

Abstract
During the fusion of the influenza virus to the host cell, bending of the HA2 chain of hemagglutinin into a hairpin-shaped structure in a pH-dependent manner facilitates the fusion of the viral envelope and the endosomal membrane. To characterize the structural and dynamical responses of the hinge region of HA2 to pH changes and examine the role of a conserved histidine in this region (the hinge histidine), we have performed an extensive set of molecular dynamics (MD) simulations of 26-residue peptides encompassing the hinge regions of several hemagglutinin subtypes under both neutral and low pH conditions, modeled by the change of the protonation state of the hinge histidine. More than 70 sets of MD simulations (collectively amounting to 25.1 μs) were performed in both implicit and explicit solvents to study the effect of histidine protonation on structural dynamics of the hinge region. In both explicit and implicit solvent simulations, hinge bending was consistently observed upon the protonation of the histidine in all the simulations starting with an initial straight helical conformation, whereas the systems with a neutral histidine retained their primarily straight conformation throughout the simulations. Conversely, the MD simulations starting from an initially bent conformation resulted in the formation of a straight helical structure upon the neutralization of the hinge histidine, whereas the bent structure was maintained when the hinge histidine remained protonated. Finally, mutation of the hinge histidine to alanine abolishes the bending response of the peptide altogether. A molecular mechanism based on the interaction of the hinge histidine with neighboring acidic residues is proposed to be responsible for its role in controlling the conformation of the hinge. We propose that this might present a common mechanism for pH-controlled structural changes in helical structures when histidines act as the pH sensor.
Introduction
Influenza viruses, as well as several other enveloped viruses, require low pH conditions to transfer their genome to the host cell, because the fusion of the viral envelope with the host cell membrane is largely catalyzed by acidic conditions (1). The virion will be endocytosed after docking to the host cell, and the low pH of the endosome initiates the process of membrane fusion (1), which is induced by structural changes in hemagglutinin (HA), a major glycoprotein on the surface of the viral envelope (2,3).
The x-ray crystal structures of the ectodomain of HA (bromelain-digested HA, BHA) characterized native HA as a homotrimer extending 135 Å in length (4–6). It is thought that every monomer must first be cleaved into two subunits, HA1 and HA2 with 328 and 221 residues, respectively, to transform into a functional unit (7,8). HA1 forms the globular head and includes the receptor-binding site (9). HA2 includes the fusion peptide, a 20–30 residue segment that makes the fusion-promoting subunits of fusion proteins, and is responsible for direct binding to the host cell membrane (10,11). HA has two important tasks:
-
1.
Recognizing and docking to the proper host cell, a task mediated by HA1; and
-
2.
Membrane fusion that is facilitated primarily by structural changes in HA2 (12,13).
Several crystal structures of HA obtained under different conditions have provided hints on some of the conformational changes involved in the fusogenic activity of the protein (Fig. 1). The trimeric HA structure at neutral pH consists of a central helical stem formed by the HA2 chains from the three monomers that are bent at position 63–72, and are covered by the three HA1 chains (14). HA1 chains detach from each other during the early response to acidic conditions. The subsequent low pH conformational changes of HA2 consist of two stages:
-
1.
Formation of a needle-shaped structure and exposure of the N-terminal fusion peptide for insertion into the endosomal membrane whereas HA2 remains connected to the viral envelope from the C-terminus (1,15,16) (B2 in Fig. 1); and
-
2.
Rebending of HA2 at a different hinge region (17,18) that consists of residues 106–111 (the focus of this study), which turns HA2 into a hairpin-shaped structure that brings the viral envelope close to the endosomal membrane (19–21) (B3 in Fig. 1).
The HA1 subunit, and the transmembrane and cytosolic domains of HA2, are not required for membrane fusion (22). A bent conformation for HA, also referred to as the “fusogenic structure” (15), was crystallized from thermolysin-digested BHA under low pH conditions. Several other structural studies have also reported a bent conformation for the region between residues 106 and 111 for the fusogenic state of the protein (21,23–25). Whereas these structures have established the nature of major conformational changes involved in the activation of HA, the mechanism by which acidification of the environment results in such structural changes remains elusive.
Figure 1.
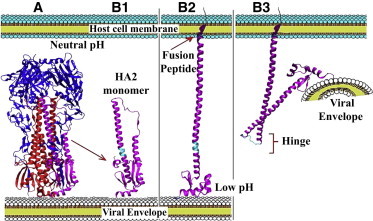
HA overall structure and functionally relevant conformational transitions. (A) Complete homotrimeric structure of HA including HA1 (blue) and HA2 (red; one HA2 chain highlighted in magenta). (B1–B3) A model for key structural changes in HA2 during the fusion process. (Cyan) Hinge region. (B1) HA2 monomer at neutral pH, with the fusion peptide locked in the HA2 core. (B2) Early low pH response including the release of the fusion peptide and its linking to the host cell membrane. (B3) Late low pH response, the bending of HA2 at the hinge (cyan), thereby placing the viral envelope very close to the host cell membrane.
Different subtypes of HA show variations in their primary sequences. The newest HA antigen subtype from fruit bats in 2012 (26) brings the total number of influenza virus HA subtypes to 17. These subtypes can be divided into two distinct groups based on the location of a conserved histidine in the HA2 hinge region (27,28), which is the focus of our study. Based on our results, we refer to this as the “hinge histidine” throughout the article. In the first group (H1, H2, H5, H6, H8, H9, H11, H12, H13, H16, and H17 subtypes), the conserved histidine is at position 111, whereas in the second group (H3, H4, H7, H10, H14, and H15 subtypes), this conserved histidine is at position 106 (25) (Table 1).
Table 1.
Sequences of the hinge peptide shown for representatives from each subtype (H1–H17), highlighting the conserved position of the histidine in the His111 and His106 groups of HA subtypes
| Residue | 95 | 96 | 97 | 98 | 99 | 100 | 101 | 102 | 103 | 104 | 105 | 106 | 107 | 108 | 109 | 110 | 111 | 112 | 113 | 114 | 115 | 116 | 117 | 118 | 119 | 120 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| His111 | H1 | N | A | E | L | L | V | L | L | E | N | E | R | T | L | D | F | H | D | S | N | V | R | N | L | Y | E |
| H2 | N | A | E | L | L | V | L | M | E | N | E | R | T | L | D | F | H | D | S | N | V | K | N | L | Y | D | |
| H5 | N | V | E | L | L | V | L | M | E | N | E | R | T | L | D | F | H | D | S | N | V | N | N | L | Y | D | |
| H6 | N | A | E | L | L | V | L | L | E | N | E | R | T | L | D | L | H | D | A | N | V | K | N | L | Y | E | |
| H8 | N | A | E | L | L | V | L | L | E | N | E | R | T | L | D | L | H | D | A | N | V | K | N | L | Y | E | |
| H9 | N | A | E | L | L | V | L | L | E | N | Q | K | T | L | D | E | H | D | A | N | V | N | N | L | Y | N | |
| H11 | N | A | Q | L | L | V | L | L | E | N | E | K | T | L | D | L | H | D | S | N | V | R | N | L | H | E | |
| H12 | N | A | E | L | L | V | L | L | E | N | Q | K | T | L | D | E | H | D | A | N | V | R | N | L | H | D | |
| H13 | N | A | K | L | L | V | L | L | E | N | D | K | T | L | D | M | H | D | A | N | V | K | N | L | H | E | |
| H16 | N | A | K | L | L | V | L | L | E | N | D | R | T | L | D | L | H | D | A | N | V | R | N | L | H | E | |
| H17 | N | T | E | L | L | V | L | L | E | N | E | R | T | L | D | F | H | D | A | N | V | K | N | L | F | E | |
| His106 | H3a | N | A | E | L | L | V | A | L | E | N | Q | H | T | I | D | L | A | D | S | E | M | N | K | L | F | E |
| H4 | N | A | E | L | L | V | A | L | E | N | Q | H | T | I | D | V | T | D | S | E | M | N | K | L | F | E | |
| H7 | N | A | E | L | L | V | A | M | E | N | Q | H | T | I | D | L | A | D | S | E | M | N | R | L | Y | E | |
| H10 | N | A | E | L | L | V | A | M | E | N | Q | H | T | I | D | M | A | D | S | E | M | L | N | L | Y | E | |
| H14 | N | A | E | L | L | V | A | L | E | N | Q | H | T | I | D | V | T | D | S | E | M | N | K | L | F | E | |
| H15 | N | A | E | L | L | V | A | M | E | N | Q | H | T | I | D | L | A | D | S | E | M | N | K | L | Y | E |
The hinge region is highlighted. The subtypes simulated in this study and the hinge histidines are indicated in boldface.
