

Abstract
Deletion of the β-bulge trigger-loop results in both a switch in the preferred folding route, from the functional loop packing folding route to barrel closure, as well as conversion of the agonist activity of IL-1β into antagonist activity. Conversely, circular permutations of IL-1β conserve the functional folding route as well as the agonist activity. These two extremes in the folding-functional interplay beg the question of whether mutations in IL-1β would result in changes in the populations of heterogeneous folding routes and the signaling activity. A series of topologically equivalent water-mediated β-strand bridging interactions within the pseudosymmetric β-trefoil fold of IL-1β highlight the backbone water interactions that stabilize the secondary and tertiary structure of the protein. Additionally, conserved aromatic residues lining the central cavity appear to be essential for both stability and folding. Here, we probe these protein backbone-water molecule and side chain-side chain interactions and the role they play in the folding mechanism of this geometrically stressed molecule. We used folding simulations with structure-based models, as well as a series of folding kinetic experiments to examine the effects of the F42W core mutation on the folding landscape of IL-1β. This mutation alters water-mediated backbone interactions essential for maintaining the trefoil fold. Our results clearly indicate that this perturbation in the primary structure alters a structural water interaction and consequently modulates the population of folding routes accessed during folding and signaling activity.
Introduction
In β-trefoil proteins, conserved aromatic residues lining the central cavity are common (1) and appear to be essential for both stability and folding. For the six-stranded β-barrel protein interleukin 1-β (IL-1β), deviations of β-strands away from optimal hydrogen-bonding geometry require bridging waters to maintain hydrogen-bonding interactions between neighboring β-strands (2), which alter the hydrogen-bonding network of the IL-1β trefoil structure and are responsible for many of the effects we observe in the folding, stability, and cooperativity (1,3). This suggests that structural waters buried within the protein cavity are essential components for both efficient folding and transmission of function (4–10).
The β-barrel of IL-1β is a geometrically stressed structure (11) that uses three water-protein interactions as structural lynchpins, pinning the cap to the β-barrel (Fig. 1, A and B). Analysis of structural water interactions using available structures of core mutants revealed a rearrangement of the backbone hydrogen-bonding characteristics for the F42W mutant (PDB ID 1L2H). In the first trefoil subunit, β-strands 1–4, we observe interactions between water and residues L10, L18, and V40. Previous studies (12–14) also detected the presence of two waters in the first trefoil, linking L18→L10 and V40→L10 (donor→acceptor). Hydration observed in the crystal structure of the F42W IL-1β mutant, where the introduction of aromatic residues within the hydrophobic core of the molecule resulted in changes to the side-chain hydrogen-bonding interactions (1) (Fig. 1 C), agrees with observations based on NMR data (15), showing that the structural water no longer interacts with V40. A schematic of the W42-L10-L18 water interactions is depicted in Fig. 1 C. The side-chain amide nitrogen of F42 is 2.93 Å from the nearest water oxygen, consistent with the expected N-Hε···O bond length of 2.8 Å (16). The adaptation in the hydration of the β-trefoil fold in the F42W sequence variant illustrates the robustness of the structural waters. However, the resilience of water interactions argues for the overall stability of the macroscopic average structure, consistent with the experimental observation where these mutations did not result in significant changes in stability (17). Furthermore, it is possible and probable that the populations of the native state ensembles of the sequence variants differ. Conformational flexibility or increased sampling within the native state ensemble may occur as previous folding kinetic studies showed that aromatic residues around the internal hydrophobic cavity of IL-1β play essential roles in folding (1,17), as folding of the F42 mutant was dramatically different compared to wild-type (WT) (17).
Figure 1.
The structural waters involved in IL-1β. (A) The three-dimensional NMR structure of IL-1β (PDB ID: 6I1B). The six β-barrel strands are in blue, whereas the six β-strands composing the cap are in red. The waters are depicted in the space-filling format as gray spheres. Structural waters and respective interactions discussed are highlighted as black spheres. (B) A two-dimensional representation of the pseudo-threefold symmetric β-trefoil fold. The β-strands are numbered, and the position of structural waters (black) and their H-bond partners are indicated. The trefoil units are numbered I–III. (C) Schematic of the WT V40-L10-L18 (upper panel) W42-L10-L18 (lower panel) water interactions. The mutant IL-1β interactions were taken from the crystal structure of the F42W mutant IL-1β sequence variant (1L2H (1)). There are two waters in between the three residues leading to the following interpretation from their model: Water 1: HN-L10→O-WAT1, Hε -W42→O-WAT1. Water 1: HN-L18→O-WAT2, H-WAT2→ O′-L10.
Most proteins have a folding landscape low in energetic frustration, with few traps. Consequently, the topology of the native fold is the key factor in determining the folding mechanism (18). Energetically unfrustrated structure-based potentials have successfully been used to simulate the initial folding intermediate and preferred folding route of WT IL-1β (19) where interactions within the central trefoil subunit (β-strands 5–10) dominate the folding landscape, with additional influences coming from β-strand 4 (residues 40, 42, 43) (20). This route is most prevalently populated experimentally as it ensures the proper packing and orientation of the trigger loop in IL-1β that is key in the functional role of the protein (20–23). Perturbation events at any site on the protein surface (or within the interior) do not create new pathways for this protein but only shift the preexisting ensemble of pathways (22,24). Thus, although the folding of IL-1β progresses through population of a single, stable, and easily detected intermediate, characterized by the dominant functional loop-packing route (central trefoil β-strands nucleating first), additional studies have confirmed that multiple distinct routes (and thus intermediate species) to native both computationally (25,26) and experimentally (22,27) exist for IL-1β, where the initial nucleating β-strands differ from the WT dominant route (22). This begged the question of whether mutations in this protein would result in altered populations in folding routes and function.
