

Abstract
In neurons, the type 3 deiodinase (D3) inactivates thyroid hormone and reduces oxygen consumption, thus creating a state of cell-specific hypothyroidism. Here we show that hypoxia leads to nuclear import of D3 in neurons, without which thyroid hormone signaling and metabolism cannot be reduced. After unilateral hypoxia in the rat brain, D3 protein level is increased predominantly in the nucleus of the neurons in the pyramidal and granular ipsilateral layers, as well as in the hilus of the dentate gyrus of the hippocampal formation. In hippocampal neurons in culture as well as in a human neuroblastoma cell line (SK-N-AS), a 24 h hypoxia period redirects active D3 from the endoplasmic reticulum to the nucleus via the cochaperone Hsp40 pathway. Preventing nuclear D3 import by Hsp40 knockdown resulted an almost doubling in the thyroid hormone-dependent glycolytic rate and quadrupling the transcription of thyroid hormone target gene ENPP2. In contrast, Hsp40 overexpression increased nuclear import of D3 and minimized thyroid hormone effects in cell metabolism. In conclusion, ischemia/hypoxia induces an Hsp40-mediated translocation of D3 to the nucleus, facilitating thyroid hormone inactivation proximal to the thyroid hormone receptors. This adaptation decreases thyroid hormone signaling and may function to reduce ischemia-induced hypoxic brain damage.
Introduction
Insufficient O2 availability triggers adaptive responses that are initiated by the accumulation of hypoxia-inducible transcriptional factors (HIFs), which decreases the rate of oxidative metabolism (Semenza, 2003; Majmundar et al., 2010). Thyroid hormone induces a cell-specific transcriptional profile that accelerates metabolism (Wu and Koenig, 2000; Zhang and Lazar, 2000; Bianco et al., 2005) and thus could be targeted during adaptation to hypoxia. The current paradigm of thyroid hormone action holds that deiodinase-mediated intracellular changes in thyroid hormone activation or deactivation determine metabolic effects, regardless of plasma hormone concentrations (Gereben et al., 2008). For example, once thyroxine (T4) crosses the plasma membrane and diffuses toward the cell nucleus, the type 2 deiodinase (D2) can convert it to the active form T3, thus enhancing the transcriptional effects of thyroid hormone and accelerating metabolism (Dentice et al., 2005; Watanabe et al., 2006). In contrast, the type 3 deiodinase (D3) pathway is thought to inactivate T3 intracellularly, and this depletion of T3 terminates thyroid hormone-driven metabolic events (Freitas et al., 2010). These deiodinase-mediated control pathways for thyroid hormone action are active in the brain, with the glial D2 generating most T3 in the CNS and the neuronal D3 inactivating both T4 and T3 (Gereben et al., 2008; Mohácsik et al., 2011).
D3 is a dimeric thyoredoxin fold-containing membrane-anchored selenoprotein (Callebaut et al., 2003) that is transcriptionally induced by HIF-1α (Simonides et al., 2008). In different models of ischemic and/or hypoxic conditions in the myocardium, liver, and skeletal muscle there is ectopic D3 expression (Peeters et al., 2003; Simonides et al., 2008). The hypoxia-HIF-1α-D3 pathway is also functional in the brain, where induction of D3 expression was detected as early as 1 h after unilateral ligation of the internal carotid artery (Freitas et al., 2010). When this is modeled in vitro, in the setting of the SK-N-AS cells, hypoxia-induced D3 expression dampens T3-mediated gene expression (Freitas et al., 2010). The adaptive nature of this response is illustrated by a marked T3-dependent acceleration of metabolic rate shortly after D3 activity is inhibited (Simonides et al., 2008). This indicates that the D3 pathway is a key element controlling neuronal thyroid hormone signaling as part of a broader HIF-1α-dependent adaptive gene network (Gereben et al., 2008; Huang and Bianco, 2008; Dentice and Salvatore, 2011), with important consequences for the expression of T3-responsive genes and the metabolic rate.
Here we report that D3 redirection and accumulation in the cell nucleus is part of the hypoxia-induced reduction in thyroid hormone signaling. Inactivation of thyroid hormone in the nucleus, physically closer to where thyroid hormone receptors (TRs) are located, constitutes an advantageous mechanism to dampen thyroid hormone signaling and reduce metabolism. Studies of directed D3 glycosylation indicated that D3 is a type I integral membrane protein that, given normal O2 availability, is predominantly transported through the Golgi system and sorted to the plasma membrane. On the other hand, hypoxia results in D3 being imported to the nucleus directly from the endoplasmic reticulum (ER) via a mechanism that depends on the cochaperone Hsp40.
Materials and Methods
Animals
Adult male Sprague Dawley rats were housed under standard environmental conditions (light between 06:00 A.M. and 6:00 P.M., temperature 22 ± 1°C, rat chow and water available ad libitum). Animal protocols followed the European Communities Council Directive of 24 November 1986 (86/609/EEC) and, when appropriate, were reviewed and approved by the Animal Welfare Committee at the Institute of Experimental Medicine, Budapest. Experiments with embryonic rat hippocampal neurons were reviewed and approved by the University of Miami Institutional Animal Care and Use Committee.
Induction of brain ischemia by surgical occlusion of middle cerebral artery
Cerebral ischemia in adult male Sprague Dawley rats was induced (n = 10) by a transient 60 min occlusion of the right middle cerebral artery (MCAO) under Nembutal anesthesia (60 mg/kg, i.p.), as previously described (Longa et al., 1989; Matucz et al., 2004; Freitas et al., 2010). Sham-operated animals (n = 10) underwent the same surgical procedure, but no filament was advanced to the internal carotid artery. Intraischemic neurological deficit was confirmed by detecting gait abnormalities characterized by circling or moving to the left in animals with successful MCAO. After 60 min of ischemia, animals were perfused with 4% acrolein plus 2% PFA in PBS (50 ml) followed by 2% PFA in PBS (150 ml). Brains were postfixed in 2% PFA overnight and six sets of 40-μm-thick sections were cut from each brain with a vibratome.
