

Abstract
Loss of retinoschisin (RS1) in Rs1 knock-out (Rs1–KO) retina produces a post-photoreceptor phenotype similar to X-linked retinoschisis in young males. However, Rs1 is expressed strongly in photoreceptors, and Rs1–KO mice have early reduction in the electroretinogram a-wave. We examined light-activated transducin and arrestin translocation in young Rs1–KO mice as a marker for functional abnormalities in maturing rod photoreceptors. We found a progressive reduction in luminance threshold for transducin translocation in wild-type (WT) retinas between postnatal days P18 and P60. At P21, the threshold in Rs1–KO retinas was 10-fold higher than WT, but it decreased to <2.5-fold higher by P60. Light-activated arrestin translocation and re-translocation of transducin in the dark were not affected. Rs1–KO rod outer segment (ROS) length was significantly shorter than WT at P21 but was comparable with WT at P60. These findings suggested a delay in the structural and functional maturation of Rs1–KO ROS. Consistent with this, transcription factors CRX and NRL, which are fundamental to maturation of rod protein expression, were reduced in ROS of Rs1–KO mice at P21 but not at P60. Expression of transducin was 15–30% lower in P21 Rs1–KO ROS and transducin GTPase hydrolysis was nearly twofold faster, reflecting a 1.7- to 2.5-fold increase in RGS9 (regulator of G-protein signaling) level. Transduction protein expression and activity levels were similar to WT at P60. Transducin translocation threshold elevation indicates photoreceptor functional abnormalities in young Rs1–KO mice. Rapid reduction in threshold coupled with age-related changes in transduction protein levels and transcription factor expression are consistent with delayed maturation of Rs1–KO photoreceptors.
Introduction
Retinoschisin protein (RS1; Swiss-Prot accession number NP_000321.1) is expressed in all neurons except horizontal cells in the developing and adult mouse retina but most prominently in photoreceptor inner segments (IS), in which it is localized to the external plasma membrane (Takada et al., 2004). The role of RS1 in photoreceptor function has not been identified, but mutations in the retinoschisin gene cause a retinal dystrophy in young males known as X-linked retinoschisis (Sauer et al., 1997) characterized by delamination of internal retinal structure and consequent reduction in retinal signaling (Manschot, 1972) as measured by the reduced b-wave of the electroretinogram (ERG). The features of this disease and the presence of a discoidin domain in retinoschisin have led to the generally accepted notion that it functions as an adhesion protein to maintain retinal architecture (for review, see Molday et al., 2012). The retinoschisin knock-out (Rs1–KO) mouse has a similar phenotype but also has a reduced photoreceptor a-wave and reduced rod outer segment (ROS) length as early as 1 month, and photoreceptor loss is apparent by 4 months (Kjellstrom et al., 2007).
Light-activated movement of phototransduction proteins between photoreceptor compartments involves major aspects of rod function, including photon capture, phototransduction, and light adaptation. In the dark, the heterotrimer transducin (Gαβγ) resides in the ROS, and arrestin localizes in the inner segment (IS). When the light intensity reaches a critical threshold at which the rate of activation of transducin α (Gαt1) exceeds the rate of inactivation by GTP hydrolysis (Kerov et al., 2005; Lobanova et al., 2007), Gαt1 moves from the ROS into the IS and the cell body. This movement helps to set the dynamics of rod sensitivity and adaptation (Sokolov et al., 2002; Mendez et al., 2003; Zhang et al., 2003; Calvert et al., 2006; Lobanova et al., 2007) and may afford protection from light stress (Burns and Arshavsky, 2005). In response to light, arrestin moves in reciprocal manner from the IS to the ROS in which it quenches photoactivated rhodopsin (R*). The level of R* (Nair et al., 2005) and the strength of the transduction signal (Strissel et al., 2006) have been proposed as stimuli for arrestin translocation into the ROS.
To address the role of RS1 in photoreceptor function at an early age, we investigated the effect of the loss of Rs1 expression on transducin and arrestin translocation light activation threshold between postnatal days P21 and P60. We found that P21 Rs1–KO mice have a much higher threshold than wild-type (WT) mice for transducin translocation, but it decreases rapidly with age and is more similar to WT at P60. Arrestin translocation threshold is not affected. This suggests the possibility of a delay in maturation of phototransduction processes involved in transducin translocation in Rs1–KO photoreceptors. Consistent with this, we found that photoreceptor transcription factors CRX and NRL are reduced in Rs1–KO mice at P21 but not P60. In addition, a large proportion of the translocation threshold increase in Rs1–KO mice at P21 could be explained by a reduction in transducin levels and an increase in GTPase activity.
Materials and Methods
Animals.
This research was conducted in accordance with the Association for Research in Vision and Ophthalmology Statement on the Use of Animals in Ophthalmic and Vision Research. Rs1–KO mice and littermate control WT male mice were studied at P18, P21, P28, P35, and P60, in which P1 corresponds to the day of birth for purposes of this paper. Animals were born and reared in our laboratory derived from the founders originally described by Zeng et al. (2004). The Rs1–KO mice have been backcrossed for at least 20 generations onto C57BL/6J background and were reared on a 12 h light/dark cycle of 20 lux. Food and water were available ad libitum. Animals were weaned at 3 weeks of age. All Rs1–KO mice in the study were genotyped from tail DNA to verify that they carried the Rs1–KO construct.
Light-activated transducin and arrestin translocation by immunohistochemistry.
Dark-adapted WT and Rs1–KO mice were anesthetized with intraperitoneal administration of ketamine (80 mg/kg) and xylazine (4 mg/kg). After pupil dilation with topical 0.5% tropicamide and 0.5% phenylephrine HCl, they were placed on a heating pad (at 37°C) facing a full-field illumination (Ganzfeld bowl) and exposed to 1, 2, 6, 12, 18, 30, 60, 120, 180, or 300 scotopic candela per square meter (sc.cd/m2) of white light for 1 h unless indicated otherwise.
For immunohistochemical analysis of transducin and arrestin translocation, mice were killed immediately after light exposure with CO2, and the eyes were quickly removed and placed in 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, at 4°C for 12 h. The fixed eye tissues were rinsed in PBS and trimmed to prepare eyecups. Eyecups were embedded in 7% agarose/PBS, pH 7.4, and 100-μm-thick retinal sections were cut by Vibratome (model VT1000S; Leica Microsystems).
Single-labeling experiments were performed by incubating the sections with one of the primary antibodies overnight at 4°C in a medium containing 5% normal goat serum (G), 0.2% Triton X-100 (T), and 0.05% sodium azide (A) in PBS (GTA–PBS). The antibodies and the dilutions used were as follows: rabbit polyclonal antibody against transducin α (Gαt1; Santa Cruz Biotechnology) at 1:800; rabbit polyclonal antibody against arrestin (from Igal Gery, National Eye Institute at National Institutes of Health, Bethesda, MD) at 1:2000; and mouse monoclonal antibody against rhodopsin (RET–P1; Abcam) at 1:2500. The sections were washed with GTA–PBS and incubated with fluorescent conjugated secondary antibodies for 2 h in appropriate combinations: Alexa Fluor 488 or 568-conjugated goat anti-mouse or goat anti-rabbit IgG (Invitrogen). The typical dilutions were 1:1000. A fluorescent nuclear stain, 4′,6-diamidino-2-phenylindole (DAPI) (1:5000; Sigma-Aldrich) was also used to visualize nuclei in retinal sections. The sections were wet mounted on glass slides with Fluorogel (EM Sciences) for analysis and visualized with a confocal microscope (TCS SP-2; Leica Microsystems). Digital images (1024 × 1024 pixels, 375 × 375 μm) were acquired and processed in Photoshop (Adobe Systems). All images were taken under identical conditions of laser intensity, brightness, and contrast.
Maximum projections of 3–20 confocal z-series images taken at 1.3–2 μm steps through sections from the retinas of 66 mice (36 Rs1–KO and 30 WT) were analyzed to determine the threshold intensity for transducin translocation. Threshold was defined as the intensity at which outer plexiform layer (OPL) staining could be clearly seen across most of the retinal length in the image. These evaluations were confirmed by measurement of fluorescent intensity in images from the retinas of 52 P21 and P60 WT and Rs1–KO mice using NIH Image J software. The program was used to construct vertical plot profiles of Gαt1 antibody and DAPI (when present) stain intensity (0–255 scale) from ROS tips to the inner nuclear layer (INL). DAPI and Gαt1 stain intensity was averaged over at least 100 μm of retinal length in the same location but in separate images of each section (see Fig. 3A). Plot profiles were replotted for analysis in GraphPad Prism 6.0 for Windows (GraphPad Software). Image scanning and plotting was done without knowledge of the identity of the samples being analyzed. The luminance threshold for transducin translocation was defined as the exposure intensity at which the peak fluorescent stain intensity in the OPL relative to the peak ROS intensity (OPL/ROS) was significantly greater than the average OPL/ROS intensity in retinas of the same age and genotype exposed to 2 sc.cd/m2 or less. The position of the OPL was confirmed in most sections by the decline in the DAPI signal adjacent to the outer nuclear layer (ONL) (see Fig. 3A).
