

Abstract
Physical origin of DNA condensation by multivalent cations remains unsettled. Here, we report quantitative studies of how one DNA-condensing ion (Cobalt3+ Hexammine, or Co3+Hex) and one nonDNA-condensing ion (Mg2+) compete within the interstitial space in spontaneously condensed DNA arrays. As the ion concentrations in the bath solution are systematically varied, the ion contents and DNA-DNA spacings of the DNA arrays are determined by atomic emission spectroscopy and x-ray diffraction, respectively. To gain quantitative insights, we first compare the experimentally determined ion contents with predictions from exact numerical calculations based on nonlinear Poisson-Boltzmann equations. Such calculations are shown to significantly underestimate the number of Co3+Hex ions, consistent with the deficiencies of nonlinear Poisson-Boltzmann approaches in describing multivalent cations. Upon increasing the concentration of Mg2+, the Co3+Hex-condensed DNA array expands and eventually redissolves as a result of ion competition weakening DNA-DNA attraction. Although the DNA-DNA spacing depends on both Mg2+ and Co3+Hex concentrations in the bath solution, it is observed that the spacing is largely determined by a single parameter of the DNA array, the fraction of DNA charges neutralized by Co3+Hex. It is also observed that only ∼20% DNA charge neutralization by Co3+Hex is necessary for spontaneous DNA condensation. We then show that the bath ion conditions can be reduced to one variable with a simplistic ion binding model, which is able to describe the variations of both ion contents and DNA-DNA spacings reasonably well. Finally, we discuss the implications on the nature of interstitial ions and cation-mediated DNA-DNA interactions.
Introduction
Cation-modulated electrostatics of nucleic acids is fundamental to nucleic acids structure and function and has served as a model system of biomolecular electrostatics (1–6). Electrostatic interaction, being the strongest among noncovalent interatomic interactions, is of central importance in biology due to the prevalence of charges in all major types of biomolecules (7,8). Although the general principles of electrostatics (i.e., Coulomb forces) are well understood, biomolecular electrostatics is complicated by multiple inherent complexities including screening by ubiquitous ions, involvement of highly polar water molecules, and significant roles of entropy (i.e., thermal energy) (7–9). Despite extensive efforts in the past decades, quantitative knowledge of biomolecular electrostatics is still incomplete and many important questions remain unanswered (10).
In describing cation-modulated electrostatics of nucleic acids, quantitative agreement between experiment and theory has only been obtained for monovalent cations (11). For nucleic acids in monovalent salts (and divalent salts to some extent), mean-field theories based on the nonlinear Poisson-Boltzmann (NLPB) equations have provided reasonable explanations to measured quantities such as osmotic pressures of DNA solutions (12), cation competitions and spatial distributions around DNA (13–17), and interaction forces between freely floating DNA oligomers (18) and between parallel DNA strands beyond 1 nm surface separation (19). Recent literature has increased in theoretical sophistication by accounting for additional details such as different ion sizes (20) and DNA-DNA azimuthal angle dependence due to helical DNA-charge patterns (21). It is worth noting that linear Poisson-Boltzmann approaches also have been shown to be effective, once corrected for the nonlinear screening in the DNA vicinity by theorems such as counterion condensation (22) and charge renormalization (23). Given their underlying assumptions, the success of mean-field approaches for monovalent cations is likely attributed to the relatively weak electrostatic coupling between ions and charged surfaces and between themselves.
Meanwhile, quantitative understanding is yet to be attained for multivalent cations, especially cations with valence of three and higher. A notable example is the multivalent cation-mediated attraction between DNA helices, whereas mean-field based theories always predict repulsion (24). Physical origin of such like-charge attraction remains under debate. As the increase of ion valences results in qualitatively different electrostatic interactions, stronger electrostatic interactions between cations and DNA and/or between cations themselves are widely accepted to lead to DNA-DNA attraction. A number of theoretical models have been proposed, including cation density fluctuations between DNA helices (25–27), ion-ion correlations leading to Wigner-like lattices (28,29), strong counterion-DNA coupling (30–32), and counterion localization giving rise to ion bridging (33) or zipper-like cation-DNA charge correlation (34). Moreover, hydration force has been shown to dominate DNA-DNA interactions at DNA-DNA surface separations <1 nm, as a result of the perturbation of hydration shells (35). The role of cations is to restructure the hydration shells whose contraposition or complementarity leads to repulsive or attractive hydration force, respectively. On the other hand, computer simulations have provided detailed descriptions of DNA-ion interactions, but evaluating the underlying physical mechanisms is difficult due to varying levels of simplifications employed (36–42). The first all-atom molecular dynamics (MD) simulation of DNA-polyamine interactions was reported in 2008 by Dai et al (40). The proposed ion-bridging mechanism therein for chain-like polyamines is unlikely to be true for point-like multivalent cations. In a more recent all-atom MD study of 64 parallel DNA arrays (43), the commonly used force fields resulted in attraction between dsDNA with mono- and divalent ions. Tuning the force fields was necessary to avoid such inconsistency. It is worth noting that multivalent cation-mediated DNA condensation differs from DNA compaction by oppositely charged polyelectrolytes. Because the polyelectrolytes’ charged groups alone cannot condense DNA, the DNA compaction is understood in terms of polyelectrolyte-mediated bridging interactions (44). One example is DNA packaging by dendrimers for which the condensing force (45) and resultant structures have been characterized (46) and atomistic simulations have achieved remarkable success in describing the energetics of compaction and the roles of hydration forces (47,48). In sum, the true physical nature of multivalent DNA-condensing cations, especially compared with DNA-noncondensing cations, is the focal point of competing theories to explain the DNA-DNA attraction and consequently electrostatic interactions in the presence of multivalent cations.
