

Abstract
To investigate the relationship between a protein’s sequence and its biophysical properties, we studied the effects of more than 100 mutations in Avena sativa light-oxygen-voltage domain 2, a model protein of the Per-Arnt-Sim family. The A. sativa light–oxygen–voltage domain 2 undergoes a photocycle with a conformational change involving the unfolding of the terminal helices. Whereas selection studies typically search for winners in a large population and fail to characterize many sites, we characterized the biophysical consequences of mutations throughout the protein using NMR, circular dichroism, and ultraviolet/visible spectroscopy. Despite our intention to introduce highly disruptive substitutions, most had modest or no effect on function, and many could even be considered to be more photoactive. Substitutions at evolutionarily conserved sites can have minimal effect, whereas those at nonconserved positions can have large effects, contrary to the view that the effects of mutations, especially at conserved positions, are predictable. Using predictive models, we found that the effects of mutations on biophysical function and allostery reflect a complex mixture of multiple characteristics including location, character, electrostatics, and chemistry.
Introduction
A direct relationship exists between a protein’s sequence, structure, and function. However, the elucidation of this biophysical relationship can be difficult. Sometimes only a small portion of a protein is clearly involved in its function. In addition, proteins can have identical functions yet have less than 20–40% sequence similarity (1–3). These observations suggest that most of the protein’s structure serves as a scaffold (Fig. 1 A). In support of this view are protein-engineering studies that demonstrate that new functions such as enzymatic activity or binding interfaces can be grafted onto existing protein scaffolds (4–6). However, other studies indicate that a large fraction of the structure is necessary for function, making a global model a more appropriate description (Fig. 1 B). An analysis of the sequence covariance of PDZ domains and other proteins suggests that functional pathways permeate most of the structure (7,8). Similarly, the extremely high sequence conservation observed for actin (9), ubiquitin (10), and histones (11) across eukaryotes indicates that nearly the entire protein is functionally relevant in these proteins. Allosteric proteins with multiple conformational substates also are likely to be sensitive to mutations throughout the structure. Weinkam et al. (12) recently developed a hybrid molecular dynamics/machine-learning method that attempts to predict the impact of mutations on the allosteric equilibrium between two conformations by considering both local and global properties.
Figure 1.
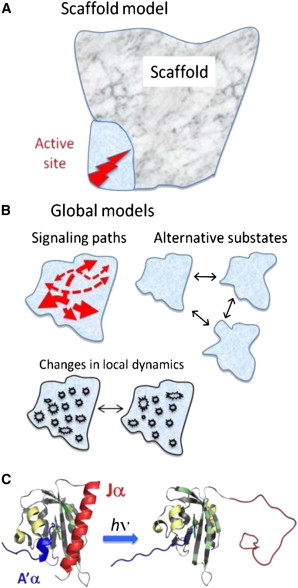
Alternative models for structure–function relationships. (A) Scaffold model in which only a small region of the protein is directly involved in function and the remainder serves as a scaffold (marble). (B) Global models in which many portions of the protein are involved in function, potentially by conducting allosteric signals through the protein with minimal change in structure, by stabilizing alternative substates, or through changes in local dynamics. (C) Conformational change of AsLOV2 upon light activation.
In principle, these two models can be distinguished by identifying the biophysical consequences of evolutionarily dissimilar mutations at multiple sites throughout the protein. When most of the structure serves as a scaffold, most substitutions are not expected to compromise function as long as the protein is structurally intact. Dissimilar substitutions in the functional regions, however, should have a strong, negative effect on function. In the global model, dissimilar substitutions at many sites across the protein are likely to be disruptive, although identifying the functional consequences may be experimentally challenging (e.g., if the protein has multiple binding conformations and partners or is susceptible to aggregation).
Many large-scale studies have selected for binding (13) or enzymatic activity (14–16), but only a few studies (17,18) have explicitly investigated how substitutions throughout a protein influence function, structure, or expression. Even fewer studies have looked at how mutations affect a conformational change or the changes in biophysical properties (19,20). This dearth presumably is due to the labor required to generate and functionally characterize individual variants. Nevertheless, we generated such a data set in our quest to understand the mechanism of light-induced conformational change in the second light–oxygen–voltage (LOV) domain of Avena sativa phototropin 1 (AsLOV2).
The AsLOV2 domain (Fig. 1 C) is a member of the Per-Arnt-Sim (PAS) superfamily. PAS-signaling domains are found in all kingdoms of life and often are a part of larger multidomain proteins. They respond to a diverse array of stimuli and generate a variety of output responses (21–25). The family has highly diverse sequences but a conserved 100–120 residue α/β-fold termed the PAS core (26). Generally, the input sensor is a ligand contained in a binding pocket located on one side of the five-stranded β-sheet, whereas the output function is mediated through the termini, typically helices, which reside on the other side of the sheet (27). The termini are highly variable, but they can undergo a conformational change when these regions activate effector domains (21,28–30).
Activation of the AsLOV2 domain occurs when the noncovalently bound flavin mononucleotide (FMN) chromophore absorbs a blue photon. The FMN forms a metastable covalent bond between its C4a atom and the sulfur on the C450 side chain. Adduct formation causes the protein to undergo a conformational change including the unfolding of the N-terminal A’α-helix. This event promotes the undocking of the C-terminal Jα-helix (30,31), and these unfolding events trigger kinase activity in the full-length phototropin (32). The flavin–cysteinyl covalent adduct spontaneously decays on a timescale of minutes and the helices refold to complete the photocycle (30,33).