Figure 3.
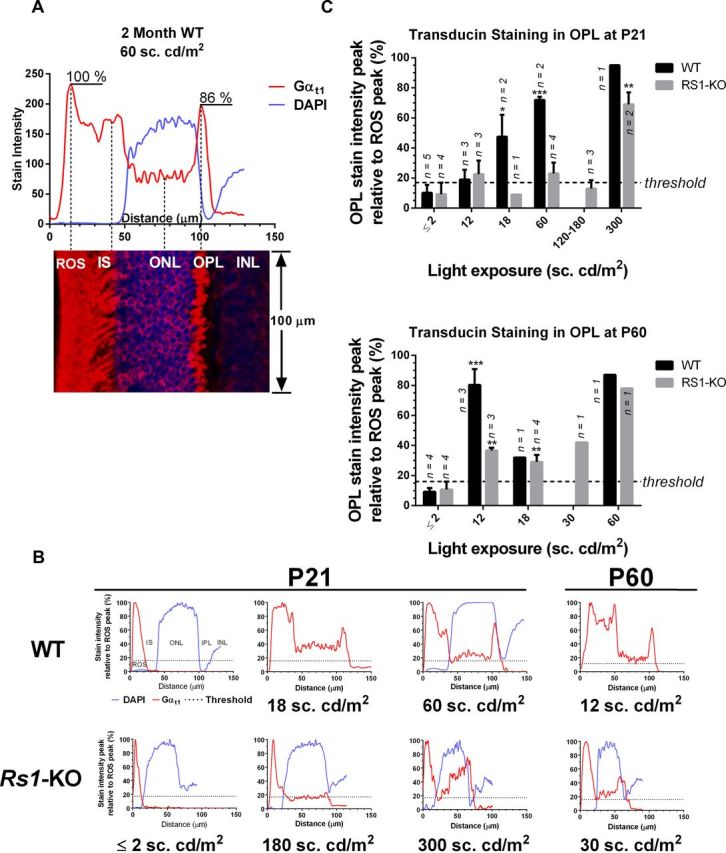
Quantification of transducin translocation by measurement of Gαt1 staining intensity. A, Plot profiles of stain intensity in retinal sections from the ROS tips to the INL in confocal images. Values are the DAPI (blue) and Gαt1 (red) stain intensity on a scale of 0–255 averaged over at least 100 μm of retinal length. DAPI and Gαt1 channels were measured separately, and the merged image is shown below the graph. B, Examples of plot profiles from WT and Rs1–KO mice replotted relative to the maximum ROS intensity in each section for comparison of OPL peak values. The threshold for transducin translocation (dashed line) was the combined average value of the OPL peak in retinas exposed to 0, 1, and 2 sc.cd/m2. The OPL peak is clearly above threshold in P21 WT at 18 and 60 sc.cd/m2, but 300 sc.cd/m2 was required in P21 Rs1–KO mice. At P60, 12 and 30 sc.cd/m2 is enough to elicit OPL peaks above threshold in WT and Rs1–KO mice, respectively. C, The average ± SE OPL peak stain intensity as a percentage of ROS peak stain intensity plotted for WT and Rs1–KO at P21 and P60 as a function of light exposure intensity. WT is significantly above threshold at 18 sc.cd/m2 in P21 mice, but among Rs1–KO mice, only those exposed to 300 sc.cd/m2 are significantly above threshold. Dashed line is OPL threshold as in B. Statistically, the OPL stain intensity in P60 Rs1–KO mice exceeds threshold at the same intensity as WT, 12 sc.cd/m2. *p < 0.05, **p < 0.01, ***p < 0.001, unpaired t test corrected for multiple comparisons using the Holm–Sidak method (GraphPad Prism 6.0 for Windows).
Retinal morphometry.
ONL thickness and ROS length were determined from 100 μm vibratome sections containing the central retina and optic nerve cut from agarose-embedded tissues processed as described above. For each genotype (Rs1–KO and WT) and for each age group (P21 and P60), eight sections were analyzed. Rhodopsin-labeled retinal sections were used to measure ROS length, and ONL thickness was measured in DAPI-stained sections. From each section, five values of ONL thickness and ROS length in five separate locations within 500 μm on each side of the optic nerve were determined and averaged. The product (ROS length × ONL thickness), which reflects the total plasma membrane area of the ROS containing c-GMP-gated channels (Machida et al., 2000), was calculated using the averages from these eight retinal sections. The length of the entire retina was measured in these same sections and compared across age and genotype. Statistical analysis by age and genotype was done using the two-way ANOVA followed by Sidak's multiple comparison test using GraphPad Prism 6.0.
Isolation of ROS and single-turnover GTPase assay.
To maintain the retina in fully dark-adapted state, all procedures were performed under infrared illumination using night-vision goggles. Retinas from dark-adapted mice were collected on ice in 8% OptiPrep (Sigma) in Ringer's buffer, pH 7.4 [in mm: 130 NaCl, 3.6 KCl, 2.4 MgCl2, 1.2 CaCl2,10 HEPES with KOH, and 0.02 EDTA plus Halt protease inhibitor cocktail (Thermo Fisher Scientific)]. The ROS from eight retinas were isolated by step gradient (10 and 18% OptiPrep in Ringer's solution) centrifugation procedure (Tsang et al., 1998). ROS were lysed by hypotonic shock in 90 μl of H2O for 10 s by vortexing and then resuspended in 100 μl of GTPase assay buffer (in mm: 10 Tris, pH 7.8, 100 NaCl, 8 MgCl2, and 1 DTT, plus Halt protease inhibitor cocktail). Single turnover Gαt GTPase assays were performed in triplicate as described previously (Cowan et al., 2000). ROS suspension in the assay buffer was bleached using a 1 ms white photoflash (Vivitar 283; Vivitar) producing ∼1% bleach done on ice shortly before each reaction. The experiment was started by mixing bleached ROS (14 μl) containing 4 μm rhodopsin with 7 μl of 0.04 μm [γ-32P]GTP, and, at predetermined times, 100 μl of 6% perchloric acid was added to the mixture to quench the reaction. The time course of 32Pi (Pi, inorganic phosphate) formation was determined by the activated charcoal assay. The blank sample was quenched by acid before the addition of GTP, and the total activity sample was incubated for 10 min to achieve complete hydrolysis of all added GTP. The rate constants of transducin GTPase activity were determined by exponential fits of the results. The results of a representative experiment from at least three similar experiments are shown.
Western blot.
Retinas or outer segment fragments from dark-adapted mice were lysed in RIPA buffer, pH 7.4 (50 mm Tris-HCl, 1% NP-40, 0.25% sodium deoxycholate, 150 mm NaCl, and 1 mm EGTA) supplemented with Halt protease inhibitor cocktail. Total protein (Pierce BCA protein assay kit; Thermo Fisher Scientific) and the levels of rhodopsin in isolated retinas or ROS were determined as described below. Retinal lysates or ROS extracts containing varying amounts of rhodopsin were run on Criterion TGX Any kD gels (Bio-Rad) and blotted onto PVDF membranes by wet transfer. The membranes were blocked in blocking buffer (LI-COR Biosciences) and later incubated overnight with one of the indicated primary antibody diluted in PBS containing 0.05% Tween 20 (PBST), pH 7.5, at 4°C. The antibodies used included the following: transducin α subunit (Gαt1, rabbit polyclonal, 1:1000); transducin β subunit (Gβ1, rabbit polyclonal, 1:2000); transducin γ subunit (Gγ1, rabbit polyclonal, 1:2000); phosphodiesterase 6γ (PDE6γ, rabbit polyclonal, 1:1000); CRX (rabbit polyclonal, 1:500); G-protein-coupled receptor kinase 1 (mouse monoclonal, 1:500); regulator of G-protein signaling 9 (RGS9, goat polyclonal, 1:400) (all from Santa Cruz Biotechnology); recoverin (rabbit polyclonal, 1:1000; Millipore); visual arrestin (rabbit polyclonal, 1:300; Sigma); phosphodiesterase 6α (PDE6α, rabbit polyclonal, 1:500; Thermo Fisher Scientific); phosducin (sheep polyclonal, 1:2500; from Maxim Sokolov, Department of Ophthalmology, West Virginia University Eye Institute, Center for Neuroscience, Morgantown, WV); Otx1 + Otx2 (rabbit polyclonal, 1:1000; Abcam); and NRL (rabbit polyclonal, 1:2500; from Anand Swaroop, National Eye Institute, Bethesda, MD). After overnight incubation, the membranes were rinsed three times in PBST and incubated with one of the following appropriate IRDye-conjugated secondary antibodies: IRDye 800CW conjugated goat (polyclonal) anti-rabbit IgG or IRDye 800CW conjugated donkey (polyclonal) anti-goat IgG (from LI-COR Biosciences); or Alexa Fluor 680-conjugated goat anti-mouse IgG (Invitrogen). Blots were scanned on LI-COR Odyssey Infrared Imaging System (model 9120, LI-COR Biosciences) and analyzed using Odyssey software. The results shown are from one of three similar experiments that yielded similar results.