Initial characterization of the F42W mutant highlighted altered folding kinetics that differed from WT protein and the single mutant (W120F), where F42W exhibited differences in observed refolding relaxation times (17). These changes appeared to be unique to the F42W mutant, as W120F displayed no change in relaxation rates when compared to the WT, thus the biphasic behavior of the F42W mutant during folding is related to interactions of W42.
Given our deeper understanding of the energy landscape of IL-1β where residue-specific minor energetic or geometric perturbations appear to alter the frequency of a chosen folding pathway (25,26), and recent experimental work confirming these complex predictions (21,22,27), we examined the effects of altering the hydrophobic interactions related to loop packing of the core of IL-1β with the F42W mutant and the resulting effects on the population dynamics of the previously characterized folding routes (25–27). We use a series of folding kinetic experiments, including pulse-labeling hydrogen-deuterium exchange (HDX) combined with electrospray ionization-mass spectrometry (ESI-MS), and folding simulations with structure-based models (SBM) (19,28,29), to characterize the changes observed in folding (17) as a result of this mutation. Our results show that although a folding intermediate containing significant secondary structure is populated by this variant, similar to that observed for WT, the longevity of the intermediate state was noticeably altered. Strikingly, the F42W mutant native state is formed on two different timescales, quite distinct from WT IL-1β (17). This supports the presence of multiple folding routes for IL-1β and indicates that a population shift in the routes can be modulated by subtle changes in water-backbone as well as side-chain interactions. Taken together, the delicate relationship between side-chain interactions, geometric stress, and route population is demonstrated by showing how an amino acid substitution, and the subsequent changes to structural backbone interactions, can help navigate the complex energy landscape during folding.
Materials and Methods
Expression, purification, and sample preparation
Site-directed mutagenesis, expression, and purification were performed to obtain F42W mutant IL-1β protein as previously described (17).
Pulse-labeling HDX
The F42W mutant protein was concentrated to 4 mg/ml and prepared as previously described (21–23). Refolding time points were collected from 20 msec to 2.5 s using a Biologic QFM5 quench flow, whereas manual mixing was carried out for time points greater than 2.5 s as previously described (23). Control reactions were carried out as previously described (23). All samples were then concentrated to 2.5 ml using a Centriprep10 (Amicon, Cork, Ireland) and the buffer exchanged to 50 mM ammonium acetate pH 5.0 using a PD10 column (Pharmacia, Buckinghamshire, UK). The samples were then concentrated to ∼0.5 mg/ml with a Centricon10 and stored at –20°C until further analysis.
Double-jump kinetic assay
Mutant IL-1β protein was unfolded in 2.4 M guanindinium hydrochloride (Gdn-HCl)/50 mM ammonium acetate pH 5.0 with a protein concentration of 1.2 mg/ml and refolded to 0.4 M Gdn-HCl by a 1:6 dilution into 50 mM ammonium acetate. The protein was allowed to refold for various times before an unfolding jump with a 1:2 dilution of 6 M Gdn-HCl. The change in fluorescent signal was followed over a time course of 1000 s. The final protein concentration in the reaction was 0.1 mg/ml with a Gdn-HCl concentration of 3.2 M. The data were obtained on the Applied Photophysics (Surry, UK) SX-17 MV similar to previously described techniques (17,22). All traces exhibited two exponential phases of 10 and 95 s. The amplitudes were determined from a global fit of the data with the time constants linked. The resulting amplitudes were then plotted as a function of delay time and fit to a single exponential.
1-anilino-8-naphthalene sulfonate (ANS) binding assay
Refolding of the F42W mutant in the presence of ANS was carried out in 50 mM ammonium acetate, pH 5.0, and 25°C as previously described (22). The protein was unfolded in 2.2 M Gdn-HCl and allowed to equilibrate overnight. Refolding jumps to final protein concentrations of 0.55, 0.1, and 0.2 mg/ml were determined in the presence of 0.2, 0.4, and 0.6 M Gdn-HCl with an ANS concentration of 100 mM. Because all studies were performed with a final protein concentration of 0.2 mg/ml the relaxation times were determined at this protein concentration.
Simulations with structure-based models (SBMs)
Simulations were performed with structure-based models of IL-1β. The WT model, coarse-grained at the Cα level, was built as in (25,26) from structure 6I1B, and using the same contact map, comprising 422 residue contacts. The form of the model is identical with the exception of the tertiary contact potential (28), which does not affect the observed mechanism. Water is not directly represented in the model, but effective solvent interactions are included by construction. In the region around the structural waters in trefoil unit I, the model contains contacts between residues Leu-10, Leu-18, Val-40, and Phe-42 that correspond to interactions mediated by hydrogen bonds made by the two water molecules (Fig. 1). Assuming that the shorter hydrogen bonds formed by the displaced structural water in the F42W mutant are also stronger, we have modeled the effect of the mutation by strengthening the contact between residues Leu-10 and Phe-42 by a factor of three. Simulations of the WT and mutant models were done with a customized version of Gromacs (30). Umbrella sampling was applied to a continuous analog of the number of native contacts Q to sample across the whole reaction coordinate and especially in the transition region. Simulations for both systems were performed at a constant reduced temperature of T = 1.13, close to both their respective equilibrium folding temperatures. Analysis of the folding routes in the WT and mutant SBMs identified structures with 210 to 220 formed contacts, corresponding to Q = 0.5, which were at the same time at the bottom of the applied umbrella potential, thus no umbrella force was acting upon them. The structures obtained from both the WT and the F42W mutant models were collected separately and clustered by structural similarity, expressed through the measure of Qmf defined in (31), using the UPGMA algorithm implemented in the neighbor program of the phylip package (32). Inspection of the resulting trees with the programs, Dendroscope (33) and VMD (34), confirmed that the two major subtrees correspond to the three available folding routes previously described for WT IL-1β (functional loop-packing, backtracking, and barrel-closing routes) (25,26). By counting structures in each subtree that originated from the WT or mutant model, relative weights of the two routes in both systems could be determined. Joint clustering of the structures from both WT and mutant models together confirmed the results.