For H3 subtype, both human and avian variants, denoted, respectively, as H3a and H3b in the text, have been simulated. The sequence shown in the table is for H3a, with the sequence of H3b being different only at position 111, where Thr replaces Ala.
According to the crystal structures, two major conformations exist for the HA2 hinge region: straight and bent. The straight conformation is primarily an α-helical structure (A and B1 and B2 in Fig. 1), whereas, under low pH conditions, the hinge region is found to form a bent structure (B3 in Fig. 1). The trigger pH for the conformational changes has been reported to be between 4.8 (29) and 6 (30). Some studies have determined that even higher pH ranges (5.8–6.4) might be sufficient to initiate the structural changes (31,32). It has been shown experimentally that the pH required for half-maximal activity of lipid mixing with erythrocyte ghosts, a process mediated by hemagglutinin on the surface of the red blood cells, was 5.5 (33). The pH required for half-maximal activity of the HA protein from the influenza virus X-31 was determined to be 5.2 (34).
Histidine is the only amino acid with a pKa value close to the pH of acid-mediated membrane fusion of HA. The imidazole ring of histidine has a pKa of ∼6.0, and overall, the amino acid has a pKa of 6.5 in water (35). Therefore, low-pH-mediated conformational changes of proteins within physiologically relevant conditions can be effectively triggered by the protonation of one or more histidine residues in their structures (36–38). The involvement of histidines in HA activation was hypothesized in 2006 by Kampmann et al. (37), who proposed a central role for histidines in initiating the structural transition of proteins mediating the viral membrane fusion (the histidine-switch hypothesis). Evidence in support of the histidine-switch hypothesis has been provided for several groups of viruses that enter the host cell in a pH-dependent manner (35,36,38–48).
Despite accumulating evidence pointing at potential involvement of histidine side chains in functionally relevant conformational changes of HA, such a structural role has not been studied directly. Furthermore, and more importantly, no mechanism at a molecular level has been offered as to how modulation of the protonation state of histidines might be coupled to conformational changes in the influenza virus HA. To address these questions, this study was designed to investigate the role of a conserved histidine in the hinge region of HA2.
Molecular dynamics (MD) simulations have been previously used to study the HA molecule, both for the full-length form (6,21,37) and the individual segments (e.g., for the fusion peptide (49)). In this study, we focus on the pH-dependent conformational changes that might be mediated specifically by the HA2 hinge region. MD simulations were performed on the hinge region of HA2 (represented by a short peptide consisting of the hinge region and 10 flanking residues on each side). The simulations cover several different subtypes of HA, whose structural dynamics have been studied using different starting conformations (straight or bent) and at different protonation states of the hinge histidine. The results clearly demonstrate the effective role of histidine protonation in triggering the low-pH-mediated conformational changes of HA2. We provide further support for the role of the hinge histidine as a pH sensor by repeating the simulations in a designed mutant where the hinge histidine is mutated to alanine. A detailed mechanistic picture emerges from the simulations characterizing how histidine protonation can be effectively coupled to structural changes of HA, which we propose to be also at work in general for helical elements in proteins.
Materials and Methods
Simulations systems
There are 17 known subtypes of HA from influenza virus A, differing in their amino-acid sequences. The sequences of the hinge region of all HA subtypes are compared in Table 1. HA subtypes can be divided into two main groups according to the position of a conserved histidine in the HA2 hinge region (25), which we refer to as the hinge histidine: the first group with the hinge histidine at position 106 and the second with the histidine at position 111 (37,39) (Table 1), hereby referred to as the His106 and His111 groups, respectively. Two subtypes from each group have been selected and simulated in this study, H1 and H5 from the His111 group, and human and avian H3 from the His106 group (denoted as H3a and H3b, respectively, in Table 2). For each subtype a 26-aa peptide encompassing the hinge region (six residues) flanked by an arm of 10 residues on each side (collectively referred to as the “hinge peptide” in this study) was used for the simulations.
Table 2.
The summary of the simulations reported in this study
| Models | Hemagglutinin subtype | PDB code | Simulation repeats | Collective length of simulations | Histidine position | Acidity | Starting conformation | Final conformation | Solvation system |
|---|---|---|---|---|---|---|---|---|---|
| M1 | H1 | 1RUZ | 10 | 4.5 μs | 111 | Neutral | Straight | Straight | Implicit |
| Acidic | Straight | Bent | Implicit | ||||||
| M2 | H5 | 2FK0 | 10 | 3.4 μs | 111 | Neutral | Straight | Straight | Implicit |
| Acidic | Straight | Bent | Implicit | ||||||
| M3 | H3b | 1MQM | 10 | 3.2 μs | 106 | Neutral | Straight | Straight | Implicit |
| Acidic | Straight | Bent | Implicit | ||||||
| M4 | H3a | 1HGF | 10 | 3.1 μs | 106 | Neutral | Straight | Straight | Implicit |
| Acidic | Straight | Bent | Implicit | ||||||
| M5 | H3a | 1HTM | 10 | 3.1 μs | 106 | Neutral | Bent | Straight | Implicit |
| Acidic | Bent | Bent | Implicit | ||||||
| M6 | H3b | 1QU1 | 10 | 3.2 μs | 106 | Neutral | Bent | Straight | Implicit |
| Acidic | Bent | Bent | Implicit | ||||||
| M7 | H3a | 1HGF | 4 | 1.2 μs | 106 | Neutral | Straight | Straight | Explicit |
| Acidic | Straight | Bent | Explicit | ||||||
| M8 | H1 | 1RUZ | 4 | 1.3 μs | 111 | Neutral | Straight | Straight | Explicit |
| Acidic | Straight | Bent | Explicit | ||||||
| M9 | H1 | 1RUZ | 5 | 1.2 μs | H111A | N/A | Straight | Straight | Implicit |
| M10 | H3a | 1HTM | 3 | 0.9 μs | H106A | N/A | Bent | Straight | Implicit |
Several crystallographic structures have been solved for influenza HA and deposited in the Protein Data Bank (50). These can be structurally divided into two distinct groups: those capturing the hinge region in a straight helical conformation, and those in which the hinge region is bent. The initial straight structures used here are: 1RUZ for H1 (Model 1, M1) (51,52); 2FK0 for H5 (Model 2, M2) (53); 1MQM for H3b (Model 3, M3) (54); and 1HGF for H3a (Model 4, M4) (9). There are two crystallographic bent structures for HA solved under acidic conditions, both for the H3 subtype from the His106 group, namely, 1HTM (15) for H3a and 1QU1 (20) for H3b. The 26-residue hinge peptides for both bent structures were included in the simulations here as Models 5 (M5) and 6 (M6), respectively.