D3 immunohistochemistry and electron microscopy
Immunohistochemical detection of D3 was performed with a rabbit polyclonal antiserum (1 μg/ml; NBP1-05767B; Novus Biologicals) as previously described (Freitas et al., 2010). Briefly, a set of sections from five ischemic and control brains were processed in pairs in the same staining jars. After elimination of the excess aldehyde and endogenous peroxidase activity, sections were cryoprotected with 15% (15 min), followed by 30% sucrose (12 h) in PBS. Sequential freeze-thaw cycles were performed three times on liquid nitrogen to permeabilize sections. Finally, 2% normal horse serum was applied (20 min) to prevent nonspecific antibody binding. The pretreated sections were incubated in the primary antibody for 36 h at 4°C, followed by biotinylated donkey anti-rabbit secondary antibody (1:1000, Jackson ImmunoResearch Laboratories) for 2 h and finally, peroxidase-conjugated ABC (1:3000, Vector Laboratories) for 1.5 h. The immunoreactive sites were visualized with 2.5% 3,3-diaminobenzidine and 0.15% nickel ammonium sulfate dissolved in Tris buffer, pH 7.6. Finally the immunoreaction product was silver-gold intensified (Kalló et al., 2001). The sections were treated then with 1% osmium tetroxide for 60 min and 2% uranyl acetate (prepared in 70% ethanol) for 40 min and then dehydrated in an ascending series of ethanol and propylene oxide. Flat embedding was performed in TAAB 812 medium epoxy resin between a pair of glass microscope slides precoated with liquid release agent (Electron Microscopy Sciences). The resin was allowed to polymerize at 56°C for 2 d. The embedded section was photographed and the dentate gyrus was cut into semithin (0.5–1 μm) and ultrathin sections (50–60 nm) with a Leica Ultracut UCT ultramicrotome. Semithin sections were mounted, stained with 1% toluidine blue, and coverslipped with DPX (Fluka). The ultrathin sections were mounted onto Formvar-coated single slot grids, contrasted with 2% lead citrate and examined with a Jeol-100C transmission electron microscope. Specificity controls for this antibody were performed as previously described (Freitas et al., 2010).
D3, Hsp40 costaining in brain
After MCAO for 1 h followed by 4 h reperfusion rats were perfused with 4% PFA. Thirty-micrometer thin sections were cut and brain slides were costained for D3 and Hsp40. Incubation with α-D3(CT; C terminus) and α-Hsp40 (ab78437) was performed for 2 d at 4°C followed by α-rabbit-biotin and streptavidin-Alexa Fluor488 signal amplification in case of D3 (green) and anti-mouse-impress and tyramide-rhodamine signal amplification detection for Hsp40 (red). After costaining brain slides were incubated with Hoechst for staining of nuclei (blue).
Primary hippocampal neuronal culture
Dissociated embryonic day 18 rat primary hippocampal neurons were prepared similar to as previously described (Banker and Goslin, 1998), with the exception of an additional 10 min enzymatic digestion step using trypsin. Cells were plated onto glass coverslips coated with poly-l-lysine (P2636) (Sigma-Aldrich) and grown in Neurobasal medium supplemented with B27SM1 (StemCell Technologies) at 37°C in 5% CO2. Neurons were cultured for 2–3 weeks before using for immunofluorescence.
Enzymes and antibodies
Endo H was purchased from New England Biolabs. D3 antibodies, α-D3(NT; N terminus) (NB110-96414), α-D3(CT) (NBP1-05767) were purchased from Novus Biologicals, DNAJB1(Hsp40) (ab69402, ab78437), γ-tubulin (ab11316), nuclear lamin (ab8984), Bip (ab21685) antibodies were purchased from Abcam, Na+-K+-ATPase antibody (a6F) was from Developmental Studies Hybridoma Bank (Iowa City, IA), TRα (600–401-Α38) antibody was from Rockland Immunochemicals, and mouse α-FLAG antibody (F7425) was purchased from Sigma-Aldrich.
Cell culture and measurement of O2 consumption and the rate of glycolysis
SK-N-AS cells were maintained in DMEM containing 10% FBS as described before (Freitas et al., 2010). Two days before the baseline oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) measurement, 15 × 103 SK-N-AS cells per well were transfected with either a control, or an Hsp40 shRNA or a GFP-Hsp40 expressing constructs (described above) using a 4D-Nucleofector system (Lonza) with solution SF and pulse program DN-100 and according to the manufacturer's recommendations. After 16 h transfection, the culture medium was replaced with DMEM containing 10% charcoal-stripped fetal bovine serum and, after a period of 24 h, OCR and ECAR (a parameter that reflects the glycolysis rate) of SK-N-AS cells were measured with an XF-96 extracellular flux analyzer (Seahorse Bioscience) (da-Silva et al., 2007) and set as baseline. Subsequently, a 50 nm T3 was added to the medium and SK-N-AS cells were incubated for an additional 24 h period, and at the end new OCR and ECAR measurements were performed.
DNA constructs and primers
D3 plasmids containing wild-type and FLAG-tagged Cys mutant have been previously described (Curcio-Morelli et al., 2003a). For glycosylation experiments, ER-specific NKT or Golgi-specific YTPPP glycosylation motif was fused to the N or C terminus of previously described FLAG-tagged D3 Cys mutant constructs following a procedure used for D2 earlier (Zeöld et al., 2006). Hsp40 (DNAJB1) construct was obtained from Addgene (vector no.19495). The DNAJB1shRNA vector (clone TRCN0000008793 sequence—CGACGGAAAGAGCATTCGA from position 616 of DNAJB1 (NM_006145.1) was purchased from Open Biosystems. The primer sequences used were as follows. GAPDH (forward: 5′-ATGGGGAAGGTGAAGGTCG-3′, reverse: 5′-GGGGTCATTGATGGCAACAATA-3′), ENPP2 (EctoNucleotide Pyrophosphatase/Phosphodiesterase 2) (forward: 5′-ACTCCGTGAAGGCAAAGAGA-3′, reverse: 5′-CAAGATCCGGAGATGTTGGT-3′).
Quantitative PCR.
mRNA was isolated from SK-N-AS using RNeasy kit (Qiagen). cDNA was synthesized using a High Capacity cDNA reverse transcription kit (Applied Biosystems). Semiquantitative PCR was done by My-IQ two-color real-time PCR detection system (Bio-Rad). The IQ-5 optical system software and the starting quantity method were used to determine relative gene expression data.
D3 immunoprecipitation.
Transfection of plasmids into HEK-293 and CHO cells was performed with Lipofectamine 2000 (Invitrogen) or FuGENE 6 (Roche) by the manufacturer's protocols. Transfection efficiency was monitored by cotransfecting pmax-GFP plasmid (Lonza). After transfection, 500 μg of a 0.5% Triton X-100 extract was incubated with α-FLAG antibodies overnight at 4°C with slow shaking. Immunoblotting of D3 was performed with rabbit α-D3 primary antibodies (0.5 μg/ml; NBP1-05767, NB110-96414, Novus Biologicals) and α-rabbit infrared secondary antibodies conjugated with IRD-dye were from (LI-COR Biosciences). Infrared immunoblotting detection was performed using the Odyssey imaging system (LI-COR Biosciences) (Curcio-Morelli et al., 2003b).
D3 activity assay.