On the other hand, quantitative measurements of cation behaviors in the DNA-DNA attraction regime are largely lacking, precluding stringent tests of theories of electrostatic interactions. Previous studies of DNA-condensing cations have determined their bulk threshold concentrations to condense DNA and the effects of monovalent cations and anions (49–52). Three commonly studied cations, Cobalt3+ Hexammine (Co3+Hex), Spermidine3+, and Spermine4+, were shown to have sub-mM threshold concentrations (49,53–55) and monovalent cations raise the threshold concentration as expected from ion competition. The spatial distribution around DNA of a DNA-condensing cation, Co3+Hex, has been measured below its threshold concentration (56). It has also been shown that DNA condensation is accompanied by an additional binding of multivalent cations and appears as an abrupt all or none process (54,57,58). More broadly, a wide array of experimental techniques, including isothermal titration calorimetry (53), light scattering (59,60), osmotic stress method (61), fluorescence microscopy (58), and nuclear magnetic resonance (62), have been applied to investigate the cation-induced DNA compaction process and DNA-cation interactions; however, much less is known about the ions within condensed DNA (in contrast to the ions in the bulk), although it is these ions that mediate the attraction between DNA helices. Such knowledge will be needed to identify the gaps in our understanding and elucidate the peculiarities of multivalent cations.
We have thus directly probed the interstitial ions in spontaneously condensed double-stranded DNA arrays by measuring the numbers of two competing ions (Co3+Hex and Mg2+) under a variety of ionic conditions in the bath solution. There are two previous studies on the ionic contents in cation-condensed DNA arrays to our knowledge. Teif (63) used isotope labeling to determine the number of Spermidine3+ ions needed to condense DNA arrays and Todd et al. (54) measured the competition between Co3+Hex and Spermidine3+ to determine their relative binding constant. Both previous studies are different from ours because either no ion competition was studied or the competition between two DNA-condensing ions was probed. Although Co3+Hex condenses DNA and Mg2+ does not, their behaviors likely differ qualitatively in the attractive regime and how they compete can provide insight into the physics of DNA-DNA attraction. Additionally, the previous studies measured only one type of ion in DNA arrays, whereas we have systematically quantified the numbers of both Co3+Hex and Mg2+ ions in DNA arrays. Furthermore, we measured the DNA-DNA spacings in the same samples, a parameter required for theoretical calculations. We found that the measured partitions of Co3+Hex are higher than predicted from mean-field approaches once the effect of ion pairing is corrected. Remarkably, the partition of the interstitial ions was observed to uniquely determine the DNA-DNA spacing, whereas there are two independent ion concentrations in the bath solution (Mg2+ and Co3+Hex). We further show that a phenomenological ion binding model is able to describe all experimental data reasonably well, resulting in a reduced solution parameter, [Mg2+]/[Co3+Hex]2/3.
Materials and Methods
High-molecular-weight genomic DNA (>10 kilo basepairs) was purified from adult chicken erythrocytes as previously described (64). Salts of Co3+Hex Chloride and MgCl2 were purchased from Sigma (St. Louis, MO) and used as received. Buffer of 2 mM pH 7.5 Tris·Cl was used in all solutions unless noted otherwise; the relatively low concentration was chosen to minimize the competition from the buffering ions. Each sample was first prepared by condensing ∼300 μg DNA into a liquid crystalline (LC) pellet by 2 mM Co3+Hex. The LC-DNA pellet was then equilibrated for 2 weeks in 1 ml of each studied salt condition with one interim buffer exchange. The pellet’s DNA-DNA spacing was then measured by x-ray diffraction (XRD) at 20°C with an in-house XRD setup (3 × 107 photons/s, beam size of 0.8 × 0.8 mm) where radiation damage is negligible.
Each DNA pellet was recovered after XRD and reequilibrated in the original buffer for another week. The pellet was then picked up and care was taken to remove the equilibration buffer by pressing the pellet between weigh papers and wicking with a kimwipe. The pellet was then dissolved in 1 ml buffer of 1.5 M NaCl with gentle thermal agitation at 55°C and daily vortexing for 2 weeks. Once the LC-DNA pellet is fully dissolved, ultraviolet-visible (UV-vis) absorption measurements were first carried out to determine the concentrations of Co3+Hex (at 475 nm, an extinction coefficient of 0.056/mM/cm) and DNA (at 260 nm, 50 μg/ml for optical density of 1.0). Inductively coupled plasma atomic emission spectroscopy (ICP-AES) was then used to simultaneously measure the numbers of Co, Mg, and P (from the DNA) atoms in the same samples on an Optima 7300DV (Perkin Elmer, Waltham, MA). ICP-AES data were collected in the axial mode with five replicates per sample. Measurements were made in the linear range of the instrument at five different wavelengths for Co, four different wavelengths for Mg, and two different wavelengths for P, and then the concentrations from all respective wavelengths were averaged. Calibration was provided by 10 linearly spaced control samples of Co, Mg, and P that spanned the entire region of concentrations. Four independent measurements using two independent calibrations were averaged to obtain the final concentrations for each set of samples. For the samples with 0 mM MgCl2 in the bath solution, the detected trace amount of Mg (<0.3% of P atoms) is treated as the baseline from the dissolvation buffer and subtracted from all data. Standard errors were determined statistically from these measurements.
To address the possible complication of incomplete buffer removal from the DNA pellet, test samples with only one type of cation were measured to verify that our experiment protocol leads to minimal excess buffer that is well below 20% of the LC-DNA pellet volume (∼1 μl). Importantly, the excess buffer, if carried by the DNA pellet, contributes little to the total numbers of ions measured, especially for Co3+Hex. This is because the effective Co3+Hex concentration in the pellet (∼140–700 mM) is at least 28–700 times that in the buffer (1–5 mM) under the studied conditions. The uncertainty introduced by the excess buffer is thus <1% for Co3+Hex. The relative uncertainty for Mg2+ could be significant when very few Mg2+ ions exist in the DNA pellet, however the Mg2+ concentration in the buffer is necessarily low and its absolute contribution should be small. This is also supported by the experimental data reported herein.
Results and Discussion
Experimental characterizations of spontaneously condensed DNA arrays
The multivalent-cation-condensed LC-DNA phase consists of locally hexagonally ordered DNA arrays (Fig. 1 a) made up of DNA, ions, and solvent. The system can be defined by a few variables, the interaxial DNA-DNA spacing (d), the number of Co3+Hex ions (nCo), the number of Mg2+ ions (nMg), and the number of Cl− ions (nCl). Here, we normalize the ion numbers by the number of DNA phosphates and do not explicitly consider the number of water molecules as no external osmotic pressure is applied on the DNA array. The interaxial spacing d is determined by XRD as shown in Fig. 1 b for LC-DNA samples under a constant [Co3+Hex] of 2 mM and varied [Mg2+]s from 0 to 30 mM. The DNA-DNA spacing is given by 2π/Qpeak × 2/√3, where 2/√3 accounts for the local hexagonal packing and Qpeak is obtained by peak fitting with a Lorentzian function. From Fig. 1 b, we observe that 2 mM Co3+Hex holds the DNA array in tight spacings ∼27.8 Å for [Mg2+]s up to 25 mM, and then an abrupt expansion occurs at 25–30 mM [Mg2+] consistent with a hexagonal to cholesteric phase transition (50). At [Mg2+] >30 mM the pellet dissolves and XRD cannot be applied.