The first phase of the study consists of simulations of models with an initially straight helical conformation (M1–M4 in Table 2). The second phase includes simulations of the models with an initially bent conformation (M5 and M6 in Table 2). The above-described systems were first simulated using an implicit solvent model, as discussed below. To test the sensitivity of the results to the choice of the solvent model, one subtype from each of the His106 and His111 groups was also simulated using explicit solvent simulations (M7 and M8, using the same initial structures as in M4 and M1, respectively). In addition, to confirm the role of the hinge histidine in controlling of the hinge region, the hinge histidine was mutated to an alanine and additional simulations performed. The M9 model is a mutant hinge peptide with an initially straight structure of the H1 subtype (same structure as in the M1 model), and the M10 model is the mutant hinge peptide with an initially bent structure of the H3a subtype (same structure as in the M5 model). The summary of all the simulation systems, along with the conditions and major outcome of each simulation, is provided in Table 2.
To mimic the effect of low pH, the hinge histidine was doubly protonated and positively charged (38). This model is based on the predicted pKa value of the hinge histidine (in the full-length molecule) to be ∼5.7 for H1 subtype (from the His111 group) and 5.2 for H3a subtype (from the His106 group). KARLSBERG+ (55,56) and H++ (57,58) web servers were used for the pKa prediction and gave similar results (with a difference of only 0.2–0.4 in pKa). In our model, the imidazole ring in the histidine side chain carries a proton only on Nε (HSE) at neutral pH, whereas an excess proton is added to Nδ (HSP) in low pH simulations. Each of the peptide models was constructed with either neutral (HSE) or protonated (HSP) histidines, emulating the neutral and acidic environments, respectively. The residue identifier in the HA2 chain of the H1 subtype (PDB:1RUZ) starts from 501. To simplify the comparison to the other models, the residue numbering for the hinge peptide of H1 was shifted from 595–620 to 95–120.
Simulation protocols
MD simulations were performed using NAMD 2.8 (59) with the CHARMM27 force field (60,61). The time step was set to 1 fs. For each system, the protocol included energy minimization, thermalization, equilibration, and production steps. The systems were energy-minimized for 2000 steps using the conjugate gradient algorithm (62). The number of energy minimization steps was verified to be sufficient based on the calculation of mean velocity and total energy converging during the first 2 ns of the following MD simulations (63). The systems were thermalized over a period of 310 ps by means of temperature reassignment (increasing the temperature by 1 K every 1 ps), followed by a 1-ns equilibration at 310 K (64). During the equilibration and production phases of the simulations, temperature was maintained at 310 K using a Langevin thermostat with a damping coefficient of 1/ps (65). Atomic coordinates were stored for analysis every 1 ps. Simulations were performed using two solvent models, implicit and explicit, as described below.
For the implicit solvent simulations, we used the generalized Born implicit solvent model (66,67) with a cutoff of 30 Å for nonbonded interactions. Five sets of simulations for each neutral/protonated (histidine) system were performed, each lasting for at least 300 ns. Some of the simulations were extended to up to 1 μs to ensure that the behavior captured within the first 300 ns did not change over longer timescales (see Table 2 for details).
For the explicit solvent simulations one model from each of the His106 and His111 groups (M7 and M8, respectively) was tested. Four sets of independent MD simulations were performed for each model. These simulations were performed each for 300 ns during which the structural change (or stability) was monitored. In the case of the simulations in which a conformational change took place too close to the 300-ns time point, we further extended the simulation time to ensure the stability of the final conformation. The TIP3 model (68,69) was used for water. Long-range electrostatic interactions were computed with the particle-mesh Ewald method (70). A Langevin piston was used to maintain the pressure at 1 atm. The peptide models were initially positioned at the center of a water box with at least 10 Å padding on each side. The systems were neutralized with the proper number of Na+ ions. The system size was ∼11,000 atoms.
Analysis
The software VMD (71) was used to visualize and analyze the simulation trajectories. Root-mean-square deviation (RMSD) of the backbone heavy atoms, radius of gyration (Rg) of the peptide, and secondary structure probability of individual residues in all models were calculated and compared. The STRIDE package (72) was used to determine the secondary structure of individual residues. The angle between the two arms of the hinge peptide was used to characterize the hinge bending (named as the bending angle, θ, in this study; see Fig. 2). The Cα atoms of the second last residue from each end of the peptide (positions 96 and 119, respectively) was used as the beginnings of the arm vectors, and the backbone nitrogen (N) atoms of the sixth residue from each end were selected as the end point of the vectors (positions 100 and 115, respectively).
Figure 2.
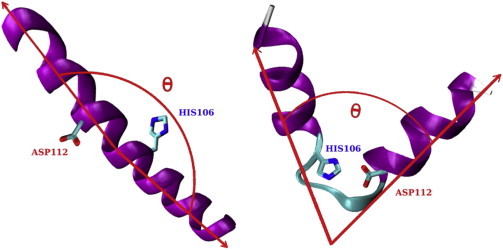
Hinge peptide model used for the simulations reported in this study in straight (left) and bent (right) conformations. The example is taken from the His106 group. The bending angle (θ) is defined between two helical arms of the hinge peptide. The hinge histidine is shown explicitly. (Magenta) Helical and (cyan) turn regions.
To better establish the molecular mechanism underlying the observed bending upon the protonation of the hinge histidine, the interactions between the residues within the peptide sequence were calculated and compared, in terms of interaction energies and hydrogen-bond probabilities, for backbone and side-chain atoms. Interresidual hydrogen-bond probabilities and interaction energies were calculated for representative trajectories. For each residue pair, probabilities for two classes of hydrogen bonds were calculated:
-
1.
Hydrogen bonds between the side-chain atoms (side-chain to side-chain), and
-
2.
Hydrogen bonds between the backbone atoms (backbone-to-backbone).
A maximum distance of 3.5 Å between the donor (D) to acceptor (A) heavy atoms, and a D–H–A angle that deviates <30° from linearity, were used to define hydrogen bonds. The probabilities were calculated for select systems representing the His106 and His111 groups and in both neutral and protonated hinge histidine cases. These included Model M1 from the His111 group and Model M4 from the His106 group (first and fourth columns in Fig. 3), and using one trajectory from the protonated set and one from the neutral set for each model. Interresidual interaction energies were calculated for each pair of residues (summing over the interaction energies between any atom from one residue and any atom from the other residue) without considering the solvent/ion and entropic effects. The last 10 ns of the trajectories described above (for the hydrogen-bond probabilities) were used for the interaction energy estimations.
Figure 3.
