

Abstract
Femtosecond laser optoporation is a powerful technique to introduce membrane-impermeable molecules, such as DNA plasmids, into targeted cells in culture, yet only a narrow range of laser regimes have been explored. In addition, the dynamics of the laser-produced membrane pores and the effect of pore behavior on cell viability and transfection efficiency remain poorly elucidated. We studied optoporation in cultured cells using tightly focused femtosecond laser pulses in two irradiation regimes: millions of low-energy pulses and two higher-energy pulses. We quantified the pore radius and resealing time as a function of incident laser energy and determined cell viability and transfection efficiency for both irradiation regimes. These data showed that pore size was the governing factor in cell viability, independently of the laser irradiation regime. For viable cells, larger pores resealed more quickly than smaller pores, ruling out a passive resealing mechanism. Based on the pore size and resealing time, we predict that few DNA plasmids enter the cell via diffusion, suggesting an alternative mechanism for cell transfection. Indeed, we observed fluorescently labeled DNA plasmid adhering to the irradiated patch of the cell membrane, suggesting that plasmids may enter the cell by adhering to the membrane and then being translocated.
Introduction
Femtosecond laser optoporation, a technique in which a transient perforation in a cell membrane is made using focused ultrashort laser pulses, has emerged as a powerful tool for introducing foreign genetic material into targeted cells and has been applied to a variety of cell types in vitro. In previous studies (1–8), researchers optoporated cells using high-repetition rate trains (∼80 MHz) of low-energy (∼1 nJ) near-infrared (800 nm) laser pulses that were focused at high numerical aperture (0.8–1.4 NA), or directed Bessel beams (9) to the membrane of targeted cells for short durations (4–250 ms). In this regime, nonlinear absorption of laser energy in the focal volume is thought to lead to the production of a low-density electron plasma that causes disruption of chemical bonds, release of free electrons, and (potentially) localized heating (10). Over many pulses, these effects cumulatively cause a transient disruption of the cell membrane that has been used to allow exogenous dyes (2,3), macromolecules (11), gold nanoparticles (5), or DNA plasmids (1–4) to enter the cell. With optimized laser parameters, the pore in the cell membrane reseals for an appreciable number of cells, leaving the cells intact and healthy (6,12). Femtosecond optoporation has also been used for cell-targeted gene transfection in goldfish retina explants (8).
The possibility of translating femtosecond laser optoporation from an in vitro to an in vivo setting offers great potential as a technique for studying the pathogenesis of complex diseases in model organisms and exploring the genetic regulation of cell fate and behavior. Few existing approaches for in vivo transfection offer the possibility of physically targeting individual cells, although single-cell transfection has been achieved in vivo using single-cell electroporation with a patch electrode (13). Using femtosecond optoporation to permeabilize a cell deep inside strongly scattering tissue, such as the rodent neocortex, would require an increase in the laser energy to make up for scattering losses and deliver sufficient intensity to the focal volume to drive nonlinear absorption. However, the average laser power that can be used is bounded by the thermal damage threshold of the tissue, limiting the utility of this approach in vivo. One solution to this problem is to use lower-repetition-rate laser sources, or even single pulses, which would allow one to substantially increase the pulse energy while maintaining low average laser powers. The mechanism for membrane damage and pore formation, however, is likely to be different when cumulative effects between successive pulses are no longer a factor (10). For irradiation with a small number of pulses at a low repetition rate, higher pulse energies will likely be necessary and will result in plasmas of high electron density that produce a large thermoelastic stress in the focal volume and lead to the formation of transient cavitation bubbles with diameters of hundreds of nanometers to micrometers (10,14,15). The bubbles, in turn, would lead to nanoscale dissection of the membrane. Previous studies in synthetic membranes suggest that the pores that are formed may be as much as an order of magnitude smaller than the cavitation bubbles that cause them (16). Disruption with this ablative mechanism has been used previously to damage blood vessels in the brain of rodents as a means of modeling ischemic (17,18) and hemorrhagic (17,19) microstrokes. However, this ablative mechanism has not been explored for cell optoporation and transfection, and optimal parameters in this irradiation regime need to be determined in vitro to establish its feasibility for future in vivo work. In addition, a more quantitative understanding of the membrane pore size that is produced for different laser parameters, as well as the influence of pore size and dynamics on subsequent cell viability and transfection efficiency, would be of great benefit to the field. Finally, although expression of exogenous DNA after optoporation has been extensively studied, the mechanisms for DNA plasmid entry have not been considered explicitly and may not be as simple as diffusion through the laser-produced pore.
In this work, we explored femtosecond optoporation in cultured cells across two different laser irradiation regimes: irradiation with millions of low-energy pulses, as has been studied in previous work, and irradiation with two higher-energy pulses. By monitoring the efflux of a small intracellular dye after optoporation and later measuring cell viability, we determined that the acute resealing of membrane pores is a reliable indicator of long-term cell viability. We measured pore formation and cell viability as a function of laser energy for both irradiation regimes, found optimal parameters for cell-targeted DNA transfection, and demonstrated ∼30% transfection efficiencies for both irradiation regimes. We estimated the size of the laser-produced pore from our data on dye efflux and found that pore size was the determining factor for cell viability for both low-energy, high-repetition-rate pulse trains and higher-energy, isolated pulses. Surprisingly, we found that large membrane pores closed faster than small pores in cells that resealed, suggesting that active mechanisms likely play a role in membrane resealing. Finally, we found that fluorescently labeled DNA plasmid accumulates over time on the cell membrane after optoporation with two higher-energy pulses, suggesting the possibility of a novel mechanism beyond diffusion for plasmid entry into the cell. This work builds a quantitative understanding of pore creation after optoporation across two laser irradiation regimes and sets the stage for future optoporation of targeted cells in vivo using low-repetition-rate lasers.