In addition to the equilibrium simulations, using the same structure-based potentials, we performed kinetic folding runs of the WT and mutant models. A total of 125 unfolded structures were taken from five different runs at T = 1.5 for each model. Each of these structures was refolded into five independent runs at T = 1.0 for a total of 625 refolding attempts for either WT or mutant. The refolding temperature lies ∼10% below Tf, which is the largest degree of undercooling practical in this system because of increasing local trapping.
Stimulation of A549 cells with IL-1β
A549 epithelial lung carcinoma cells were cultured in Ham’s F12K medium supplemented with 2 mM L-glutamine, 1.5 g/L sodium bicarbonate, and 10% fetal bovine serum (Invitrogen, Carlsbad, CA) and incubated at 37°C with 5% CO2. Before the assay, 25,000 cells were seeded per well in a 96-well plate, and allowed to adhere to the plate for 15 h at 37°C, 5% CO2. WT and F42W/W120F protein concentrations were determined by Bradford assay and subsequently normalized to 2 μg/ml. The cells were stimulated with varying amounts of WT or F42W/W120F IL-1β for 4 h, and the supernatant collected. The supernatant was diluted 1:20 with media and stored at 4°C overnight. The level of secreted IL-6 was measured by enzyme-linked immunosorbent assay and quantified against IL-6 standards (eBioscience, San Diego, CA).
Results
To date, details of the complex nature of the folding mechanism of IL-1β have been effectively characterized through the combined use of computer simulations (25,26,35) and experimentation (20,22,23,27). More recently, greater insight regarding route selection and population dynamics of IL-1β has been gained from the partnership of these two approaches, detailing, an interplay between folding and function on a complex folding landscape with multiple routes available for folding (21,22,36). In this study, in vitro experiments and SBMs were coupled to elucidate the role of structured-water backbone interactions in modulating the folding landscape of IL-1β.
Computer simulations modeling altered structural water interactions
For equilibrium simulations of IL-1β, the dominant component of the equilibrium distribution of structures in the transition region in the WT model corresponds to the functional route (packing of the central trefoil β-strands). A second minor route consists of the barrel-closure and backtracking routes (both require the terminal ends to coalesce first). In the mutant model, the population of these minor termini-related routes is increased at the expense of the functional route (Fig. 2). Although a preference for either of these routes cannot be identified, the shift away from the functional route is significant (80% population for WT, 68% population for F42W, respectively) (Fig. 2, A and B). Furthermore, the distribution in the simulations below Tf, under strong refolding conditions, are more reflective of experimental conditions (i.e., strong refolding/unfolding), and reveal a similar increase in the fraction of barrel-closure structures for the mutant model, where the difference between WT and mutant is greater than in the equilibrium simulations (Fig. 2, B and C). It is interesting to note that the behavior in the undercooling runs is quite different from that of the equilibrium data, suggesting strong kinetic effects on the folding mechanism for these proteins. As both sets of simulations show a clear difference in populations of folding routes in the F42W mutant, the question now becomes, can these population changes be experimentally observed given the dominance of the functional folding route population?
Figure 2.
The simulated multiple folding routes for the F42W IL-1β mutant. (A) Contact maps highlighting the different available folding routes for the F42W mutant. The functional loop-packing route (purple, right), where contacts from the central and C-terminal trefoil units form first, is the dominant population for the WT and mutant IL-1β. For the mutant protein, this route is less populated as a result of the altered water interaction. The barrel-closure route (green, left), where the N- and C-termini first come together, thereby closing the β-barrel, is more populated as a result of the altered contacts. (B) A bar graph representing the shift in route distribution seen in simulated equilibrium experiments as a result of the change in side-chain and water interaction for the mutant protein. (C) Results of SBM modeling below Tf for WT (gray) and mutant (blue) models. Colored traces represent the mean fraction formed contact interactions in the N-terminal region. At Q = 0.5 (dashed vertical line), the mutant shows a substantial gain in N-terminal contacts. Errors are smaller than symbols.
Altered folding mechanism monitored optically
Experimentally, the refolding of WT IL-1β is characterized by the fast formation of an intermediate followed by the slow formation of the native state. Previous studies have shown that the refolding reaction is devoid of a burst phase and that the intermediate and native species exhibit relaxation times that are different from one another (23). Formation of the IL-1β intermediate can also be monitored by ANS as the fluorophore allows for optical detection of this structure (37–40). In particular, for IL-1β, ANS binds to the hydrophobic region of the protein associated with the kinetic intermediate and the geometrically stressed functional loop-packing region (β-strands 5–10, part of β-strand 4) (22,41). Using this fluorescent probe, refolding in the presence of ANS was employed to assess the formation of the intermediate in the F42W mutant. Fig. 3 shows the refolding kinetics as monitored by ANS fluorescence. The process exhibits an increase in fluorescence, followed by a biexponential decay in the fluorescence intensity. The kinetic behavior of the F42W mutant (Fig. 3, inset) differs from that of WT IL-1β where release of ANS is noticeably faster than previously determined for WT IL-1β (22). This distinct difference in release of ANS provides a signature signal for an altered mechanism of folding (22) for the mutant protein. The kinetic behavior for F42W was adequately fit to three phases with relaxation times of 31 msec, 2.2 s, and 18.4 s.