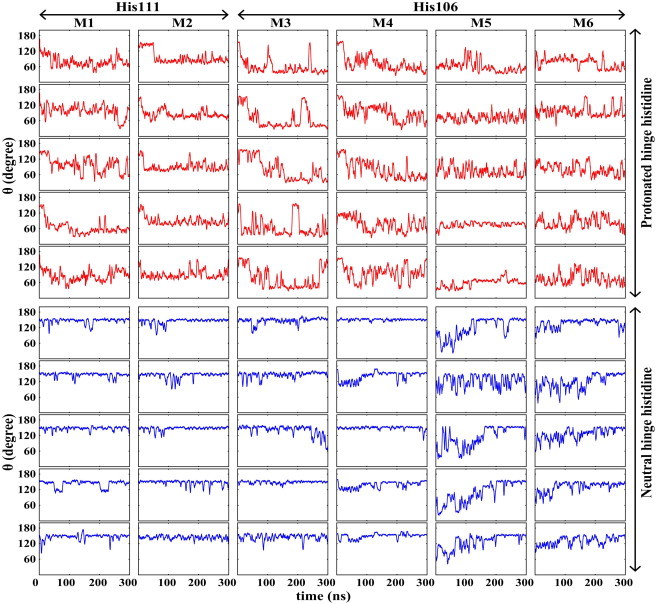
Time series of the bending angle along 10 independent simulations of Models M1–M6. The top five rows (red) represent peptides with a protonated histidine and the bottom five rows (blue) represent peptides with a neutral hinge histidine, over the first 300 ns of the trajectories of each model. (Columns 1 and 2) M1 and M2 systems are from the His111 group, whereas M3–M6 systems represent systems from the His106 group. A running-average window of 3 ns has been used.
Results and Discussion
The results presented below demonstrate the key role of the hinge histidine protonation in pH-dependent conformational changes of HA2 based on a set of MD simulations collectively amounting to 25.1 μs. The results will be presented in the following four sections:
1. The bending of initially straight peptides in response to the protonation of the hinge histidine;
2. Straightening of initially bent peptides when the hinge histidine was neutralized;
3. The effect of substitution of the hinge histidine with a neutral residue; and
4. A mechanistic analysis of the effect of histidine protonation on the hinge region.
Bending of helical peptides at low pH
All implicit solvent MD simulations starting with an initially straight helical structure show consistent hinge bending in models with a protonated hinge histidine, whereas the straight conformation is largely retained in models with a neutral hinge histidine. In the following, we will describe the behavior of the models from the His111 and His106 groups separately:
His111 group
In the His111 group simulations (M1 and M2 models), the hinge region starts to bend within the first 10 ns of the simulation after the protonation of the hinge histidine. These two models reach maximal bending after ∼25 and 20 ns, respectively. M1 models are found to be more bent (θ < 60°) in comparison to the M2 models (θ < 100°), but the latter models exhibited fewer fluctuations in the bending angle (between 40 and 90° in the M1 models, and between 60 and 110° in the M2 models; see Fig. 3). The degree of conformational changes of the peptides at low pH is also evident from RMSD and Rg analyses. The RMSD calculation shows major changes from the initial conformation, increasing to ∼10.0 and 7.0 Å in M1 and M2 models, respectively, after 10 ns (see Fig. S1 in the Supporting Material). The drop in Rg indicates a decrease in the molecular dimensions; Rg dropped from 13.0 Å in the beginning of the simulation (after the initial 1 ns of equilibration) to ∼10.0 (see Fig. S2) in both M1 and M2 models.
In contrast to low pH simulations, no significant bending was observed in the simulations of the initially straight peptides with a neutral hinge histidine (HSE111), and the structure remained linear during the simulations (average θ ≈ 150°, Fig. 3). The RMSD and Rg values stayed relatively constant throughout the simulations at ∼3.5 and 13.1 Å, respectively (see Fig. S1 and Fig. S2).
Explicit solvent MD simulations were performed for one of the His111 subtypes, namely the M8 model with either a neutral or protonated hinge histidine (each simulation was repeated four times). The M8 model peptide uses the same initial straight structure as in the M1 model. The results are consistent with the larger data set obtained from the implicit solvent simulations described above. The M8 models with a protonated hinge histidine reach a bent structure in ∼190–225 ns and remain bent throughout the rest of the simulations, which were extended to 400 ns (M8 in Fig. 4), thereby confirming the stability of the bent structure in the presence of a protonated histidine (θ < 90°). In contrast, no significant bending was observed for the peptide with a neutral hinge histidine after 570 ns of explicit solvent simulations (M8 in Fig. 4).
Figure 4.
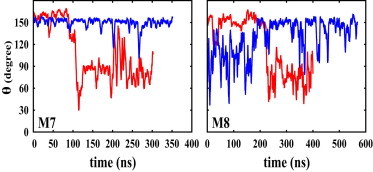
Time series of the bending angle (θ) from the explicit solvent simulations with a protonated/neutral (red/blue) hinge histidine. A running-average window of 3 ns has been used.
His106 group
Despite the different position of the hinge histidine in this group, the implicit solvent simulations of the peptides were from the His106 group (M3 and M4). All models starting from a straight conformation showed bending after 25–30 ns when the hinge histidine was modeled as a protonated, charged species (low pH). The hinge, however, was found to be more flexible in these models, compared to the His111 group, with the bending angle θ exhibiting a range of fluctuation between 40 and 170°. The RMSD reaches a value of ∼11.0 Å in M3 models and ∼7.3 Å in M4 models with a dramatic increase coinciding the bending of the peptide. Rg was also decreased in all models from an initial value of ∼13.0 Å (after the initial 1 ns of equilibration) to 9–10 Å in 50–60 ns and remained constant until the end of the simulations.
In contrast to the low pH simulations, there was no significant change in the bending angle during the simulations of the initially straight hinge peptides with a neutral hinge histidine. The peptide retains its near straight helical conformation with an average θ of 150° (M3 and M4 in Fig. 3). The RMSD remained relatively low (3.75 Å) and the Rg was stable at ∼13 Å throughout the simulations, indicating the stability of the straight conformation at neutral pH.
Explicit solvent simulations were performed for at least 300 ns for the M7 model that uses the same initial straight helical structure as in the M4 model under either neutral or low pH conditions with each simulation repeated 4 times. Peptide bending started after 85–115 ns upon the protonation of the hinge histidine, whereas no significant bending was observed with a neutral hinge histidine in at least 300 ns.
The conformational response of the hinge peptide in implicit and explicit solvent simulations is very similar; the peptide adopts a bent conformation when the hinge histidine is protonated, as characterized by the bending angle θ, and the secondary structure of the hinge residues indicating a turn conformation. The bending in explicit water simulations is naturally associated with a larger lag (∼10× larger than that of the implicit solvent simulations), due to the slower relaxation in the presence of explicit water molecules.
Straightening of the bent peptide at neutral pH
In this phase of the study, the reversibility of the bending induced by the protonation of the hinge histidine was tested. The simulations described in this section were initiated from an initially bent conformation of the hinge peptide and the conformational response to the protonation state of the hinge histidine examined. The simulations consistently either resulted in straightening of the peptide upon neutralization of the hinge histidine, or preserved its bent conformation when the hinge histidine was protonated. As mentioned in Materials and Methods, two crystal structures of bent HA2 models obtained under acidic conditions (M5 and M6 models) were used for this part of the study.
The starting conformation in both models was bent with a turn secondary structure assigned to residues 106–111 (the residue 106 is the hinge histidine). A large fluctuation in the bending angle, θ, was observed during the first 150–200 ns of both M5 and M6 simulations with a protonated hinge histidine that eventually converged to θ = 62 ± 9° in both models after 200 ns (Fig. 3). The observed stability of the bent conformation was confirmed by the RMSD and Rg calculations, which resulted in values close to those obtained from low pH simulations in other models, namely, at ∼7 and 11 Å, respectively (see Fig. S1 and Fig. S2). Secondary structure analysis also indicates a mostly turn state for the hinge residues.