Materials and Methods
Cell culture
Optoporation experiments were performed using adherent Chinese hamster ovarian (CHO) cells that were grown to a subconfluent monolayer in 35-mm glass-bottomed culture dishes coated with poly-d-lysine (MatTek, Ashland, MA) and placed in a humidified incubator with 5% CO2/95% air at 37°C. Growth media solutions were made using advanced Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA), 1% GlutaMAX solution (Invitrogen, Carlsbad, CA), 1% penicillin-streptomycin (Invitrogen, Carlsbad, CA), and 50 μg/mL gentamicin (Invitrogen, Carlsbad, CA). During optoporation and imaging experiments, the cells were placed in media consisting of DMEM, 1% sodium pyruvate (Invitrogen), 1% GlutaMAX solution, 1% penicillin-streptomycin, and 50 μg/mL gentamicin. It was necessary to image cells in this solution to avoid optical absorption of emitted fluorescence in the normal growth media by the pH indicator.
Two-photon excited fluorescence microscopy
CHO cells were imaged using a custom-built two-photon excited fluorescence (2PEF) microscope with two simultaneous fluorescent detection channels. Images were acquired using a Ti:Sapphire laser (MIRA-HP; Coherent, Santa Clara, CA) with 100-fs pulse duration, a central wavelength at 800 or 850 nm depending on the species being excited, 10-nm bandwidth, and a repetition rate of 76 MHz. A 20× magnification, 1.0 numerical aperture (NA) water-immersion microscope objective (Zeiss, Thornwood, NY) was used for all imaging. Fluorescence emission from green fluorescent molecules (green fluorescent protein (GFP), calcein-AM, fluorescein) was collected through a 517-nm bandpass filter with 65-nm bandwidth, and a 645-nm bandpass filter with a 70-nm bandwidth was used for red-emitting dyes (ethidium bromide, CellTrace calcein red-orange AM). Image series were acquired during optoporation, and three-dimensional (3D) image stacks with 0.5-μm spacing in the axial direction were attained at baseline and after optoporation. Image processing and analysis were performed using ImageJ and custom scripts written in MATLAB (The MathWorks, Natick, MA).
Cell membrane disruption using femtosecond laser systems
Targeted membrane disruption was performed using two laser systems. The first was a low-repetition-rate, high-pulse-energy Ti:Sapphire regenerative amplifier with 100-fs pulse duration, 1-kHz repetition rate, and 800-nm central wavelength (Legend-USP; Coherent, Santa Clara, CA). A polarizing beamsplitter cube was used to introduce this beam into the 2PEF microscope so that the pulses were focused at the center of the imaging field and in the 2PEF imaging plane, enabling real-time monitoring of optoporation dynamics. Laser energy incident on the cells was controlled by neutral density filters, and a fast mechanical shutter limited the number of pulses incident on each cell to two pulses. We also optoporated cells using pulses from the 76-MHz laser used for 2PEF imaging. The laser beam from the Ti:Sapphire oscillator was split so that one beam could be used for 2PEF imaging, while the second was aligned, as above, to focus at the center of the imaging field and in the imaging plane. Laser energy was varied using a half-waveplate/polarizing beamsplitter pair, and exposure time was controlled to be 50 ms, corresponding to 3.8 × 106 pulses, with a mechanical shutter. In both cases, a mechanical stage with submicrometer motion control in three dimensions (Newport, Irvine, CA) enabled precise targeting of the laser focus to the edge of the plasma membrane of the cell.
CHO cell labeling with calcein dyes
We labeled CHO cells for 2PEF imaging using calcein-AM or CellTrace calcein red-orange AM (Invitrogen, Carlsbad, CA). Initially, calcein dyes are nonfluorescent due to an ester-bond-attached group that both quenches fluorescence and makes the dye cell-membrane-permeable. After entering functional cells, the intracellular esterases cleave the quenching group, producing a hydrophilic, membrane-impermeable fluorescent label that remains in the cytoplasm of the cell. We added 5 μL of a 1 mg/mL calcein-AM or CellTrace calcein red-orange AM dissolved in dimethyl sulfoxide (DMSO) to 10 mL of phosphate-buffered saline (PBS). The growth media was siphoned and 2 mL of dye solution was added to each culture dish and incubated at room temperature for 45 min. To reduce background fluorescence, a washout step was performed in which the dye solution was removed and replaced with culture media before imaging. For each assay, calcein-AM or CellTrace calcein red-orange AM was used to complement ethidium bromide or GFP labeling, respectively. These pairings allowed for simultaneous excitation at a single wavelength in each case, with effective spectral separation of fluorescence emissions from the two labels.
Monitoring efflux of calcein dye after optoporation
We evaluated the dependence of optoporation efficiency on laser parameters by monitoring the efflux of calcein dye from the cell after laser irradiation of the membrane. For calcein-AM, we used an 850-nm wavelength for 2PEF imaging. Dye efflux was quantified from images using custom scripts in MATLAB. To determine the fluorescence intensity as a function of time, which is directly proportional to dye concentration inside the cell, we outlined the targeted cell by hard thresholding to create a mask and calculated the average fluorescence intensity of the pixels within this mask over time. The mean background pixel intensity was subtracted from all data points. The baseline fluorescence was determined from the average across multiple frames taken before cell targeting. We established a detection threshold for dye efflux based on the fluctuations in fluorescence of controls cells (N = 83). Irradiated cells with a decrease in fluorescence intensity of more than twice the SD of the fluctuations in control cells were defined as permeabilized.
Assessing cell viability after optoporation
In addition to the use of calcein-AM to monitor dye efflux, cells were restained with 4 μM of calcein-AM 90 min after optoporation as a cell viability assay, since calcein-AM only becomes fluorescent after being cleaved by active intracellular esterases. In addition, 4 μM of ethidium bromide (EtBr) was added simultaneously to assess membrane integrity. Cells that exhibited increased fluorescence after calcein-AM restaining and excluded the membrane-impermeable, nucleic-acid-binding EtBr were interpreted to have intact plasma membranes that had resealed after optoporation and to have active intracellular enzymes. These cells were considered to be alive and functional.