SK-N-AS cells were treated with 1% O2 (hypoxia) or 20% O2 (normoxia) for 24 h in a hypoxia chamber as previously described (Simonides et al., 2008). Next, cells were homogenized with a ball homogenizer (Isobiotec) with a10 μm clearance and centrifuged at 800 × g to collect crude nuclear (N) fraction. The supernatant was further centrifuged at 200,000 × g, and the membranous pellet (M) collected for the D3 assay. D3 activity was measured as previously described (Simonides et al., 2008). For nuclear T3, T2 profile experiment, SK-N-AS cells were treated with 12 h of normoxia and hypoxia, at 12 h 4.5 μCi of 125I-T3 was added and additional 12 h of normoxia and hypoxia was given respectively. Then the cells were collected, homogenized and cell nucleus was isolated by 800 × g centrifugation. The T3 and T2 in the nucleus was analyzed by Acquity UPLC (Waters).
Immunofluorescence of D3.
SK-N-AS cells were grown on chamber slides and treated with hypoxic or normoxic conditions for 24 h. Next, cells were fixed with 4% paraformaldehyde for 10 min and then permeabilized with 0.1% Triton X-100 for 10 min. Slides were then blocked with 3% BSA and incubated with various primary antibodies overnight at 4°C followed by 1 h incubation with secondary antibodies Alexa Fluor 488 and 568 (Invitrogen). Images were obtained using Nikon Eclipse C1 or Nikon A1R confocal microscope. Colocalization analysis was done using NIS-Element AR software (Nikon Instruments). In the case of primary neurons, fully differentiated neurons grown on coverslips were fixed in 4% paraformaldehyde and 4% sucrose for 45 min at room temperature. After 10 min of 0.1% Triton permeabilization, the coverslip were blocked by 0.2% fish skin gelatin, 0.1% saponin. For quantitation of nuclear localization, 40 cells from each duplicate slides were counted after scanning by confocal microscope (Nikon Eclipse C1).
Isolation of subcellular membrane fractions
Cells were collected, resuspended in PBS solution with Complete-mini EDTA-free protease inhibitor (Roche) at 4°C. Cells were homogenized by 10 passages through a ball homogenizer (Isobiotec) at 10 μm clearance. Homogenates were centrifuged for 10 min at 800 × g. The resulting nuclear (N′) pellet was washed twice with ice-cold PBS by centrifugation, while the supernatant fraction, containing cytosolic membranes and the cytosol, was further centrifuged at 200,000 × g for 1 h, and the pellet was designated the membrane (M) fraction. When cellular fraction is not used for activity assay, Nuclear (N) and non-nuclear supernatant (NN) was prepared using nonionic detergent Triton X-100. SK-N-AS cells were treated with 0.5% Triton X-100 at 4°C and centrifuged at 800 × g for 10 min to isolate nuclear (N) fraction. The supernatant after 800 × g centrifugation which contain Triton X-100 soluble membranes and cytosol was designated as non-nuclear supernatant (NN).
Results
An analysis of D3 immunoreactivity after 1 h MCAO indicates not only an increase in D3 immunoreactivity in the ischemic hippocampus, but also that there were significant changes in D3 distribution in the ipsilateral hippocampal formation (Fig. 1A). The most pronounced D3 immunoreactivity was seen in the nuclei of the pyramidal and granular layers and the hilus of the hippocampal formation (Fig. 1B,C).
Figure 1.

Increased nuclear D3 labeling in the ipsilateral hippocampus of rats with MCAO. A, Boxes showing the areas of the hippocampal formation in a Nissl-stained section of an intact animal, which were comparatively investigated on the contra- and ipsilateral sides, respectively (B-box 1 b vs b′ and C-box 2 c vs c′) after MCAO (ipsilateral). Scale bars, 1000 μm. B, D3 immunoreactivity in the perikaryal layers of CA1 (CA1/SP) and the upper blade of the dentate gyrus (box 1). Scale bar, 100 μm. C, D3 immunoreactivity in the CA4/hilus (CA4/H) and the lower blade of dentate gyrus (DG (lb)) (box 2). Scale bar, 100 μm. D, E, High-power light microscopic images of nuclear D3 labeling in the contra- and ipsilateral cells of the DG (lb) (D, d vs d′) and the CA4/H (E, e vs e′) revealed by the α-D3(CT) antibody. Scale bar, 20 μm. F–H, A toluidine blue-stained semithin section (F) and an ultrathin section labeled with silver-gold enhanced nickel-diaminobenzidine (G, H) show D3 in the granular cells of the ipsilateral dentate gyrus. The labeling (black arrowheads) forms clusters in the cell nucleus (N) and the cytoplasm (Cp), as shown at high power. In H, the nuclear membrane is marked by white arrowheads. Scale bars: F, 5 μm; G, 2 μm; H, 400 nm. A, Alveus; CA, cornu Ammonis; H, hilus; HS, hippocampal sulcus; PL, polymorphic layer; SG, stratum granulosum; SL/M, stratum lacunosum/moleculare; SL, stratum lucidum; SM, stratum moleculare; SO, stratum oriens; SP, stratum pyramidale; SR, stratum radiatum.
Under higher-power light microscopy, D3 immunoreactivity appeared as silver grains at both the contra and ipsilateral sides of the dentate gyrus and the hilus (Fig. 1D,E). However, the labeling was more intense in cell nuclei of the upper blade on the ipsilateral dentate gyrus (Fig. 1D) and the CA4/hilus (Fig. 1E). Nuclear D3 immunoreactivity was also identified in semi- and ultrathin sections of ipsilateral granule cells (Fig. 1F,G). The majority of the silver grains resided within the cell nucleus and was associated with nuclear chromatin (Fig. 1F,G). Few grain clusters also appeared in the cytoplasm, often in association with the nuclear membrane (Fig. 1H). Similar subcellular distribution of D3 was also observed in the dark neurons, cells that experience dramatic compaction of ultrastructural elements due to the hypoxic stress (data not shown) (Kövesdi et al., 2007).
To investigate hypoxia-induced nuclear D3 accumulation and its impact on thyroid hormone signaling, we examined SK-N-AS cells with αD3(CT) and αD3(NT) antisera, the latter directed against an epitope that is near the transmembrane domain. In cells kept under normoxic conditions, the use of αD3(CT) identified D3 in the cytoplasm and in the nuclear compartment (Fig. 2A), compatible with the in vivo observations made in the rat brain with the same antiserum (Fig. 1). This is also documented by partial colocalization of D3 with nuclear lamin (Fig. 2B). At the same time, the use of αD3(NT) identified D3 in the nucleus and in the plasma membrane (data not shown), as previously described in other normoxic cell models using a similar αD3(NT) antibody (Baqui et al., 2003). The presence of D3 in the nucleus of SK-N-AS cells was confirmed by Western blotting of nuclear fractions (Fig. 2C). That this was not the result of ER contamination of the nuclear fraction was defined by the negligible signal of the ER marker BiP in the nuclear fraction (Fig. 2C). In these cells, D3 enzymatic activity was found predominantly in the fraction containing cellular membranes (Fig. 2D) with less D3 activity distributed to the nuclear fraction (Fig. 2D).