To investigate the allosteric mechanism between adduct formation and the unfolding events, we characterized more than 100 variants of AsLOV2. Mutated sites included the evolutionarily conserved positions in AsLOV2 and three other flavin-containing LOV domains, AsLOV1, Vvd from Neurospora crassa, and YtvA from Bacillus subtilis, which share 36–73% sequence identity (Fig. 2 A). Mutations were intended to be mildly or strongly disruptive, including large aliphatic to alanine and aromatic to aliphatic substitutions, and of differing charge or polarity to test specific hypotheses. Our results indicate that AsLOV2 does not fit into a standard model of protein function but one in which chemically dissimilar mutations can both positively and negatively impact a protein’s function, indicating that the functional landscape is plastic.
Figure 2.
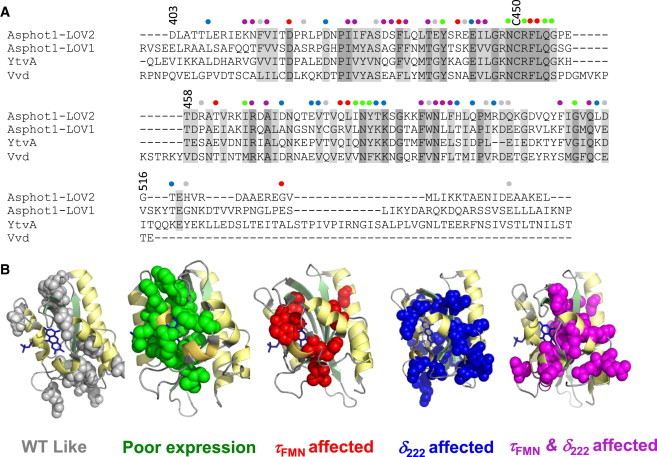
AsLOV2 conservation and mutations. (A) Sequence alignment in AsLOV1, AsLOV2, YtvA, and Vvd (dark gray, identical; light gray, similar). (B) Consequences of mutations mapped onto the AsLOV2 structure.
Materials and Methods
Cloning, expression, and purification
A construct of AsLOV2 (residues 404–560) with an N-terminal His6-Gβ1 tag was used. Mutations were created using standard site-directed mutagenesis techniques and verified by sequencing. As previously described (30), all proteins were expressed in BL21 (DE3) Codon Plus cells (Invitrogen, Carlsbad, CA) grown in LB at 37°C until an OD600 of 0.6. The His6-Gβ1 tag was removed using tobacco etch virus protease. The final protein contained residues GEF on the N-terminus and G on the C terminus as cloning artifacts. Proteins were purified on a Sephadex S100 size exclusion column (GE Healthcare, Tyrone, PA), and if the Abs280nm/Abs447nm ratio differed greatly from ∼2.7, proteins were further purified using anion exchange chromatography.
Spectroscopy
Circular dichroism studies were performed on a Jasco J-715 spectrometer (Jasco, MD) using ∼1 μM protein in 50-mM NaH2PO4, 200-mM NaCl, pH 7 at 22°C using a 1-cm pathlength cuvette. Averages were taken of the first 10–20 data points and last 10–20 data points, depending on the noise, to calculate a δ222-value. Ultraviolet-visual spectra were acquired using a Hewlett Packard diode array with a 1-cm pathlength cuvette. Samples were illuminated using a 40-W white LED (Model BT DWNLT A, The LED Light, Carson City, NV) for 30 s and the absorbance at the λmax, usually 448 nm, was measured every 1–30 s depending on the photorecovery rate. The data were fit to a single exponential decay using Origin software (OriginLab, Northampton, MA). NMR experiments were performed on a Varian Inova 600 MHz spectrometer with a cryoprobe at 25°C. Proteins were concentrated to 100–500 μM in 10% D2O. Data were processed using NMRPipe (34) and analyzed using SPARKY (35). NMR experiments were run using standard pulse sequences available in Varian BioPack, 15N-1H heteronuclear single quantum coherence (HSQC) (gNhsqc).
Statistical analyses
The R package randomForest (36) was used to implement the Random Forest algorithm using 10,000 trees per run with two variables randomly sampled at each tree split. We here define a significant effect on τFMN as a deviation of ≥ 22 s from wild-type AsLOV2 and a significant effect on δ222 as a deviation of ≥ 0.075 units from wild-type AsLOV2. Classifier accuracy was determined using the identical thresholds for the predicted values. Receiver operating characteristic area under the curves was determined by plotting the true positive rate versus the false positive rate as a function of prediction values versus expected binary effects using the package ROCR (37).
Results and Discussion
Structural and functional effects
Most large-scale mutational analyses screen for certain properties that are enhanced compared to the wild-type protein. Our data set is different in that we characterized each individual variant biophysically according to whether the substitution affected the photocycle, the conformational change, or severely compromised expression. We also looked at NMR spectra of 26 variants to assay for compromised structure. The photocycle lifetime, τFMN, is determined using ultraviolet-visual spectroscopy, A448nm, the absorbance at λ = 448 nm. For wild-type AsLOV2, τFMN is 80 ± 2 s at 293K (30,38). The lifetime is limited by deprotonation of the N5 on the flavin (39), which can be affected by solvent exposure or by reducing the stability of the N5-hydrogen bond. Other factors affecting the lifetime include steric destabilization of the adduct conformation or electronic effects that destabilize the reduced state (33).
The majority of photocycles measured in LOV domains have lifetimes at or greater than AsLOV2’s value of 80 s (33,40). Longer activation could be detrimental through the overexpression of genes, but shorter activation could be even more so as some genes may be underexpressed or not expressed at all. Phototropin-containing LOV domains should provide a fitness advantage in low-light and high-light situations (41–43). This reasoning suggests that shorter photocycle lifetimes would generally result in less fitness.