ERG recordings.
Full-field scotopic ERGs were recorded from male Rs1–KO mice and male WT littermates at P21 and P60. Recordings were obtained only once from each mouse. Animals were dark adapted overnight and prepared under red dim light. Mice were anesthetized with intraperitoneal administration of ketamine (80 mg/kg) and xylazine (4 mg/kg). Pupils were dilated with topical 0.5% tropicamide and 0.5% phenylephrine HCl. Body temperature was kept at 37°C with a heating pad. ERGs were recorded from both eyes simultaneously, but only left eye traces were used for analysis in the majority of the mice (32 of 36). After application of 1% tetracaine topical corneal anesthesia, gold wire recording loops were placed on the cornea with a drop of methylcellulose for corneal hydration. Gold wires on the sclera of each eye at the limbus were used as reference electrodes, and the ground electrode was attached to the tail. Scotopic ERG responses were elicited using single flashes from a xenon discharge source (Grass Photic Stimulator PS33 Plus; AstroMed) from −4.9 to +0.6 log cd · s/m2 in 0.5-log steps. Responses were computer averaged with stimulus intervals of 3 to 180 s depending on stimulus intensity. Stimuli were presented in a Ganzfeld bowl that provided uniform full-field illumination, and stimulus intensity was attenuated with neutral density Wratten filters (Eastman Kodak). Responses were amplified at 2000 or 5000 gain and filtered using a 0.1 Hz to 1 kHz bandpass.
Studies of the a-wave in mouse (Hetling and Pepperberg, 1999), cat and monkey (Robson and Frishman, 1998), and human (Hood and Birch, 1993) have shown that ERG b-wave and oscillatory potentials can begin to intrude on the rod contribution to the a-wave beginning at post-flash time of 10 ms. Hence, we measured a-wave amplitude at 8 ms after flash onset to assess the photoreceptor contribution. Intensity series measurements were made on nine WT mice per age group, and on 11 and 7 Rs1–KO mice at P21 and P60, respectively.
Whole-retina rhodopsin content, regeneration rate, and photon capture.
All mice were dark adapted for 16 h from 6:00 P.M. to 10:00 A.M., and all pre-bleach and post-bleach procedures were performed under infrared illumination. Mice were killed by CO2 asphyxiation, and retinas were removed by extrusion through a slit in the cornea and frozen at −80°C (Winkler, 1972). This method of retinal isolation was originally developed for recording the rat whole retinal ERG in vitro, including the photoreceptor potential. It has been used successfully for biochemical assays, and especially rhodopsin quantification, in a variety of mouse and rat studies (Penn and Anderson, 1987; Sieving et al., 2001; Wenzel et al., 2001; Haruta et al., 2009; Skeie et al., 2011) and was shown to yield >98% of the total retinal rhodopsin in a mouse model of retinal degeneration (Fulton et al., 1982). Rhodopsin from single retinas was extracted by disruption in 0.5 ml of 1% emulphogene in 50 mm Tris-acetate buffer, pH 6.5, incubation for 1 h at 4°C, and centrifugation at 14,000 × g for 15 min. The supernatant was scanned from 700 to 400 nm using a Lambda 45 UV/Vis spectrophotometer (PerkinElmer Life and Analytical Sciences), bleached in 1200 lux for 10 min, and then scanned again. Rhodopsin quantity per retina was calculated using the difference spectrum maximum absorbance, a 1 cm path length, and an extinction coefficient of 4.2 × 104 mol−1 (Shichi et al., 1969). Statistical analysis of dark-adapted and steady-state bleach rhodopsin content by age and genotype was done by two-way ANOVA, followed by Sidak's multiple comparison test using GraphPad Prism 6.0 for Windows.
To compare the ability of Rs1–KO and WT retinas to activate transducin through photon capture by rhodopsin, we determined the rhodopsin regeneration rate in vivo in Rs1–KO and WT mice in the dark after exposure to steady illumination at an intensity and duration consistent with the transducin translocation experiments. The rate of formation of bleached rhodopsin is the rate of photon capture, and, under steady-state conditions, in which the rate of regeneration is the same as the rate of bleach, the regeneration rate can be used to calculate the rate of photon capture (Penn and Williams, 1986). First-order kinetics have been widely used to describe the regeneration rate of rhodopsin in rats (Tansley, 1931; Dowling, 1963; Penn and Williams, 1986) and mice (Wenzel et al., 2001), although other models have been proposed based on data from humans (Alpern and Krantz, 1981; Lamb and Pugh, 2004), cats (Ripps et al., 1981), and rodents (Lamb and Pugh, 2004). In addition, the kinetics of regeneration may be different under anesthesia (Perlman, 1978; Keller et al., 2001). We used the first-order kinetic model to allow comparison with previous mouse studies. In this model, the regeneration rate of rhodopsin is described by the first-order rate equation r = −d[B]/dt = kr × [B], where r is the regeneration rate, B is bleached rhodopsin, and kr is the regeneration rate constant. A plot of ln[B] is fit by a straight line with a slope equal to −kr. In our experiments, the proportion of rhodopsin in the bleached state at time (t) in the dark after exposure, (R0 − Rt)/R0), where R0 is the average dark-adapted rhodopsin level for the genotype and age of mice being tested, and Rt is the average rhodopsin level measured at time (t), was substituted for [B], and kr was calculated as the slope of the linear regression fit to the values of ln((R0 − Rt)/R0) plotted against time in the dark (Penn and Williams, 1986).
For light exposure, unanesthetized mice were individually housed in clear plastic cages inside a well-ventilated light-tight box and exposed to diffuse white light for 1 h at 60 sc.cd/m2 (45–70 sc.cd/m2 in the horizontal direction) provided by cool white fluorescent bulbs. One hour is sufficient time to reach a steady-state level of rhodopsin, because it was shown previously that the level of bleach in B6/129S(N2) mice exposed to 5,000 lux did not change between 10 min and 1 hour exposure (Wenzel et al., 2001). After exposure, mice were killed by CO2 at various times in the dark, and their retinas removed for rhodopsin measurement. For time 0 (steady-state bleach level), retinas were removed in the dark and frozen within 3 min after the lights were turned off. Rhodopsin in these samples was measured as described above for dark-adapted samples, and plotting and statistical comparison of rate plots was done using GraphPad Prism 6.0 for Windows.
Results
Light-driven transducin translocation
After extended dark adaptation, Gαt1 immunoreactivity was restricted to the ROS of both WT and Rs1–KO retinas. One hour of exposure to 60 sc.cd/m2 triggered transducin translocation from ROS to the IS, ONL, and OPL in WT retina (Fig. 1). The transducin translocation threshold for P60 WT mice was 12 sc.cd/m2 for complete distribution of Gαt1 into the IS and the OPL (Fig. 2), consistent with the previously reported value of 10 sc.cd/m2 in adult C57BL/6 mice (Lobanova et al., 2007). The average OPL/ROS Gαt1 stain intensity signal in mice exposed to 12 sc.cd/m2 was significantly greater than threshold (p < 0.001, n = 3; Fig. 3B,C). P21 WT retinas required higher light intensity: 12 sc.cd/m2 caused no transducin translocation, weak staining of transducin was observed in the OPL at 18 sc.cd/m2, and staining was observed uniformly across the OPL at 30 sc.cd/m2 (Fig. 2). The OPL/ROS Gαt1 stain intensity was significantly above threshold in WT retinas exposed to 18 sc.cd/m2 (p < 0.05, n = 2) but not in those exposed to 12 sc.cd/m2 (n = 3; Fig. 3B,C). Threshold was even higher at P18, because 30 sc.cd/m2 was required to produce detectable staining in the OPL (data not shown) of WT mice.