Figure 2.

Catalytically active D3 is present in the cell nucleus. A, Immunofluorescence of normoxic SK-N-AS cells stained with α-D3(CT) antibody. B, Same as in A except that α-nuclear lamin antibody was also used for staining and images were overlaid. C, Western blotting with α-D3(NT) antibody or α-BiP antibody. SK-N-AS cells homogenized with the ball homogenizer (Isobiotec). Nuclear pellet was obtained after centrifugation at 800 × g and washing. Supernatant was centrifuged at 200,000 × g to yield the membrane pellet. D, D3 activity in nuclear or membrane fractions shown in C; data are the mean ± SEM; n = 2 per group; experiments were repeated 3 times.
To gain further insight into the mechanisms leading to hippocampal D3 redistribution we next examined SK-N-AS cells kept under hypoxic conditions for 0–24 h (Fig. 3A). Whereas there was clear upregulation of D3 during hypoxia, the confocal analysis of the nuclear compartment with D3 and nuclear lamin indicated that by 16 h of hypoxia there was an increase in nuclear D3 content, which was maximal by 24 h (Fig. 3A). Notably, there was a dramatic increase in nuclear HIF-1α (Fig. 3B) and a clear redistribution of D3, which predominated in the nuclear compartment (Fig. 3B). Simultaneously, there was a ∼3-fold increase in nuclear D3 activity (Fig. 3C) whereas total membrane D3 remained unaffected (Fig. 3D), illustrating that the nuclear D3 is catalytically active even after its hypoxia-induced import to the nucleus. This phenomenon was also observed in hippocampal synaptically active cultured neurons (Fig. 3E), in which a 24 h hypoxia period doubled D3 localization (Fig. 3F). Given that ischemia also leads to glucose deprivation in the brain, we also looked at whether glucose availability interfered with hypoxia-induced nuclear D3 accumulation. However, D3 accumulated in the nucleus of hypoxic neurons in response to hypoxia even when cells grown in glucose-free medium (Fig. 4A).
Figure 3.

Time-dependent hypoxia-induced nuclear D3 import. A, Immunofluorescence of normoxic SK-N-AS cells processed with α-D3(CT) antibody, α-nuclear lamin antibody, and image overlay. Cells were transferred to a hypoxic chamber (1% O2) and processed for immunofluorescence 8, 16, or 24 h later. B, Western blotting of the indicated SK-N-AS cellular fractions using α-HIF1α, α-nuclear lamin, α-Bip, and α-D3(CT) antibodies; hypoxia lasted for 24 h. N is nucleus (800 g pellet), NN is non-nuclear supernatant prepared in 0.5% Triton X-100 after 800 × g centrifugation; N′ is nucleus (800 g pellet), M is membrane pellet prepared by centrifugation only, after homogenization by ball homogenizer, no detergent was used. α-Nuclear lamin, α-Bip antibodies were used as loading control for nuclear (N′) and membrane (M) fraction, respectively. C, D, D3 activity in nuclear (N′) and membrane (M) fractions prepared by centrifugation only; data are the mean ± SEM; n = 2 per group; experiments were repeated 3 times. E, F, Nuclear localization of D3 under hypoxia in primary hippocampal neuronal cells. Quantification of cells that showed nuclear localization of D3 was done by using a confocal scanning microscope.
Figure 4.

T3/T2 levels in the nucleus during hypoxia and effect of glucose deprivation on D3 under hypoxia. A, SK-N-AS cells were incubated with high glucose (4.5 g/L) versus glucose-free medium for 24 h under hypoxia; nuclear localization of D3 was examined by immunofluorescence using α-D3(CT) antibody. B, C, Nuclear T3 and T2 profiles of normoxic (B) or hypoxic (C) SK-N-AS cells. D, Relative concentrations of T3 and T2 in the nucleus of SK-N-AS under normoxia and hypoxia. E, Immunoprecipitation of D3 was done in HEK-293 cells transiently expressing flag-tagged D3-Cys and TRα. IN, 20% of total input; IgG, immunoprecipitated by IgG; Flag, immunoprecipitated by α-FLAG. The presence of TRα was examined by Western blot with α-TRα antibody.
The analysis of nuclear 125I-T3 and 125I-T2 supports the idea that nuclear D3 plays a role by dampening thyroid hormone signaling via inactivation of T3 to T2. In hypoxic SK-N-As cells there was a reduction of nuclear T3 by ∼30% whereas T2 nuclear content almost doubled (Fig. 4B–D). Given these clear-cut results, it is unlikely that D3 plays a direct role in transcriptional control, independent of T3. For example, transiently expressed D3 failed to coimmunoprecipitate with TRα in HEK-293 cells (Fig. 4E).
D3 traffics through the ER and Golgi systems and reaches the plasma membrane where it undergoes endocytosis and recycling with the early endosomes (Baqui et al., 2003). Thus, to ascertain whether hypoxia induces D3 sorting to the nuclear compartment as a result of direct import from the ER or after trafficking through the Golgi complex and/or plasma membrane, we mapped the source of nuclear D3 by analyzing the D3 glycosylation patterns in CHO cells transiently expressing different FLAG-tagged D3 containing sequences that direct N-link (NKT) or O-link (YTPPP) glycosylation fused to the N or C termini of D3 (Yoshida et al., 1997; Pedrazzini et al., 2000) (Fig. 5A). To interpret these studies, we first established that only in lysates (Fig. 5B) or immunoprecipitates (Fig. 5C) of cells transiently expressing N-link-NH2-D3 was there a slightly higher molecular weight of ENDO-H-sensitive D3 band, indicating ER D3 glycosylation. Second, we established that only in lysates of cells transiently expressing O-link-NH2-D3 was there a slightly higher molecular weight D3 band, indicating Golgi D3 glycosylation (Fig. 5D,E). In both N-link and O-link fusion constructs, no higher molecular weight D3 bands were visualized when the sequences were fused to the C terminus of the D3 molecule (Fig. 5B,C,E), indicating that, similar to the other deiodinases, D3 is a type I integral membrane protein.
Figure 5.