Conversely, in the models with a neutral hinge histidine, the hinge peptide adopted a straight helical conformation after 150–170 ns (Fig. 3). In M5 models, the bending angle was increased to a stable value (average θ ≈ 150°) after 120–150 ns. Retuning to the straight conformation resulted in RMSD increase to ∼10–11 Å and Rg increase to ∼13 Å in both M5 and M6 models, with the latter comparable to other models with straight helical conformations.
The results show that the process of hinge bending is reversible upon neutralization of the hinge histidine. Kampmann et al. (37) also proposed that conformational changes in fusion proteins induced by histidine protonation might be reversible. The changes of the bending angle during all 10 sets of the simulations for models M1–M6 (five independent simulations for each protonation state of each model) are compared in Fig. 3, in which the M1–M4 simulations were initiated from a straight conformation, whereas M5 and M6 were initiated from a bent conformation.
Mutation of the hinge histidine
To further verify that the above described pH-controlled conformational changes are related to the histidine protonation, the hinge histidine in the M1 and M5 models was mutated to an alanine, and several independent simulations were performed on the mutants. Specifically, the two mutated cases studied here included the M9 model with an initially straight conformation, and the M10 model with an initially bent conformation. In contrast to the protonated wild-type simulations, no bending was observed for the M9 mutant in 300-ns production runs. The peptide largely maintained its straight helical conformation, with the bending angle indicating a close to linear conformation (θ > 155°). The RMSD was ∼2.9 Å, which is even lower than the wild-type peptide with a neutral hinge histidine (M9 in Fig. 5). In the M10 simulations, the peptide changed from an initially bent structure to a straight helix in 28–40 ns and remained stable for the remainder of the simulations (M10 in Fig. 5). The phenotype observed experimentally for the H106A/T111A double mutant of HA2 is consistent with our observations (43); other than a delay in structural changes, no fusion activity was observed for this mutant.
Figure 5.
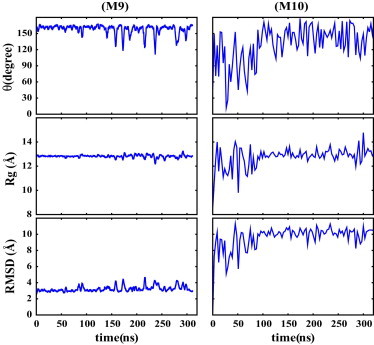
The bending angle (θ), Rg, and RMSD of the mutant models with the hinge histidine substituted with an alanine, starting from straight (M9) and bent (M10) conformations.
Mechanistic analysis
Several experimental and computational studies have proposed a role for histidine residues in controlling the conformational changes of the proteins involved in pH-dependent membrane fusion of viruses (73,74) as well as in nonviral proteins (45,75–77). Using NMR measurements, Sancho et al. (35) reported the role of histidine protonation in secondary structural changes by replacing a single histidine in bacterial ribonuclease A (Barnase). Mueller et al. (39) performed 80 ns of MD simulations and observed conformational changes in a flaviviral (dengue virus type 2) fusion protein (E protein) in response to systematic protonation of all 22 histidine residues present in the protein, whereas no changes were observed in the model with neutral histidines after 60 ns of simulation. Prakash et al. (78) reported a switching role for histidine protonation at positions 144 and 317 in E protein of the dengue virus by a series of short MD simulations (20 ns). Chen et al. (36) experimentally mutated His17 in the HA1 cleavage site and suggested a role for histidine protonation in acid-induced structural transitions of HA. Later, the histidines at positions 18 and 37 in HA1 and at positions 106, 111, and 147 in HA2 have been suggested to cooperatively regulate the structural transitions of HA (37).
The main goal of this study was to determine whether a conserved histidine in the hinge region of HA2 could act as the pH sensor to detect the acidic environment of the endosome and initiate the proper conformational response at HA. We demonstrated this potential by neutralizing the hinge histidine in the models with an initially bent conformation and observing the relaxation of the hinge region into a straight helical conformation. The observed phenomenon is indicative of the role of a neutral histidine in conserving the helical structure of the hinge at physiological pH, possibly by making standard n → n + 4 helical hydrogen bonds. The behavior of the simulated mutant peptide with the hinge histidine substituted by an alanine also confirms that, in the absence of a charged side chain at this position, the standard helical backbone hydrogen bonds are dominant and stabilize the helical structure.
Although there are two major different HA groups according to the position of the hinge histidine, in both groups this residue is proximal to several negatively charged residues within the sequence, e.g., E103, E105, D109, and D112 in M1 (His111), and E103, D109, and D112 in M4 (His106). The protonation of the hinge histidine promotes its attractive interaction with these residues, as reflected by considerably stronger interresidual interaction energies presented in Fig. 6), which result in similar bending behaviors in both His106 and His111 groups.
Figure 6.
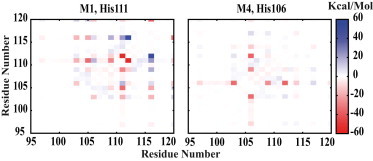
The change in the interresidual interaction energies (in kcal/mol) accompanying the bending of the peptide induced by the protonation of the hinge histidine for M1 (left) and M4 (right) models. The change is calculated by subtracting the interaction energies for the protonated hinge histidine model from those for the neutral hinge histidine model (see Materials and Methods). (Red/blue) Stronger/weaker interactions between a residue pair.
Analysis of hydrogen-bond patterns better explains this behavior. Upon the protonation of the hinge histidine, in both His106 and His111 groups, the standard helical (backbone) hydrogen bonds were found to be broken between most residues around the hinge histidine (top panels in Fig. 7), whereas new contacts form between the side chains of the peptide, particularly between the side chains of the charged residues with opposite charges (bottom panels in Fig. 7). In the M4 (His106) model, the protonated histidine itself forms contacts with the neighboring acidic residues, including E103, D109, D112, and E114. In the M1 (His111) model, several contacts form between the positively charged residues H111, R106, and R116 and the negatively charged residues such as E103, D109, and D112. Whereas the protonation of the hinge histidine is the key event controlling the overall structure of the peptide, upon the formation of a bent structure the reduced distance between the two helical arms allows for the formation of additional salt bridges that further stabilize a bent configuration. For instance, E103 and R116, which form a relatively strong contact, can do so only in a bent peptide.
Figure 7.
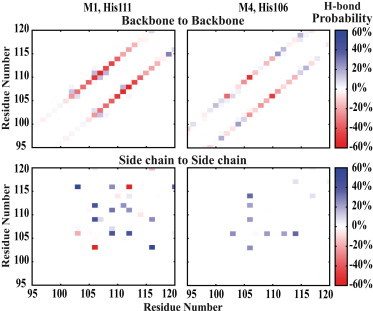
The change in the interresidual hydrogen-bond probabilities (in percentage) accompanying the bending of the peptide induced by the protonation of the hinge histidine for M1 (left) and M4 (right) models. (Top and bottom panels) Backbone and side-chain hydrogen bonding probabilities, respectively. The change is calculated by subtracting the hydrogen bond probabilities for the protonated hinge histidine model from those for the neutral hinge histidine model (see Materials and Methods). (Red/blue) Decrease/increase in the chance for H-bond formation between a residue pair.
