. 1990;187:385–397. doi: 10.1016/0014-2999(90)90366-e. [DOI] [PubMed] [Google Scholar]
- Gittis AH, Hang GB, LaDow ES, Shoenfeld LR, Atallah BV, Finkbeiner S, Kreitzer AC. Rapid target-specific remodeling of fast-spiking inhibitory circuits after loss of dopamine. Neuron. 2011;71:858–868. doi: 10.1016/j.neuron.2011.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA. Physiology of the normal and dopamine-depleted basal ganglia: insights into levodopa pharmacotherapy. Mov Disord. 2008;23(Suppl 3):S560–S569. doi: 10.1002/mds.22020. [DOI] [PubMed] [Google Scholar]
- Håkansson K, Pozzi L, Usiello A, Haycock J, Borrelli E, Fisone G. Regulation of striatal tyrosine hydroxylase phosphorylation by acute and chronic haloperidol. Eur J Neurosci. 2004;20:1108–1112. doi: 10.1111/j.1460-9568.2004.03547.x. [DOI] [PubMed] [Google Scholar]
- Haycock JW, Haycock DA. Tyrosine hydroxylase in rat brain dopaminergic nerve terminals: multiple-site phosphorylation in vivo and in synaptosomes. J Biol Chem. 1991;266:5650–5657. [PubMed] [Google Scholar]
- Heien ML, Wightman RM. Phasic dopamine signaling during behavior, reward, and disease states. CNS Neurol Disord Drug Targets. 2006;5:99–108. doi: 10.2174/187152706784111605. [DOI] [PubMed] [Google Scholar]
- Hooks MS, Kalivas PW. The role of mesoaccumbens–pallidal circuitry in novelty-induced behavioral activation. Neuroscience. 1995;64:587–597. doi: 10.1016/0306-4522(94)00409-x. [DOI] [PubMed] [Google Scholar]
- Jenner P. Molecular mechanisms of l-DOPA-induced dyskinesia. Nat Rev Neurosci. 2008;9:665–677. doi: 10.1038/nrn2471. [DOI] [PubMed] [Google Scholar]
- Jung MY, Skryabin BV, Arai M, Abbondanzo S, Fu D, Brosius J, Robakis NK, Polites HG, Pintar JE, Schmauss C. Potentiation of the D2 mutant motor phenotype in mice lacking dopamine D2 and D3 receptors. Neuroscience. 1999;91:911–924. doi: 10.1016/s0306-4522(98)00705-2. [DOI] [PubMed] [Google Scholar]
- Kapfhamer D, Berger KH, Hopf FW, Seif T, Kharazia V, Bonci A, Heberlein U. Protein phosphatase 2a and glycogen synthase kinase 3 signaling modulate prepulse inhibition of the acoustic startle response by altering cortical M-type potassium channel activity. J Neurosci. 2010;30:8830–8840. doi: 10.1523/JNEUROSCI.1292-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MA, Rubinstein M, Phillips TJ, Lessov CN, Burkhart-Kasch S, Zhang G, Bunzow JR, Fang Y, Gerhardt GA, Grandy DK, Low MJ. Locomotor activity in D2 dopamine receptor-deficient mice is determined by gene dosage, genetic background, and developmental adaptations. J Neurosci. 1998;18:3470–3479. doi: 10.1523/JNEUROSCI.18-09-03470.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel RA, Turnbull DH, Blanquet V, Wurst W, Loomis CA, Joyner AL. Two lineage boundaries coordinate vertebrate apical ectodermal ridge formation. Genes Dev. 2000;14:1377–1389. [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Hetzel A, Häckel O, Jones I, Liss B, Roeper J. Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron. 2008;57:760–773. doi: 10.1016/j.neuron.2008.01.022. [DOI] [PubMed] [Google Scholar]
- Lammel S, Ion DI, Roeper J, Malenka RC. Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron. 2011;70:855–862. doi: 10.1016/j.neuron.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberger T, Parlato R, Dassesse D, Westphal M, Casanova E, Turiault M, Tronche F, Schiffmann SN, Schütz G. Expression of Cre recombinase in dopaminoceptive neurons. BMC Neurosci. 2007;8:4. doi: 10.1186/1471-2202-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L'Hirondel M, Chéramy A, Godeheu G, Artaud F, Saiardi A, Borrelli E, Glowinski J. Lack of autoreceptor-mediated inhibitory control of dopamine release in striatal synaptosomes of D2 receptor-deficient mice. Brain Res. 1998;792:253–262. doi: 10.1016/s0006-8993(98)00146-2. [DOI] [PubMed] [Google Scholar]
- Lindgren N, Xu ZQ, Lindskog M, Herrera-Marschitz M, Goiny M, Haycock J, Goldstein M, Hökfelt T, Fisone G. Regulation of tyrosine hydroxylase activity and phosphorylation at Ser(19) and Ser(40) via activation of glutamate NMDA receptors in rat striatum. J Neurochem. 2000;74:2470–2477. doi: 10.1046/j.1471-4159.2000.0742470.x. [DOI] [PubMed] [Google Scholar]
- Lindgren N, Xu ZQ, Herrera-Marschitz M, Haycock J, Hökfelt T, Fisone G. Dopamine D(2) receptors regulate tyrosine hydroxylase activity and phosphorylation at Ser40 in rat striatum. Eur J Neurosci. 2001;13:773–780. doi: 10.1046/j.0953-816x.2000.01443.x. [DOI] [PubMed] [Google Scholar]