D3 with glycosylation motifs and its glycosylation. A, D3 constructs fused with FLAG epitope or glycosylation sequences as indicated. B, Constructs containing N-glycosylation site with different orientation (a1, a2) were transfected into CHO cells. The cell lysates were analyzed by Western blot using α-FLAG antibody; GFP was used as a control for transfection. C, The cell lysates used in B were immunoprecipitated by α-FLAG antibody. The immunoprecipitate from a2 was treated with EndoH. D, D3 constructs without (b1) and with (b2) O-glycosylation motif as indicated. E, The constructs b1 and b2 were transfected into CHO cells and total cell lysates were analyzed by Western blot using α-FLAG antibody. F, For N-glycosylation, constructs a0 and a2 were transfected into CHO cells. After 24 h of hypoxia treatment, cells were lysed in 0.5% Triton X-100; after 800 × g centrifugation, resulting nuclear (N) and non-nuclear (NN) supernatant fractions were analyzed by Western blot using α-FLAG antibody. G, For O-glycosylation, constructs b1 and b2 were transfected into CHO cells, treated with 24 h of hypoxia, and lysed in 0.5% Triton X-100. After 800 × g centrifugation, nuclear (N) and non-nuclear (NN) supernatant fractions were analyzed by Western blot using α-FLAG antibody as in F.
Next, based on the knowledge of D3's topology, we looked at glycosylated D3 in the nucleus of hypoxic CHO cells transiently expressing N-link-NH2- or O-link-NH2-D3 fusion constructs. Remarkably, N-link-NH2-D3 but not O-link-NH2-D3 could be found in the nucleus of hypoxic cells (Fig. 5F,G), indicating that hypoxia induces nuclear D3 accumulation based on direct recruitment from the ER. It is notable that under hypoxic conditions O-link glycosylated D3 was absent even from postnuclear supernatants (NN) (Fig. 5G), confirming that hypoxia shifts ER D3 to the nucleus, minimizing its trafficking through the Golgi system.
The data suggest that trafficking through the Golgi system commits D3 to the plasma membrane, whereas hypoxia accelerates D3 import into the nuclear compartment. To learn more about this mechanism we used αD3(CT) in pull down experiments and tested for coimmunoprecipitation of known nuclear transporting proteins, a strategy that led to the identification of a D3-interacting protein: DNAJB1, a member of the Hsp40 family of proteins (Li et al., 2009). Next, we verified that this interaction occurs in mammalian cells by using transiently expressed FLAG-D3 in HEK-293 cells to pull down endogenously expressed Hsp40 (Fig. 6A). Hsp40 is a hypoxia-inducible cochaperone (Fig. 6B) that regulates complex formation between Hsc70 and client proteins by binding and delivering specific proteins to Hsc70 or by stabilizing Hsc70-polypeptide complexes (Fan et al., 2003; Qiu et al., 2006). Hsp40-directed client proteins to Hsc70 is known to mediate the nuclear import of glucocorticoid receptors (Yang and DeFranco, 1994), HIV-1 preintegration complex (Agostini et al., 2000) and karyopherin α (Agostini et al., 2000). Moreover, the Hsp40 family member DNAJB6 is also involved in nuclear import of HIV-2 preintegration complex (Cheng et al., 2008) and localization of T-Cell quiescence protein, Schlafen-1 in the nucleus (Zhang et al., 2008). In fact, coimmunoprecipitation experiments revealed a potential interaction between FLAG-D3 and Hsc70 as well (Fig. 6A). Furthermore, immunofluorescence studies revealed under normoxia that D3 and Hsp40 coexist in close proximity in the cell (Fig. 6C) and both accumulate in the nuclear compartment after 24 h of hypoxia (Fig. 6D), as quantified in (Fig. 6E). Notably, Hsp40-D3 interaction was also documented in vivo during ischemia by immunohistochemistry in CA1 hippocampal region (Fig. 6F). The fact that D3 interacts and colocalizes with the cochaperone Hsp40 and also interacts with Hsc70 indicates that nuclear D3 import is likely to be mediated by these two proteins.
Figure 6.

Hsp40 interacts with and colocalizes with D3. A, HEK-293 cells transiently expressing FLAG-tagged D3 were lysed with Triton X-100; samples were immunoprecipitated with IgG or α-FLAG antibody and analyzed by Western blotting with different antibodies as indicated; IN is 10% of input, IP is immunoprecipitate pellet. B, Western blotting of SK-N-AS cell sonicates with α-Hsp40 antibody; hypoxia lasted for 24 h. C, Immunofluorescence of normoxic SK-N-AS cells processed with α-D3(CT) and α-Hsp40 antibodies and image overlay with two different magnifications. D, Same as in C, except that cells were treated with 24 h of hypoxia instead of 24 h of normoxia. E, Quantification of D3 or Hsp40 colocalization with the DAPI signal in the nucleus; also shown in the far right column, colocalization of D3 and Hsp40, p < 0.001. F, Colocalization of D3 with Hsp40 in cell nucleus of neuron in the rat hippocampal CA1 region. D3, Green; Hsp40, red; nucleus, blue (Hoechst). Scale bar, 5 μm.
To gain further insight into the mechanism of D3 import into the nucleus, SK-N-AS cells transiently expressing Hsp40 kept under normoxic conditions were analyzed for nuclear D3 expression by immunofluorescence. Expression of Hsp40 alone increased D3 nuclear import (Fig. 7A) as evidenced by a ∼3.5-fold increase in colocalization with DAPI (Fig. 7B). Next, to confirm that Hsp40 mediates nuclear D3 import, shRNA vectors targeting Hsp40 was used in SK-N-AS cells, reducing its levels by >90% (Fig. 7C,D). Remarkably, under these conditions of Hsp40 knockdown, hypoxia-induced nuclear D3 import was markedly reduced (Fig. 7E,F) without changes in D3 expression (data not shown), indicating that Hsp40 is critical for the shift in D3 trafficking to the nucleus.
Figure 7.

Hsp40 mediates hypoxia-induced D3 nuclear import. A, SK-N-AS cells electroporated with a GFP-Hsp40 expression plasmid (Addgene 19495) were stained with both α-D3(CT) antibody and DAPI. Overlay is shown on the third row. Small panel on lower right shows GFP-Hsp40-expressing cells. B, Colocalization between D3 and DAPI signal from GFP-Hsp40-expressing cells was analyzed by Pearson's coefficients using NIS-AR software (Nikon Instruments); data are presented as mean ± SEM, n = 20. C, SK-N-AS cells stably expressing either a scrambled (left) or a shRNA plasmid targeting endogenous Hsp40 mRNA molecules (right). KD, Knockdown. D, Hsp40 protein levels as analyzed by Western blot of SK-N-AS cells from C. shRNA construct is specific for DNAJB1 and DNAJB1 (Hsp40) antibody used in this experiment (ab78437) does not cross-react with other J-domain proteins such as YDJ2. In E and F, SK-N-AS cells stably expressing the Hsp40 shRNA were exposed for 24 h to normoxic (E) or hypoxic (F) condition and then stained with DAPI and α-D3(CT) antibody as in A. For C–F, stable SK-N-AS cell lines expressing a control or an Hsp40 shRNA plasmid were generated by lentiviral transduction coupled with 2 weeks of puromycin (1 μg/ml) selection.