The extent of allosteric conformational change is determined using the circular dichroism signal at λ = 222 nm, θ222, with the fractional change defined as δ222 = (θ222,dark − θ222,lit)/θ222,dark. For the WT protein, δ222 = 0.30 units (representative kinetic spectra in Fig. S3 in the Supporting Material). This parameter mostly reflects the unfolding of the ∼20 residue Jα-helix (∼0.15 units) and the ∼6 residue A’α-helix (∼0.05 units) (30,44). The source of the remaining 0.10 units is unknown and may be due to a change in the conformation of the β-sheets and the other three helices. A reduction in the δ222-value likely is because of less unfolding of the two helices upon illumination, resulting from either a decrease in helicity in the dark state or an increase in helicity in the lit state.
To maximize the chance of finding functionally significant positions in the protein, we introduced dissimilar mutations at evolutionarily conserved sites with the intent of disrupting AsLOV2’s function. The degree of dissimilarity of the substitutions was quantified according to BLOSUM62 (45) and SIFT scores (46). BLOSUM62 scores reflect the amino acid substitution frequency observed in many proteins. SIFT scores are generated for a single sequence alignment, in our case, for PAS domains. BLOSUM62 scores reflect canonical amino acid substitutability, and SIFT scores include information specific to PAS domains. BLOSUM62 values range from −4.00 to 11.00, with negative scores given to less likely substitutions. SIFT scores range from 0.00 to 1.00, with scores less than or equal to 0.05 predicted to be deleterious. In our set of the 93 single-point mutants, 62 had a BLOSUM62 score below 1.00 and another partially disjoint set of 62 had SIFT scores below 0.05. Intriguingly, the correlation between these two metrics is negligible (Fig. 3 and Table S1). Potentially, the divergence in LOV domains input (e.g., FMN or flavin adenine dinucleotide) and output modes (e.g., different amino and carboxy helices) going to different effectors produces this poor correlation.
Figure 3.
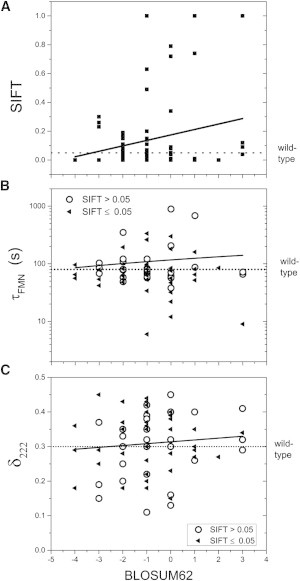
Effects of point mutations on photocycle times and conformational change. (A) BLOSUM62 and SIFT scores are poorly correlated (black line). (B and C) Effects as a function of BLOSUM62 score.
Of the 105 total variants, the 17 that expressed poorly are at 13 evolutionarily conserved positions. These substitutions, which are located in or near the FMN binding pocket, likely alter FMN binding because of destabilization of the binding pocket or the loss of interactions with the flavin. Sixty-five mutants affect either or both τFMN and δ222 (Fig. 4 and Table S1) with the overall pattern partially explainable by location within the protein (Fig. 2 B) rather than the BLOSUM62 or SIFT scores, which have little if any correlation with either τFMN or δ222 (Fig. 3, B and C). Only substitutions of the adduct-forming C450 completely prevented photocycling.
Figure 4.

Effects of function altering single-point mutations mapped onto the secondary structure. Single stars indicate conserved sequence similarity in the LOV family and two stars indicate complete or near complete sequence conservation.
To sample how mutations affected the structural integrity of proteins, we acquired 15N-1H HSQC spectra of a diverse set of 26 variants (representative NMR spectra in Fig. S4). In each spectrum, the majority of peaks with chemical shift differences were near the site of the mutation, which is expected given the change in the local chemical environment. Twenty-one variants have a WT-like 15N-1H HSQC NMR spectrum (Fig. 4). For 4 variants, their HSQC spectra are similar to the spectrum of AsLOV2ΔJα, a construct lacking the Jα-helix. This similarity suggests that this helix is undocked in these variants in the dark, but the remainder of the protein is otherwise intact. The final variant, L493A, a rather benign substitution (BLOSUM62 = −1; SIFT = 0.5), has a spectrum with the majority of the peaks not overlapping with either the wild-type or AsLOV2ΔJα. However, L493A still photocycles (τFMN = 121 s) but with minimal conformational change (δ222 = 0.11), suggesting a significant structural perturbation. Regardless, this was the only variant of the 26 examined with a measurably compromised structure, and the effects are essentially unpredictable based on either SIFT or BLOSUM62.
Twenty-seven substitutions (26%) significantly affected the photocycle time (τFMN < 50 s or τFMN > 110 s), and 55 (52%) altered the conformational change (δ222 < 0.25 or δ222 > 0.35). No obvious correlation exists between these two properties (Fig. 3 A). For the eight substitutions that only affected τFMN, their side chains are located near the chromophore but distal to the A’α- and Jα-helices. The complementary pattern is observed for 30 of 36 mutations that affected only δ222; their side chains are not adjacent to the chromophore and reside either on or near the Jα-helix and the A’α-helix, or they face outward on the β-sheet. The 21 (20%) substitutions that affected both τFMN and δ222 are generally on or located near the central β-sheet and the two helices. Another 21 (20%) substitutions that have near wild-type properties are located throughout the whole protein. These mutations are generally located toward the surface of the protein, with some potentially in functionally significant positions contacting the A’α-helix, the Jα-helix, and the chromophore.
Structure–function and conservation relationships
More than 75% of the variants have an effect on either or both τFMN and δ222. Effects are found at both evolutionarily conserved and nonconserved sites. The most sensitive sites tend to be in the anticipated regions, for example, near the chromophore or the A’α- and Jα-helices. Because effects are found throughout the protein, we conclude that most of the protein does not function as an inert scaffold. Potentially, this result reflects the small size of the protein coupled with its multiple functionalities related to ligand binding and conformational change. As a result, the average substitution is likely to be located in or near functional regions. Nevertheless, effects also are found for substitutions distal to these moieties (Fig. 2 B).