Measurement of volume change after optoporation
To assess changes in cell volume after optoporation, we transfected CHO cells with GFP and acquired 2PEF image stacks before and after optoporation. GFP-labeled cells were used instead of cells labeled with calcein-AM to minimize efflux of cytoplasmic fluorescent molecules after optoporation. Because of the larger molecular size and radius of gyration of GFP, we observed very little change in fluorescence after optoporation in GFP-labeled cells (data not shown). Cells were transfected with pAAVeGFP via Lipofectamine 2000 24 h before optoporation. Approximately 2 μg of DNA plasmid was added to 750 μL of OPTI-MEM (Invitrogen, Carlsbad, CA), and 5 μL of the Lipofectamine solution was added to 750 μL of OPTI-MEM in a separate tube. The two solutions were then combined, mixed gently, and incubated at room temperature for 20 min before the solution was added to the cells. 3D 2PEF image stacks, with 0.5 μm spacing between sections, were attained at baseline and at 30 s and 5 min after membrane irradiation. Volume analysis was performed using Volocity Imaging Software (PerkinElmer, Waltham, MA). The cell of interest was selected by hard thresholding and the cell volume was reconstructed from the 3D image stack. Images of the volume projections were created using OsiriX imaging software. Volume measurements were made in cells in which a hole was produced and the membrane resealed, and volume changes of nontargeted, control cells were assessed at the same time points.
Movement of labeled DNA plasmid after optoporation
To track DNA plasmid movement after optoporation, we monitored the movement of fluorescently labeled DNA plasmids in the extracellular solution after cell membrane irradiation. We used 20 μg/ml Label IT Plasmid Delivery Control (Mirus Bio, Madison, WI) labeled with fluorescein (approximately one fluorescein molecule for every 20–60 bp of DNA plasmid). The 2.7 kb plasmid was not capable of expressing protein due to the locations of fluorescein binding. Fluorescein-labeled plasmid was imaged using 800-nm excitation. The cells were labeled with CellTrace calcein red-orange. The 2PEF imaging laser intensity had to be increased to image the labeled plasmid, which caused some photobleaching of the calcein red-orange dye. We quantified the intensity of labeled plasmid on the targeted patch of the plasma membrane after optoporation by monitoring the average fluorescence intensity over time. Two control experiments were performed. In the first control, localized increases in signal after optoporation due to autofluorescence were examined at the location of membrane irradiation in the absence of labeled plasmid. In the second control, labeled plasmid accumulation in nontargeted cells was measured to establish a detection threshold for plasmid accumulation at the membrane of targeted cells.
Transgene expression
To transfect single cells using laser-based optoporation, we prepared DNA plasmids from transfected Escherichia coli cells using a Midiprep DNA purification system (Qiagen, Valencia, CA). A 4.7-kb pAAVeGFP plasmid that coded for GFP was dissolved in clear growth media solution at a concentration of 10 μg/ml and applied to cell culture plates for the duration of the optoporation session. CellTrace calcein red-orange AM was used to label the CHO cells, and 800-nm excitation was used for 2PEF imaging. We used femtosecond laser ablation to place distinguishing marks in the polystyrene culture dish underneath the targeted cells to facilitate later identification of specific cells. After each optoporation session, the plasmid solution was siphoned, normal growth media was applied, and the cells were stored in a humidified incubator at 37°C. Cells were reimaged using either 2PEF or wide-field fluorescence microscopy 24 and 48 h after optoporation to monitor expression of GFP. To rule out double counting of successfully transfected cells due to cell division, targeted cells were spatially separated by at least 100 μm. Two cells expressing GFP in close proximity (e.g., <20 μm apart) 48 h after irradiation were considered a single transfection event followed by cell division. To ensure the plasmid was functional, a Lipofectamine transfection reagent was used as a positive control. Negative controls were obtained by leaving a solution containing plasmid and clear growth media in a cell culture dish for the length of the optoporation session without targeting the cells.
Statistics
Statistical comparisons were made using a two-tailed Fisher’s exact test (see Figs. 1 h and 5 h). Results were considered significant when p < 0.05. All error bars shown in bar graphs (Figs. 1 h, 2, a–d, and 3 d; also see Figs. 5 h and 6 f) represent 95% confidence intervals and were calculated using a binomial distribution. The boxplots (Figs. 1, i and j, 3, b and c, and 4 d) display the 25th to 75th percentile (box), the median (red line), and data within 1.5 times the interquartile range from the 25th to 75th percentile (whiskers). Outliers were defined as points that fell outside the range denoted by the whiskers and are identified with a red + through the datapoint. Means were calculated without statistical outliers and are indicated by an orange x on the box plots. All means and SDs reported in this work do not include statistical outliers.
Figure 1.
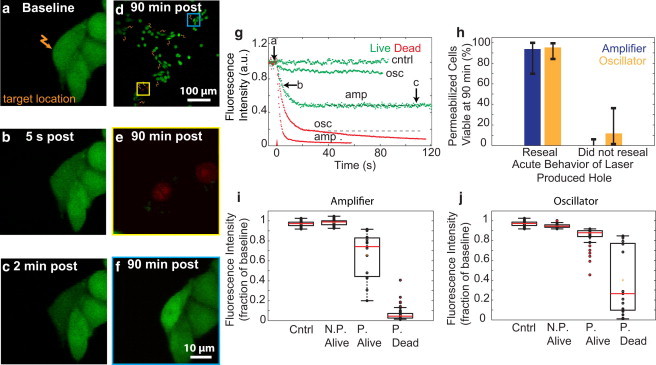
Pore formation and cell viability after femtosecond laser irradiation. (a–c) CHO cells were irradiated with femtosecond pulses (two pulses, 14 nJ) at the plasma membrane (a, arrow), and calcein fluorescence intensity (green) was monitored over time (b and c). (d) Multiple spatially separated cells were irradiated (arrows). (e) Application of calcein-AM and EtBr (red) 90 min after optoporation produced EtBr-labeled nuclei in dead cells. (f) Live cells showed no EtBr labeling and exhibited an increase in calcein fluorescence. (g) Fluorescence intensity was monitored as a function of time for cells targeted with either amplifier (amp; two pulses, 1 kHz) or oscillator (osc; 3.8 × 106 pulses, 1 MHz) sources at various energies. Cells with final intensities approaching the noise floor failed to reseal (red), whereas cells with stabilized, constant intensity regained membrane integrity (green). The labeled arrows along the green trace representing amplifier optoporation correspond to the fluorescence intensity at time points of the images in a–c. (h) Cell viability at 90 min for holes that did or did not reseal acutely for optoporation with each laser source. (i and j) Fluorescence intensity after optoporation, expressed as a fraction of baseline intensity, was compared against cell permeabilization (P, permeabilized; N.P., not permeabilized) and viability for cells irradiated with the amplifier (i) or the oscillator (j). Cells with an intensity drop of 2 SDs below baseline intensity were considered permeabilized.