To test whether this shift in cell distribution of D3 is biologically relevant we studied SK-N-AS cells and looked at the effect of hypoxia and/or Hsp40 knockdown on the expression of ENPP2, a bona fide T3-responsive gene in these cells (Freitas et al., 2010). In fact, 12 h hypoxia reduced T3 induction of this gene by ∼30% (Fig. 8A). In contrast, Hsp40 knockdown resulted in the opposite phenotype, amplifying the T3 induction of ENPP2 by ∼3-fold (Fig. 8A). This demonstrates that T3 signaling is substantially enhanced by disruption of the nuclear import of D3 in hypoxic SK-N-AS cells. This corollary was further strengthened by studies of the T3-induced metabolic rate in SK-N-AS cells (Fig. 8B). Whereas during the first 24 h Hsp40 knockdown did not affect the T3-induced O2 consumption rate, it promptly amplified the T3 induction of the glycolytic rate by almost doubling it in the same period of time (Fig. 8B). At the same time, transient expression of Hsp40 led to the opposite effect, with reductions of ∼20% in the T3 induction of both O2 consumption and glycolytic rate (Fig. 8C). These findings strongly indicate that nuclear D3 import rapidly limits the transcriptional T3 footprint and its downstream metabolic actions.
Figure 8.
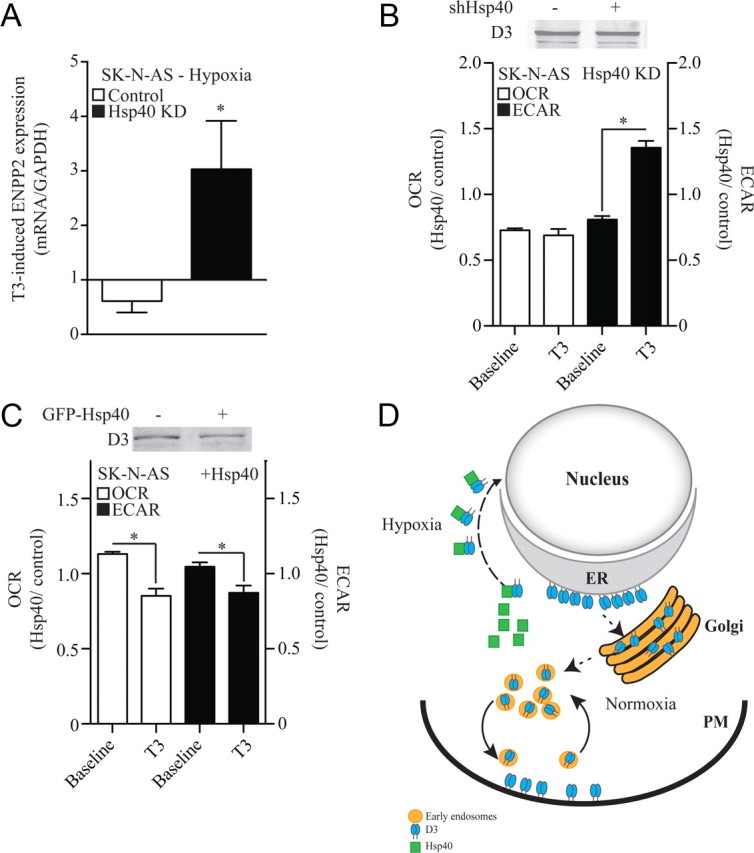
Nuclear D3 import dampens thyroid hormone signaling and slows down cellular metabolism. A, Effect of Hsp40 knockdown (KD) on the T3 inducibility of ENPP2 gene expression in hypoxic SK-N-AS cells; hypoxia and 50 nm T3 treatments were simultaneous and lasted for 24 h. Results are presented as a ratio between the T3 induction during hypoxia and the T3 induction during normoxia. B, OCR and ECAR (glycolysis) of SK-N-AS cells transiently expressing a control or an Hsp40 shRNA; 15,000 cells/well were plated and baseline values for both OCR and ECAR were measured as previously described (Simonides et al., 2008). Results are expressed as the OCR or ECAR ratio between Hsp40 KD and the control constructs at two different times: baseline and after treatment with 50 nm T3 for 24 h; data are presented as mean ± SEM; n = 30 per group; *p < 0.01 versus baseline reading by Student's t test. C, Same as in B except that studies were performed in cells transiently expressing a GFP-Hsp40. Both in B and C, D3 protein level did not change. D, Model explaining how O2 availability and Hsp40 regulate D3 subcellular localization. When O2 is available, ER-born D3 reaches the Golgi system and plasma membrane (PM), where it is internalized and recycled. During hypoxia, there is Hsp40-mediated nuclear import of D3, thus placing D3 physically closer to the thyroid hormone receptor.
Discussion
D3 prevents T4 activation and terminates thyroid hormone action by inactivating T3 once both molecules cross the plasma membrane and diffuses in the intracellular compartment toward the cell nucleus (Gereben et al., 2008). The net amount of T3 that ends up in the nucleus and eventually binds to its receptors i.e., TRα and/or TRβ, modifies thyroid hormone signaling by defining a transcriptional footprint that is cell-specific (Wu and Koenig, 2000; Zhang and Lazar, 2000). That hypoxia markedly induces D3 is remarkable, dampening thyroid hormone signaling and slowing down O2 consumption (Simonides et al., 2008). That at the same time trafficking of active D3 is redirected to the nucleus is novel and provides the first evidence that thyroid hormone metabolism occurs in the nuclear compartment, physically closer to where TR-mediated gene transcriptional control takes place (Fig. 8D). It is conceivable that the presence of D3 in the nucleus explains the fast changes in O2 consumption observed in SK-N-AS cells after D3 is inactivated by iopanoic acid (Simonides et al., 2008), thus providing a much tighter regulation of nuclear T3 content at times that rapid decrease in thyroid hormone signaling could be advantageous, such as ischemia and hypoxia. This mechanism is physiologically relevant as it was documented in pyramidal and granular neurons in vivo in the hippocampal formation of rats during ischemia (Fig. 1).
D3 was originally described as a plasma membrane protein that is internalized and recycled via the early endosomes (Baqui et al., 2003). The present study reveals a much more complex intracellular trafficking of D3 that affects thyroid hormone signaling and is regulated by O2 availability, with two alternative routes after it is synthesized in the ER: (1) trafficking toward the Golgi system and the plasma membrane whenever O2 is available or (2) nuclear import when O2 availability is inadequate (Fig. 8D). D3 is a client of the Hsp40 pathway, which is conceivably sensitive to the redox state of the cell via two conserved redox-sensitive cysteine residues in Hsp40 (Ago et al., 2008). Because nuclear D3 retains catalytic activity and is physically closer to TRs, this mechanism promotes profound depletion of T3 (Fig. 4B–D) that rapidly slows down metabolism.