Our second major finding is that most mutations have only modest effects on τFMN and δ222. No single mutation completely eliminates the photocycle except for ones at the adduct-forming C450 position (although the functional properties are unknown for the 17 poorly expressing substitutions that were not otherwise characterized). This tolerance argues that the light-triggered conformational change does not result from a single mechanism. Rather, multiple mechanisms are probably involved in relaying the signal from the FMN to the A’α- and Jα-helices. Some mechanisms may involve a conformational change in the β-sheet (47,48).
Our third notable finding is that ∼60% of the substitutions have biophysical effects in the direction opposite of what we anticipated. The photocycle and conformational change of AsLOV2 require an architecture with an extensive set of interactions along with a binding pocket that prevents access of solvent to the FMN so that it can form a long-lived adduct. In turn, the adduct generates a signal that is transduced across the protein, resulting in a conformational change via the A’α-helix and potentially involving the β-sheets as well. Accordingly, we anticipated that most of our substitutions should have reduced the photocycle time by destabilizing the excited, metastable state of the chromophore and/or the magnitude of the conformational change by disrupting a signalling pathway.
Unexpectedly, most mutations either lengthen the photocycle or increase the magnitude of the conformational change. We appreciate that τFMN and δ222 are biophysical properties of the isolated AsLOV2 domain, whereas the actual biological function of phototropin 1 is related to phosphorylation and phototropism. Nevertheless, most characterized phototropin LOV domains have a minute-long photocycle (40) and mechanisms of conformational change have been validated in vivo (32,49). These results support our contention that photocycling times and conformational change are properties critical to AsLOV2’s biological function, and altering these properties can compromise fitness. Therefore, AsLOV2 does not appear to fit well into the second structure–function paradigm in which dissimilar substitutions are often disruptive.
A protein’s active site generally is conserved across its family (50). In LOV domains, however, conserved residues appear to play a larger role in stability than function. All 17 mutations that poorly expressed are substitutions at conserved positions and have SIFT scores below 0.05. On average, substitutions at conserved positions have a bigger influence on the τFMN than on δ222, suggesting that the photocycle lifetime is under stronger selection pressure than the conformational change (Fig. 4 and Fig. 5). The six substitutions that most accelerated the photocycle are at conserved sites. Similarly, the majority of the substitutions that decelerated the photocycle are at conserved sites, although the three substitutions having the largest effects on photocycle lifetime are at the nonconserved N414 position. However, conservation is generally a poor predictor for the effect of a substitution on δ222, with the largest effects mostly occurring at nonconserved sites. Therefore, sequence conservation generally is an inaccurate reporter of the functional significance of sites in AsLOV2.
Figure 5.
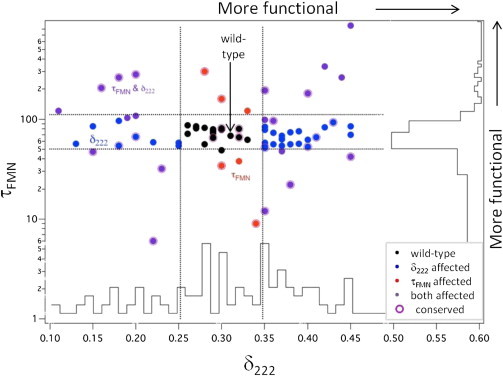
Comparison of τFMN and δ222 of mutant proteins. The distribution for τFMN (vertical histogram) is narrower than for δ222 (horizontal). Dotted lines denote our boundaries for wild-type behavior.
Among the conserved residues, we identified an unusual three-residue β-bulge that is conserved across all PAS domains with known structures and comes from all kingdoms of life (27). The billion-year conservation of this motif located at the amino terminus of the Bβ-strand (I427-I428-F429 in AsLOV2) sharply contrasts with the extensive diversity in PAS function and the type and location of ligands within PAS domains. Suspecting that this region would be functionally sensitive, we extensively mutated it. We found only moderate effects, except for a charged I428D substitution that expressed poorly. We tentatively conclude that the highly conserved bulge structure is a legacy that is difficult to evolve away without compromising foldability. This underscores our finding that conservation is not an accurate predictor of function in AsLOV2.
Of the 74 single-residue mutations that expressed, 44 are at positions that can be hydrogen-bond donors or acceptors. Using a molecular dynamics relaxed structure of AsLOV2 derived from PDB ID: 2V1B (30), we found that only 23 of the side chains form intraprotein hydrogen bonds. Of these 23 residues, 13 (57%) have an effect on ultraviolet or circular dichroism when mutated to a non–hydrogen-bonding residue. The presence or absence of a hydrogen bond on the wild-type side chain is a mediocre predictor of an effect. However, this deficiency does not imply that all hydrogen bonds are of equal importance. We anticipated that side-chain hydrogen bonding would play a greater functional role than what we observed as changes in hydrogen-bonding patterns have been suggested to be a significant factor in the mechanism of light-activated conformational change in LOV2 (51–53) and photoactive yellow protein (PYP) (17).
We divided the residues into seven chemical categories, polar (Gln and Asn), positive (Arg, Lys, and His), negative (Asp, Glu, and Tyr), large hydrophobic (Ile, Leu, Met, Phe, Trp), small hydrophobic (Ala and Val), (Pro and Gly), and (Thr, Ser, Cys). When polar residues are substituted, 67% have an effect on conformational change. Mutations to polar residues also affect the photocycle length 67% of the time. These results point to the general importance of electrostatics in the protein’s function.