Figure 1.
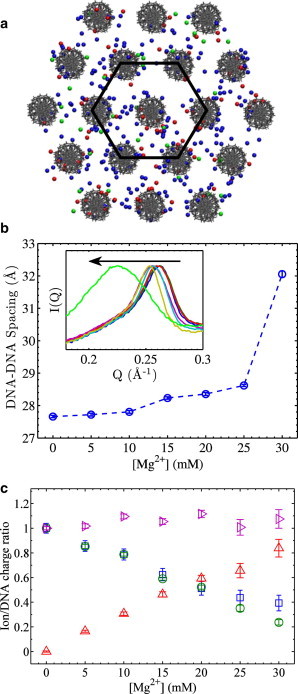
Illustrations of the studied system of DNA arrays and representative experimental data. (a) Cartoon of an ordered DNA array and its interstitial ions, Co3+Hex (red), Mg2+ (blue), and Cl− (green). The hexagonal geometry is indicated by a hexagon drawn in black. Atomic radii are chosen to facilitate visualization only. Another view of the DNA array is shown in Fig. S1. (b) The DNA-DNA spacing as a function of Mg2+ concentration with constant 2 mM Co3+Hex in the bath solution. Inset shows the XRD peak profiles with the arrow pointing to increasing [Mg2+]s, indicating a hexagonal to cholesteric phase transition at the highest [Mg2+]. (c) The ion/DNA charge ratios as a function of [Mg2+] measured from the same samples shown in (b). The charge ratio of Co3+Hex/DNA (3 nCo) was measured by both UV-vis (□) and ICP-AES (○), showing good agreement. The charge ratio of Mg2+/DNA (2 nMg) was measured by ICP-AES only (Δ). Their sum , 3 nCo + 2 nMg, is very close to unity, indicating that Cl− ions are essentially completely excluded by the DNA arrays under studied conditions. This also supports that minimal amounts of excess buffer were carried in the measured DNA pellets.
The measured numbers of the interstitial Co3+Hex and Mg2+ ions are shown in Fig. 1 c in the form of ion/DNA charge ratios (i.e., 3 nCo, 2 nMg). As expected from ion competition, the fraction of Co3+Hex in the DNA array decreases upon increasing [Mg2+] in the bath solution. For the Co3+Hex/DNA charge ratios (3 nCo) measured by both UV-vis and ICP-AES, good agreement was found. Slightly larger 3 nCo values were obtained by UV-vis at high [Mg2+]s, which is attributed to the susceptibility of the UV-vis method to baseline variations when [Co3+Hex] is low. ICP-AES data are thus regarded more reliable and used in all subsequent analysis. ICP-AES also has the advantage of measuring Mg2+ in addition to Co3+Hex. Although our current instrumentation is not able to directly measure the co-ion Cl−, it is possible to derive its number nCl via the charge neutrality condition 3 nCo + 2 nMg–ncl = 1. However, we observe that the 3 nCo + 2 nMg values are practically unity for all measured samples (Fig. 1 c and other data discussed later), indicating a negligible amount of Cl− in DNA arrays (nCl ≈ 0) due to its electrostatic exclusion. The condition of 3 nCo + 2 nMg = 1 thus holds for our studied conditions and simplifies our subsequent analysis. Taken all together, our XRD and ICP-AES measurements provide quantitative details of DNA arrays that can be directly compared with theoretical predictions.
Comparisons of the numbers of ions between experiments and mean-field theories
The well-defined states of DNA arrays allow direct comparisons with quantitative model predictions. Although it is preferred to compare with theories that predict DNA-DNA attraction, such theories often involve resource-demanding computations or their predictions are sometimes of qualitative nature. As a first step, here we choose to compare with two commonly used theoretical models based on the NLPB equations. The first is the primitive cylindrical cell model (CCM) (65) where DNA is treated as a 20-Å-diameter cylinder with a uniform charge density and ions are treated by density distributions. The NLPB equations are then solved within a cylindrical cell of diameter set by the measured DNA-DNA spacing, under the boundary condition of zero electric field at the outer boundary. As the actual DNA array is hexagonal, the cylindrical cell overestimates the DNA density by ∼10%. We verified that CCM calculations are insensitive to the DNA-DNA spacing and that a 10% change of the cell volume leads to negligible changes (see Fig. S2 in the Supporting Material). The second model takes into account the atomic structure of double-stranded DNA and solves the NLPB equations using the program Adaptive Poisson-Boltzmann Solver (APBS) (66). To represent the system of DNA arrays, hexagonal arrays of 19 DNA helices (24 basepairs of random sequences) are generated with the measured DNA-DNA spacings. Fig. 1 a shows the view down the axial direction and another view is shown in Fig. S1. The box for the ABPS calculation has dimensions of 225 × 225 × 150 Å, considerably larger than the size of the array (∼120 × 120 × 80 Å). The grid size is 0.45 Å and has been verified to be sufficient (Fig. S3). All ions are treated as spheres of 2-Å radius, which is used to determine the ion accessible volume during the APBS calculations (66). Uses of different ion radii lead to relatively small changes in the APBS results and do not affect the conclusions of our study (Fig. S4).
