

Abstract
Background
Considerable evidence suggests that coagulation proteases (TF/FVIIa/FXa/thrombin) and their target protease activated receptors (PAR-1/PAR-2) play important roles in myocardial ischemia-reperfusion (I-R) injury. We hypothesized that localized inhibition of TF/FVIIa on the membrane surfaces of ischemic cells could effectively block coagulation cascade and subsequent PAR-1/PAR-2 cell signaling, thereby protecting the myocardium from I-R injury.
Objectives
We recently developed an annexin V-Kunitz inhibitor fusion protein (ANV-6L15) that could specifically bind to anionic phospholipids on the membrane surfaces of apoptotic cells and efficiently inhibit the membrane-anchored TF/FVIIa. In this study, we investigated the cardioprotective effect of ANV-6L15 in a rat cardiac I-R model in comparison to that of hirudin.
Methods
Left coronary artery occlusion was maintained for 45 minutes followed by 4 hours of reperfusion in anesthetized Sprague Dawley rats. One minute before or 2 minutes after coronary ligation, rats received an i.v. bolus injection of ANV-6L15 (2.5 to 250 μg/kg), vehicle, or hirudin by bolus injection and continuous infusion.
Results and Conclusions
ANV-6L15 dose-dependently reduced infarct size by up to 87 %, and decreased plasma levels of cardiac troponin I, TNF-α, and sICAM-1, by up to 97 %, 96 %, and 66 %, respectively, with little impact on the coagulation parameters. ANV-6L15 also ameliorated hemodynamic derangements, attenuated neutrophil infiltration and reduced TUNEL-(+) apoptotic cardiomyocytes. Hirudin was less efficacious even at supra-clinical dose. ANV-6L15 confers exceptionally potent cardioprotection and is a promising drug candidate for prevention of myocardial I-R injury.
Keywords: Annexin V, Ischemia-reperfusion injury, Cardioprotective agents, Apoptosis, Receptors, Proteinase-Activated
Introduction
Myocardial ischemia restricts blood supply to the heart, resulting in numerous metabolic changes including cessation of aerobic metabolism, accumulation of lactate and H+, progressive depletion of ATP, and increased osmolar load. These changes are associated with contractile dysfunction and electrocardiographic alterations [1]. Reperfusion after prolonged periods of coronary occlusion predisposes the myocardium to a spectrum of reperfusion-associated pathologies, including myocardial stunning, microvascular dysfunction, no-reflow, ventricular arrhythmia, contraction band necrosis and cardiomyocyte death [2–5].
Many biochemical and pathological mechanisms contribute to myocardial I-R injury. Experimental animal studies have shown that therapeutic interventions and pharmacological treatments targeting various biological pathways during myocardial I-R can reduce infarct size by up to about 50% [6]. These interventions include ischemic conditioning, therapeutic hypothermia, and pharmaceutical agents such as antioxidants, ion channel blockers, anti-inflammatory agents, metabolic modulators, adenosine, atrial natriuretic peptide, magnesium, NO donors, protein kinase inhibitors, and inhibitors of mitochondria permeability transition pore opening [5–7]. Unfortunately, despite demonstration of cardioprotection by numerous therapeutic interventions in animal studies, translation of these results into human clinical application has largely remained unsuccessful [5–7].
Accumulating evidence suggests that activation of the tissue factor (TF) pathway and cell signaling by coagulation proteases via their target protease activated receptors (PAR-1 and PAR-2 in particular) plays an important role in the pathogenesis of myocardial I-R injury. TF is prominently expressed on cardiomyocytes, with the highest amount in the left ventricular myocardium [8–9]. In rabbit coronary artery ligation models, at-risk areas of the myocardium showed increased TF expression in the coronary vasculature and the sarcolemma of cardiomyocytes during post-ischemic reperfusion [10]. Blockade of TF pathway by anti-TF monoclonal antibody [10] or active site-blocked FVIIa (FVIIai) [11] and functional inhibition of thrombin activity by hirudin [10] reduced infarct size. Treatment with a selective PAR-1 antagonist (SCH79797) before or during ischemia reduced myocardial necrosis following I-R in rats [12]. Mouse deficient in PAR-2 had reduced infarct size after I-R compared with wild-type littermates [13]. Taken together, these results suggest that multiple coagulation proteases (TF/FVIIa, FXa, and thrombin, in particular) and their target receptors (PAR-1 and PAR-2) appear to play critical roles in the pathogenesis of I-R injury. Effective control of TF pathway of coagulation might confer cardioprotection against myocardial I-R injury.