To quantify whether amino acid conservation can predict function in AsLOV2, we examined the correlation between a mutation’s τFMN and δ222 value with its SIFT and BLOSUM62 scores (Fig. 3 and Table S1). Although no significant correlation was observed between either score and τFMN or δ222, some trends emerged. For example, mutations that were or were not predicted to be deleterious according to SIFT had a mean difference in τFMN of only 26 and 18 s, respectively. Unfavorable mutations, with BLOSUM62 scores below 1.00, had a median effect on δ222 of 0.07 units, whereas the mutations with positive scores had a median effect of 0.03 units. Only 3 of the 12 mutations with positive BLOSUM62 scores changed δ222 by at least 0.05 units, whereas 28 of the 47 mutations with BLOSUM62 scores below 0.00 (∼60%) changed δ222 by this amount, although the majority increased this quantity (18 increased δ222 and 10 decreased δ222). This difference indicates that unfavorable BLOSUM62 scores are enriched with mutations that positively affect δ222.
To examine whether we could predict τFMN or δ222 levels from our basic feature data (original and mutated residue, residue position, SIFT and BLOSUM62 scores), we implemented the Random Forests algorithm to construct predictive classifiers and assess the proportion of variance explained by each variable (36,54). The Random Forests algorithm is a machine-learning technique that uses thousands of independent decision trees to perform classification or regression by building trees from sampling random subsets of all available variables (36). Leveraging our feature data alone, our Random Forests classifier for δ222 was 69% accurate at predicting interactions that affect δ222 and had an area under the receiver operating characteristic curve of 0.84 (Fig. S2 A). The Pearson correlation coefficient (r) between predicted and experimental δ222 values was 0.91 (Fig. S2 B). Our classifier for τFMN was 77% accurate at predicting interactions that affect τFMN and had an area under the receiver operating characteristic curve of 0.85 (Fig. S2 D). The r-value between predicted and experimental τFMN values was 0.90 (Fig. S2 E). These observations are predominantly because of the smaller proportion of mutations that have an effect on τFMN. Therefore, the classifier was considerably more specific (accurate at correctly predicting mutations that would not affect τFMN) than sensitive. The BLOSUM62 score and mutated amino acid features are the most important features for classifying δ222, and the original and mutated amino acid features are the most important for classifying τFMN (Fig. S2, C and F). The reliability of our predictions is comparable to similar predictive models that have used considerably denser feature data to predict mutation-induced protein stability changes (55). However, our classifiers explained only 39% of the total variance in τFMN effects (Fig. S2 D) and could not accurately predict δ222 effects (Fig. S2 B). The relative contributions of SIFT scores are minor to predicting the effect of a mutation on τFMN or δ222, further supporting the notion that sequence conservation is an inaccurate reporter of the functional significance in PAS domains.
As compared with any other factor we examined, negative BLOSUM62 scores were the best predictor of mutational effects. Potentially, this scoring system accurately represents changes in residue character (charge, sterics, and other chemical properties) because this score takes into account the character of both the wild-type and substituted residues. Mutations with negative BLOSUM62 scores have >60% chance of having an effect on allosteric conformational change and photocycle length. Overall, BLOSUM62 scores are good predictors; however, the inclusion of more specific terms such as polarity or hydrophobicity may be beneficial.
Comparison with other PAS domains
In contrast to the typical selection study that identifies only winners, for example, tighter binders or more active enzymes (56,57), only a few studies have measured function for point mutations throughout the protein. In one such study, Hoff and coworkers (17) performed a global alanine scan on PYP, another PAS domain. Their results were similar to ours in that ∼60% of the substitutions had modest effects. This finding led them to conclude that PYP combines robustness with a high degree of evolvability. This conclusion is supported by our data for AsLOV2, suggesting that it is a common property of PAS domains and may explain their widespread use in a variety of contexts across multiple kingdoms.
Using an analysis of sequence covariation among PAS domains, Halabi et al. (19) identified two distinct functional sectors in LOV2, the loop connecting the Jα-helix to PAS core and the residues around the chromophore. However, the allosteric communication in LOV2 is between adduct formation and the unfolding Jα-helix. Hence, one may have expected these two regions to be part of the same functional sector. The failure to identify the allostery using sequence covariation probably can be explained by PAS domains having evolved to have different carboxy terminal output-signaling motifs as well as different binding pockets for different ligands (e.g., FMN or flavin adenine dinucleotide).
Freddolino et al. (52) applied clustering methods to their molecular dynamics for both lit- and dark-state trajectories of AsLOV2. They found that A’α-helix motions correlate with motions of the Aβ- and Bβ-strands, and that these motions play an important role in the initial dissociation of the Jα-helix, a finding supported by our current and previous findings (30). The authors also propose that a G528A mutation should stabilize Jα-helix docking. Although this mutation does not increase δ222, it does decrease conformational fluctuations in an AsLOV2–TrpR photoswitch (44). Furthermore, the G528A mutation decreases τFMN by approximately twofold, suggesting that mutations in the N-terminus of the Jα-helix can form an interaction network with the FMN. The molecular dynamics analysis suggests a role for the tilting of the Iβ-strand. Previously, we found the μsecond–millisecond dynamics of the whole Iβ are poorly correlated with the dynamics of A’α, Hβ, and Jα, the three structural elements whose dynamics are most correlated with undocking of the Jα-helix (30). However, the Iβ-sheet could play a more nuanced role in photoactivated conformational change, as the β-sheets are shown by our analysis to be sensitive to mutations. A more detailed study is needed to determine the role of the β-sheets in photoexcitation and helix undocking.
Comparison with other proteins
With regards to other proteins, when a large mutational library of TEM-1 β-lactamase was screened for ampicillin resistance, the wild-type amino acid was found at only 16% of the positions (18). These residues are mostly located in the active site or on the binding surface, although some are scattered throughout the protein. A selection study using the DNA repair enzyme 3-methyladenine DNA glycosylase calculated a 34% probability that a random substitution would reduce the activity of the enzyme below a particular level (15). Using a β-galactosidase-coupled assay of DNA binding by λ Cro, Pakula et al. (58) found that stability and DNA binding were altered in one third and one sixth of random mutations, respectively. Conserved and catalytically crucial residues are approximately twofold less substitutable than residues in the rest of the protein. These studies agree with our study in that many substitutions do not have a negative effect.