The CCM and APBS calculations make quantitative predictions of the ion/DNA charge ratios (3 nCo, 2 nMg, and –nCl). Both models yield the electrostatic potential distributions, which in turn give the ion density profiles via the Boltzmann probability function and the total number of each ionic type via integration. For the APBS calculation where a finite atomic model of DNA arrays is used, to avoid the edge effects, only the core volume of the DNA array is analyzed by selecting the inner hexagonal region as indicated in Fig. 1 a (solid line) and the middle 12 basepairs along the axial direction. We verified that summing all ion/DNA charge ratios gives unity as expected from charge neutrality. We then directly compare the experimental values and theoretical predictions. Fig. 2, a and b, show the changes of 3 nCo and 2 nMg (in the DNA array) at fixed Co3+Hex concentrations and varied Mg2+ concentrations (in the bath solution). The CCM and the APBS results overlap with each other nearly exactly. Their predictions of nCl < 0.01 for all studied conditions (except a few at highest [Mg2+]s) are consistent with experimental observations of nCl ≈ 0. At 1 and 2 mM Co3+Hex (Fig. 2, a and b), both model predictions appear to explain the 3 nCo and 2 nMg curves fairly well. This may be somewhat surprising because both the CCM and APBS models do not predict DNA condensation, whereas the studied DNA arrays are spontaneously condensed. However, at 5 mM Co3+Hex (Fig. 2, a and b), both models considerably overestimate the 3 nCo values and consequently underestimate the 2 nMg, as 3 nCo + 2 nMg = 1 practically holds for both model calculations and experiments. These observations are qualitatively in contrast to the expectation that DNA-condensing cations (e.g., Co3+Hex) interact with DNA more strongly than mean-field theoretical predictions (e.g., the CCM and APBS models).
Figure 2.
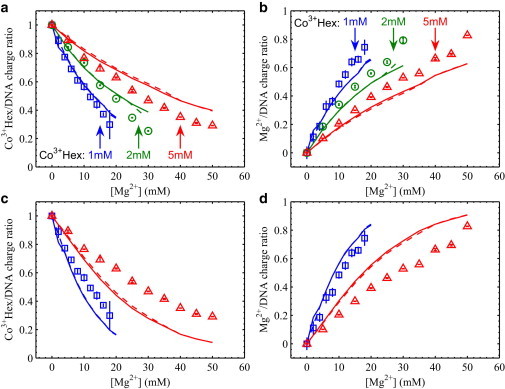
Measured ion/DNA charge ratios in the Co3+Hex-Mg2+ competition series compared with theoretical calculations. Symbols are experimental data at fixed [Co3+Hex]s of 1 mM (□), 2 mM (○), and 5 mM (Δ) and varied [Mg2+]s as the x axis. Lines in matched colors are the theoretical predictions under the same conditions from the CCM (solid) and APBS (dashed) models. (a and b) Show the Co3+Hex/DNA charge ratio (3 nCo) and the Mg2+/DNA charge ratio (2 nMg), respectively, where all Co3+Hex ions are assumed to be completely dissociated in the trivalent state for the CCM and APBS calculations. (c and d) Show the same experimental data as in (a and b), respectively, where the effect of ion pairing between Co3+Hex and Cl− pairing is considered for the CCM and APBS calculations. The dissociation constant Kd is 20 mM. The curves from the 2 mM Co3+Hex series are not shown to reduce data cluttering in the plots. Note that the large error bars (and slight deviation from charge neutrality) at the highest [Mg2+] for each series are due to the sample loss when handling these loosely packed DNA arrays.
To investigate the unexpected outcomes from the comparisons with mean-field theories, we next consider one effect that is known to be significant for multivalent cations at high concentrations, ion pairing (49). In this context, ion pairing refers to the association of trivalent Co3+Hex ions with Cl− anions giving rise to effectively divalent Co3+HexCl− ions, as a result of strong electrostatic attraction. The pairings of divalent cations and anions (i.e., Mg2+ and Cl−, Co3+HexCl− and Cl−) are not considered as we have chosen to study relatively low ion concentrations (maximum [Mg2+] of 50 mM, maximum [Co3+Hex] of 5 mM). For the ion paring reaction Co3+Hex + Cl− ↔ Co3+HexCl−, the dissociation constant Kd (from right to left) is defined as [Co3+Hex][Cl−]/[Co3+HexCl−] and was reported to be ∼20 mM (50). Although we have chosen to work with low Co3+Hex concentrations (1–5 mM), ion paring still has significant effects on the concentration of trivalent Co3+Hex ions in the bath solution. For example, in the presence of 20 mM MgCl2, a nominal 1 mM Co3+HexCl3 gives 0.32 mM (32%) trivalent species and a nominal 5 mM gives 1.4 mM (28%). Given such dramatic changes, it is somewhat surprising that the measured 3 nCo + 2 nMg values are very close to unity, which suggests that the Co3+Hex ions within the DNA arrays are predominantly trivalent species. We speculate that the presumably bulky divalent Co3+HexCl− ions compete poorly against Co3+Hex or Mg2+ ions and are thus largely excluded from the DNA array. If otherwise, we would expect the measured 3 nCo + 2 nMg values to be significantly greater than unity.
As a result of ion pairing, the substantial decreases in the actual Co3+Hex ion concentrations lead to different predictions by the CCM and APBS models, noting that the divalent Co3+HexCl− ions are not considered for reasons previously mentioned. As shown in Fig. 2, c and d, both models show to underestimate the numbers of Co3+Hex ions for all data series by as large as 40% and consequently overestimate the numbers of Mg2+ ions. This underestimation of the Co3+Hex competition is qualitatively different from the observations without the ion pairing correction (Fig. 2, a and b). We like to note that it is possible to match the experimental data with the CCM and APBS model predictions if an arbitrary Kd is used, e.g., a value of Kd = 120 mM was found to be able to give excellent agreements (Fig. S5), though the physical meaning of this is unclear. Overall, although we are not able to implement more sophisticated calculations, our measurement and analysis nonetheless quantify the deficiencies of mean-field approaches and can serve as quantitative tests of modern theories of DNA-DNA attraction.
Relations between the DNA-DNA spacings and the numbers of interstitial ions
Increasing the [Mg2+] in the bath solution not only changes the partition of Co3+Hex and Mg2+ ions in the DNA array, but also expands the DNA array before eventually dissolving it (Fig. 1 b). The smallest measurable Co3+Hex/DNA charge ratios (3 nCo) correspond to the minimum charge fractions of Co3+Hex that incur net DNA-DNA attraction. Interestingly, for all nominal Co3+Hex concentrations studied (Fig. 2 a), essentially the same minimal 3 nCo value of ∼0.2 (20% of DNA charges) is observed. This is not entirely expected as it is generally believed that ∼70–90% of the DNA charge needs to be neutralized by DNA-condensing ions for attraction to occur (24), e.g., 87% for Spermidine in 10 mM NaCl (53), 67% for Co3+Hex in 10 mM NaCl (53), and 80% for Spermine in 10 mM Tris buffer (55). Our measurement shows that this number is much smaller, i.e., as low as ∼20% of DNA charge neutralization by Co3+Hex is needed for condensation. This suggests against the need for significant cation-cation correlation to mediate DNA-DNA attraction. Rather, it may be explained because each Co3+Hex ion mediates a finite amount of attractive force independently and that ions mediate DNA-DNA forces additively, though concurrent measurements of DNA-DNA attractive forces would be needed for additional proof. Nonetheless, this observation does not contradict previous studies because the other 80% DNA charge is neutralized by Mg2+ in this study, whereas previous studies used low salts of monovalent ions. A different minimal 3 nCo value is expected if Na+ is used instead.