Figure 1.

The light-dependent distribution of transducin α-subunit (Gαt1) in WT mouse retina. In normal mouse retina, Gαt1 distributes in the ROS layer in the dark and moves from the ROS to the IS, ONL, and OPL after light exposure (60 sc.cd/m2).
Figure 2.
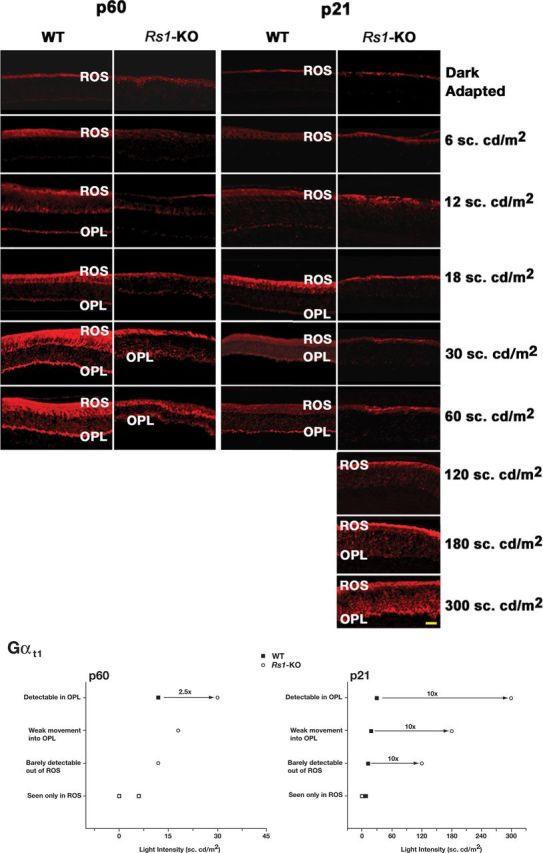
Light-intensity-dependent transducin translocation in WT and Rs1–KO mice at P21 and P60. Animals were either dark adapted or exposed to 1 h of light of different intensities (indicated on the right). Comparison of Gαt1 distribution in WT and Rs1–KO mice retinas shows that, in P21 and P60 WT retinas, Gαt1 translocates from ROS and distributes into OPL at much lower light intensities compared with Rs1–KO retinas. Immunofluorescence of Gαt1 in P21 Rs1–KO retinas shows persistent staining only in the ROS. Only exposure to very bright light (180–300 sc.cd/m2) caused Gαt1 distribution into the OPL. The inset shows the light intensities at which movement of Gαt1 out of the ROS and into the OPL was detected in P21 and P60 Rs1–KO and WT mice. All slides were visualized using the same microscope settings. Scale bar, 20 μm.
Rs1–KO mice required higher light intensity for transducin translocation than age-matched WT mice at both P60 and P21; substantial movement from ROS into the OPL of Rs1–KO retinas at P60 occurred at 30 sc.cd/m2. In stain intensity plot profiles, however, the average Gαt1 signal was significantly greater than threshold at 12 sc.cd/m2 (p < 0.01, n = 3) and 18 sc.cd/m2 (p < 0.01, n = 4), although the amplitude of the signal was much lower than WT. At P28 and P35, however, translocation was incomplete at 60 sc.cd/m2, with a reduced level of Gαt1 staining in an irregular pattern extending into the IS (data not shown). The movement of transducin from ROS into IS was barely detectable at 120 sc.cd/m2 in P21 Rs1–KO mice, but some transducin staining was clearly seen at 180 sc.cd/m2 and was detectable by image scanning (Fig. 3B). However, the combined average signal intensity in retinas exposed to 120–180 sc.cd.m2 (n = 3) was not significantly above threshold (Fig. 3C). Unambiguous detection of transducin in the OPL occurred by 300 sc.cd/m2 at P21 (Fig. 2), and the OPL/ROS intensity ratio was significantly greater than threshold (p < 0.01, n = 2) and similar to WT (Fig. 3C). Overall, transducin translocation in adult P60 Rs1–KO retinas required <2.5-fold higher light intensity compared with P60 WT retinas, but at P21, Gαt1 movement into the OPL was seen in Rs1–KO retinas only at 10-fold higher light intensity compared with P21 WT retinas. Image scanning for stain intensity showed similar results but with slightly lower thresholds for WT at P21 and for Rs1–KO at P60, so that the thresholds were ≥10-fold different at P21 and <2.5-fold different at P60 (Fig. 3C). This was not merely a delay in the response, because exposing for 3 h to 60 sc.cd/m2 also failed to cause transducin translocation in P21 Rs1–KO retinas (data not shown).
Arrestin translocation in light and re-translocation of transducin and arrestin in the dark
Unlike transducin, arrestin staining was not seen in the ROS in the dark in either WT or Rs1–KO (P21, Fig. 4; P60, data not shown). After exposure to 2 sc.cd/m2 for 1 h, arrestin moved into the ROS of both WT and Rs1–KO, although there were subtle differences in protein localization in the dark-adapted retinas and retinas exposed to 1 sc.cd/m2: arrestin was prominent in the ONL and IS of WT retinas, but arrestin immunolabeling of Rs1–KO retinas was mainly in the ONL and not in the IS. At 1 sc.cd/m2, very sparse arrestin staining of the ROS was barely visible in WT after 1 h and not detectable at all in Rs1–KO. Thus, the threshold light intensity for arrestin translocation for both WT and Rs1–KO mice at P21 was between 1 and 2 sc.cd/m2. At 6 sc.cd/m2, arrestin labeling was totally confined to the ROS in both lines. The migration to the ROS in light quenches R* (Burns and Arshavsky, 2005), ultimately serving as a biochemical feedback to limit light-driven rhodopsin activity and transduction.
Figure 4.

Arrestin movement in response to light in P21 WT and Rs1–KO mice. In the dark, arrestin immunostaining was confined to the OPL, ONL, and IS in WT mice. After exposure to light intensity of 1 sc.cd/m2 for 1 h, arrestin was concentrated in the IS compartment, and very sparse staining was detected in the ROS. Light of 2 sc.cd/m2 was able to mobilize arrestin from the IS to the ROS. In Rs1–KO retina, the threshold light intensity for arrestin translocation was the same as WT retina. However, at light intensity below the threshold, arrestin redistribution in the inner retina was confined mainly in the ONL and OPL, and none was detected in the IS of Rs1–KO mice. Scale bar, 15 μm.
The extinction of light activates the dark recovery phase that leads to the re-translocation of arrestin and transducin back into their respective compartments: transducin moves from the IS to the ROS, whereas arrestin re-translocates from the ROS to the IS (Sokolov et al., 2002). Because transducin in P21 Rs1–KO retinas translocated at a higher light intensity (300 sc.cd/m2) than P21 WT retinas (Fig. 2), we verified that transducin would move back into ROS in the dark after exposure to this intensity. In WT and Rs1–KO mice kept in the dark for 4 h, the majority of transducin had moved from the OPL, ONL, and IS to the ROS layer (Fig. 5A,B). Re-translocation of arrestin in the dark from ROS to IS also occurred normally in P21 WT and Rs1–KO retinas after preexposure to 6 sc.cd/m2 (Fig. 5C,D). These results demonstrate that the loss of RS1 protein did not impair the axial diffusion of transducin and arrestin between the photoreceptor compartments.
Figure 5.

Re-translocation of Gαt1 (rows A and B) and arrestin (rows C and D) in P21 WT and Rs1–KO mice retinas. Mice were dark adapted overnight (12 h), exposed to light (for 1 h), and then placed in the dark (for 4 h). Gαt1 partially re-translocated into the ROS when mice were replaced in the dark for 4 h after exposure to 30 sc.cd/m2 (WT, row A) or 300 sc.cd/m2 (Rs1–KO, row B) sufficient to cause translocation from the ROS into the OPL. Arrestin moved from the IS in dark to the ROS in the light (6 sc.cd/m2) and then back into OPL in the dark in WT (row C) and Rs1–KO (row D) retinas. As in Figure 4, arrestin was not detected in the IS of Rs1–KO retina after 12 h dark adaptation. Scale bar, 22 μm.