Studies on other proteins have found different behavior. Palzkill and coworkers noted a positive correlation between tolerance to substitution and solvent accessibility with buried residues being less tolerant (18) for results on λ repressor (59), lac repressor (60), T4 lysozyme (61), and the f1 gene V protein (62). A directed evolution screen of cytochrome P450 BM3 variants for increased hydroxylation of small alkanes observed effects at a few sites distal to the functional region (63). The difference between the results we see found and those of other systems may be attributed to the function involving only a small fraction of the protein or the conformational change may be less and more localized. These studies also highlight that the interpretation of the effects of distal mutations could benefit from having structural information or additional information regarding the dynamics and excited state(s) rather than relying on the structure of the ground state alone.
The concept of a localized active site has been useful for understanding protein structure–function relationships. However, our and other data suggest that mutations at many sites in a protein can alter its function. As a result, the protein mutational landscape is plastic, allowing for the evolution of novel functions. Furthermore, the observation that many putative deleterious substitutions throughout AsLOV2 only mildly influence function suggests that allosteric communication can be generated by multiple mechanisms.
Conclusion
We performed one of the few large-scale functional analyses of individual mutations throughout a protein, in particular, one undergoing an allosteric conformational change. Despite our intention to introduce disruptive substitutions, most substitutions have modest or no effect on function, and many even appear more functional in that they either lengthen the photocycle (i.e., stabilize the metastable lit state) or increase the magnitude of the conformational change. Completely conserved residues can have minimal effect, whereas substitutions at nonconserved sites can have large effects. These data suggest that a PAS domain’s allosteric function involves a more complex interplay of residues throughout the protein than can be described by either the standard scaffold or global models of protein function. Further studies are required to assess whether our LOV2 results are generalizable to other proteins, in particular those undergoing pervasive conformational changes.
Acknowledgments
J.P.Z., C.A., and A.R.F. performed the experiments. J.P.Z, C.A., A.R.F., R.H., and T.R.S. analyzed the data. J.P.Z, C.A., A.R.F., R.H., and T.R.S. wrote the manuscript.
The authors declare no competing financial interests.
The authors thank J. Thornton, D. Strickland, M. Glotzer, and members of our group for helpful discussions and comments on the article.
This work was supported by research and training grants from the U.S. National Institutes of Health (GM088668 to T.R.S. and M. Glotzer, 5T32GM007183-34 to B. Glick and T32 GM07197 to L. Rothman-Denes) and the Chicago Biomedical Consortium with support from The Searle Funds at The Chicago Community Trust (to T.R.S., M. Glotzer, and E. Weiss).
Supporting Material
References
- 1.Addou S., Rentzsch R., Orengo C.A. Domain-based and family-specific sequence identity thresholds increase the levels of reliable protein function transfer. J. Mol. Biol. 2009;387:416–430. doi: 10.1016/j.jmb.2008.12.045. [DOI] [PubMed] [Google Scholar]
- 2.Attwood T.K., Eliopoulos E.E., Findlay J.B. Multiple sequence alignment of protein families showing low sequence homology: a methodological approach using database pattern-matching discriminators for G-protein-linked receptors. Gene. 1991;98:153–159. doi: 10.1016/0378-1119(91)90168-b. [DOI] [PubMed] [Google Scholar]
- 3.Jensen L.J., Ussery D.W., Brunak S. Functionality of system components: conservation of protein function in protein feature space. Genome Res. 2003;13:2444–2449. doi: 10.1101/gr.1190803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gilbreth R.N., Esaki K., Koide S. A dominant conformational role for amino acid diversity in minimalist protein–protein interfaces. J. Mol. Biol. 2008;381:407–418. doi: 10.1016/j.jmb.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koide A., Gilbreth R.N., Koide S. High-affinity single-domain binding proteins with a binary-code interface. Proc. Natl. Acad. Sci. USA. 2007;104:6632–6637. doi: 10.1073/pnas.0700149104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wojcik J., Hantschel O., Koide S. A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain. Nat. Struct. Mol. Biol. 2010;17:519–527. doi: 10.1038/nsmb.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerek Z.N., Ozkan S.B. Change in allosteric network affects binding affinities of PDZ domains: analysis through perturbation response scanning. PLoS Comput. Biol. 2011;7:e1002154. doi: 10.1371/journal.pcbi.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petit C.M., Zhang J., Lee A.L. Hidden dynamic allostery in a PDZ domain. Proc. Natl. Acad. Sci. USA. 2009;106:18249–18254. doi: 10.1073/pnas.0904492106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheterline P., Clayton J., Sparrow J. Actin. Protein Profile. 1995;2:1–103. [PubMed] [Google Scholar]