Figure 5.
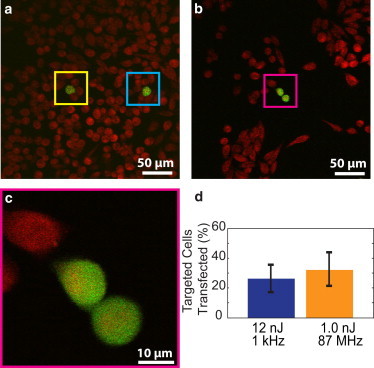
Transfection of cells with oscillator and amplifier laser pulses. Cells were targeted in media containing plasmids coding for eGFP. (a–c) Cells were targeted with either two 12 nJ pulses from the amplifier (a) or 3.8 × 106 1.0-nJ pulses from the oscillator (b and c). All cells were labeled using calcein red-orange (red) with transfected cells expressing GFP (green). To avoid double counting due to cell division after transfection (b and c), cells were sparsely targeted (a). (d) Transfection efficiencies were determined for both laser sources and were not statistically different.
Figure 2.
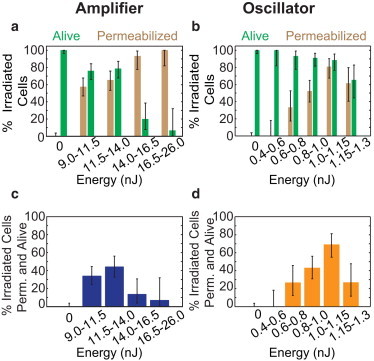
Cell viability and permeabilization probability as a function of irradiation energy. (a and b) The percentage of cells that were permeabilized and the percentage of cells that were viable after irradiation were considered separately, across a range of energies, for pulses from the amplifier (a) and oscillator (b). (c and d) Percentage of cells that were both permeabilized and viable after 90 min as a function of irradiation energy for amplifier (c) and oscillator (d) irradiation.
Figure 3.
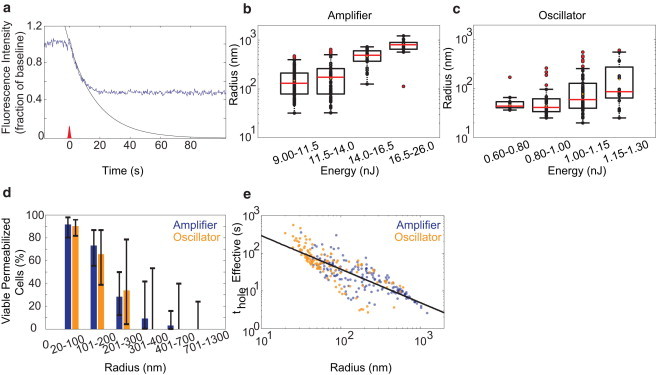
Quantification of membrane pore size and opening time. (a–c) Fitting (black curve) of the initial decay in intracellular fluorescence intensity (blue trace) after optoporation (a) was used to determine the initial radius of the membrane pore across a range of energies for both amplifier (b) and oscillator (c) pulses. (d) Plot of cell viability as a function of pore radius for both amplifier and oscillator irradiation. (e) Plot of the effective time the pore was open, thole, as a function of pore size.
Figure 4.
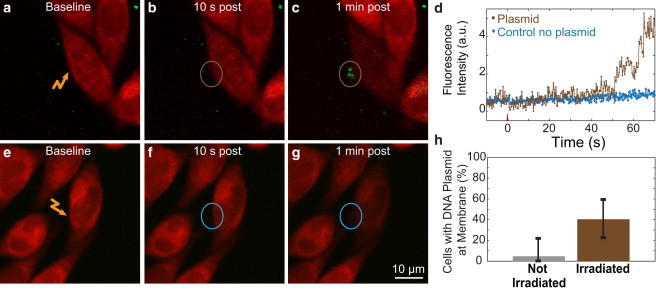
Accumulation of DNA plasmid on the irradiated section of the cell membrane. (a–c) Fluorescently labeled DNA plasmid (green) accumulated at the site of irradiation (two 12 nJ pulses from the amplifier) along the plasma membrane of cells (labeled with calcein red-orange (red). (d–f) No increase in fluorescence intensity at the targeted site was observed in cells irradiated without labeled plasmid present. (g) Accumulation of labeled plasmid as a function of time for cells irradiated with and without labeled plasmid present. (h) The fraction of cells that exhibited accumulation of labeled plasmid at the membrane for cells that were (irradiated) or were not (not irradiated) optoporated with amplified pulses.
Results
Imaging dye efflux allowed quantification of pore creation and cell viability after optoporation
To identify optimal laser parameters for membrane pore creation, we tightly focused femtosecond laser pulses on the membrane of CHO cells labeled with calcein-AM and monitored the efflux of dye over time using 2PEF (Fig. 1, a–c). We used two different exposure conditions: two pulses from a 1-kHz repetition-rate pulse train with pulse energies between 9 and 26 nJ (amplifier; Fig. 1, a–c), or ∼3.8 × 106 pulses from a 76-MHz pulse train with pulse energies between 0.4 and 1.3 nJ (oscillator). For both exposure conditions, cells that were permeabilized exhibited an exponential decrease in fluorescence intensity immediately after targeting, indicative of pore creation (Fig. 1 g). The fluorescence intensity then either stabilized, indicating resealing of the pore, or continued to decrease, suggesting that the laser-created pore did not reseal (Fig. 1 g). Nontargeted cells served as controls and exhibited stable fluorescence intensity over time.