As the DNA-DNA spacing is the equilibrium distance between DNA strands that balance the attractive and repulsive forces, it is of interest to examine its dependence on the interstitial ions modulating the forces. Fig. 3 a shows the DNA-DNA spacing as a function of [Mg2+] for each fixed nominal [Co3+Hex]. Qualitatively resembling trends are observed for all curves: the hexagonal to cholesteric phase transition at ∼29.5 Å and the complete dissolution at ∼33.8 Å. We then show the DNA-DNA spacing as a function of the measured Co3+Hex/DNA charge ratio in Fig. 3 b; remarkably, all series collapse onto one single curve. It also becomes clear that the hexagonal-cholesteric phase transition occurs at 3 nCo ∼0.3 and the DNA array dissolves at 3 nCo ∼0.2. The convergence indicates that the DNA-DNA spacing is practically solely determined by the partition of the interstitial ions, though this may not be surprising because it is the interstitial ions that mediate the forces. Given that nCl ≈ 0 and 3 nCo + 2 nMg = 1, the state of the DNA array (d, nCo, nMg, and nCl) is thus determined by only one independent variable.
Figure 3.
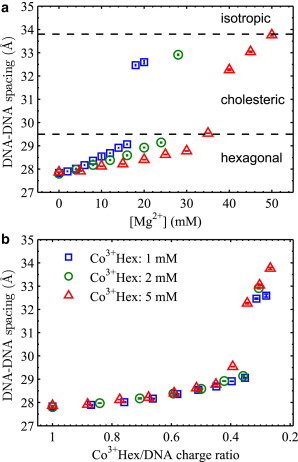
Measured DNA-DNA spacings in the Co3+Hex-Mg2+ competition series shown in Fig. 2. (a) The DNA-DNA spacings as a function of the nominal [Mg2+] in the bath solution. The nominal Co3+Hex concentrations in the bath solution for all data series are given in the legend in (b). The dashed lines delimit the regions for different structure phases as labeled. (b) The same DNA-DNA spacings as in (a) as a function of the corresponding measured Co3+Hex/DNA charge ratio, 3 nCo. Note that the x axis is plotted in the reverse direction.
Relations between the numbers of interstitial ions and the bath ion concentrations: a phenomenological ion binding model
In our experiment, as there is free exchange of ions and solvent between the DNA array and the bath solution, the chemical potentials of all ions (μCo, μMg, and μCl) are equal between the two phases. It is the chemical potentials of ions that are directly varied via their concentrations in the bath solution and the changes of d, nCo, nMg, and nCl in the DNA array are measured at constant temperature and pressure. For the ionic bath solution constrained by charge neutrality, two independent variables exist in the case of three ionic species with chemical potentials μCo, μMg, and μCl. However, previous discussions based on Fig. 3 suggest one independent variable for the state of the DNA array. Thus, it may be possible to reduce the variables of the bath solution in determining the state of the DNA array. For the benefit of simplicity, we consider a phenomenological ion binding model (IBM) treating the DNA array as composed of binding sites with a constant electrostatic potential φ. As the bath solution sets the ion chemical potentials by μCo = ln([Co3+Hex]) and μMg = ln([Mg2+]), the ion binding equilibrium is established when the total free energy is unchanged by swapping δ number of Mg2+ with 2/3δ number of Co3+Hex between the two phases (due to charge neutrality). Because φ is assumed to be constant, no net change of electrostatic energies is incurred during the virtual swap of equal amounts of ion charges. Only the changes of the ion entropies need to be considered. Taking the entropy of bound (i.e., interstitial) ions in the form of ln(nion) (note that nion is defined as the ion/DNA-phosphate number ratio), we obtain,
| (1) |
Here, ξ is a constant to account for offsets due to the different choices of units for nion vs. [ion] and the likely different nature of reference states between in the bath solution and the DNA array. Given that 3 nCo + 2 nMg = 1, this indicates that nCo and nMg are uniquely determined by a single reduced variable of the bath solution, [Mg2+]/[Co3+Hex]2/3. It should be noted that Rouzina and Bloomfield (67) have derived a more detailed model additionally considering the change of electrostatic potential due to ion exchange. The IBM model is used here for its simplicity and because the correction due to the varying electrostatic potentials is small (67).
With ξ as the fitting parameter for each series, the measured Co3+Hex/DNA charge ratios (3 nCo) are fitted with the IBM as shown in Fig. 4 a. Note that the concentrations of trivalent Co3+Hex ions are corrected for the effect of ion pairing using Kd = 20 mM and that the divalent Co3+HexCl- ions are considered to be excluded from the DNA array and do not compete. Fig. 4 a shows excellent fits to all curves, which is rather remarkable given the simplistic nature of the IBM and the use of only one fitting parameter for each series. Fig. 4 b shows the measured Mg2+/DNA charge ratios along the 1–3 nCo values calculated from the IBM fits. Good agreements are observed as expected from charge neutrality for both the measurements and the IBM. The same data and IBM fits are shown as a function of [Mg2+]/[Co3+Hex]2/3 in Fig. 4, c and d. Although we expect all curves to exactly collapse onto one single curve based on the IBM, the convergences of the Co3+Hex/DNA and Mg2+/DNA ratios are clear, but nonperfect. Likewise, similar levels of convergence of the DNA-DNA spacing are attained and shown in Fig. S6. The reason for these nonexact overlaps in Fig. 4, c and d is the slightly different ξ values obtained from the IBM fits, e.g., 2.68, 2.71, and 2.72 for the [Co3+Hex] series of 1, 2, and 5 mM, respectively. As the ξ values should be the same for all curves, this reflects deviations from ideal IBM behaviors, as well as the need for more sophisticated models. One possible consideration beyond the IBM is the change of the apparent ion binding energy with the increase of ionic strength. The logic is that the increase of nCo, upon the increase of ionic strength at a fixed [Mg2+]/[Co3+Hex]2/3, slightly contracts the DNA array (Fig. S6), which likely favors the binding of Co3+Hex more than Mg2+ (as indicated in Fig. S2 b). Nonetheless, the nearly universal dependence of the ion partitions on the single reduced variable of [Mg2+]/[Co3+Hex]2/3 is evident. As the IBM only considers the entropy of mixing of ions, its validity again disfavors the presence of significant ion-ion correlations. The analogy of ion binding sites in the IBM may be explained by the confinement of cations by the local molecular fields on DNA surfaces. On the whole, given the need for the fitting parameter ξ, the IBM provides qualitative explanations of the trends of ion competition, but lacks quantitative power to predict absolute ion partitions.