Figure 3.
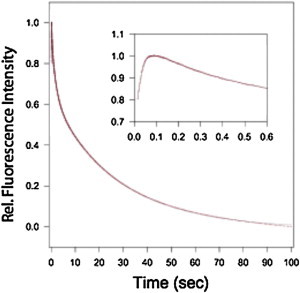
Plot of the relative fluorescence versus time for the refolding of the mutant in the presence of ANS. The refolding data are in red, whereas the fit is shown in black. The process exhibits an increase in fluorescence, followed by a biexponential decay in the fluorescence intensity. The inset shows the first 600 msec of the folding reaction. The kinetic behavior of the F42W mutant differs from that of WT IL-1β where release of ANS is noticeably faster than that previously determined for WT IL-1β (22) in the initial phase of folding (inset).
Shift in the population ensemble for the F42W mutant protein
Characterization of the folding populations using the refolding of the F42W mutant sequence variant was carried out by HDX pulse labeling and analyzed by ESI-MS. As a necessary prerequisite to folding studies, the number of protons stable to solvent exchange, which can serve as probes for folding, was measured. A comparison of the ESI-MS spectra of the protonated and deuterated control samples for the +10 charge state is shown in Fig. 4 A. The difference between the two peaks corresponds to 48 Da; therefore, the mutant has 48 probes with which to follow the folding process.
Figure 4.
(A) Plot of the mass difference between the unfolded and folded F42W IL-1β mutant protein. The masses differ by 48 Da, which is due to the trapping of stable deuterium atoms in the folded state of the protein. The mass/charge ratio corresponds to the +10 charge peak. (B) Plot of the hydrogen-exchange data for the F42W mutant from 20 msec to 300 s after the initiation of folding. The decay of the unfolded population with the increase of an intermediate is evident along with the formation of the native state. The time is plotted in milliseconds on a logarithmic scale where the mass/charge ratio corresponds to the +10 charge peak. (C) Plot of the population versus time for the folding of F42W/W120F. The populations of U, I, and N are indicated by open triangles, solid circles, and open circles, respectively. The lines represent the fit of the data to a parallel pathway model where the intermediate folds to the native state by two discrete paths. The dashed line represents the various populations for WT IL-1β as represented in (23).
The refolding reaction was followed from 0 to 300 sec. with a combination of rapid quench and manual-mixing techniques. Time points earlier than 2.5 sec were determined by rapid quench, whereas those greater than 5 sec were collected by manual mixing, as previously described (23). Fig. 4 B shows the full time course of the folding reaction. Early time points indicate no significant protection from exchange. The production of an intermediate species with 22 amides protected from exchange is evident, as is the subsequent formation of native protein. The native state does not appear before the intermediate species and has an increase in mass of 26 Da over this intermediate. Amide protection obtained for the mutant is similar to the WT protein in that the intermediate shows protection of 20, whereas the native traps 24 deuterons. Rate of intermediate formation is similar to that observed for WT intermediate formation (23).
For clarity, the population data of the U (unfolded), I (intermediate), and N (native) species over time are plotted as a two-dimensional projection in Fig. 4 C. In WT, the intermediate species is highly populated and long-lived where the intermediate builds to a maximal value at 200 msec and starts to decay around 20 s (23) (Fig.4 C, dashed lines). In the case of the F42W mutant, the folding intermediate reaches a maximal population within 200 msec and then starts to decay after 1 s, a stark contrast when compared to WT (Fig.4 C) (23). The shorter-lived intermediate population for the F42W mutant has a faster turnover to native from the intermediate species when compared to WT IL-1β. Faster turnover of the F42W intermediate indicates a shift in the population dynamics, where alteration of the backbone-water (1) and packing interactions contribute to the observed changes in the intermediate species, as a greater population of native is present earlier in folding, compared to WT (Fig. 4 C). We hypothesize that this faster turnover of the intermediate is a result of destabilization of the strained topology (14), as more fluid backbone dynamics lead to a shift in the route populations as functional-folding strain is reduced, and therefore a single route is no longer dominant in folding.
Double-jump assay demonstrates multiple routes to native
A conventional experiment to probe for the presence of multiple populations during folding is the double-jump kinetic assay (Fig. 5 A). For WT IL-1β, the slow rate of unfolding and broadness of the intermediate state is not amenable to this particular experiment, but mutants of the protein that unfold faster than WT can be analyzed. To verify the observed shift in the route population and experimentally confirm multiple routes to native during folding, a series of double-jump assays were performed on the F42W mutant where the protein was allowed to refold over a time course from 18 msec to 200 s before the initiation of the unfolding jump (Fig. 5 A). The extent of formation of N at any time during folding was assayed by the rate as well as amplitude of the fast phase of unfolding. The amplitudes determined for each relaxation phase were then plotted and fit to a single exponential as shown in Fig. 5 B. The data for native state formation from the double-jump assay indicate two different relaxation times leading to native formation, giving direct experimental evidence of the bifurcation of the folding pathway as multiple routes to the native state ensemble are observed (Fig. 5 B).
Figure 5.