Ninety minutes after targeting, the cells were stained with additional calcein-AM and with EtBr. The cells were then reimaged and scored for cell viability (Fig. 1 d). EtBr uptake and unchanged calcein-AM fluorescence (Fig. 1 e) indicated cell death, whereas EtBr exclusion and increased calcein-AM fluorescence (Fig. 1 f) suggested both an intact plasma membrane and functional esterase activity inside the cell, indicating a viable cell. All cells that were negative for EtBr also exhibited an increase in calcein-AM fluorescence. For both of the laser exposure conditions, >90% of cells that resealed acutely were viable at 90 min (Fig. 1 h). The total amount of dye efflux was higher for cells killed by permeabilization as compared with cells that survived for both amplifier (Fig. 1 i) and oscillator (Fig. 1 j) irradiation. However, permeabilized cells that remained viable after amplifier irradiation showed greater dye efflux, on average, than permeabilized, viable cells irradiated with the oscillator, and cells killed by amplifier irradiation showed much more dye efflux than those killed by oscillator irradiation (Fig. 1, i and j). Because we found such a strong relationship between acute membrane resealing and cell viability at 90 min (Fig. 1 h; p = 1.0 × 10−13 for amplifier, p = 3.2 × 10−10 for oscillator, Fisher’s exact test), we considered cells that resealed acutely as viable in subsequent analyses.
The threshold for membrane permeabilization with amplifier irradiation was 9.5 nJ and the percentage of permeabilized cells increased to 100% at the highest energies used, although cell viability decreased sharply for pulse energies higher than 14 nJ (Fig. 2 a). For the oscillator pulses, our threshold for permeabilization of 0.65 nJ agreed well with previous reports (2,3). The percentage of permeabilized cells increased more gradually with increasing energy for the oscillator pulses as compared with the amplifier irradiation, whereas cell viability decreased significantly only at the highest pulse energies used (Fig. 2 b). The fraction of cells that were successfully permeabilized and remained viable reached a maximum of 45% for ∼12 nJ pulse energy for the amplifier irradiation (Fig. 2 c), and reached 70% for 1.0 nJ pulse energy for the oscillator irradiation (Fig. 2 d).
Modeling of dye efflux after optoporation
A simple diffusion-based model was used to estimate laser-produced hole radii in both laser regimes. Briefly, we considered a laser-produced hole of radius r in the cell membrane that was much smaller than the cell diameter. We further assumed that the hole radius was constant in time while the pore was open, and then rapidly closed. This model predicted the initial decrease in dye concentration, ϕ, inside the cell to be an exponential decay (see Supporting Material):
| (1) |
where ϕ0 is the initial concentration of dye within the cell and τ is the diffusive time constant for the hole. For viscosity, η0, dye molecule radius of gyration, Rg, membrane thickness, e, cell volume, Vcell, temperature, T, and Boltzmann constant, kB, τ is given by (see Supporting Material):
| (2) |
We fit the initial decay of fluorescence (linearly proportional to dye concentration) after optoporation to Eq. 1 (Fig. 3 a) and used Eq. 2 to determine the initial hole radius. We further estimated an effective hole opening time, thole, which was taken as the time it would take for the concentration to decay to the final value observed after membrane resealing, ϕ∞, assuming a constant hole radius while the hole was open, i.e.,
| (3) |
In both laser regimes, hole radii increased with increasing laser energy and holes produced with the 1-kHz laser were larger, on average, than holes produced with the 76-MHz laser (Fig. 3, b and c, respectively). In the optimal energy range for the amplifier pulses where the greatest percentage of targeted cells were permeabilized and viable (∼12 nJ), the hole radii were 160 ± 115 nm (mean ± SD). For the oscillator pulses at optimal energy (∼1.0 nJ), the holes had radii of 77 ± 55 nm. Hole radius was a strong predictor of cell viability for both amplifier and oscillator pulses (Fig. 3 d).
We found phenomenologically that the approximate duration the pore was open, thole, scaled as a power law with the initial radius (Fig. 3 e):
| (4) |
where a and γ were determined by a least-squares fit. We found a = 4.0 ± 3 × 104 s⋅nm-γ and γ = −1.6 ± 0.2, where r was measured in nanometers. In cells that remained viable, larger holes (∼500 nm radius) were observed to reseal between 1 and 10 s, whereas smaller holes (∼30 nm) could persist longer than 100 s (Fig. 3 e). Cell volume was also measured after irradiation, with a transient increase in cell volume seen due to the formation of a protrusion at the irradiation site (see Fig. S1).
DNA plasmid accumulated on the plasma membrane of cells targeted with amplified laser pulses
To understand the movement of DNA plasmids after optoporation, we irradiated cells in the presence of fluorescently labeled DNA plasmid. Cells targeted with the amplifier exhibited increased fluorescence at the site of membrane irradiation, suggesting membrane accumulation of DNA at the irradiated location (Fig. 4, a–c). In comparison, cells irradiated by amplified pulses in the absence of labeled plasmid showed negligible increase in fluorescence (Fig. 4, e–g). Plasmid accumulation at the plasma membrane began ∼20–60 s after membrane irradiation (Fig. 4 d). Forty percent of cells (12 of 30) irradiated in the presence of labeled DNA plasmid exhibited a localized increase in fluorescence at the irradiated site on the membrane, whereas only 4% of cells that were not irradiated showed an increase (Fig. 4 h; p = 0.004, Student’s t-test). The signal/noise ratio of 2PEF imaging of the labeled plasmid was not sufficient to resolve plasmid entry into the cell. In experiments using oscillator irradiation for optoporation, we observed a localized increase in cell autofluorescence at the target location on the plasma membrane, which precluded similar measurements of DNA plasmid movement after oscillator-based optoporation.
Single cells were transfected using both laser systems
Single cells were optoporated in the presence of a DNA plasmid that coded for the production of GFP, and expression was assessed at 24 and 48 h after optoporation. Both single cells (Fig. 5 a, amplifier example) and cells that had divided after optoporation (Fig. 5, b and c, oscillator example) were observed after single-cell transfection for each laser system. In all cases, cells that had divided after optoporation were counted as only one transfected cell. When the optimal pulse energies determined above (Fig. 5 d) were used, the transfection efficiency (defined as the percentage of targeted cells that both survived and later expressed GFP) was ∼25% and 30% for the amplified and oscillator laser, respectively. The spontaneous transfection without laser irradiation was negligibly low (<1 in 106 cells).