During myocardial ischemia, increased intracellular Ca2+ and reduced ATP level cause the inhibition of aminophospholipid translocase activity, leading to the externalization of phosphatidylserine (PS) and phosphatidylethanolamine (PE) on the membrane surfaces of ischemic cardiomyocytes and endothelial cells [14,15]. In experimental coronary artery ligation models, PS/PE exposure occurred within 5 minutes of ischemia and remained exposed for over 6 hours [16]. PS/PE-exposed membranes on the ischemic cells could promote the assembly of extrinsic tenase (TF/FVIIa), intrinsic tenase (FVIIIa/FIXa), and prothrombinase (FVa/FXa), leading to initiation/amplification/propagation of coagulation cascade. We developed a series of 2-domain recombinant fusion proteins, each consisting of a human annexin V (ANV) domain linked to a Kunitz protease inhibitor (KPI) domain, abbreviated ANV-KPIs [17]. ANV is a 35 kDa protein with strong binding affinity (Kd ~nM range) for PS/PE-exposed membranes, KPIs are 6–7 kDa serine protease inhibitors that inhibit various coagulation proteases. ANV-KPIs were capable of binding specifically to PS/PE-exposed membranes (site of coagulation cascade and thrombogenesis) via their ANV domain. After docking onto the membrane surfaces, juxtaposition of the KPI domain with the active site of the enzyme/cofactor assemblies resulted in highly efficient inhibition of the coagulation complexes. Among our ANV-KPI series, ANV-6L15 efficiently inhibited the TF/VIIa complex on the surface of the PS/PE-exposed membrane, with an in vitro anticoagulant potency ~1200% that of the full-length human tissue factor pathway inhibitor (TFPI) in TF-induced plasma clotting assay [17]. We speculated that specific localization of ANV-6L15 to the PS/PE-exposed membrane surfaces of ischemic cells could greatly enhance the inhibition of the membrane-associated TF/FVIIa, thereby preventing generation of coagulation proteases and subsequent cell signaling via PAR-1/PAR-2. In this study, we investigated the cardioprotective effects of ANV-6L15 in a rat myocardial I-R model in comparison to hirudin, a leach-derived direct thrombin inhibitor.
Materials and Methods
Recombinant ANV-6L15
Escherichia coli BL21(DE3)pLysS and the expression vector pET20b(+) (Novagen, Madison, WI, USA) were used for the expression of ANV-6L15 and the recombinant protein was purified and characterized as described before [17].
Myocardial I-R injury model
Male Sprague Dawley rats weighing 230–300 g were used for this study. The animal protocol was approved by the Animal Care and Use Committee of Chang Gung Memorial Hospital-Keelung. All animals received humane care in compliance with the Guide for the Care and Use of Laboratory Animals (US National Institutes of Health publication No. 85-23, revised 1996). Ten rats were used in each group. Myocardial I/R was induced by ligation of the left coronary artery according to the method of Wang et al. [18]. Briefly, under anesthesia (isoflurane inhalation), the right femoral artery was cannulated (PRS-LS2, Scisense Inc. Canada) for hemodynamic measurements with a P-V conductance system (Millar Instruments, Inc.; Huston, TX) coupled to a PowerLab/4SP A/D converter (AD Instruments; Mountain View, CA) and a personal computer, and the right femoral vein used for drug administration. Blood samples were drawn at various time points. Prothrombin time (PT) and activated partial thromboplastin time (aPTT) were measured with a coagulometer (Coatron M1, TECO GmbH, Germany). Under ventilation with isoflurane (induced with 3–4% and maintained with 1.5–2.5%), the heart was exposed via a left thoracotomy performed at the fifth intercostal space, followed by encircling the left descending coronary artery with a 6–0 prolene suture-snare about 7 mm from its origin at the base of the heart. Regional left ventricular ischemia was produced by tightening the snare and occluding the coronary artery for 45 minutes. The coronary snare was then released to allow 4 hours of reperfusion as confirmed by visualizing an epicardial hyperemic response. After 4 hours of reperfusion, all animals were sacrificed with an intravenous bolus of concentrated pentobarbital. The myocardial tissue was isolated and processed for calculation of infarct size and histological analysis.