Photoreceptor morphology and rhodopsin content
Transducin translocation threshold in WT retinas showed a gradual reduction between P18 and P60, and there was a large elevation in threshold in P21 Rs1–KO mice, followed by rapid decline to near WT levels by P60. This suggested a delay in maturation of photoreceptor function, and we wondered whether there was a similar delay in maturation of photoreceptor morphology. Our previous study indicated shorter ROS compared with WT at 4 weeks, increasing to WT length by 4 months of age (Kjellstrom et al., 2007). In the P21 Rs1–KO retina, ONL thickness was 76% of WT at P21 (p < 0.0001) and 82% of WT at P60 (p < 0.05). Rs1–KO ROS length was 68% of WT at P21 (p < 0.01) but was not significantly different from WT at P60 (Fig. 6). The ROS length in Rs1–KO increased significantly by 53% between P21 and P60 (p < 0.05). WT ROS length increased 28%, but the increase was not significant. Neither age nor genotype had a significant effect on length of the retina in sections from P21 and P60 mice (n = 4–8 per group).
Figure 6.

Age-related changes in photoreceptor morphology in WT and Rs1–KO mice. Top, Compared with WT retinas, ONL width was thinner in Rs1–KO retina at both P21 (p < 0.001) and P60 (p < 0.05). A significant (p < 0.05) thinning of the ONL occurred in WT mice from P21 to P60, which was not seen in Rs1–KO retinas. Bottom, ROS in Rs1–KO were shorter compared with WT at P21 (p < 0.05), but their length increased significantly between P21 and P60 (p < 0.05) when Rs1–KO and WT were not significantly different. Error bars indicate mean ± SE. n = 8 mice for all histological measurements. *p < 0.05, ***p < 0.001, two-way ANOVA and Sidak's multiple comparison test (GraphPad Prism 6.0 for Windows).
Whole retina rhodopsin levels were significantly lower in P21 Rs1–KO than in P21 WT retinas (p < 0.0001; Table 1). They increased 70% by P60 in Rs1–KO (p < 0.0001) and 51% in WT (p < 0.0001). Although rhodopsin levels were still lower than WT at P60 (p < 0.0001), like ROS length, the increase between P21 and P60 tended to be greater in Rs1–KO than in WT. Changes in transducin translocation threshold, ROS length, and rhodopsin during this period suggest a delay in the maturation of Rs1–KO photoreceptors.
Table 1.
Rhodopsin content and photon capture
| P21 |
P60 |
|||
|---|---|---|---|---|
| WT | Rs1–KO | WT | Rs1–KO | |
| Molecules × 1014 | ||||
| Whole retina | ||||
| Dark adapted (R0) | 2.35 ± 0.17 (n = 6) | 0.98 ± 0.16** (n = 6) | 3.55 ± 0.10 (n = 8) | 1.67 ± 0.09** (n = 8) |
| Unbleached steady state (Rss) | 1.08 ± 0.32 (n = 6) | 0.82 ± 0.20 (n = 4) | 1.51 ± 0.66 (n = 12) | 0.92 ± 0.22* (n = 6) |
| Bleached steady state (Bss = R0 − Rss) | 1.27 | 0.16 | 2.04 | 0.75 |
| Regeneration rate constant (kr)/min | 0.012 ± 0.001 (n = 12) | 0.048 ± 0.020* (n = 11) | 0.008 ± 0.001 (n = 19) | 0.011 ± 0.004 (n = 21) |
| Photons/s/retina ((kr/60) × Bss) × 1010 | 2.54 | 1.28 | 2.72 | 1.38 |
| Molecules × 106/μm ROS | ||||
| Whole retina/(ROS × ONL) | ||||
| Dark adapted (R0) | 1.25 | 1.00 | 1.69 | 1.18 |
| Bleached steady state (Bss) | 0.67 | 0.15 | 0.97 | 0.53 |
| Photons/s/μm ROS ((kr/60) × Bss) × 102 | 1.35 | 1.20 | 1.29 | 0.98 |
Measured values = mean ± SD. R0, Rhodopsin in mice after 16 h dark adaptation; Rss, rhodopsin present after exposure of mice to 1 h of 60 sc.cd/m2; Bss, rhodopsin bleached after exposure of mice to 1 h of 60 sc.cd/m2, calculated from (average R0 − individual mouse Rss); kr, regeneration rate constant of rhodopsin in the dark [same in whole retina and whole retina/(ROS × ONL), calculated from the slope of the linear regression fit to ln((R0 − Rt)/R0), where Rt, rhodopsin at time (t minutes) in the dark after exposure to 1 h of 60 sc.cd/m2]; ROS, average rod outer segment length (micrometers) from Figure 6; ONL, estimated number of photoreceptor cells (average ONL width/average ONL width of P60 WT) * 6.4 × 106 cells in adult WT C57BL/6 (Jeon et al., 1998); Whole retina/(ROS/ONL), whole retina rhodopsin values normalized by cell number and ROS length.
*p < 0.05;
**p < 0.0001, significantly different from WT (two-way ANOVA, Bonferroni's multiple comparisons).
Although whole retinal rhodopsin levels in P21 Rs1–KO retinas were only ∼40% of WT, an estimate of rhodopsin density in ROS [whole retina rhodopsin/(ROS length × ONL cell number)] indicated that it was 80% of WT and did not change substantially between P21 and P60 (Table 1).
Expression levels of photoreceptor-specific transcriptional factors in Rs1–KO retinas
In mice, functional maturation of rod photoreceptors continues during the postnatal period for 2–4 weeks after the final mitosis and is attributed to the consecutive waves of gene expression patterns of NRL, CRX, nR2E3, and Otx2 that eventually lead to structurally and functionally mature rod photoreceptors and continue to be expressed at high levels in the mature retina (Levine et al., 2000; Swaroop et al., 2010). Thus, we wondered whether differences in the time course of changes in transducin translocation threshold and ROS length reflect a delay in rod maturation controlled by transcription factors. In quantitative immunoblot analysis of retinal extracts from Rs1–KO and WT retinas, we observed that the expression of Nrl and CRX was downregulated by 50 and 25%, respectively, in Rs1–KO retinas at P21, but their expressions levels were comparable with WT at P60 (Fig. 7). These observations demonstrate that the regulation of the rod-specific transcriptional network controlling the expression of transduction and other ROS proteins is altered during maturation in the retinas of Rs1–KO mice. We also assessed the expression of Otx2, a homeobox gene product essential not only for rod photoreceptor cell fate determination but also in the morphological and electrophysiological maturation of bipolar cells (Koike et al., 2007). We found that the expression of Otx2 was downregulated by 30% at P21 and 40% at P60 in Rs1–KO retinas (Fig. 7).
Figure 7.

Expression levels of photoreceptor-specific transcription factors. Retinal protein extracts (∼10 μg of total protein) from WT and Rs1–KO mice at ages P21 and P60 were Western blotted and probed for each of the transcription factor indicated on the left with specific antibodies, followed by anti-tubulin antibody. Expression levels were quantified using Odyssey software and normalized to α-tubulin. At P21, all of these transcription factor levels were lower (by 25–50%) in Rs1–KO mice. However, at P60, both NRL and CRX levels were comparable with that collected from WT retinas. Replicate gels were run for each antibody tested.
Transduction protein expression and GTPase activity
The alteration in transcription factor levels at P21 but not P60 in Rs1–KO mice support the idea that the time course of photoreceptor maturation is altered. Changes in protein expression related to the maturational delay in the rod-specific transcriptional network could alter processes involved in phototransduction and explain the effects on the transducin translocation threshold. The key factor determining transducin translocation threshold is the local concentration of activated Gαt1 generated by phototransduction after rhodopsin bleaching and its inactivation by GTP hydrolysis (Lobanova et al., 2007). We explored whether the quantity of activated transducin during rhodopsin bleaching is altered in Rs1–KO retinas as a result of the availability of transducin or the rate of inactivation by measuring the dark-adapted levels of transducin and RGS9 and the rate of GTP hydrolysis after light exposure. Western blot analysis of retinal extracts normalized by rhodopsin (5 pmol) showed marginal differences in expression profiles of photo transduction proteins between WT and Rs1–KO mice retinas (results not shown). However, quantitative immunoblot analysis of extracts of purified outer segments revealed that transducin levels were consistently decreased relative to rhodopsin by 15–30% in Rs1–KO mice at P21 (Fig. 8A,C). Whereas PDEα and PDEγ were slightly elevated relative to rhodopsin in Rs1–KO photoreceptors, RGS9, the GTPase-accelerating protein (GAP) for Gαt1, was elevated in Rs1–KO photoreceptors 1.7- to 2.5-fold above WT (Fig. 8B,D). These differences between WT and Rs1–KO retinas were not observed at P60 (data not shown). Consistent with this observation, we found that the rate of GTP hydrolysis by transducin in ROS of Rs1–KO mice was nearly twofold higher than that in WT retinas at P21 but indistinguishable from WT at P60 (Fig. 8E,F). The increased inactivation rate in Rs1–KO mice at P21 results in a shorter lifetime of activated transducin, which could shift the light intensity threshold for transducin translocation to higher intensity by reducing the amount of activated transducin present during exposure.