Secondary structure analysis of individual residues (averaged over 300 ns of the simulations) in models with a protonated hinge histidine shows a decrease in the helical probability and an increase of turn and coil probabilities of the peptide at positions 107–112. In contrast, an almost completely helical structure is obtained for all the residues in a peptide with a neutral hinge histidine (Fig. 8). Kampmann et al. (37) suspected that hydrogen bonds involving the hinge histidine as an acceptor might be perturbed upon the protonation of this residue and that this event might disrupt the helical structure.
Figure 8.
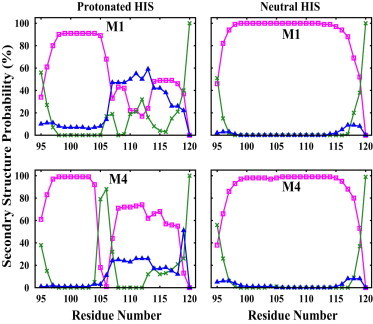
Secondary structure probabilities of individual residues in the hinge peptide: helical (pink), turn (green), and coil (blue), for peptides with a protonated/neutral (left/right) hinge histidine, averaged over the first 300 ns of the simulations of M1 and M4 models.
Residues 106–111 of HA2 are located at a key position for the conformational changes required for the formation of the fusion needle, and at a critical location in the post-fusion structure where they form the trimer of hairpins at acidic pH (21). The results of this study, however, clearly show that the protonation of the hinge histidine (His106 or His111) in HA2 can effectively change the conformation of the hinge peptide from a straight state to a bent one, whereas a neutral hinge histidine is needed for inducing or maintaining the straight structure of the hinge. Whereas previous experimental and computational studies suspected the involvement of histidine residues in conformational changes of HA2, our results strongly show the role of a specific histidine in switching the conformational changes of HA2.
The mechanism proposed in this study can be further verified experimentally by mutagenesis studies focusing on the effect of the removal of the hinge histidine, and possibly recovering the effect by reintroducing this side chain at a slightly different position. As described before, we believe that salt-bridge interactions responsible for the formation of the bent structure are not too sensitive to the exact position of the hinge histidine, and we speculate that any position within the hinge region should be able, in principle, to accommodate the hinge histidine. Furthermore, such mutagenesis studies can also investigate the role of acidic residues partnering with the hinge histidine in producing the bent structure, although in this case multiple mutations might be needed because more than one acidic residue seem to contribute to the effect.
Conclusion
The conformational response of HA to low pH conditions is key to its proper function and to efficient viral entry into the host cell. Nevertheless, given the complexity of this multistep process, the molecular details of the process are largely unknown. Whereas the involvement of histidine side chains have been suspected, no single histidine has been identified to act as the main pH sensor, and more importantly, the dynamics and the mechanism of such histidine-mediated effect were completely uncharacterized.
In this study, we try to address one of the steps involved in the HA-mediated fusion of the influenza virus to the host cell membrane, namely bending of the HA2 chain at a hinge region characterized crystallographically. To test the involvement of a conserved histidine within the hinge region, which we dubbed the hinge histidine due to its key role in acid-mediated bending, 73 sets of simulations were performed on six classes of crystallographic structures (straight or bent) of the HA2 hinge regions from various HA subtypes amounting to a total of 25.1 μs. The protonation state of the hinge histidine was changed in different simulations and the conformational response of the hinge peptide analyzed. In all the simulations starting from an initial straight helical conformation, the hinge adopts a bent conformation when the hinge histidine is protonated, whereas no significant deviation from a linear structure is observed with a neutral hinge histidine.
Conversely, neutralization of the hinge histidine results in straightening of initially bent structures, whereas the protonation of this conserved side chain retains the bent structure of the hinge. These results indicate a significant triggering effect for the protonation of the hinge histidine on bending of HA2 during the fusion process, suggesting that the conserved hinge histidine (His106 or His111) might play the role of a pH sensor, at least in one of the major acid-induced structural changes in HA.
In this study, we have focused on the hinge peptide, isolated from the rest of the protein, to focus on the problem and allow for better sampling. Whereas we expect that the pH-dependent switching mechanism suggested by these simulations may very likely be relevant to the full-length structure, the conformational landscape of the hinge peptide can be clearly influenced by its environment. The next natural step is to investigate whether and/or how the bending/straightening conformational changes of the hinge peptide might be influenced by the presence of the rest of the protein. As of this writing, we are expanding the scope of this research project to study the phenomenon within the context of the full-length protein.
Based on the results of the simulations, we offer a mechanistic picture in which interaction between a protonated histidine in a helix (in response to pH drop) and neighboring acidic side chains that remain negatively charged at mild acidic environments can result in bending of the helix. Given its mechanistic robustness, such a phenomenon can be efficiently exploited in other helical proteins where a major conformational change is induced by acidification through a change in the protonation state of histidine side chains.
Acknowledgments
We acknowledge the Research and Technology Affairs of Golestan University of Medical Sciences, Gorgan, Iran, for their support.
This research is supported by National Institutes of Health grants No. R01-GM086749, No. U54-GM087519, and No. P41-GM104601. All simulations were performed using computer time on the XSEDE resources of the National Science Foundation (grant No. MCA06N060) as well as the Campus Cluster of the Computational Science and Engineering Program at the University of Illinois at Urbana-Champaign.