(A) Schematic of the double jump experiment used to identify multiple folding routes. For the F42W mutant, the protein was amenable to the assay that probed for multiple routes to native, as the observed rate of unfolding is noticeably faster than WT (17). Cyan represents the functional-relevant 90’s loop and β-bulge. The two routes that are available can easily be monitored optically. These are the barrel-closure and functional-loop packing routes. (B) Plot of the fluorescence amplitudes as a function of time for F42W mutant IL-1β. The amplitudes were determined from the double jump assay where the unfolded protein was refolded to 0.4 M Gdn-HCl for the period of time shown on the abscissa. The protein was subsequently unfolded and the amplitudes for each kinetic phase determined. The data indicate two different relaxation times leading to native formation and is experimental evidence of the bifurcation of the folding pathway and multiple routes to the native state ensemble.
Reduced activity in mutant protein compared to WT
IL-1β has been shown to enhance the expression of IL-6 in several different cell types (43,44). IL-1β activity, thus, can be monitored by the changes in expression of IL-6 in stimulated cells in vitro. Changes in the secretion of IL-6 as a result of IL-1β mutation are observed for the F42 mutation (Fig. 6), where the mutant shows reduced stimulation of IL-6, compared to WT. The reduced up-regulation of IL-6 suggests that altered packing of the core reduces signal transmission as a result of the modified hydration interaction.
Figure 6.
Plot indicating reduced signaling activity for the F42 mutant. IL-1β has been shown to enhance the expression of IL-6. Changes in the secretion of IL-6 as a result of IL-1β mutation are observed for the F42 mutation (blue), where the mutant shows reduced stimulation of IL-6 (blue), compared to WT (black).
Discussion
Packing of the hydrophobic core
The hydrophobic core of WT IL-1β contains a large cavity, calculated from the crystallographic structure to be ∼88 Å3 (1). Although Phe-42 and Trp-42 have similar placements, the side chain of the Trp-42 extends further into the cavity than that of the Phe-42, reducing the total cavity volume. Interestingly, instead of the backbone amide of Trp-42 participating in hydrogen-bonding interactions, as occurs with Val-40 forming an interstrand hydrogen bond to Cys-8 in WT, the indole amide of Trp-42 participates in hydrogen bonding to a structural water molecule (Fig. 1, B and C) (1). The change in backbone bonding of a structural water molecule in the mutant (Fig. 1 C) (1) may improve the packing of the functional loops with little added geometric strain. Although the reduction of cavity volume and swapped hydrogen bonding has had little effect on protein stability (17), these consequences confirm that core packing and geometric strain are rate limiting in the folding of IL-1β. In the case of this F42W mutant, the presumed reduction of strain in the molecule increases the rate of unfolding (17) and may contribute to more efficient and dynamic folding as a result of the change in the hydrophobic core.
In WT IL-1β, folding is initiated via the dominant functional route (β-strands 5–10, part of β-strand 4), where early collapse of the central trefoil region is observed experimentally, as detected by quench-flow mass spectroscopy and HDX-NMR spectroscopy (20,23). Partially folded protein, at times, needs to backtrack, where initial native contacts loosen to allow for packing of strained barrel strands that include pinning down the cap to the β-barrel, before reaching the final native fold (26,27). Based on HDX pulse labeling NMR spectroscopy, the barrel-closure route (folding initiated by β-strand 1 and 12, i.e., the terminal ends) will dominate the folding landscape of IL-1β if geometric stress imparted by packing of functional relevant loops is reduced or eliminated (22). Here, we speculate that interactions within the intermediate between residues in β-strand 4 (Val-40, Phe-42, Ser-43), contribute to route selection/switching. For the F42W mutant protein, altering and strengthening of the mutant-water contacts computationally (Fig. 2), and changes in the backbone water interaction experimentally (Fig. 1 C (1)), along with subtle alterations to the overall cavity volume of the mutant protein (1,45), shift the sampling between the available routes (Figs. 4 and 5) and signaling efficacy (Fig. 6). The extension of the hydrogen-bonding network between β-strand 1 and β-strand 4 as a result of the distinct changes in backbone interaction switching from the backbone N of Val-40 to the indole ring N of Trp-42 (Fig. 1 C (1)), allows for a subtle relaxation of the constraint imposed by packing of the β-bulge functional loop, located between β-strand 4 and β-strand 5. As previously shown, reducing geometric strain within the IL-1β molecule contributes significantly to route selection/switching (22). Thus, the less-strained mutation results in an experimentally observed shift from a single dominant folding route to a more dynamic folding landscape as the native state is achieved much faster (Fig. 4 C) and via multiple routes (Fig. 5) for the F42W mutant, compared to WT IL-1β.
In addition to packing of the hydrophobic core, backbone water interactions in proteins may do more than stabilize secondary and tertiary structure; they may also have functional roles. For IL-1β, which binds to the interleukin type I receptor using two large distinct docking surfaces designated A and B, located on either end of the IL-1β molecule (46) (Fig. 1 A), mutations at site A propagates binding and structural changes to site B via the hydrogen-bonding network (47). Simulation data outline the coupling mechanism further with the hypothesis that hydration in IL-1β can thermodynamically enhance hydrogen bond networks and promote coupling across long distances, which in turn allows kinetic control of transmission of signal (48). This hydration coupling not only is important for biological function (47) (Fig. 6), but, as demonstrated here, may also significantly affect folding and population dynamics.