From a broad perspective, the induction of heat shock proteins plays an important role in neuronal protection during ischemia or neurodegeneration (Brown, 2007). Thus, it is likely that the Hsp40-induced nuclear import of D3 is part of this much broader neuroprotective mechanism against hypoxia, which leads to localized hypothyroidism. In fact, in a model of ischemia reperfusion similar to the one used here, systemically hypothyroid rats had insignificant neurological deficit, smaller area of infarct, significantly lower markers of oxidative stress and necrosis. In addition, only in the hypothyroid brain markers of antioxidant activity and mitochondrial integrity were preserved but not in the euthyroid controls (Rastogi et al., 2006). Thus, the present results indicate that a localized D3-mediated decrease in thyroid hormone signaling provides an adaptive advantage during brain ischemia and reperfusion.
The present studies also clearly demonstrate that D3 is a type I integral membrane protein with its active center inside the cell. A consensus exists that D3 has a single transmembrane domain that plays a role in homodimerization (Curcio-Morelli et al., 2003a; Sagar et al., 2008). However, data were conflicting as to whether D3 was a type I or type II integral membrane protein. Earlier we concluded on the basis of selective biotinylation studies that transiently expressed FLAG-tagged D3 is a type II integral membrane protein (Baqui et al., 2003). However, later it was found that MCT8-mediated transport of thyroid hormone across the plasma membrane enhances D3-catalyzed T3 inactivation, indicating that D3-mediated T3 degradation is an intracellular event, compatible with D3 assuming a type I topology (Friesema et al., 2006). The latter is indeed supported by the present findings given that glycosylated D3 was only observed when the glycosylation sequences were fused to the NH2 terminus of D3.
In conclusion, the present studies indicate that D3-mediated thyroid hormone inactivation occurs in the cell nucleus under hypoxic conditions. D3 is a client of the Hsp40-Hsc70 pathway known to direct other proteins to the cell nucleus and is conceivably sensitive to the redox state of the cell. Because nuclear D3 retains catalytic activity and is physically much closer to TRs, this mechanism promotes profound localized hypothyroidism, as evidenced by the jump is O2 consumption a few hours after D3 activity is inhibited in SK-N-AS cells (Simonides et al., 2008). Thus, nuclear import of D3 is an adaptive mechanism to rapidly dampen thyroid hormone signaling, minimizing cell metabolism and ischemia-induced hypoxic brain damage.
Footnotes
This work was supported by NIH Grants DK65055, NS061348, and HD057521, Hungarian Scientific Research Fund Grant OTKA K81226, the Medical Research Council (ETT Grant 02-121/2009), the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 259772 and 245009, and the János Bolyai Research Scholarship of the Hungarian Academy of Sciences. The excellent technical help of Andrea Juhász and Barna László is gratefully acknowledged. We thank H. Kampinga for GFP-Hsp40 construct (Addgene 19495).
The authors declare no competing financial interests.
References
- Ago T, Liu T, Zhai P, Chen W, Li H, Molkentin JD, Vatner SF, Sadoshima J. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell. 2008;133:978–993. doi: 10.1016/j.cell.2008.04.041. [DOI] [PubMed] [Google Scholar]
- Agostini I, Popov S, Li J, Dubrovsky L, Hao T, Bukrinsky M. Heat-shock protein 70 can replace viral protein R of HIV-1 during nuclear import of the viral preintegration complex. Exp Cell Res. 2000;259:398–403. doi: 10.1006/excr.2000.4992. [DOI] [PubMed] [Google Scholar]
- Banker GA, Goslin K. Culturing nerve cells. Cambridge, MA: MIT; 1998. pp. 339–370. [Google Scholar]
- Baqui M, Botero D, Gereben B, Curcio C, Harney JW, Salvatore D, Sorimachi K, Larsen PR, Bianco AC. Human type 3 iodothyronine selenodeiodinase is located in the plasma membrane and undergoes rapid internalization to endosomes. J Biol Chem. 2003;278:1206–1211. doi: 10.1074/jbc.M210266200. [DOI] [PubMed] [Google Scholar]
- Bianco AC, Maia AL, da Silva WS, Christoffolete MA. Adaptive activation of thyroid hormone and energy expenditure. Biosci Rep. 2005;25:191–208. doi: 10.1007/s10540-005-2885-6. [DOI] [PubMed] [Google Scholar]
- Brown IR. Heat shock proteins and protection of the nervous system. Ann N Y Acad Sci. 2007;1113:147–158. doi: 10.1196/annals.1391.032. [DOI] [PubMed] [Google Scholar]
- Callebaut I, Curcio-Morelli C, Mornon JP, Gereben B, Buettner C, Huang S, Castro B, Fonseca TL, Harney JW, Larsen PR, Bianco AC. The iodothyronine selenodeiodinases are thioredoxin-fold family proteins containing a glycoside hydrolase clan GH-A-like structure. J Biol Chem. 2003;278:36887–36896. doi: 10.1074/jbc.M305725200. [DOI] [PubMed] [Google Scholar]
- Cheng X, Belshan M, Ratner L. Hsp40 facilitates nuclear import of the human immunodeficiency virus type 2 Vpx-mediated preintegration complex. J Virol. 2008;82:1229–1237. doi: 10.1128/JVI.00540-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio-Morelli C, Gereben B, Zavacki AM, Kim BW, Huang S, Harney JW, Larsen PR, Bianco AC. In vivo dimerization of types 1, 2, and 3 iodothyronine selenodeiodinases. Endocrinology. 2003a;144:937–946. doi: 10.1210/en.2002-220960. [DOI] [PubMed] [Google Scholar]
- Curcio-Morelli C, Zavacki AM, Christofollete M, Gereben B, de Freitas BC, Harney JW, Li Z, Wu G, Bianco AC. Deubiquitination of type 2 iodothyronine deiodinase by von Hippel-Lindau protein-interacting deubiquitinating enzymes regulates thyroid hormone activation. J Clin Invest. 2003b;112:189–196. doi: 10.1172/JCI18348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da-Silva WS, Harney JW, Kim BW, Li J, Bianco SD, Crescenzi A, Christoffolete MA, Huang SA, Bianco AC. The small polyphenolic molecule kaempferol increases cellular energy expenditure and thyroid hormone activation. Diabetes. 2007;56:767–776. doi: 10.2337/db06-1488. [DOI] [PubMed] [Google Scholar]
- Dentice M, Salvatore D. Local impact of thyroid hormone inactivation: deiodinases: the balance of thyroid hormone. J Endocrinol. 2011;209:273–282. doi: 10.1530/JOE-11-0002. [DOI] [PubMed] [Google Scholar]