- Lindgren N, Usiello A, Goiny M, Haycock J, Erbs E, Greengard P, Hokfelt T, Borrelli E, Fisone G. Distinct roles of dopamine D2L and D2S receptor isoforms in the regulation of protein phosphorylation at presynaptic and postsynaptic sites. Proc Natl Acad Sci U S A. 2003;100:4305–4309. doi: 10.1073/pnas.0730708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Chefer VI, Shippenberg TS, Hjelmstad GO, Fields HL. Kappa opioids selectively control dopaminergic neurons projecting to the prefrontal cortex. Proc Natl Acad Sci U S A. 2006;103:2938–2942. doi: 10.1073/pnas.0511159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel P, Leo D, Fulton S, Bérard M, Trudeau LE. Role of Kv1 potassium channels in regulating dopamine release and presynaptic D2 receptor function. PLoS One. 2011;6:e20402. doi: 10.1371/journal.pone.0020402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melmed S. Update in pituitary disease. J Clin Endocrinol Metab. 2008;93:331–338. doi: 10.1210/jc.2007-1409. [DOI] [PubMed] [Google Scholar]
- Mercuri NB, Saiardi A, Bonci A, Picetti R, Calabresi P, Bernardi G, Borrelli E. Loss of autoreceptor function in dopaminergic neurons from dopamine D2 receptor deficient mice. Neuroscience. 1997;79:323–327. doi: 10.1016/s0306-4522(97)00135-8. [DOI] [PubMed] [Google Scholar]
- Mosharov EV, Sulzer D. Analysis of exocytotic events recorded by amperometry. Nat Methods. 2005;2:651–658. doi: 10.1038/nmeth782. [DOI] [PubMed] [Google Scholar]
- Paladini CA, Robinson S, Morikawa H, Williams JT, Palmiter RD. Dopamine controls the firing pattern of dopamine neurons via a network feedback mechanism. Proc Natl Acad Sci U S A. 2003;100:2866–2871. doi: 10.1073/pnas.0138018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera A, Alberti I, Martín AB, Narváez JA, de la Calle A, Moratalla R. Molecular phenotype of rat striatal neurons expressing the dopamine D5 receptor subtype. Eur J Neurosci. 2002;16:2049–2058. doi: 10.1046/j.1460-9568.2002.02280.x. [DOI] [PubMed] [Google Scholar]
- Rouge-Pont F, Usiello A, Benoit-Marand M, Gonon F, Piazza PV, Borrelli E. Changes in extracellular dopamine induced by morphine and cocaine: crucial control by D2 receptors. J Neurosci. 2002;22:3293–3301. doi: 10.1523/JNEUROSCI.22-08-03293.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D. Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci. 2001;21:5916–5924. doi: 10.1523/JNEUROSCI.21-16-05916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz Y, Schmauss C, Sulzer D. Altered dopamine release and uptake kinetics in mice lacking D2 receptors. J Neurosci. 2002;22:8002–8009. doi: 10.1523/JNEUROSCI.22-18-08002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz Y, Benoit-Marand M, Gonon F, Sulzer D. Presynaptic regulation of dopaminergic neurotransmission. J Neurochem. 2003;87:273–289. doi: 10.1046/j.1471-4159.2003.02050.x. [DOI] [PubMed] [Google Scholar]
- Simon HH, Saueressig H, Wurst W, Goulding MD, O'Leary DD. Fate of midbrain dopaminergic neurons controlled by the engrailed genes. J Neurosci. 2001;21:3126–3134. doi: 10.1523/JNEUROSCI.21-09-03126.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Lindskog M, Ledent C, Parmentier M, Greengard P, Fredholm BB, Fisone G. Regulation of the phosphorylation of the dopamine- and cAMP-regulated phosphoprotein of 32 kDa in vivo by dopamine D1, dopamine D2, and adenosine A2A receptors. Proc Natl Acad Sci U S A. 2000;97:1856–1860. doi: 10.1073/pnas.97.4.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirotta E, De Mei C, Iitaka C, Ramos M, Holmes D, Borrelli E. Unraveling the role of dopamine receptors in vivo: lessons from knockout mice. In: Neve KA, editor. The dopamine receptors. 2nd edition. New York: Humana; 2010. pp. 303–322. [Google Scholar]
- Ungless MA, Magill PJ, Bolam JP. Uniform inhibition of dopamine neurons in the ventral tegmental area by aversive stimuli. Science. 2004;303:2040–2042. doi: 10.1126/science.1093360. [DOI] [PubMed] [Google Scholar]
- Usiello A, Baik JH, Rougé-Pont F, Picetti R, Dierich A, LeMeur M, Piazza PV, Borrelli E. Distinct functions of the two isoforms of dopamine D2 receptors. Nature. 2000;408:199–203. doi: 10.1038/35041572. [DOI] [PubMed] [Google Scholar]
- van Os J, Kapur S. Schizophrenia. Lancet. 2009;374:635–645. doi: 10.1016/S0140-6736(09)60995-8. [DOI] [PubMed] [Google Scholar]
- Welter M, Vallone D, Samad TA, Meziane H, Usiello A, Borrelli E. Absence of dopamine D2 receptors unmasks an inhibitory control over the brain circuitries activated by cocaine. Proc Natl Acad Sci U S A. 2007;104:6840–6845. doi: 10.1073/pnas.0610790104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Doyon WM, Clark JJ, Phillips PE, Dani JA. Controls of tonic and phasic dopamine transmission in the dorsal and ventral striatum. Mol Pharmacol. 2009;76:396–404. doi: 10.1124/mol.109.056317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Zhang L, Liang Y, Siapas AG, Zhou FM, Dani JA. Dopamine signaling differences in the nucleus accumbens and dorsal striatum exploited by nicotine. J Neurosci. 2009;29:4035–4043. doi: 10.1523/JNEUROSCI.0261-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]