Route population shift observed in folding kinetics
Initial characterization of the F42W mutant protein highlighted folding kinetics that were different from WT protein and the single mutant (W120F), where F42W exhibited differences in observed relaxation times for refolding (17). As W120F displayed no change in relaxation rates when compared to the WT, the biphasic behavior of F42W during folding was interpreted as an alteration in the kinetic barrier from the U to I to N transitions (17) related to side-chain interactions of Trp-42. Probing the effects of the altered side-chain interactions through simulation revealed that, indeed, there is a shift in the population dynamics for the mutant protein (Fig. 2) despite the smoothness of the energy landscape in the SBMs that tend to minimize perturbations. Interestingly, experimentally, it has been recently determined that one can probe the various routes to native IL-1β with ANS binding, where the different routes to native yield distinctive observed changes in ANS release upon intermediate formation (22). Folding by the barrel-closure route has a distinctly different signal by ANS compared to the functional loop-packing route that dominates the folding of WT IL-1β (22). The trace indicated in Fig. 3, where release of the fluorophore and the subsequent decay in signal is significantly faster in the initial phase of the folding reaction (i.e., first 5 s), than that of WT protein, is strikingly similar to that observed for the bulge-deletion construct (22), suggesting a shift in route selection. This population shift is attributed to an increased population of the barrel-closure route for the formation of the native mutant protein.
Multiple routes lead to the native state
HDX pulse labeling experiments performed indicate that the relaxation time for the intermediate formation is similar between the F42W mutant and WT proteins (Fig. 4). However, with F42W, the kinetic barrier for the transition from intermediate to native state apparently can be crossed by more than one route, as indicated by the double-jump assay (Fig. 5 B). We interpret this as a significant change in the population distribution of the various routes of folding for IL-1β. Hence, the reduced kinetic barrier of the intermediate ensemble and its turnover to native state are an indication of a redistribution of the population landscape. Consistent with both the SBM simulations (Fig. 2) and the ANS refolding experiment (Fig. 3), the population shift resulted in more of the barrel-closure route being populated for the mutant. Additionally, the turnover of the intermediate to native is increased, as highlighted by the decreased longevity (broadness) of the intermediate population as viewed by ESI-MS (Fig. 4 C). All this is linked to the change in the side-chain interaction where the decrease in hydrophobicity and a subtle alteration in cavity volume in the local environment for Trp-42 allow for increased flexibility from reduced geometric strain and greater observed heterogeneity in the multiple available routes. Furthermore, these changes translate into reduced activity, demonstrating the interplay between folding and function (Fig. 6).
Conclusion
From both computational and experimental techniques, the kinetic behavior of the F42W mutant of IL-1β indicates the delicate balance of interactions involved in protein folding. The results described here agree with our view of protein folding-function interplay proceeding via multiple competing pathways on a funneled energy landscape. Using the geometrically strained IL-1β molecule as a model system, the altered packing of the bulkier hydrophobic side chain of Trp-42 into the hydrophobic core of the β-barrel clearly modulates route selection (Fig. 7). The subtle difference of the Trp-42 structural water interaction relative to WT Val-40 leads to an indirect geometric rearrangement of structure, where the larger aromatic side chain of Trp-42 packs into the hydrophobic core, swapping the backbone hydrogen bonding, perturbing the interstrand packing between β-strand 1 and β-strand 4 and, ultimately, the cavity volume of the protein. These differences, similar to a switch on a train track, propagate into changes in the folding mechanism that lead to a shift in route population (Figs. 2, 4, and 7). Shifting of route populations can be more demonstrative, with dominant routes (22) or transition states (49) completely switching, with similar functional implications (22). For the water-interaction changing mutation illustrated here, the change is reflected in an increase in alternate routes, allowing for greater experimentally observed heterogeneity in folding routes and subsequent changes in signaling activity. Taken together, subtle changes in the local environment (β-strand 4 interactions) related to function (packing of the functional loops) translate into larger changes in folding (increased route heterogeneity) as the drive for efficient folding, an unperturbed native state, and functional signaling intersect.
Figure 7.
Schematic representing the effects of the altered cavity and structural interactions of the Trp-42 mutation on folding route selection for IL-1β. Similar to a switch in a train track, the change in backbone hydrogen-bonding interaction and cavity volume for Trp-42 (red) modulates the selection of routes for the folding of IL-1β, through the altered interactions effecting β-strand 4 (yellow). For the F42W mutant, switching between the two routes (green and purple) leads to more efficient folding.
Acknowledgments
We thank members of both laboratories, in particular Ellinor Haglund and Joanna Sulkowska, for discussion and review of this work. We also thank Peter Wright for use of the quench-flow instrument.
This work was supported by National Institutes of Health grants R01 GM054038 (P.A.J., D.T.C., M.R.) and by the Center for Theoretical Biological Physics sponsored by the National Science Foundation (NSF) grants PHY-1308264 and MCB-1214457 (H.L., J.N.O.). J.N.O. is a CPRIT Scholar in Cancer Research sponsored by the Cancer Prevention and Research Institute of Texas.