- Dentice M, Bandyopadhyay A, Gereben B, Callebaut I, Christoffolete MA, Kim BW, Nissim S, Mornon JP, Zavacki AM, Zeöld A, Capelo LP, Curcio-Morelli C, Ribeiro R, Harney JW, Tabin CJ, Bianco AC. The Hedgehog-inducible ubiquitin ligase subunit WSB-1 modulates thyroid hormone activation and PTHrP secretion in the developing growth plate. Nat Cell Biol. 2005;7:698–705. doi: 10.1038/ncb1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan CY, Lee S, Cyr DM. Mechanisms for regulation of Hsp70 function by Hsp40. Cell Stress Chaperones. 2003;8:309–316. doi: 10.1379/1466-1268(2003)008<0309:mfrohf>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas BC, Gereben B, Castillo M, Kalló I, Zeöld A, Egri P, Liposits Z, Zavacki AM, Maciel RM, Jo S, Singru P, Sanchez E, Lechan RM, Bianco AC. Paracrine signaling by glial cell-derived triiodothyronine activates neuronal gene expression in the rodent brain and human cells. J Clin Invest. 2010;120:2206–2217. doi: 10.1172/JCI41977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesema EC, Kuiper GG, Jansen J, Visser TJ, Kester MH. Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in intracellular metabolism. Mol Endocrinol. 2006;20:2761–2772. doi: 10.1210/me.2005-0256. [DOI] [PubMed] [Google Scholar]
- Gereben B, Zavacki AM, Ribich S, Kim BW, Huang SA, Simonides WS, Zeöld A, Bianco AC. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev. 2008;29:898–938. doi: 10.1210/er.2008-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SA, Bianco AC. Reawakened interest in type III iodothyronine deiodinase in critical illness and injury. Nat Clin Pract Endocrinol Metab. 2008;4:148–155. doi: 10.1038/ncpendmet0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalló I, Butler JA, Barkovics-Kalló M, Goubillon ML, Coen CW. Oestrogen receptor beta-immunoreactivity in gonadotropin releasing hormone-expressing neurones: regulation by oestrogen. J Neuroendocrinol. 2001;13:741–748. doi: 10.1046/j.1365-2826.2001.00708.x. [DOI] [PubMed] [Google Scholar]
- Kövesdi E, Pál J, Gallyas F. The fate of “dark” neurons produced by transient focal cerebral ischemia in a non-necrotic and non-excitotoxic environment: neurobiological aspects. Brain Res. 2007;1147:272–283. doi: 10.1016/j.brainres.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Li J, Qian X, Sha B. Heat shock protein 40: structural studies and their functional implications. Protein Pept Lett. 2009;16:606–612. doi: 10.2174/092986609788490159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matucz E, Móricz K, Gigler G, Simó A, Barkóczy J, Lévay G, Hársing LG, Jr, Szénási G. Reduction of cerebral infarct size by non-competitive AMPA antagonists in rats subjected to permanent and transient focal ischemia. Brain Res. 2004;1019:210–216. doi: 10.1016/j.brainres.2004.05.098. [DOI] [PubMed] [Google Scholar]
- Mohácsik P, Zeöld A, Bianco AC, Gereben B. Thyroid hormone and the neuroglia: both source and target. J Thyroid Res. 2011;2011:215718. doi: 10.4061/2011/215718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrazzini E, Villa A, Longhi R, Bulbarelli A, Borgese N. Mechanism of residence of cytochrome b(5), a tail-anchored protein, in the endoplasmic reticulum. J Cell Biol. 2000;148:899–914. doi: 10.1083/jcb.148.5.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters RP, Wouters PJ, Kaptein E, van Toor H, Visser TJ, Van den Berghe G. Reduced activation and increased inactivation of thyroid hormone in tissues of critically ill patients. J Clin Endocrinol Metab. 2003;88:3202–3211. doi: 10.1210/jc.2002-022013. [DOI] [PubMed] [Google Scholar]
- Qiu XB, Shao YM, Miao S, Wang L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol Life Sci. 2006;63:2560–2570. doi: 10.1007/s00018-006-6192-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastogi L, Godbole MM, Ray M, Rathore P, Pradhan S, Gupta SK, Pandey CM. Reduction in oxidative stress and cell death explains hypothyroidism induced neuroprotection subsequent to ischemia/reperfusion insult. Exp Neurol. 2006;200:290–300. doi: 10.1016/j.expneurol.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Sagar GD, Gereben B, Callebaut I, Mornon JP, Zeöld A, Curcio-Morelli C, Harney JW, Luongo C, Mulcahey MA, Larsen PR, Huang SA, Bianco AC. The thyroid hormone-inactivating deiodinase functions as a homodimer. Mol Endocrinol. 2008;22:1382–1393. doi: 10.1210/me.2007-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- Simonides WS, Mulcahey MA, Redout EM, Muller A, Zuidwijk MJ, Visser TJ, Wassen FW, Crescenzi A, da-Silva WS, Harney J, Engel FB, Obregon MJ, Larsen PR, Bianco AC, Huang SA. Hypoxia-inducible factor induces local thyroid hormone inactivation during hypoxic-ischemic disease in rats. J Clin Invest. 2008;118:975–983. doi: 10.1172/JCI32824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, Schoonjans K, Bianco AC, Auwerx J. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- Wu Y, Koenig RJ. Gene regulation by thyroid hormone. Trends Endocrinol Metab. 2000;11:207–211. doi: 10.1016/s1043-2760(00)00263-0. [DOI] [PubMed] [Google Scholar]
- Yang J, DeFranco DB. Differential roles of heat shock protein 70 in the in vitro nuclear import of glucocorticoid receptor and simian virus 40 large tumor antigen. Mol Cell Biol. 1994;14:5088–5098. doi: 10.1128/mcb.14.8.5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A, Suzuki M, Ikenaga H, Takeuchi M. Discovery of the shortest sequence motif for high level mucin-type O-glycosylation. J Biol Chem. 1997;272:16884–16888. doi: 10.1074/jbc.272.27.16884. [DOI] [PubMed] [Google Scholar]
- Zeöld A, Pormüller L, Dentice M, Harney JW, Curcio-Morelli C, Tente SM, Bianco AC, Gereben B. Metabolic instability of type 2 deiodinase is transferable to stable proteins independently of subcellular localization. J Biol Chem. 2006;281:31538–31543. doi: 10.1074/jbc.M604728200. [DOI] [PubMed] [Google Scholar]
- Zhang J, Lazar MA. The mechanism of action of thyroid hormones. Annu Rev Physiol. 2000;62:439–466. doi: 10.1146/annurev.physiol.62.1.439. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yang Z, Cao Y, Zhang S, Li H, Huang Y, Ding YQ, Liu X. The Hsp40 family chaperone protein DnaJB6 enhances Schlafen1 nuclear localization which is critical for promotion of cell-cycle arrest in T-cells. Biochem J. 2008;413:239–250. doi: 10.1042/BJ20071510. [DOI] [PubMed] [Google Scholar]