Figure 4.
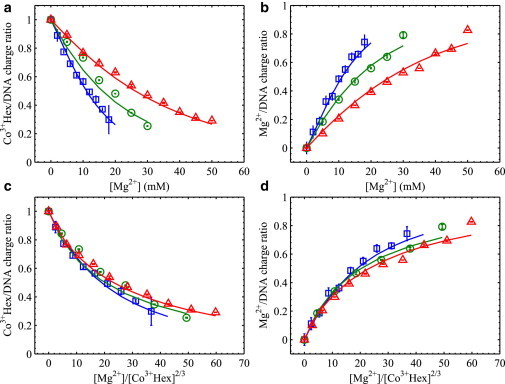
Measured ion/DNA charge ratios and fits using the IBM. Symbols show experimental data under fixed nominal [Co3+Hex]s of 1 mM (□), 2 mM (○), and 5 mM (Δ) in the bath solution (the same as in Fig. 2). Solid lines in matched colors are the IBM fits as described in the text. (a and b) Show the Co3+Hex/DNA charge ratio and Mg2+/DNA charge ratio as a function of [Mg2+] in the bath solution, respectively. (c and d) Show the same data in (a and b), respectively, but as a function of the [Mg2+]/[Co3+Hex]2/3 in the bath solution.
Conclusion
In conclusion, we have carried out quantitative measurement and analysis of the partitions of DNA-condensing and non-DNA-condensing cations in spontaneously condensed DNA arrays. The availability of such data in the attractive regime has led to several interesting observations. First, mean-field calculations based on NLPB equations (considering DNA either as uniform cylinders or atomic structures) underestimate the fraction of Co3+Hex and overestimate that of Mg2+. Importantly, our study quantifies the discrepancies and provides valuable data sets to test advanced theories of electrostatics. Second, DNA-DNA attraction can be mediated when as low as ∼20% of the DNA charge is neutralized by Co3+Hex ions (the rest by Mg2+ ions), contrary to the generally accepted 70–90% neutralization of the DNA charge by DNA-condensing ions. Third, the competition between Co3+Hex and Mg2+ ions in the DNA array can be qualitatively explained by a minimalistic ion binding model whose predictions are derived from one reduced variable of the bath solution, which in turn determines the state of the DNA array (d, nCo, nMg, and ncl ≈ 0). These observations suggest interesting implications on the relationship between DNA-DNA forces and ion partitions, and the origin of such behaviors may be explained by the spatial placement of interstitial ions. Efforts in addressing these questions are underway.
Acknowledgments
We thank Donald Rau for experimental advice and stimulating discussions.
This work was supported by the George Washington University and the Gettysburg College.
Contributor Information
Xiangyun Qiu, Email: xqiu@gwu.edu.
Kurt Andresen, Email: kandrese@gettysburg.edu.
Supporting Material
References
- 1.Gelbart W.M., Bruinsma R.F., Parsegian V.A. DNA-inspired electrostatics. Phys. Today. 2000;53:38–44. [Google Scholar]
- 2.Solis F.J., de la Cruz M.O. Flexible polymers also counterattract. Phys. Today. 2001;54:71–72. [Google Scholar]
- 3.Kornyshev A.A., Olson W. From DNA-inspired physics to physics-inspired biology. J. Phys. Condens. Matter. 2010;22:410301. doi: 10.1088/0953-8984/22/41/410301. [DOI] [PubMed] [Google Scholar]
- 4.Wong G.C.L., Pollack L. Electrostatics of strongly charged biological polymers: ion-mediated interactions and self-organization in nucleic acids and proteins. Annu. Rev. Phys. Chem. 2010;61:171–189. doi: 10.1146/annurev.physchem.58.032806.104436. [DOI] [PubMed] [Google Scholar]
- 5.Cherstvy A.G. Electrostatic interactions in biological DNA-related systems. Phys. Chem. Chem. Phys. 2011;13:9942–9968. doi: 10.1039/c0cp02796k. [DOI] [PubMed] [Google Scholar]
- 6.Teif V.B., Bohinc K. Condensed DNA: condensing the concepts. Prog. Biophys. Mol. Biol. 2011;105:208–222. doi: 10.1016/j.pbiomolbio.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 7.Dong F., Olsen B., Baker N.A. Computational methods for biomolecular electrostatics. Methods Cell Biol. 2008;84:843–870. doi: 10.1016/S0091-679X(07)84026-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Honig B., Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 9.Strey H.H., Podgornik R., Parsegian V.A. DNA–DNA interactions. Curr. Opin. Struct. Biol. 1998;8:309–313. doi: 10.1016/s0959-440x(98)80063-8. [DOI] [PubMed] [Google Scholar]
- 10.Ren P.Y., Chun J.H., Baker N.A. Biomolecular electrostatics and solvation: a computational perspective. Q. Rev. Biophys. 2012;45:427–491. doi: 10.1017/S003358351200011X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ben-Yaakov D., Andelman D., Podgornik R. Beyond standard Poisson-Boltzmann theory: ion-specific interactions in aqueous solutions. J. Phys. Condens. Matter. 2009;21:1–11. doi: 10.1088/0953-8984/21/42/424106. [DOI] [PubMed] [Google Scholar]
- 12.Raspaud E., da Conceiçao M., Livolant F. Do free DNA counterions control the osmotic pressure? Phys. Rev. Lett. 2000;84:2533–2536. doi: 10.1103/PhysRevLett.84.2533. [DOI] [PubMed] [Google Scholar]
- 13.Andresen K., Das R., Pollack L. Spatial distribution of competing ions around DNA in solution. Phys. Rev. Lett. 2004;93:248103. doi: 10.1103/PhysRevLett.93.248103. [DOI] [PubMed] [Google Scholar]
- 14.Das R., Mills T.T., Pollack L. Counterion distribution around DNA probed by solution X-ray scattering. Phys. Rev. Lett. 2003;90:188103. doi: 10.1103/PhysRevLett.90.188103. [DOI] [PubMed] [Google Scholar]