Figure 8.

Quantitative immunoblot analyses (Odyssey imaging system; LI-COR) of key phototransduction protein subunits, transducin α (Gαt1), RGS9, PDE6α, andPDE6γ, in dark-adapted outer segment extracts from P21 WT and Rs1–KO mice (A–D). Transducin α levels relative to rhodopsin were 15–30% lower in Rs1–KO mice than in WT. RGS9, the GAP for Gαt, was 1.7- to 2.5-fold higher in Rs1–KO than in WT. Both PDE6α and PDE6γ protein levels were marginally elevated in Rs1–KO retinas. Replicate gels were run for each antibody tested. The time course of phosphate formation during hydrolysis of [γ-32P]GTP by Gt* in ROS of WT and Rs1–KO mice at P21 (E) and P60 (F). In single-turnover GTPase activity measurements in isolated ROS, GTP hydrolysis by transducin was nearly twofold higher in Rs1–KO than in WT at P21, resulting in a shorter lifetime of activated transducin in Rs1–KO ROS. The rates were not different at P60. The data were fitted to a single-phase exponential decay curve using GraphPad Prism (GraphPad Software).
The ERG and phototransduction
To assess the in vivo light activation of transducin, we used the dark-adapted ERG a-wave, which is attributable to the entire rod transduction cascade leading to changes in the rod circulating dark current (Penn and Hagins, 1972; Baylor et al., 1984; Breton et al., 1994; Pugh et al., 1998). At a stimulus intensity of +0.6 log cd · s/m2, the mean ERG a-wave maximum amplitude was reduced significantly in Rs1–KO mice at both ages compared with WT (P21, 63%, p < 0.0001; P60, 50%, p = 0.0006; unpaired t test). However, when mean ERG a-wave amplitude values at 8 ms for Rs1–KO and WT mice at P21 and P60 were normalized by the corresponding mean dark-adapted whole retina rhodopsin content of each group, the V-log I response curves plotted for intensities up to +0.6 log cd · s/m2 showed nearly complete overlap (Fig. 9). In addition, the plot in Figure 9 (inset) shows that the maximum a-wave mean amplitude at 8 ms recorded from WT and Rs1–KO mice is linearly proportional to (ROS length × ONL width) as we described previously for a quite different model of photoreceptor degeneration, the P23H rhodopsin rat (Machida et al., 2000). These findings confirm that the phototransduction process in Rs1–KO outer segments remains intact even with major morphological changes, implying that the elevation in transducin translocation threshold is not attributable to a deficiency in transducin activation.
Figure 9.

Intensity–response series of scotopic ERG a-wave amplitudes in P21 and P60 WT and Rs1–KO mice measured at 8 ms after the flash. Mean ± SE amplitudes (microvolts) were normalized by the average dark-adapted whole retina rhodopsin content for each category of mice. The response curves for all groups of mice were essentially superimposed up to the maximum stimulus intensity of +0.6 log cd · s/m2. A linear correlation between the mean a-wave amplitude at 8 ms (+0.6 log cd · s/m2) and the product (ROS × ONL) calculated from the ONL widths and ROS lengths graphed in Figure 6 is displayed in the inset.
Rhodopsin bleaching, regeneration, and photon capture
Transducin translocation threshold was monitored under conditions of constant illumination in which the activation of transducin depends on the continual bleach and recovery of rhodopsin. Although whole retinal rhodopsin levels in P21 Rs1–KO retinas were only ∼40% of WT, the local concentration of rhodopsin in ROS, calculated by normalizing by ROS length and an estimate of ONL cell number, was 80% of WT (Table 1). Calculation of the rate of rhodopsin turnover from the level of bleach and the regeneration rate constant measured at steady state under constant illumination indicated that photon capture was only slightly reduced in P21 Rs1–KO mice, nor was there a substantial change with age. This analysis was done under the steady-state illumination, as were the transducin translocation experiments, and indicate that Rs1–KO photoreceptors do not have a defect in rhodopsin kinetics that would lead to substantially reduced levels of activated transducin.
Discussion
RS1 implicated in timely rod maturation
The progressive decrease in luminance threshold for transducin translocation in WT mice from P18 to P21 to P60 indicates that changes in the sensitivity of transducin translocation are part of normal rod maturation. Three observations in this study suggest that the loss of Rs1 expression causes a delay in this maturation process: (1) there was an exaggerated, 10-fold, elevation of threshold in Rs1–KO mice at P21, which decreased to <2.5-fold above WT by P60; (2) there was a significant increase in ROS length in Rs1–KO mice after P21, when length normally has already reached adult levels in WT mice (LaVail, 1973) and rats (Fulton et al., 1995), going from 68% of WT (p < 0.05) to not significantly different from WT at P60; and (3) Rs1–KO mice had reduced levels of the transcription factors NRL and CRX at P21 but not P60. NRL and CRX regulate the expression of photoreceptor transduction proteins and are cooperatively involved in photoreceptor development but continue to have high expression levels in mature retina and help maintain rod homeostasis (Swaroop et al., 2010). This would suggest that delayed maturation of transducin translocation threshold in Rs1–KO mouse photoreceptors is a functional consequence of perturbations in the rod photoreceptor development specific gene regulatory network proteins and possibly directly mediated by delayed maturation in phototransduction processes.
RS1 loss alters maturation-dependent photo transduction processes
The delayed development of threshold sensitivity of transducin translocation in Rs1–KO mice was linked to phototransduction by alterations in transducin and RGS9 levels and GTPase hydrolysis rate at P21, which were not seen at P60. We found a 15–30% reduction in transducin level relative to rhodopsin and a nearly twofold higher GTPase hydrolysis rate, which was consistent with the 1.7- to 2.5-fold higher RGS9 level. The critical factor affecting the threshold for transducin translocation is the level of Gαt1 produced by the activation/inactivation cycle during light exposure (Lobanova et al., 2007). Therefore, the decreased transducin and shorter lifetime of activated transducin in Rs1–KO mice at P21 could account for ∼60% of the tenfold elevation in transducin translocation threshold. Because these differences were found only at P21 and not at P60, we can conclude they had a major contribution to the shift in translocation threshold to higher light intensity in young Rs1–KO mice and its reduction with age. Thus, the elevated threshold in P21 Rs1–KO mice is a consequence of a delay in maturation of rod phototransduction processes and an indication of the consequences of the functional differences in phototransduction between immature and mature rods. From this perspective, our calculation of the degree of threshold elevation explained by GTPase activity and transducin levels has to be considered a very rough estimate, because the meaning of these changes in immature photoreceptors may be very different from more mature WT rods.
There may also have been modifications to elements of translocation other than those involved directly in phototransduction. Transducin translocation is thought to occur by diffusion in an energy-independent manner and not by active transport. We know from the ERG a-wave normalized by rhodopsin level that the phototransduction response, which involves diffusion controlled interactions with other transduction proteins within the disc (Calvert et al., 2001), was not impaired. However, factors that affect the axial diffusion and/or interdisc transfer of Gαt1 GTP (Kerov and Artemyev, 2011) would be more likely to play a role in transducin translocation. Also, although protein movement between inner and outer segment through the connecting cilium was apparently not restricted because re-translocation was not affected, movement within the proximal cell body and binding to targets in these compartments could be altered.