- 10.Catic A., Ploegh H.L. Ubiquitin—conserved protein or selfish gene? Trends Biochem. Sci. 2005;30:600–604. doi: 10.1016/j.tibs.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Postberg J., Forcob S., Lipps H.J. The evolutionary history of histone H3 suggests a deep eukaryotic root of chromatin modifying mechanisms. BMC Evol. Biol. 2010;10:259. doi: 10.1186/1471-2148-10-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weinkam P., Chen Y.C., Sali A. Impact of mutations on the allosteric conformational equilibrium. J. Mol. Biol. 2013;425:647–661. doi: 10.1016/j.jmb.2012.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fowler D.M., Araya C.L., Fields S. High-resolution mapping of protein sequence–function relationships. Nat. Methods. 2010;7:741–746. doi: 10.1038/nmeth.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thyme S.B., Jarjour J., Baker D. Exploitation of binding energy for catalysis and design. Nature. 2009;461:1300–1304. doi: 10.1038/nature08508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo H.H., Choe J., Loeb L.A. Protein tolerance to random amino acid change. Proc. Natl. Acad. Sci. USA. 2004;101:9205–9210. doi: 10.1073/pnas.0403255101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexandrova A.N., Röthlisberger D., Jorgensen W.L. Catalytic mechanism and performance of computationally designed enzymes for Kemp elimination. J. Am. Chem. Soc. 2008;130:15907–15915. doi: 10.1021/ja804040s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Philip A.F., Kumauchi M., Hoff W.D. Robustness and evolvability in the functional anatomy of a PER-ARNT-SIM (PAS) domain. Proc. Natl. Acad. Sci. USA. 2010;107:17986–17991. doi: 10.1073/pnas.1004823107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang W., Petrosino J., Palzkill T. Amino acid sequence determinants of beta-lactamase structure and activity. J. Mol. Biol. 1996;258:688–703. doi: 10.1006/jmbi.1996.0279. [DOI] [PubMed] [Google Scholar]
- 19.Halabi N., Rivoire O., Ranganathan R. Protein sectors: evolutionary units of three-dimensional structure. Cell. 2009;138:774–786. doi: 10.1016/j.cell.2009.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reynolds K.A., McLaughlin R.N., Ranganathan R. Hot spots for allosteric regulation on protein surfaces. Cell. 2011;147:1564–1575. doi: 10.1016/j.cell.2011.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zoltowski B.D., Schwerdtfeger C., Crane B.R. Conformational switching in the fungal light sensor Vivid. Science. 2007;316:1054–1057. doi: 10.1126/science.1137128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McIntosh B.E., Hogenesch J.B., Bradfield C.A. Mammalian Per-Arnt-Sim proteins in environmental adaptation. Annu. Rev. Physiol. 2010;72:625–645. doi: 10.1146/annurev-physiol-021909-135922. [DOI] [PubMed] [Google Scholar]
- 23.Krell T., Lacal J., Ramos J.L. Bacterial sensor kinases: diversity in the recognition of environmental signals. Annu. Rev. Microbiol. 2010;64:539–559. doi: 10.1146/annurev.micro.112408.134054. [DOI] [PubMed] [Google Scholar]
- 24.Henry J.T., Crosson S. Ligand-binding PAS domains in a genomic, cellular, and structural context. Annu. Rev. Microbiol. 2011;65:261–286. doi: 10.1146/annurev-micro-121809-151631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morais Cabral J.H., Lee A., Mackinnon R. Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell. 1998;95:649–655. doi: 10.1016/s0092-8674(00)81635-9. [DOI] [PubMed] [Google Scholar]
- 26.Hefti M.H., Françoijs K.-J., Vervoort J. The PAS fold. A redefinition of the PAS domain based upon structural prediction. Eur. J. Biochem. 2004;271:1198–1208. doi: 10.1111/j.1432-1033.2004.04023.x. [DOI] [PubMed] [Google Scholar]
- 27.Möglich A., Ayers R.A., Moffat K. Structure and signaling mechanism of Per-ARNT-Sim domains. Structure. 2009;17:1282–1294. doi: 10.1016/j.str.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harigai M., Yasuda S., Kataoka M. Amino acids in the N-terminal region regulate the photocycle of photoactive yellow protein. J. Biochem. 2001;130:51–56. doi: 10.1093/oxfordjournals.jbchem.a002961. [DOI] [PubMed] [Google Scholar]
- 29.Ng C.A., Hunter M.J., Vandenberg J.I. The N-terminal tail of hERG contains an amphipathic α-helix that regulates channel deactivation. PLoS ONE. 2011;6:e16191. doi: 10.1371/journal.pone.0016191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zayner J.P., Antoniou C., Sosnick T.R. The amino-terminal helix modulates light-activated conformational changes in AsLOV2. J. Mol. Biol. 2012;419:61–74. doi: 10.1016/j.jmb.2012.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halavaty A.S., Moffat K. N- and C-terminal flanking regions modulate light-induced signal transduction in the LOV2 domain of the blue light sensor phototropin 1 from Avena sativa. Biochemistry. 2007;46:14001–14009. doi: 10.1021/bi701543e. [DOI] [PubMed] [Google Scholar]
- 32.Harper S.M., Christie J.M., Gardner K.H. Disruption of the LOV-Jalpha helix interaction activates phototropin kinase activity. Biochemistry. 2004;43:16184–16192. doi: 10.1021/bi048092i. [DOI] [PubMed] [Google Scholar]
- 33.Zoltowski B.D., Vaccaro B., Crane B.R. Mechanism-based tuning of a LOV domain photoreceptor. Nat. Chem. Biol. 2009;5:827–834. doi: 10.1038/nchembio.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delaglio F., Grzesiek S., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 35.Kneller D.G., Kuntz I.D. UCSF Sparky: an NMR display, annotation and assignment tool. J. Cell. Biochem. 1993;53:254. [Google Scholar]
- 36.Breiman L. Random Forests. Mach. Learn. J. 2001;45:5–32. [Google Scholar]
- 37.Sing T., Sander O., Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21:3940–3941. doi: 10.1093/bioinformatics/bti623. [DOI] [PubMed] [Google Scholar]