- 15.Pabit S.A., Qiu X., Pollack L. Both helix topology and counterion distribution contribute to the more effective charge screening in dsRNA compared with dsDNA. Nucleic Acids Res. 2009;37:3887–3896. doi: 10.1093/nar/gkp257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bai Y., Chu V.B., Doniach S. Critical assessment of nucleic acid electrostatics via experimental and computational investigation of an unfolded state ensemble. J. Am. Chem. Soc. 2008;130:12334–12341. doi: 10.1021/ja800854u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korolev N., Lyubartsev A.P., Nordenskiöld L. Competitive binding of Mg2+, Ca2+, Na+, and K+ ions to DNA in oriented DNA fibers: experimental and Monte Carlo simulation results. Biophys. J. 1999;77:2736–2749. doi: 10.1016/s0006-3495(99)77107-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiu X., Kwok L.W., Pollack L. Measuring inter-DNA potentials in solution. Phys. Rev. Lett. 2006;96:138101. doi: 10.1103/PhysRevLett.96.138101. [DOI] [PubMed] [Google Scholar]
- 19.Podgornik R., Rau D.C., Parsegian V.A. The action of interhelical forces on the organization of DNA double helices - fluctuation-enhanced decay of electrostatic double-layer and hydration forces. Macromolecules. 1989;22:1780–1786. [Google Scholar]
- 20.Chu V.B., Bai Y., Doniach S. Evaluation of ion binding to DNA duplexes using a size-modified Poisson-Boltzmann theory. Biophys. J. 2007;93:3202–3209. doi: 10.1529/biophysj.106.099168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanduc M., Dobnikar J., Podgornik R. Counterion-mediated electrostatic interactions between helical molecules. Soft Matter. 2009;5:868–877. [Google Scholar]
- 22.Manning G.S., Ray J. Counterion condensation revisited. J. Biomol. Struct. Dyn. 1998;16:461–476. doi: 10.1080/07391102.1998.10508261. [DOI] [PubMed] [Google Scholar]
- 23.Trizac E., Bocquet L., von Grunberg H.H. Alexander’s prescription for colloidal charge renormalization. Langmuir. 2003;19:4027–4033. [Google Scholar]
- 24.Bloomfield V.A. DNA condensation by multivalent cations. Biopolymers. 1997;44:269–282. doi: 10.1002/(SICI)1097-0282(1997)44:3<269::AID-BIP6>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 25.Ha B.Y., Liu A.J. Effect of non-pairwise-additive interactions on bundles of rodlike polyelectrolytes. Phys. Rev. Lett. 1998;81:1011–1014. [Google Scholar]
- 26.Kjellander R., Marcelja S. Correlation and image charge effects in electric double-layers. Chem. Phys. Lett. 1984;112:49–53. [Google Scholar]
- 27.Oosawa F. Interaction between parallel rodlike macroions. Biopolymers. 1968;6:1633–1647. [Google Scholar]
- 28.Shklovskii B.I. Wigner crystal model of counterion induced bundle formation of rodlike polyelectrolytes. Phys. Rev. Lett. 1999;82:3268–3271. [Google Scholar]
- 29.Rouzina I., Bloomfield V.A. Macroion attraction due to electrostatic correlation between screening counterions 1. Mobile surface-adsorbed ions and diffuse ion cloud. J. Phys. Chem. 1996;100:9977–9989. [Google Scholar]
- 30.Naji A., Netz R.R. Attraction of like-charged macroions in the strong-coupling limit. Eur Phys J E Soft Matter. 2004;13:43–59. doi: 10.1140/epje/e2004-00039-x. [DOI] [PubMed] [Google Scholar]
- 31.Tan Z.J., Chen S.J. Ion-mediated nucleic acid helix-helix interactions. Biophys. J. 2006;91:518–536. doi: 10.1529/biophysj.106.084285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Y.G., Weeks J.D. Local molecular field theory for effective attractions between like charged objects in systems with strong Coulomb interactions. Proc. Natl. Acad. Sci. USA. 2006;103:7560–7565. doi: 10.1073/pnas.0600282103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de la Cruz M.O., Belloni L., Drifford M. Precipitation of highly-charged polyelectrolyte solutions in the presence of multivalent salts. J. Chem. Phys. 1995;103:5781–5791. [Google Scholar]
- 34.Kornyshev A.A., Leikin S. Electrostatic zipper motif for DNA aggregation. Phys. Rev. Lett. 1999;82:4138–4141. [Google Scholar]
- 35.Stanley C., Rau D.C. Evidence for water structuring forces between surfaces. Curr. Opin. Colloid Interface Sci. 2011;16:551–556. doi: 10.1016/j.cocis.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linse P. Structure, phase stability, and thermodynamics in charged colloidal solutions. J. Chem. Phys. 2000;113:4359–4373. [Google Scholar]
- 37.Boroudjerdi H., Kim Y.W., Serr A. Statics and dynamics of strongly charged soft matter. Phys. Rep. 2005;416:129–199. [Google Scholar]
- 38.Guldbrand L., Jonsson B., Linse P. Electrical double-layer forces: a Monte-Carlo Study. J. Chem. Phys. 1984;80:2221–2228. [Google Scholar]
- 39.Gronbech-Jensen N., Mashl R.J., Gelbart W.M. Counterion-induced attraction between rigid polyelectrolytes. Phys. Rev. Lett. 1997;78:2477–2480. [Google Scholar]
- 40.Dai L., Mu Y.G., van der Maarel J.R.C. Molecular dynamics simulation of multivalent-ion mediated attraction between DNA molecules. Phys. Rev. Lett. 2008;100:118301. doi: 10.1103/PhysRevLett.100.118301. [DOI] [PubMed] [Google Scholar]
- 41.Luan B., Aksimentiev A. DNA attraction in monovalent and divalent electrolytes. J. Am. Chem. Soc. 2008;130:15754–15755. doi: 10.1021/ja804802u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Allahyarov E., Gompper G., Lowen H. Attraction between DNA molecules mediated by multivalent ions. Phys. Rev. E. 2004;69:041904. doi: 10.1103/PhysRevE.69.041904. [DOI] [PubMed] [Google Scholar]
- 43.Yoo J.J., Aksimentiev A. Improved parametrization of Li+, Na+, K+, and Mg2+ ions for all-atom molecular dynamics simulations of nucleic acid systems. J. Phys. Chem. Lett. 2012;3:45–50. [Google Scholar]