Arrestin versus transducin translocation
The pathology in the Rs1–KO retina alters the threshold for light-activated transducin translocation out of the ROS but has no effect on arrestin translocation into the ROS in the light. To our knowledge, this is the first time that translocation of these two proteins has been investigated simultaneously in an animal model of photoreceptor pathology, and it shows that pathology can affect these mechanisms in a separable manner. This is consistent with independent pathways for at least the activation of transducin and arrestin translocation. Photoactivation of rhodopsin is requisite for both, but the level of activation required and downstream signaling mechanisms leading to activation, as well as binding partners that facilitate diffusion, are probably different (Slepak and Hurley, 2008; Elsaesser et al., 2010; Orisme et al., 2010; Satoh et al., 2010; Kerov and Artemyev, 2011). Transducin translocation is triggered at intensity levels beyond rod saturation and occurs by diffusion that depends on the concentration gradient of free Gαt1, which in turn is set by the rate of Gαt1 activation and inactivation. In comparison, very low levels of bleach near threshold (1.5–3.0%), (Strissel et al., 2006) trigger a disproportionately large amount of arrestin translocation, leading to the idea that transduction-mediated signaling is required for triggering arrestin movement to ROS. This may include accessory signaling pathways, such as G-protein-linked phospholipase C (Orisme et al., 2010; Satoh et al., 2010). Extrapolating from our 54% rhodopsin bleach in WT mice exposed for 1 h at 60 sc.cd/m2, the proportion of bleached rhodopsin at 2 sc.cd/m2 would be ∼1.8%, which agrees well with Strissel et al. (2006). However, others showed that arrestin translocation depends on the rate of accumulation of bleached rhodopsin that can bind arrestin (Nair et al., 2005; Slepak and Hurley, 2008) and propose that arrestin movement is controlled by the availability of binding partners in the ROS (bleached rhodopsin) and the cell body (e.g., microtubules) (Slepak and Hurley, 2008). Our results appear not to be consistent with this hypothesis, because Rs1–KO photoreceptors had a much lower density of bleached rhodopsin in the ROS than WT under the same level of continuous illumination (Table 1), yet both had the same arrestin translocation threshold. Conversely, signaling via the phototransduction cascade as measured by the ERG a-wave whether normalized by rhodopsin or (ROS × ONL) (Fig. 9) was the same in Rs1–KO and WT retinas. The rate of bleached rhodopsin generation at steady state was similar in both genotypes as a result of differences in the rhodopsin regeneration rate constant, and this may be the critical factor determining the availability of binding sites for arrestin movement into the ROS, not the amount of bleached rhodopsin per se. When comparing mice that have similar rhodopsin kinetics, bleach level is probably an accurate way of evaluating the stoichiometry between photons and arrestin translocation.
Transcription factors, cell fate, and the Rs1–KO inner retinal phenotype
Our results demonstrate that loss of Rs1 gene function has a significant impact on the expression of photoreceptor transcription factor network genes, and morphological and functional defects in young (P21) Rs1–KO mice are consistent with changes in transduction proteins regulated by these genes. Although these and other transcription factors also help determine photoreceptor cell fate, commitment to cell fate occurs before the onset of Rs1 expression [P7 (Takada et al., 2004)] and does not appear to be altered in Rs1–KO mice. The reduction in Otx2 expression, a homeobox gene product essential for rod photoreceptor cell fate determination and also for morphological and electrophysiological maturation of bipolar cells (Koike et al., 2007), at P21 and P60 is consistent with long-term deficiency in OPL synaptic proteins (Takada et al., 2006), which is accompanied by a relatively larger decrease in the bipolar cell ERG b-wave than a-wave in the Rs1–KO mouse (Kjellstrom et al., 2007).
Footnotes
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute on Deafness and Other Communication Disorders, and the National Eye Institute. We thank Robert N. Fariss and Maria M. Campos for technical assistance with confocal imaging and immunohistochemistry and Maria C. Santos for assistance with animal breeding and experiments.
References
- Alpern M, Krantz DH. Visual pigment kinetics in abnormalities of the uvea-retinal epithelium interface in man. Invest Ophthalmol Vis Sci. 1981;20:183–203. [PubMed] [Google Scholar]
- Baylor DA, Nunn BJ, Schnapf JL. The photocurrent, noise and spectral sensitivity of rods of the monkey Macaca fascicularis. J Physiol. 1984;357:575–607. doi: 10.1113/jphysiol.1984.sp015518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton ME, Schueller AW, Lamb TD, Pugh EN., Jr Analysis of ERG a-wave amplification and kinetics in terms of the G-protein cascade of phototransduction. Invest Ophthalmol Vis Sci. 1994;35:295–309. [PubMed] [Google Scholar]
- Burns ME, Arshavsky VY. Beyond counting photons: trials and trends in vertebrate visual transduction. Neuron. 2005;48:387–401. doi: 10.1016/j.neuron.2005.10.014. [DOI] [PubMed] [Google Scholar]
- Calvert PD, Govardovskii VI, Krasnoperova N, Anderson RE, Lem J, Makino CL. Membrane protein diffusion sets the speed of rod phototransduction. Nature. 2001;411:90–94. doi: 10.1038/35075083. [DOI] [PubMed] [Google Scholar]
- Calvert PD, Strissel KJ, Schiesser WE, Pugh EN, Jr, Arshavsky VY. Light-driven translocation of signaling proteins in vertebrate photoreceptors. Trends Cell Biol. 2006;16:560–568. doi: 10.1016/j.tcb.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Cowan CW, Wensel TG, Arshavsky VY. Enzymology of GTPase acceleration in phototransduction. Methods Enzymol. 2000;315:524–538. doi: 10.1016/s0076-6879(00)15865-3. [DOI] [PubMed] [Google Scholar]
- Dowling JE. Neural and photochemical mechanisms of visual adaptation in the rat. J Gen Physiol. 1963;46:1287–1301. doi: 10.1085/jgp.46.6.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsaesser R, Kalra D, Li R, Montell C. Light-induced translocation of Drosophila visual Arrestin2 depends on Rac2. Proc Natl Acad Sci U S A. 2010;107:4740–4745. doi: 10.1073/pnas.0906386107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton AB, Manning KA, Baker BN, Schukar SE, Bailey CJ. Dark-adapted sensitivity, rhodopsin content, and background adaptation in pcd/pcd mice. Invest Ophthalmol Vis Sci. 1982;22:386–393. [PubMed] [Google Scholar]
- Fulton AB, Hansen RM, Findl O. The development of the rod photoresponse from dark-adapted rats. Invest Ophthalmol Vis Sci. 1995;36:1038–1045. [PubMed] [Google Scholar]
- Haruta M, Bush RA, Kjellstrom S, Vijayasarathy C, Zeng Y, Le YZ, Sieving PA. Depleting Rac1 in mouse rod photoreceptors protects them from photo-oxidative stress without affecting their structure or function. Proc Natl Acad Sci U S A. 2009;106:9397–9402. doi: 10.1073/pnas.0808940106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetling JR, Pepperberg DR. Sensitivity and kinetics of mouse rod flash responses determined in vivo from paired-flash electroretinograms. J Physiol. 1999;516:593–609. doi: 10.1111/j.1469-7793.1999.0593v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood DC, Birch DG. Light adaptation of human rod receptors: the leading edge of the human a-wave and models of rod receptor activity. Vis Res. 1993;33:1605–1618. doi: 10.1016/0042-6989(93)90027-t. [DOI] [PubMed] [Google Scholar]
- Jeon CJ, Strettoi E, Masland RH. The major cell populations of the mouse retina. J Neurosci. 1998;18:8936–8946. doi: 10.1523/JNEUROSCI.18-21-08936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C, Grimm C, Wenzel A, Hafezi F, Rem é C. Protective effect of halothane anesthesia on retinal light damage: inhibition of metabolic rhodopsin regeneration. Invest Ophthalmol Vis Sci. 2001;42:476–480. [PubMed] [Google Scholar]
- Kerov V, Artemyev NO. Diffusion and light-dependent compartmentalization of transducin. Mol Cell Neurosci. 2011;46:340–346. doi: 10.1016/j.mcn.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerov V, Chen D, Moussaif M, Chen YJ, Chen CK, Artemyev NO. Transducin activation state controls its light-dependent translocation in rod photoreceptors. J Biol Chem. 2005;280:41069–41076. doi: 10.1074/jbc.M508849200. [DOI] [PubMed] [Google Scholar]