References
- 1.Adamek D.H., Guerrero L., Caspar D.L. Structural and energetic consequences of mutations in a solvated hydrophobic cavity. J. Mol. Biol. 2005;346:307–318. doi: 10.1016/j.jmb.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 2.Murzin A.G., Lesk A.M., Chothia C. Principles determining the structure of beta-sheet barrels in proteins. I. A theoretical analysis. J. Mol. Biol. 1994;236:1369–1381. doi: 10.1016/0022-2836(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 3.Covalt J.C., Jr., Roy M., Jennings P.A. Core and surface mutations affect folding kinetics, stability and cooperativity in IL-1 beta: does alteration in buried water play a role? J. Mol. Biol. 2001;307:657–669. doi: 10.1006/jmbi.2001.4482. [DOI] [PubMed] [Google Scholar]
- 4.Park S., Saven J.G. Statistical and molecular dynamics studies of buried waters in globular proteins. Proteins. 2005;60:450–463. doi: 10.1002/prot.20511. [DOI] [PubMed] [Google Scholar]
- 5.Tashiro M., Stuchebrukhov A.A. Thermodynamic properties of internal water molecules in the hydrophobic cavity around the catalytic center of cytochrome c oxidase. J. Phys. Chem. B. 2005;109:1015–1022. doi: 10.1021/jp0462456. [DOI] [PubMed] [Google Scholar]
- 6.Schobert B., Brown L.S., Lanyi J.K. Crystallographic structures of the M and N intermediates of bacteriorhodopsin: assembly of a hydrogen-bonded chain of water molecules between Asp-96 and the retinal Schiff base. J. Mol. Biol. 2003;330:553–570. doi: 10.1016/s0022-2836(03)00576-x. [DOI] [PubMed] [Google Scholar]
- 7.Fischer S., Verma C.S. Binding of buried structural water increases the flexibility of proteins. Proc. Natl. Acad. Sci. USA. 1999;96:9613–9615. doi: 10.1073/pnas.96.17.9613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olano L.R., Rick S.W. Hydration free energies and entropies for water in protein interiors. J. Am. Chem. Soc. 2004;126:7991–8000. doi: 10.1021/ja049701c. [DOI] [PubMed] [Google Scholar]
- 9.Petrone P.M., Garcia A.E. MHC-peptide binding is assisted by bound water molecules. J. Mol. Biol. 2004;338:419–435. doi: 10.1016/j.jmb.2004.02.039. [DOI] [PubMed] [Google Scholar]
- 10.Fischer S., Smith J.C., Verma C.S. Dissecting the vibrational entropy change on protein/ligand binding: burial of a water molecule in bovine pancreatic trypsin inhibitor. J. Phys. Chem. B. 2001;105:8050–8055. [Google Scholar]
- 11.Veerapandian B., Gilliland G.L., Poulos T.L. Functional implications of interleukin-1 beta based on the three-dimensional structure. Proteins. 1992;12:10–23. doi: 10.1002/prot.340120103. [DOI] [PubMed] [Google Scholar]
- 12.Clore G.M., Bax A., Gronenborn A.M. Identification and localization of bound internal water in the solution structure of interleukin 1 beta by heteronuclear three-dimensional 1H rotating-frame Overhauser 15N-1H multiple quantum coherence NMR spectroscopy. Biochemistry. 1990;29:5671–5676. doi: 10.1021/bi00476a004. [DOI] [PubMed] [Google Scholar]
- 13.Finzel B.C., Clancy L.L., Einspahr H.M. Crystal structure of recombinant human interleukin-1 beta at 2.0 A resolution. J. Mol. Biol. 1989;209:779–791. doi: 10.1016/0022-2836(89)90606-2. [DOI] [PubMed] [Google Scholar]
- 14.Veerapandian B. Structure and function of interleukin-1, based on crystallographic and modeling studies. Biophys. J. 1992;62:112–115. doi: 10.1016/S0006-3495(92)81796-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rudolph M.G., Kelker M.S., Wilson I.A. Use of multiple anomalous dispersion to phase highly merohedrally twinned crystals of interleukin-1beta. Acta Crystallogr. D Biol. Crystallogr. 2003;59:290–298. doi: 10.1107/s0907444902021704. [DOI] [PubMed] [Google Scholar]
- 16.Gorbitz C. Hydrogen-bond distances and angles in the structures of amino acids and peptides. Acta Crystallogr. B. 1989;45:390–395. [Google Scholar]
- 17.Heidary D.K., Jennings P.A. Three topologically equivalent core residues affect the transition state ensemble in a protein folding reaction. J. Mol. Biol. 2002;316:789–798. doi: 10.1006/jmbi.2001.5270. [DOI] [PubMed] [Google Scholar]
- 18.Bryngelson J.D., Onuchic J.N., Wolynes P.G. Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 19.Clementi C., Jennings P.A., Onuchic J.N. How native-state topology affects the folding of dihydrofolate reductase and interleukin-1beta. Proc. Natl. Acad. Sci. USA. 2000;97:5871–5876. doi: 10.1073/pnas.100547897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varley P., Gronenborn A.M., Clore G.M. Kinetics of folding of the all-beta sheet protein interleukin-1 beta. Science. 1993;260:1110–1113. doi: 10.1126/science.8493553. [DOI] [PubMed] [Google Scholar]
- 21.Capraro D.T., Gosavi S., Jennings P.A. Folding circular permutants of IL-1β: route selection driven by functional frustration. PLoS ONE. 2012;7:e38512. doi: 10.1371/journal.pone.0038512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Capraro D.T., Roy M., Jennings P.A. β-Bulge triggers route-switching on the functional landscape of interleukin-1β. Proc. Natl. Acad. Sci. USA. 2012;109:1490–1493. doi: 10.1073/pnas.1114430109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heidary D.K., Gross L.A., Jennings P.A. Evidence for an obligatory intermediate in the folding of interleukin-1 beta. Nat. Struct. Biol. 1997;4:725–731. doi: 10.1038/nsb0997-725. [DOI] [PubMed] [Google Scholar]