Supporting Material
References
- 1.Palese P., Shaw M.L. Orthomyxoviridae: the viruses and their replication. In: Knipe D.M., Howley P.M., editors. 5th Ed. Vol. II. Lippincott Williams and Wilkins; Philadelphia, PA: 2007. pp. 1647–1690. (Fields Virology). [Google Scholar]
- 2.Chernomordik L.V., Frolov V.A., Zimmerberg J. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol. 1998;140:1369–1382. doi: 10.1083/jcb.140.6.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wiley D.C., Skehel J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 1987;56:365–394. doi: 10.1146/annurev.bi.56.070187.002053. [DOI] [PubMed] [Google Scholar]
- 4.Wilson I.A., Skehel J.J., Wiley D.C. Structure of the hemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature. 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 5.Eisen M.B., Sabesan S., Wiley D.C. Binding of the influenza A virus to cell-surface receptors: structures of five hemagglutinin-sialyloligosaccharide complexes determined by x-ray crystallography. J. Virol. 1997;232:19–31. doi: 10.1006/viro.1997.8526. [DOI] [PubMed] [Google Scholar]
- 6.Madhusoodanan M., Lazaridis T. Investigation of pathways for the low-pH conformational transition in influenza hemagglutinin. Biophys. J. 2003;84:1926–1939. doi: 10.1016/S0006-3495(03)75001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gething M.J., Skehel J.J., Waterfield M. Cloning and DNA sequence of double-stranded copies of hemagglutinin genes from H2 and H3 strains elucidates antigenic shift and drift in human influenza virus. Nature. 1980;287:301–306. doi: 10.1038/287301a0. [DOI] [PubMed] [Google Scholar]
- 8.Fang R., Min Jou W., Fiers W. Complete structure of A/duck/Ukraine/63 influenza hemagglutinin gene animal virus as progenitor of human H3 Hong Kong 1968 influenza hemagglutinin. Cell. 1981;25:315–323. doi: 10.1016/0092-8674(81)90049-0. [DOI] [PubMed] [Google Scholar]
- 9.Sauter N.K., Hanson J.E., Wiley D.C. Binding of influenza virus hemagglutinin to analogs of its cell-surface receptor, sialic acid: analysis by proton nuclear magnetic resonance spectroscopy and x-ray crystallography. Biochemistry. 1992;31:9609–9621. doi: 10.1021/bi00155a013. [DOI] [PubMed] [Google Scholar]
- 10.Tatulian S.A., Tamm L.K. Secondary structure, orientation, oligomerization, and lipid interactions of the transmembrane domain of influenza hemagglutinin. J. Biochem. 2000;39:496–506. doi: 10.1021/bi991594p. [DOI] [PubMed] [Google Scholar]
- 11.Tamm L.K., Han X., Lai A.L. Structure and function of membrane fusion peptides. Biopolymers. 2002;66:249–260. doi: 10.1002/bip.10261. [DOI] [PubMed] [Google Scholar]
- 12.Danieli T., Pelletier S.L., White J.M. Membrane fusion mediated by the influenza hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J. Cell Biol. 1996;133:559–569. doi: 10.1083/jcb.133.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamilton B.S., Whittaker G.R., Daniel S. Influenza virus-mediated membrane fusion: determinants of hemagglutinin fusogenic activity and experimental approaches for assessing virus fusion. Viruses. 2012;4:1144–1168. doi: 10.3390/v4071144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Melikyan G.B., Lin S., Cohen F.S. Amino acid sequence requirements of the transmembrane and cytoplasmic domains of influenza virus hemagglutinin for viable membrane fusion. Mol. Biol. Cell. 1999;10:1821–1836. doi: 10.1091/mbc.10.6.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bullough P.A., Hughson F.M., Wiley D.C. Structure of influenza hemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 16.Fontana J., Cardone G., Steven A.C. Structural changes in influenza virus at low pH characterized by cryo-electron tomography. J. Virol. 2012;86:2919–2929. doi: 10.1128/JVI.06698-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wharton S.A., Calder L.J., Wiley D.C. Electron microscopy of antibody complexes of influenza virus hemagglutinin in the fusion pH conformation. EMBO J. 1995;14:240–246. doi: 10.1002/j.1460-2075.1995.tb06997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gruenke J.A., Armstrong R.T., White J.M. New insights into the spring-loaded conformational change of influenza virus hemagglutinin. J. Virol. 2002;76:4456–4466. doi: 10.1128/JVI.76.9.4456-4466.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bodian D.L., Yamasaki R.B., Kuntz I.D. Inhibition of the fusion-inducing conformational change of influenza hemagglutinin by benzoquinones and hydroquinones. J. Biochem. 1993;32:2967–2978. doi: 10.1021/bi00063a007. [DOI] [PubMed] [Google Scholar]
- 20.Chen J., Skehel J.J., Wiley D.C. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA2 subunit to form an N cap that terminates the triple-stranded coiled coil. Proc. Natl. Acad. Sci. USA. 1999;96:8967–8972. doi: 10.1073/pnas.96.16.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu R., Wilson I.A. Structural characterization of an early fusion intermediate of influenza virus hemagglutinin. J. Virol. 2011;85:5172–5182. doi: 10.1128/JVI.02430-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim C.S., Epand R.F., Chernomordik L.V. The final conformation of the complete ectodomain of the HA2 subunit of influenza hemagglutinin can by itself drive low pH-dependent fusion. J. Biol. Chem. 2011;286:13226–13234. doi: 10.1074/jbc.M110.181297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gething M.J., Doms R.W., White J. Studies on the mechanism of membrane fusion: site-specific mutagenesis of the hemagglutinin of influenza virus. J. Cell Biol. 1986;102:11–23. doi: 10.1083/jcb.102.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorieau J.L., Louis J.M., Bax A. The complete influenza hemagglutinin fusion peptide adopts a tight helical hairpin arrangement at the lipid/water interface. Proc. Natl. Acad. Sci. USA. 2010;107:11341–11346. doi: 10.1073/pnas.1006142107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ha Y., Stevens D.J., Wiley D.C. H5 avian and H9 swine influenza virus hemagglutinin structures: possible origin of influenza subtypes. EMBO J. 2002;21:865–875. doi: 10.1093/emboj/21.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tong S., Li Y., York I.A. A distinct lineage of influenza A virus from bats. Proc. Natl. Acad. Sci. USA. 2012;109:4269–4274. doi: 10.1073/pnas.1116200109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Air G.M. Sequence relationships among the hemagglutinin genes of 12 subtypes of influenza A virus. Proc. Natl. Acad. Sci. USA. 1981;78:7639–7643. doi: 10.1073/pnas.78.12.7639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Russell R.J., Gamblin S.J., Skehel J.J. H1 and H7 influenza hemagglutinin structures extend a structural classification of hemagglutinin subtypes. J. Virol. 2004;325:287–296. doi: 10.1016/j.virol.2004.04.040. [DOI] [PubMed] [Google Scholar]
- 29.Skehel J.J., Bayley P.M., Wiley D.C. Changes in the conformation of influenza virus hemagglutinin at the pH optimum of virus-mediated membrane fusion. Proc. Natl. Acad. Sci. USA. 1982;79:968–972. doi: 10.1073/pnas.79.4.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maeda T., Ohnishi S. Activation of influenza virus by acidic media causes hemolysis and fusion of erythrocytes. FEBS Lett. 1980;122:283–287. doi: 10.1016/0014-5793(80)80457-1. [DOI] [PubMed] [Google Scholar]
- 31.DuBois R.M., Zaraket H., Russell C. Acid stability of the hemagglutinin protein regulates H5N1 influenza virus pathogenicity. PLoS Pathog. 2011;7:e1002398. doi: 10.1371/journal.ppat.1002398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reed M.L., Bridges O.A., Yen H.L. The pH of activation of the hemagglutinin protein regulates H5N1 influenza virus pathogenicity and transmissibility in ducks. J. Virol. 2010;84:1527–1535. doi: 10.1128/JVI.02069-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Remeta D.P., Krumbiegel M., Blumenthal R. Thermal denaturation of influenza virus and its relationship to membrane fusion. J. Biochem. 2002;41:2044–2054. [Google Scholar]
- 34.Steinhauer D.A., Martin J., Wiley D.C. Morphological changes and fusogenic activity of influenza virus hemagglutinin. Proc. Natl. Acad. Sci. USA. 1996;93:12873–12878. doi: 10.1073/pnas.93.23.12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sancho J., Serrano L., Fersht A.R. Histidine residues at the N- and C-termini of α-helixes: perturbed pKas and protein stability. J. Biochem. 1992;31:2253–2258. doi: 10.1021/bi00123a006. [DOI] [PubMed] [Google Scholar]