- 38.Nash A.I., Ko W.-H., Gardner K.H. A conserved glutamine plays a central role in LOV domain signal transmission and its duration. Biochemistry. 2008;47:13842–13849. doi: 10.1021/bi801430e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alexandre M.T.A., Arents J.C., Kennis J.T.M. A base-catalyzed mechanism for dark state recovery in the Avena sativa phototropin-1 LOV2 domain. Biochemistry. 2007;46:3129–3137. doi: 10.1021/bi062074e. [DOI] [PubMed] [Google Scholar]
- 40.Kasahara M., Swartz T.E., Briggs W.R. Photochemical properties of the flavin mononucleotide-binding domains of the phototropins from Arabidopsis, rice, and Chlamydomonas reinhardtii. Plant Physiol. 2002;129:762–773. doi: 10.1104/pp.002410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jarillo J.A., Gabrys H., Cashmore A.R. Phototropin-related NPL1 controls chloroplast relocation induced by blue light. Nature. 2001;410:952–954. doi: 10.1038/35073622. [DOI] [PubMed] [Google Scholar]
- 42.Kagawa T., Sakai T., Wada M. Arabidopsis NPL1: a phototropin homolog controlling the chloroplast high-light avoidance response. Science. 2001;291:2138–2141. doi: 10.1126/science.291.5511.2138. [DOI] [PubMed] [Google Scholar]
- 43.Liscum E., Briggs W.R. Mutations in the NPH1 locus of Arabidopsis disrupt the perception of phototropic stimuli. Plant Cell. 1995;7:473–485. doi: 10.1105/tpc.7.4.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Strickland D., Yao X., Sosnick T.R. Rationally improving LOV domain-based photoswitches. Nat. Methods. 2010;7:623–626. doi: 10.1038/nmeth.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henikoff S., Henikoff J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ng P.C., Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Möglich A., Moffat K. Structural basis for light-dependent signaling in the dimeric LOV domain of the photosensor YtvA. J. Mol. Biol. 2007;373:112–126. doi: 10.1016/j.jmb.2007.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kennis J.T.M., Crosson S., van Grondelle R. Primary reactions of the LOV2 domain of phototropin, a plant blue-light photoreceptor. Biochemistry. 2003;42:3385–3392. doi: 10.1021/bi034022k. [DOI] [PubMed] [Google Scholar]
- 49.Aihara Y., Yamamoto T., Nagatani A. Mutations in N-terminal flanking region of blue light-sensing light-oxygen and voltage 2 (LOV2) domain disrupt its repressive activity on kinase domain in the Chlamydomonas phototropin. J. Biol. Chem. 2012;287:9901–9909. doi: 10.1074/jbc.M111.324723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galperin M.Y., Koonin E.V. Divergence and convergence in enzyme evolution. J. Biol. Chem. 2012;287:21–28. doi: 10.1074/jbc.R111.241976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peter E., Dick B., Baeurle S.A. Mechanism of signal transduction of the LOV2-Jα photosensor from Avena sativa. Nat Commun. 2010;1:122. doi: 10.1038/ncomms1121. [DOI] [PubMed] [Google Scholar]
- 52.Freddolino P.L., Gardner K.H., Schulten K. Signaling mechanisms of LOV domains: new insights from molecular dynamics studies. Photochem. Photobiol. Sci. 2013;12:1158–1170. doi: 10.1039/c3pp25400c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Möglich A., Yang X., Moffat K. Structure and function of plant photoreceptors. Annu. Rev. Plant Biol. 2010;61:21–47. doi: 10.1146/annurev-arplant-042809-112259. [DOI] [PubMed] [Google Scholar]
- 54.Liaw A., Wiener M. Classification and regression by randomForest. R News. 2002;2:18–22. [Google Scholar]
- 55.Li Y., Fang J. PROTS-RF: a robust model for predicting mutation-induced protein stability changes. PLoS ONE. 2012;7:e47247. doi: 10.1371/journal.pone.0047247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hagemann U.B., Mason J.M., Arndt K.M. Selectional and mutational scope of peptides sequestering the Jun-Fos coiled-coil domain. J. Mol. Biol. 2008;381:73–88. doi: 10.1016/j.jmb.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 57.Huang J., Koide A., Koide S. Design of protein function leaps by directed domain interface evolution. Proc. Natl. Acad. Sci. USA. 2008;105:6578–6583. doi: 10.1073/pnas.0801097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pakula A.A., Young V.B., Sauer R.T. Bacteriophage lambda cro mutations: effects on activity and intracellular degradation. Proc. Natl. Acad. Sci. USA. 1986;83:8829–8833. doi: 10.1073/pnas.83.23.8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bowie J.U., Reidhaar-Olson J.F., Sauer R.T. Deciphering the message in protein sequences: tolerance to amino acid substitutions. Science. 1990;247:1306–1310. doi: 10.1126/science.2315699. [DOI] [PubMed] [Google Scholar]
- 60.Markiewicz P., Kleina L.G., Miller J.H. Genetic studies of the lac repressor. XIV. Analysis of 4000 altered Escherichia coli lac repressors reveals essential and non-essential residues, as well as “spacers” which do not require a specific sequence. J. Mol. Biol. 1994;240:421–433. doi: 10.1006/jmbi.1994.1458. [DOI] [PubMed] [Google Scholar]
- 61.Rennell D., Bouvier S.E., Poteete A.R. Systematic mutation of bacteriophage T4 lysozyme. J. Mol. Biol. 1991;222:67–88. doi: 10.1016/0022-2836(91)90738-r. [DOI] [PubMed] [Google Scholar]
- 62.Terwilliger T.C., Zabin H.B., Schlunk P.M. In vivo characterization of mutants of the bacteriophage f1 gene V protein isolated by saturation mutagenesis. J. Mol. Biol. 1994;236:556–571. doi: 10.1006/jmbi.1994.1165. [DOI] [PubMed] [Google Scholar]
- 63.Chen M.M.Y., Snow C.D., Arnold F.H. Comparison of random mutagenesis and semi-rational designed libraries for improved cytochrome P450 BM3-catalyzed hydroxylation of small alkanes. Protein Eng. Des. Sel. 2012;25:171–178. doi: 10.1093/protein/gzs004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.