- 24.del Sol A., Tsai C.J., Nussinov R. The origin of allosteric functional modulation: multiple pre-existing pathways. Structure. 2009;17:1042–1050. doi: 10.1016/j.str.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chavez L.L., Gosavi S., Onuchic J.N. Multiple routes lead to the native state in the energy landscape of the beta-trefoil family. Proc. Natl. Acad. Sci. USA. 2006;103:10254–10258. doi: 10.1073/pnas.0510110103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gosavi S., Chavez L.L., Onuchic J.N. Topological frustration and the folding of interleukin-1 beta. J. Mol. Biol. 2006;357:986–996. doi: 10.1016/j.jmb.2005.11.074. [DOI] [PubMed] [Google Scholar]
- 27.Capraro D.T., Roy M., Jennings P.A. Backtracking on the folding landscape of the beta-trefoil protein interleukin-1beta? Proc. Natl. Acad. Sci. USA. 2008;105:14844–14848. doi: 10.1073/pnas.0807812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lammert H., Schug A., Onuchic J.N. Robustness and generalization of structure-based models for protein folding and function. Proteins. 2009;77:881–891. doi: 10.1002/prot.22511. [DOI] [PubMed] [Google Scholar]
- 29.Noel J.K., Whitford P.C., Onuchic J.N. SMOG@ctbp: simplified deployment of structure-based models in GROMACS. Nucleic Acids Res. 2010;38(Web Server issue):W657–W661. doi: 10.1093/nar/gkq498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hess B., Kutzner C., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 31.Eastwood M.P., Wolynes P.G. Role of explicitly cooperative interactions in protein folding funnels: a simulation study. J. Chem. Phys. 2001;114:4702–4716. [Google Scholar]
- 32.Felsenstein J. PHYLIP - Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- 33.Huson D.H., Richter D.C., Rupp R. Dendroscope: an interactive viewer for large phylogenetic trees. BMC Bioinformatics. 2007;8:460. doi: 10.1186/1471-2105-8-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 35.Clementi C., Jennings P.A., Onuchic J.N. Prediction of folding mechanism for circular-permuted proteins. J. Mol. Biol. 2001;311:879–890. doi: 10.1006/jmbi.2001.4871. [DOI] [PubMed] [Google Scholar]
- 36.Gosavi S. Understanding the folding-function tradeoff in proteins. PLoS ONE. 2013;8:e61222. doi: 10.1371/journal.pone.0061222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones B.E., Jennings P.A., Matthews C.R. Development of nonpolar surfaces in the folding of Escherichia coli dihydrofolate reductase detected by 1-anilinonaphthalene-8-sulfonate binding. Biochemistry. 1994;33:15250–15258. doi: 10.1021/bi00255a005. [DOI] [PubMed] [Google Scholar]
- 38.Latypov R.F., Liu D., Raibekas A.A. Structural and thermodynamic effects of ANS binding to human interleukin-1 receptor antagonist. Protein Sci. 2008;17:652–663. doi: 10.1110/ps.073332408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ptitsyn O.B., Bychkova V.E., Uversky V.N. Kinetic and equilibrium folding intermediates. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1995;348:35–41. doi: 10.1098/rstb.1995.0043. [DOI] [PubMed] [Google Scholar]
- 40.Ptitsyn O.B., Pain R.H., Razgulyaev O.I. Evidence for a molten globule state as a general intermediate in protein folding. FEBS Lett. 1990;262:20–24. doi: 10.1016/0014-5793(90)80143-7. [DOI] [PubMed] [Google Scholar]
- 41.Finke J.M., Roy M., Jennings P.A. Aggregation events occur prior to stable intermediate formation during refolding of interleukin 1beta. Biochemistry. 2000;39:575–583. doi: 10.1021/bi991518m. [DOI] [PubMed] [Google Scholar]
- 42.Reference deleted in proof.
- 43.Kauma S.W., Turner T.T., Harty J.R. Interleukin-1 beta stimulates interleukin-6 production in placental villous core mesenchymal cells. Endocrinology. 1994;134:457–460. doi: 10.1210/endo.134.1.8275959. [DOI] [PubMed] [Google Scholar]
- 44.Tsukihara S., Harada T., Terakawa N. Interleukin-1beta-induced expression of IL-6 and production of human chorionic gonadotropin in human trophoblast cells via nuclear factor-kappaB activation. Am. J. Reprod. Immunol. 2004;52:218–223. doi: 10.1111/j.1600-0897.2004.00209.x. [DOI] [PubMed] [Google Scholar]
- 45.Voss N.R., Gerstein M. 3V: cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 2010;38(Web Server issue):W555–W562. doi: 10.1093/nar/gkq395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vigers G.P., Anderson L.J., Brandhuber B.J. Crystal structure of the type-I interleukin-1 receptor complexed with interleukin-1beta. Nature. 1997;386:190–194. doi: 10.1038/386190a0. [DOI] [PubMed] [Google Scholar]
- 47.Heidary D.K., Roy M., Jennings P.A. Long-range coupling between separate docking sites in interleukin-1beta. J. Mol. Biol. 2005;353:1187–1198. doi: 10.1016/j.jmb.2005.08.072. [DOI] [PubMed] [Google Scholar]
- 48.Somani S., Chng C.P., Verma C.S. Hydration of a hydrophobic cavity and its functional role: a simulation study of human interleukin-1beta. Proteins. 2007;67:868–885. doi: 10.1002/prot.21320. [DOI] [PubMed] [Google Scholar]
- 49.Oliveberg M., Tan Y.J., Fersht A.R. The changing nature of the protein folding transition state: implications for the shape of the free-energy profile for folding. J. Mol. Biol. 1998;277:933–943. doi: 10.1006/jmbi.1997.1612. [DOI] [PubMed] [Google Scholar]