- 36.Chen J., Lee K.H., Wiley D.C. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell. 1998;95:409–417. doi: 10.1016/s0092-8674(00)81771-7. [DOI] [PubMed] [Google Scholar]
- 37.Kampmann T., Mueller D.S., Kobe B. The role of histidine residues in low-pH-mediated viral membrane fusion. Structure. 2006;14:1481–1487. doi: 10.1016/j.str.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 38.Carneiro F.A., Stauffer F., Da Poian A.T. Membrane fusion induced by vesicular stomatitis virus depends on histidine protonation. J. Biol. Chem. 2003;278:13789–13794. doi: 10.1074/jbc.M210615200. [DOI] [PubMed] [Google Scholar]
- 39.Mueller D.S., Kampmann T., Mark A.E. Histidine protonation and the activation of viral fusion proteins. Biochemistry. 2008;36:43–45. doi: 10.1042/BST0360043. [DOI] [PubMed] [Google Scholar]
- 40.Chanel-Vos C., Kielian M. A conserved histidine in the ij loop of the Semliki Forest virus E1 protein plays an important role in membrane fusion. J. Virol. 2004;78:13543–13552. doi: 10.1128/JVI.78.24.13543-13552.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fritz R., Stiasny K., Heinz F.X. Identification of specific histidines as pH sensors in flavivirus membrane fusion. J. Cell Biol. 2008;183:353–361. doi: 10.1083/jcb.200806081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin Z.L., Zheng Y., Kielian M. Role of conserved histidine residues in the low-pH dependence of the Semliki Forest virus fusion protein. J. Virol. 2009;83:4670–4677. doi: 10.1128/JVI.02646-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thoennes S., Li Z.N., Steinhauer D.A. Analysis of residues near the fusion peptide in the influenza hemagglutinin structure for roles in triggering membrane fusion. J. Virol. 2008;370:403–414. doi: 10.1016/j.virol.2007.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barbey-Martin C., Gigant B., Knossow M. An antibody that prevents the hemagglutinin low pH fusogenic transition. J. Virol. 2002;294:70–74. doi: 10.1006/viro.2001.1320. [DOI] [PubMed] [Google Scholar]
- 45.Nordlund H.R., Hytonen V.P., Kulomaa M.S. Introduction of histidine residues into avidin subunit interfaces allows pH-dependent regulation of quaternary structure and biotin binding. FEBS Lett. 2003;555:449–454. doi: 10.1016/s0014-5793(03)01302-4. [DOI] [PubMed] [Google Scholar]
- 46.Bressanelli S., Stiasny K., Rey F.A. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J. 2004;23:728–738. doi: 10.1038/sj.emboj.7600064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stevens J., Corper A.L., Wilson I.A. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science. 2004;303:1866–1870. doi: 10.1126/science.1093373. [DOI] [PubMed] [Google Scholar]
- 48.Roussel A., Lescar J., Rey F.A. Structure and interactions at the viral surface of the envelope protein E1 of Semliki Forest virus. Structure. 2006;14:75–86. doi: 10.1016/j.str.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 49.Sammalkorpi M., Lizaridis T. Configuration of influenza hemagglutinin fusion peptide monomers and oligomers in membranes. Biochim. Biophys. Acta. 2007;1768:30–38. doi: 10.1016/j.bbamem.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 50.Berman H.M., Westbrook J., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gamblin S.J., Haire L.F., Xiao B. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science. 2004;303:838–842. doi: 10.1126/science.1093155. [DOI] [PubMed] [Google Scholar]
- 52.Yang H., Carney P., Stevens J. Structure and receptor binding properties of a pandemic H1N1 virus hemagglutinin. PLoS Currents: Influenza. 2010;22:RRN1152. doi: 10.1371/currents.RRN1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stevens J., Blixt O., Wilson I.A. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science. 2006;312:404–410. doi: 10.1126/science.1124513. [DOI] [PubMed] [Google Scholar]
- 54.Ha Y., Stevens D.J., Wiley D.C. X-ray structure of the hemagglutinin of a potential H3 avian progenitor of the 1968 Hong Kong pandemic influenza virus. J. Virol. 2003;309:209–218. doi: 10.1016/s0042-6822(03)00068-0. [DOI] [PubMed] [Google Scholar]
- 55.Kieseritzky G., Knapp E.-W.W. Optimizing pKa computation in proteins with pH adapted conformations. Proteins. 2008;71:1335–1348. doi: 10.1002/prot.21820. [DOI] [PubMed] [Google Scholar]
- 56.Rabenstein B., Knapp E.W. Calculated pH-dependent population and protonation of carbon-monoxy-myoglobin conformers. Biophys. J. 2001;80:1141–1150. doi: 10.1016/S0006-3495(01)76091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gordon J.C., Myers J.B., Onufriev A. H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005;33:W368–W371. doi: 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anandakrishnan R., Onufriev A. Analysis of basic clustering algorithms for numerical estimation of statistical averages in biomolecules. J. Comput. Biol. 2008;15:165–184. doi: 10.1089/cmb.2007.0144. [DOI] [PubMed] [Google Scholar]
- 59.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brooks B.R., Bruccoleri R.E., Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 61.Brooks B.R., Brooks C.L., Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reid J.K. On the method of conjugate gradients for the solution of large sparse systems of linear equations. In: Reid J.K., editor. Large Sparse Sets of Linear Equations. Academic Press; London, UK: 1971. pp. 231–254. [Google Scholar]
- 63.Karimian S.M.H., Izadi S., Barati Farimani A. A study on the measurement of mean velocity and its convergence in molecular dynamics simulations. Num. Methods Fluids. 2011;67:2130–2140. [Google Scholar]
- 64.Cramer C.J., Truhlar D.G. Implicit solvation models: equilibria, structure, spectra and dynamics. Chem. Rev. 1999;99:2161–2200. doi: 10.1021/cr960149m. [DOI] [PubMed] [Google Scholar]
- 65.Karplus M. Molecular dynamics simulation of biomolecules. Acc. Chem. Res. 2003;35:321–323. doi: 10.1021/ar020082r. [DOI] [PubMed] [Google Scholar]
- 66.Fan H., Mark A.E., Honig B. Comparative study of generalized Born models: protein dynamics. Proc. Natl. Acad. Sci. USA. 2005;102:6760–6764. doi: 10.1073/pnas.0408857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsui V., Case D.A. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers. 2000;56:275–291. doi: 10.1002/1097-0282(2000)56:4<275::AID-BIP10024>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 68.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 69.Zhang L.Y., Gallicchio E., Levy R.M. Solvent models for protein-ligand binding: comparison of implicit solvent Poisson and surface generalized Born models with explicit solvent simulations. Comput. Chem. 2001;22:591–607. [Google Scholar]
- 70.Darden T., York D., Pedersen L.G. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 71.Humphrey W., Dalke A., Schulten K. VMD—visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 72.Frishman D., Argos P. Knowledge-based secondary structure assignment. Proteins. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 73.Li W., Trabuco L.G., Frank J. Molecular dynamics of EF-G during translocation. Proteins. 2011;79:1478–1486. doi: 10.1002/prot.22976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Modis Y., Ogata S., Harrison S.C. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. USA. 2003;100:6986–6991. doi: 10.1073/pnas.0832193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sandoz G., Douguet D., Lesage F. Extracellular acidification exerts opposite actions on TREK1 and TREK2 potassium channels via a single conserved histidine residue. Proc. Natl. Acad. Sci. USA. 2009;106:14628–14633. doi: 10.1073/pnas.0906267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rodnin M.V., Kyrychenko A., Ladokhin A. Conformational switching of the diphtheria toxin T domain. J. Mol. Biol. 2010;402:1–7. doi: 10.1016/j.jmb.2010.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Achilonu I., Fanucchi S., Dirr H.W. Role of individual histidines in the pH-dependent global stability of human chloride intracellular channel 1. J. Biochem. 2012;51:995–1004. doi: 10.1021/bi201541w. [DOI] [PubMed] [Google Scholar]
- 78.Prakash M.K., Barducci A., Parrinello M. Probing the mechanism of pH-induced large scale conformational change in the dengue virus envelope protein using atomistic simulations. Biophysics (Oxf.) 2010;99:588–594. doi: 10.1016/j.bpj.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.