Discussion
We investigated pore dynamics and cell viability after femtosecond laser optoporation with two different laser irradiation regimes: a train of millions of low-energy pulses at high repetition rate from a Ti:Sapphire oscillator, and two higher-energy, isolated pulses from a regenerative amplifier. The mechanisms that lead to pore formation are quite different in these two regimes, with cumulative damage across many pulses arising from low-density plasma presumed to dominate in the former case, and ablative damage and cavitation bubble formation from a high-density plasma by each pulse thought to govern the latter (10). We identified optimal laser parameters for optoporation in both regimes, identified by high rates of both successful permeabilization of the membrane and cell viability. We found that cell viability after optoporation, indicated by both enzymatic function and membrane integrity at 90 min after irradiation, was completely dependent on resealing of the laser-produced pore after irradiation for both laser irradiation regimes. Using trains of low-energy pulses, we identified an optimal viability rate of ∼70% at pulse energies of ∼1 nJ. This optimal energy agrees well with that found in previous work for similar focusing conditions and exposure times (3). The 12-nJ pulse energy that was found to be optimal for isolated, higher-energy pulses is above the breakdown threshold for water at the 1.0-NA focusing used here, supporting an ablative mechanism for pore formation. In this case, a smaller fraction of targeted cells (∼45%) were successfully optoporated, and cell viability dropped rapidly with increases in pulse energy above the optimal range. Improvements in the percentage of viable porated cells have been realized with the use of structured illumination (7) and low-fluence irradiation of gold nanoparticles (20), suggesting that higher efficiency can be achieved by combining optimized laser parameters with better cell targeting.
By carefully measuring and modeling the efflux of a small dye out of optoporated cells, we quantified the size and dynamics of the laser-created pore. Although we did not attempt to characterize the pore at the nanoscale by direct measurement, the assumption that the pores are true mechanical perforations and not localized changes in permeability has been verified in red blood cells in previous work (21). For both irradiation regimes, the pore size increased with laser energy. Pore size was, on average, about twice as large at the energy associated with optimal optoporation for higher-energy, isolated pulses (∼160-nm radius) as compared with trains of low-energy pulses (∼80-nm radius). For our focusing parameters, these radii corresponded to ∼0.6 and 0.3 times the diffraction-limited laser spot size (∼250-nm radius) for the amplifier and oscillator, respectively. These holes were subdiffraction in size due to the nonlinear nature of the poration processes. Moreover, for holes formed with higher-energy pulses, the hole radii were considerably smaller than the expected size of the cavitation bubble formed at that energy. Taking our lowest energy of 9 nJ as the threshold energy for plasma formation, experimental results from Vogel et al. (15) would suggest that a pulse energy of 11.5–14 nJ would have a corresponding bubble radius of ∼5 μm, or 30 times the observed membrane hole radius. In good agreement with these observations, previous studies (16) demonstrated that the size of laser-produced holes in a polyethylene membrane was a factor of 20 times smaller than the measured cavitation bubble. Regardless of the mechanism for pore formation, for the same hole radii, the two irradiation regimes resulted in a similar percentage of cells being permeabilized and remaining viable. Collectively, these data suggest that the total area of the laser-produced pore, independently of the irradiation regime used to form it, dictated the acute resealing behavior of the pore and therefore the long-term viability of the cell. Although it may seem surprising that such enormous differences in pulse numbers and laser energies would produce pores of similar size, it should be noted that we explored in detail only a narrow window of pulse energies for both laser regimes within which we observed efficient poration and reasonable viability. Successful pore formation and cell viability effectively bound the hole size from below and above, respectively. In our experiments, we thus effectively sought laser parameters for each irradiation regime such that the very different mechanisms of pore formation yielded holes of similar size.
Interestingly, the rate for successful DNA transfection did not differ significantly between the two laser systems (∼30%). This suggests that a larger fraction of cells that were successfully optoporated with trains of low-energy pulses failed to take in a sufficient quantity of the DNA plasmid for robust transgene expression as compared with cells optoporated using isolated, higher-energy pulses. The DNA plasmid was much larger than the small-molecule dye we used to indicate membrane permeabilization, and we found that the average pore size created with trains of low-energy pulses was smaller than that produced with isolated, higher-energy pulses. Taken together, these results suggest that perhaps the pore sizes were on the small side for successful entry of DNA plasmid into the cells when they were optoporated with trains of low-energy pulses.
In any case, although the ∼30% transfection rate we measured for trains of low-energy pulses agrees well with some previous results (3,22), it is lower than those reported in other studies (1,2). However, we note that our reported efficiency includes all targeted cells, regardless of whether membrane permeabilization was achieved or whether the cell was killed. In addition, we spatially separated irradiated cells (Fig. 5 a) to prevent overestimates of the transfection efficiencies due to double counting of cells that divided posttransfection (Fig. 5 b). Cell-division events may account for the higher efficiencies reported by Stevenson et al. (2), who included all GFP-expressing cells at the endpoint in their count. If we exclude cells that were either not permeabilized or died after optoporation, our transfection rates would be 55% and 43% for the amplifier and oscillator, respectively.
Although we ignored dynamic changes in the pore radius in our model by assuming a constant radius until the pore rapidly snaps shut (i.e., a step function temporal profile), our predictions for initial dye efflux agree well with experimental data. Our model proposes diffusion as the transport driver, allowing us to make quantitative predictions about the rates of transport of different solute molecules. For example, we observed a significant efflux of calcein dye molecules, but a much more limited transport of GFP protein, which has a larger radius of gyration (data not shown). In fact, the GFP efflux was below the detection threshold for optoporation with a train of millions of low-energy pulses. In contrast, efflux of GFP was observed after optoporation with isolated, higher-energy pulses, consistent with the larger pore radius produced. From the pore size estimates based on calcein efflux, we were able to predict the amount of GFP efflux experimentally observed (data not shown), confirming that diffusion is the dominant driver of solute transport through the pore.
As mentioned above, our model is limited compared with previous models developed by Puc et al. (23) and Wang et al. (24) because it did not account for hole-resealing dynamics. However, it is difficult to incorporate a robust theory for the pore dynamics because active cellular mechanisms are likely involved in addition to passive membrane dynamics. In previous studies, Brochard-Wyart et al. (25) and Karatekin et al. (26) described the dynamic behavior of transient pores in giant unilamellar vesicles (GUVs). They achieved a complete explanation by considering the competing effects of surface tension, line tension, and relaxation of the Laplace pressure by fluid leak-out through the transient pore. Importantly, such models predict a pore lifetime that is an increasing function of initial pore size. This trend is also the case for a purely elastic medium. In contrast, we found that larger pores actually closed faster in those cells that successfully resealed and remained viable after optoporation. We note, however, that there was clearly selection bias in this result, as we only included cells that successfully resealed in this analysis. It is likely that for larger-diameter pores, only the pores that reclosed quickly were able to reclose at all without cell death.