- 44.Podgornik R. Polyelectrolyte-mediated bridging interactions. J. Polym. Sci. Pol. Phys. 2004;42:3539–3556. [Google Scholar]
- 45.Ritort F., Mihardja S., Bustamante C. Condensation transition in DNA-polyaminoamide dendrimer fibers studied using optical tweezers. Phys. Rev. Lett. 2006;96:118301. doi: 10.1103/PhysRevLett.96.118301. [DOI] [PubMed] [Google Scholar]
- 46.Evans H.M., Ahmad A., Safinya C.R. Structural polymorphism of DNA-dendrimer complexes. Phys. Rev. Lett. 2003;91:075501. doi: 10.1103/PhysRevLett.91.075501. [DOI] [PubMed] [Google Scholar]
- 47.Mills M., Orr B., Andricioaei I. Microscopic basis for the mesoscopic extensibility of dendrimer-compacted DNA. Biophys. J. 2010;98:834–842. doi: 10.1016/j.bpj.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mills M., Orr B.G., Andricioaei I. Attractive hydration forces in DNA-dendrimer interactions on the nanometer scale. J. Phys. Chem. B. 2013;117:973–981. doi: 10.1021/jp309616t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raspaud E., Durand D., Livolant F. Interhelical spacing in liquid crystalline spermine and spermidine-DNA precipitates. Biophys. J. 2005;88:392–403. doi: 10.1529/biophysj.104.040113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang J., Rau D.C. Incomplete ion dissociation underlies the weakened attraction between DNA helices at high spermidine concentrations. Biophys. J. 2005;89:1932–1940. doi: 10.1529/biophysj.105.065060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pelta J., Livolant F., Sikorav J.L. DNA aggregation induced by polyamines and cobalthexamine. J. Biol. Chem. 1996;271:5656–5662. doi: 10.1074/jbc.271.10.5656. [DOI] [PubMed] [Google Scholar]
- 52.Korolev N., Berezhnoy N.V., Nordenskiöld L. A universal description for the experimental behavior of salt-(in)dependent oligocation-induced DNA condensation. Nucleic Acids Res. 2009;37:7137–7150. doi: 10.1093/nar/gkp683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matulis D., Rouzina I., Bloomfield V.A. Thermodynamics of DNA binding and condensation: isothermal titration calorimetry and electrostatic mechanism. J. Mol. Biol. 2000;296:1053–1063. doi: 10.1006/jmbi.1999.3470. [DOI] [PubMed] [Google Scholar]
- 54.Todd B.A., Rau D.C. Interplay of ion binding and attraction in DNA condensed by multivalent cations. Nucleic Acids Res. 2008;36:501–510. doi: 10.1093/nar/gkm1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raspaud E., Olvera de la Cruz M., Livolant F. Precipitation of DNA by polyamines: a polyelectrolyte behavior. Biophys. J. 1998;74:381–393. doi: 10.1016/S0006-3495(98)77795-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Andresen K., Qiu X., Pollack L. Mono- and trivalent ions around DNA: a small-angle scattering study of competition and interactions. Biophys. J. 2008;95:287–295. doi: 10.1529/biophysj.107.123174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qiu X., Andresen K., Pollack L. Abrupt transition from a free, repulsive to a condensed, attractive DNA phase, induced by multivalent polyamine cations. Phys. Rev. Lett. 2008;101:228101. doi: 10.1103/PhysRevLett.101.228101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoshikawa K., Takahashi M., Khokhlov A.R. Large discrete transition in a single DNA molecule appears continuous in the ensemble. Phys. Rev. Lett. 1996;76:3029–3031. doi: 10.1103/PhysRevLett.76.3029. [DOI] [PubMed] [Google Scholar]
- 59.Bloomfield V.A. Static and dynamic light scattering from aggregating particles. Biopolymers. 2000;54:168–172. doi: 10.1002/1097-0282(200009)54:3<168::AID-BIP20>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 60.Ke, F. Y., Y. K. Luu, …, D. H. Liang. 2010. Characterizing DNA condensation and conformational changes in organic solvents. PLoS ONE. 5:e13308. http://www.plosone.org/article/info:doi/10.1371/journal.pone.0013308. [DOI] [PMC free article] [PubMed]
- 61.Rau D.C., Parsegian V.A. Direct measurement of the intermolecular forces between counterion-condensed DNA double helices. Evidence for long range attractive hydration forces. Biophys. J. 1992;61:246–259. doi: 10.1016/S0006-3495(92)81831-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Feigon J., Butcher S.E., Hud N.V. Solution nuclear magnetic resonance probing of cation binding sites on nucleic acids. Methods Enzymol. 2001;338:400–420. doi: 10.1016/s0076-6879(02)38230-2. [DOI] [PubMed] [Google Scholar]
- 63.Teif V.B. Ligand-induced DNA condensation: choosing the model. Biophys. J. 2005;89:2574–2587. doi: 10.1529/biophysj.105.063909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yager T.D., van Holde K.E. Dynamics and equilibria of nucleosomes at elevated ionic strength. J. Biol. Chem. 1984;259:4212–4222. [PubMed] [Google Scholar]
- 65.Deserno M., Holm C. Theory and simulations of rigid polyelectrolytes. Mol. Phys. 2002;100:2941–2956. [Google Scholar]
- 66.Baker N.A., Sept D., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rouzina I., Bloomfield V.A. Competitive electrostatic binding of charged ligands to polyelectrolytes: practical approach using the non-linear Poisson-Boltzmann equation. Biophys. Chem. 1997;64:139–155. doi: 10.1016/s0301-4622(96)02231-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.