- Kjellstrom S, Bush RA, Zeng Y, Takada Y, Sieving PA. Retinoschisin gene therapy and natural history in the Rs1h-KO mouse: long-term rescue from retinal degeneration. Invest Ophthalmol Vis Sci. 2007;48:3837–3845. doi: 10.1167/iovs.07-0203. [DOI] [PubMed] [Google Scholar]
- Koike C, Nishida A, Ueno S, Saito H, Sanuki R, Sato S, Furukawa A, Aizawa S, Matsuo I, Suzuki N, Kondo M, Furukawa T. Functional roles of Otx2 transcription factor in postnatal mouse retinal development. Mol Cell Biol. 2007;27:8318–8329. doi: 10.1128/MCB.01209-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb TD, Pugh EN., Jr Dark adaptation and the retinoid cycle of vision. Prog Retin Eye Res. 2004;23:307–380. doi: 10.1016/j.preteyeres.2004.03.001. [DOI] [PubMed] [Google Scholar]
- LaVail MM. Kinetics of rod outer segment renewal in the developing mouse retina. J Cell Biol. 1973;58:650–661. doi: 10.1083/jcb.58.3.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine EM, Fuhrmann S, Reh TA. Soluble factors and the development of rod photoreceptors. Cell Mol Life Sci. 2000;57:224–234. doi: 10.1007/PL00000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanova ES, Finkelstein S, Song H, Tsang SH, Chen CK, Sokolov M, Skiba NP, Arshavsky VY. Transducin translocation in rods is triggered by saturation of the GTPase-activating complex. J Neurosci. 2007;27:1151–1160. doi: 10.1523/JNEUROSCI.5010-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida S, Kondo M, Jamison JA, Khan NW, Kononen LT, Sugawara T, Bush RA, Sieving PA. P23H rhodopsin transgenic rat: correlation of retinal function with histopathology. Invest Ophthalmol Vis Sci. 2000;41:3200–3209. [PubMed] [Google Scholar]
- Manschot WA. Pathology of hereditary juvenile retinoschisis. Arch Ophthalmol. 1972;88:131–138. doi: 10.1001/archopht.1972.01000030133002. [DOI] [PubMed] [Google Scholar]
- Mendez A, Lem J, Simon M, Chen J. Light-dependent translocation of arrestin in the absence of rhodopsin phosphorylation and transducin signaling. J Neurosci. 2003;23:3124–3129. doi: 10.1523/JNEUROSCI.23-08-03124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molday RS, Kellner U, Weber BH. X-linked juvenile retinoschisis: clinical diagnosis, genetic analysis, and molecular mechanisms. Prog Retin Eye Res. 2012;31:195–212. doi: 10.1016/j.preteyeres.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair KS, Hanson SM, Mendez A, Gurevich EV, Kennedy MJ, Shestopalov VI, Vishnivetskiy SA, Chen J, Hurley JB, Gurevich VV, Slepak VZ. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron. 2005;46:555–567. doi: 10.1016/j.neuron.2005.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orisme W, Li J, Goldmann T, Bolch S, Wolfrum U, Smith WC. Light-dependent translocation of arrestin in rod photoreceptors is signaled through a phospholipase C cascade and requires ATP. Cell Signal. 2010;22:447–456. doi: 10.1016/j.cellsig.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn JS, Anderson RE. Effect of light history on rod outer-segment membrane composition in the rat. Exp Eye Res. 1987;44:767–778. doi: 10.1016/s0014-4835(87)80040-4. [DOI] [PubMed] [Google Scholar]
- Penn JS, Williams TP. Photostasis: regulation of daily photon-catch by rat retinas in response to various cyclic illuminances. Exp Eye Res. 1986;43:915–928. doi: 10.1016/0014-4835(86)90070-9. [DOI] [PubMed] [Google Scholar]
- Penn RD, Hagins WA. Kinetics of the photocurrent of retinal rods. Biophys J. 1972;12:1073–1094. doi: 10.1016/S0006-3495(72)86145-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman I. Kinetics of bleaching and regeneration of rhodopsin in abnormal (RCS) and normal albino rats in vivo. J Physiol. 1978;278:141–159. doi: 10.1113/jphysiol.1978.sp012297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh EN, Falsini B, Lyubarsky AL. The origin of the major rod- and cone-driven components of the rodent electroretinogram, and the effect of age and light-rearing history on the magnitude of these components. In: Williams TP, Thistle AB, editors. Photostasis and related topics. New York: Plenum; 1998. pp. 93–128. [Google Scholar]
- Ripps H, Mehaffey L, 3rd, Siegel IM. Rhodopsin kinetics in the cat retina. J Gen Physiol. 1981;77:317–334. doi: 10.1085/jgp.77.3.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson JG, Frishman LJ. Dissecting the dark-adapted electroretinogram. Doc Ophthalmol. 1998;95:187–215. doi: 10.1023/a:1001891904176. [DOI] [PubMed] [Google Scholar]
- Satoh AK, Xia H, Yan L, Liu CH, Hardie RC, Ready DF. Arrestin translocation is stoichiometric to rhodopsin isomerization and accelerated by phototransduction in Drosophila photoreceptors. Neuron. 2010;67:997–1008. doi: 10.1016/j.neuron.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer CG, Gehrig A, Warneke-Wittstock R, Marquardt A, Ewing CC, Gibson A, Lorenz B, Jurklies B, Weber BH. Positional cloning of the gene associated with X-linked juvenile retinoschisis. Nat Genet. 1997;17:164–170. doi: 10.1038/ng1097-164. [DOI] [PubMed] [Google Scholar]
- Shichi H, Lewis MS, Irreverre F, Stone AL. Biochemistry of visual pigments. I. Purification and properties of bovine rhodopsin. J Biol Chem. 1969;244:529–536. [PubMed] [Google Scholar]
- Sieving PA, Chaudhry P, Kondo M, Provenzano M, Wu D, Carlson TJ, Bush RA, Thompson DA. Inhibition of the visual cycle in vivo by 13-cis retinoic acid protects from light damage and provides a mechanism for night blindness in isotretinoin therapy. Proc Natl Acad Sci U S A. 2001;98:1835–1840. doi: 10.1073/pnas.041606498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeie JM, Tsang SH, Mahajan VB. Evisceration of mouse vitreous and retina for proteomic analyses. J Vis Exp. 2011;pii:2795. doi: 10.3791/2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slepak VZ, Hurley JB. Mechanism of light-induced translocation of arrestin and transducin in photoreceptors: interaction-restricted diffusion. IUBMB Life. 2008;60:2–9. doi: 10.1002/iub.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov M, Lyubarsky AL, Strissel KJ, Savchenko AB, Govardovskii VI, Pugh EN, Jr, Arshavsky VY. Massive light-driven translocation of transducin between the two major compartments of rod cells: a novel mechanism of light adaptation. Neuron. 2002;34:95–106. doi: 10.1016/s0896-6273(02)00636-0. [DOI] [PubMed] [Google Scholar]
- Strissel KJ, Sokolov M, Trieu LH, Arshavsky VY. Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. J Neurosci. 2006;26:1146–1153. doi: 10.1523/JNEUROSCI.4289-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaroop A, Kim D, Forrest D. Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat Rev Neurosci. 2010;11:563–576. doi: 10.1038/nrn2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada Y, Fariss RN, Tanikawa A, Zeng Y, Carper D, Bush R, Sieving PA. A retinal neuronal developmental wave of retinoschisin expression begins in ganglion cells during layer formation. Invest Ophthalmol Vis Sci. 2004;45:3302–3312. doi: 10.1167/iovs.04-0156. [DOI] [PubMed] [Google Scholar]
- Takada Y, Fariss RN, Muller M, Bush RA, Rushing EJ, Sieving PA. Retinoschisin expression and localization in rodent and human pineal and consequences of mouse RS1 gene knockout. Mol Vis. 2006;12:1108–1116. [PubMed] [Google Scholar]
- Tansley K. The regeneration of visual purple: its relation to dark adaptation and night blindness. J Physiol. 1931;71:442–458. doi: 10.1113/jphysiol.1931.sp002749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang SH, Burns ME, Calvert PD, Gouras P, Baylor DA, Goff SP, Arshavsky VY. Role for the target enzyme in deactivation of photoreceptor G protein in vivo. Science. 1998;282:117–121. doi: 10.1126/science.282.5386.117. [DOI] [PubMed] [Google Scholar]
- Wenzel A, Reme CE, Williams TP, Hafezi F, Grimm C. The Rpe65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration. J Neurosci. 2001;21:53–58. doi: 10.1523/JNEUROSCI.21-01-00053.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler BS. The electroretinogram of the isolated rat retina. Vis Res. 1972;12:1183–1198. doi: 10.1016/0042-6989(72)90106-x. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Takada Y, Kjellstrom S, Hiriyanna K, Tanikawa A, Wawrousek E, Smaoui N, Caruso R, Bush RA, Sieving PA. RS-1 gene delivery to an adult Rs1h knockout mouse model restores ERG b-wave with reversal of the electronegative waveform of X-linked retinoschisis. Invest Ophthalmol Vis Sci. 2004;45:3279–3285. doi: 10.1167/iovs.04-0576. [DOI] [PubMed] [Google Scholar]
- Zhang H, Huang W, Zhang H, Zhu X, Craft CM, Baehr W, Chen CK. Light-dependent redistribution of visual arrestins and transducin subunits in mice with defective phototransduction. Mol Vis. 2003;9:231–237. [PubMed] [Google Scholar]