Our data on pore closure strongly suggest that the passive sealing processes used to describe the pore dynamics in vesicles will be inadequate for cells. Indeed, other researchers have shown that resealing of transected neurites is a calcium-mediated, active process (27). Calcium dynamics after optoporation were studied by Baumgart et al. (28), who demonstrated that chelating calcium with EGTA prevents a rise in intracellular calcium after optoporation; however, they did not explore the relationship between calcium entry and cell viability or hole closure time. They also found that calcium entry did not depend strongly on the pulse repetition rate. A direct comparison of these observations with our study is complicated by the disparate number of pulses used even at lower repetition rates (160,000 in the previous work versus two in our study). Furthermore, intracellular calcium release has been shown (29) to be strongly dependent on the pulse repetition rate and pulse number, with lower repetition rates and fewer pulses being much less likely to induce intracellular calcium waves or reactive oxygen species.
The mechanism for DNA plasmid entry into optoporated cells has been assumed to be diffusion. Using our experimentally determined dependence of pore duration on pore size in Eq. 4, we calculated how many plasmids would diffuse into the cell as a function of pore size. For typical concentrations of the plasmid used in these experiments, our calculations suggest that just a few copies of the DNA enter the cell (Fig. S2), as compared with the thousands to millions of copies of DNA plasmid that are estimated to enter the cell using traditional techniques, such as microinjection.
Under irradiation with higher-energy, isolated pulses, we found that DNA plasmid accumulates on the plasma membrane at the irradiated site (Fig. 4). Several mechanisms may play a role in this. First, previous studies (3) have demonstrated that the cell strongly depolarizes following optoporation, thereby lowering the energy barrier for negatively charged DNA to adhere to the cell membrane. Second, the ablated patch of the cell membrane likely requires some time, perhaps long after the pore has resealed, to fully regain a normal complement of membrane proteins and to otherwise regain normal structure and function. This in turn may create a permissive environment for DNA to adhere at the irradiated site on the cell membrane. Over time, the DNA that has adhered to the cell membrane will likely be translocated, thus providing an alternative mechanism for DNA plasmid entry after optoporation. Because of signal/noise ratio limitations, we were unable to follow the DNA that adhered to the cell membrane over time to see if it was translocated into the cell or to elucidate further mechanisms of entry. It is possible that a similar accumulation of DNA at the irradiated site of the membrane occurred when we targeted cells with a train of millions of low-energy pulses, but we were unable to resolve the DNA plasmid accumulation at the membrane after irradiation in this regime due to a localized increase in autofluorescence at the target location on the membrane, which would mask the signal from DNA accumulation. In agreement with this DNA entry hypothesis, a similar mechanism was observed for electroporation-mediated DNA entry, with DNA plasmid interacting with the plasma membrane and then being translocated into the cell ∼30 min after membrane permeabilization (30). The plasmid accumulation on the membrane only occurred at the electropermeabilized side of the cell facing the cathode. Our observation of plasmid interaction with the targeted patch of the plasma membrane may suggest common mechanisms for plasmid entry in optoporation and electroporation. If delayed translocation is, in fact, a mechanism for DNA entry, this would have implications for the best choice of parameters for laser transfection. In this respect, many small irradiated patches of membrane that enable DNA localization to the cell membrane may provide higher transfection efficiencies.
In conclusion, we have built a quantitative understanding of pore creation and resealing after optoporation across two distinct regimes of irradiation with femtosecond lasers. In addition, we have identified potential mechanisms for DNA plasmid entry after optoporation that rely on DNA adherence to patches of cell membrane altered by irradiation and later translocation. Importantly, the demonstration of transfection using higher-energy, isolated femtosecond laser pulses in cultured cells sets the stage for future in vivo optoporation studies deep inside scattering tissues, where the increase in the incident pulse energy required to compensate for losses due to scattering is not confined by thermal damage at the tissue surface, as is the case when using a train of millions of low-energy pulses. These findings will be critical for the development of a minimally invasive, high-fidelity, laser-based transfection technique that can be used in vivo and has the potential to probe complex diseases through precise genetic manipulation of single cells in a living organism.
Acknowledgments
This work was supported by a grant from the National Science Foundation (No. 0846534 to C.B.S.).
Footnotes
Andrew A. Davis’s present address is Feinberg School of Medicine, Northwestern University, Chicago, Illinois
Matthew J. Farrar’s present address is Department of Neurobiology and Behavior, Cornell University, Ithaca, New York
Supporting Material
References
- 1.Tirlapur U.K., König K. Targeted transfection by femtosecond laser. Nature. 2002;418:290–291. doi: 10.1038/418290a. [DOI] [PubMed] [Google Scholar]
- 2.Stevenson D., Agate B., Dholakia K. Femtosecond optical transfection of cells: viability and efficiency. Opt. Express. 2006;14:7125–7133. doi: 10.1364/oe.14.007125. [DOI] [PubMed] [Google Scholar]
- 3.Baumgart J., Bintig W., Heisterkamp A. Quantified femtosecond laser based opto-perforation of living GFSHR-17 and MTH53 a cells. Opt. Express. 2008;16:3021–3031. doi: 10.1364/oe.16.003021. [DOI] [PubMed] [Google Scholar]
- 4.Uchugonova A., König K., Tempea G. Targeted transfection of stem cells with sub-20 femtosecond laser pulses. Opt. Express. 2008;16:9357–9364. doi: 10.1364/oe.16.009357. [DOI] [PubMed] [Google Scholar]
- 5.McDougall C., Stevenson D.J., Dholakia K. Targeted optical injection of gold nanoparticles into single mammalian cells. J Biophotonics. 2009;2:736–743. doi: 10.1002/jbio.200910030. [DOI] [PubMed] [Google Scholar]
- 6.Zeigler M.B., Chiu D.T. Laser selection significantly affects cell viability following single-cell nanosurgery. Photochem. Photobiol. 2009;85:1218–1224. doi: 10.1111/j.1751-1097.2009.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antkowiak M., Torres-Mapa M.L., Dholakia K. Application of dynamic diffractive optics for enhanced femtosecond laser based cell transfection. J Biophotonics. 2010;3:696–705. doi: 10.1002/jbio.201000052. [DOI] [PubMed] [Google Scholar]
- 8.Gu L., Mohanty S.K. Targeted microinjection into cells and retina using optoporation. J. Biomed. Opt. 2011;16:128003. doi: 10.1117/1.3662887. [DOI] [PubMed] [Google Scholar]
- 9.Rendall H.A., Marchington R.F., Dholakia K. High-throughput optical injection of mammalian cells using a Bessel light beam. Lab Chip. 2012;12:4816–4820. doi: 10.1039/c2lc40708f. [DOI] [PubMed] [Google Scholar]
- 10.Vogel A., Noack J., Paltauf G. Mechanisms of femtosecond laser nanosurgery of cells and tissues. Appl. Phys. B. 2005;81:1015–1047. [Google Scholar]
- 11.Stracke F., Rieman I., König K. Optical nanoinjection of macromolecules into vital cells. J. Photochem. Photobiol. B. 2005;81:136–142. doi: 10.1016/j.jphotobiol.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 12.Peng C., Palazzo R.E., Wilke I. Laser intensity dependence of femtosecond near-infrared optoinjection. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2007;75:041903. doi: 10.1103/PhysRevE.75.041903. [DOI] [PubMed] [Google Scholar]
- 13.Kitamura K., Judkewitz B., Häusser M. Targeted patch-clamp recordings and single-cell electroporation of unlabeled neurons in vivo. Nat. Methods. 2008;5:61–67. doi: 10.1038/nmeth1150. [DOI] [PubMed] [Google Scholar]
- 14.Schaffer C.B., Nishimura N., Mazur E. Dynamics of femtosecond laser-induced breakdown in water from femtoseconds to microseconds. Opt. Express. 2002;10:196–203. doi: 10.1364/oe.10.000196. [DOI] [PubMed] [Google Scholar]
- 15.Vogel A., Linz N., Paltauf G. Femtosecond-laser-induced nanocavitation in water: implications for optical breakdown threshold and cell surgery. Phys. Rev. Lett. 2008;100:038102. doi: 10.1103/PhysRevLett.100.038102. [DOI] [PubMed] [Google Scholar]
- 16.Vogel A., Hentschel W., Lauterborn W. Cavitation bubble dynamics and acoustic transient generation in ocular surgery with pulsed neodymium: YAG lasers. Ophthalmology. 1986;93:1259–1269. doi: 10.1016/s0161-6420(86)33576-0. [DOI] [PubMed] [Google Scholar]
- 17.Nishimura N., Schaffer C.B., Kleinfeld D. Targeted insult to subsurface cortical blood vessels using ultrashort laser pulses: three models of stroke. Nat. Methods. 2006;3:99–108. doi: 10.1038/nmeth844. [DOI] [PubMed] [Google Scholar]
- 18.Nishimura N., Rosidi N.L., Schaffer C.B. Limitations of collateral flow after occlusion of a single cortical penetrating arteriole. J. Cereb. Blood Flow Metab. 2010;30:1914–1927. doi: 10.1038/jcbfm.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosidi N.L., Zhou J., Schaffer C.B. Cortical microhemorrhages cause local inflammation but do not trigger widespread dendrite degeneration. PLoS ONE. 2011;6:e26612. doi: 10.1371/journal.pone.0026612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baumgart J., Humbert L., Meunier M. Off-resonance plasmonic enhanced femtosecond laser optoporation and transfection of cancer cells. Biomaterials. 2012;33:2345–2350. doi: 10.1016/j.biomaterials.2011.11.062. [DOI] [PubMed] [Google Scholar]
- 21.Ingle N., Mohanty S.K. Live atomic force microscopy imaging of laser microbeam assisted cellular microsurgery. Proc. SPIE. 2011;7902:L1–L7. [Google Scholar]
- 22.Schneckenburger H., Hendinger A., Schmitt M. Laser-assisted optoporation of single cells. J. Biomed. Opt. 2002;7:410–416. doi: 10.1117/1.1485758. [DOI] [PubMed] [Google Scholar]
- 23.Puc M., Kotnik T., Miklavčič D. Quantitative model of small molecules uptake after in vitro cell electropermeabilization. Bioelectrochemistry. 2003;60:1–10. doi: 10.1016/s1567-5394(03)00021-5. [DOI] [PubMed] [Google Scholar]
- 24.Wang M., Orwar O., Weber S.G. Single-cell transfection by electroporation using an electrolyte/plasmid-filled capillary. Anal. Chem. 2009;81:4060–4067. doi: 10.1021/ac900265f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brochard-Wyart F., De Gennes P.G., Sandre O. Transient pores in stretched vesicles: role of leak-out. Physica A. 2000;278:32–51. [Google Scholar]
- 26.Karatekin E., Sandre O., Brochard-Wyart F. Transient pores in vesicles. Polym. Int. 2003;52:486–493. [Google Scholar]
- 27.Spaeth C.S., Robison T., Bittner G.D. Cellular mechanisms of plasmalemmal sealing and axonal repair by polyethylene glycol and methylene blue. J. Neurosci. Res. 2012;90:955–966. doi: 10.1002/jnr.23022. [DOI] [PubMed] [Google Scholar]
- 28.Baumgart J., Bintig W., Heisterkamp A. Fs-laser-induced Ca2+ concentration change during membrane perforation for cell transfection. Opt. Express. 2010;18:2219–2229. doi: 10.1364/OE.18.002219. [DOI] [PubMed] [Google Scholar]
- 29.Iwanaga S., Smith N.I., Kawata S. Slow Ca(2+) wave stimulation using low repetition rate femtosecond pulsed irradiation. Opt. Express. 2006;14:717–725. doi: 10.1364/opex.14.000717. [DOI] [PubMed] [Google Scholar]
- 30.Golzio M., Teissie J., Rols M.P. Direct visualization at the single-cell level of electrically mediated gene delivery. Proc. Natl. Acad. Sci. USA. 2002;99:1292–1297. doi: 10.1073/pnas.022646499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.