To assess the cardio-protective effect of ANV-6L15 on myocardial I/R injury, a single bolus of ANV-6L15 in normal saline at various dosages (2.5, 5.0, 10, 50 and 250 μg/kg) was administered intravenously to rats either 1 minute before (n=10, subgroup a) or 2 minutes after (n=10, subgroup b) coronary ligation. Control rats (n=10) received same volume normal saline injections. To determine the effect of thrombin inhibition on myocardial I/R injury, rats (n=10) were treated with recombinant hirudin (lepirudin; Hoechst Marion Roussel, Kansas City, MO). Hirudin treatment began with an intravenous administration of a 1 mg/kg bolus 30 minutes before ischemia; 45 minutes later, an intravenous infusion of 1 mg/kg/hour was given for 1 hour, and then an infusion of 0.5 mg/kg/hour throughout the remaining reperfusion period. This dosing protocol has been shown to prolong the activated partial thromboplastin time (aPTT) to greater than twice baseline values throughout the period of ischemia and reperfusion [19]. Although this dosing scheme was commonly used in experimental animal studies, much lower doses (0.05–0.1 mg/kg/min continuous infusion without a loading bolus) were used for human clinical applications because of increased bleeding risks [20,21]. Thus, the dose of hirudin used in previous and current animal studies represent a supra-clinical dosage.
Determination of Infarct Size
The myocardial infarct size after 45-min ischemia and 4-h reperfusion was determined as described by Erlich et al. [10]. Briefly, at the completion of the 4-hour reperfusion period, the coronary artery was re-occluded, and 20% Evans blue dye (Sigma Chemical Co.) was injected into the right atrium to identify all perfused tissue (blue). The area of myocardium receiving its blood supply from the ligated vessel remained pink, thus demarcating the regions of the left ventricle (LV) at risk (AR) for injury. After arrest with pentobarbital, the heart was rapidly excised, weighed, and cut into 2-mm-thick cross-sections in parallel with the atrioventricular groove. The LV was isolated from the remainder of the heart and weighed. The normal LV myocardium (blue) was separated from the LV myocardial AR area for injury (pink). The AR area was then placed in a 37°C solution of 1% triphenyltetrazolium chloride (Sigma Chemical Co. St. Louis, MO) for 30 minutes, staining the viable tissue brick red and leaving the necrotic zone pale white. Red-stained (noninfarcted) tissue was separated from white-stained (infarcted; necrotic) tissue under a dissecting microscope, and each area was weighed. The percentage of LV at risk for infarction (% AR) was calculated by dividing the weight of the LV AR area by the weight of the total LV. The percentage infarct size within the area of AR was calculated by dividing the weight of necrotic tissue by the weight of the LV AR area.
Determination of plasma levels of troponin I, TNF-α and sICAM-1
The blood was collected in sodium citrate-containing tubes, centrifuged for 15 min at 4°C, 3,000g, and the plasma was stored at −80°C until assayed for cytokines. Enzyme-linked immunosorbent assays (ELISA) were performed according to the manufacturers' specifications to measure the plasma concentrations of troponin I (cTnI, Life Diagnostics, Inc., West Chester, PA), tumor necrosis factor-α (TNF-α) and intercellular adhesion molecule-1 (sICAM-1) (R&D Systems, Inc., Minneapolis, MN). The changes in plasma levels of cTnI, TNF-α, and sICAM-1 after reperfusion were expressed as percentages over the pre-ligation plasma levels of cTnI, TNF-α and sICAM-1, respectively.
PMN accumulation
Reperfused cardiac tissues were harvested and washed with PBS, embedded in OTC, and stored at −80°C. Cardiomyocytes were counterstained with Hoechst 33342 (Invitrogen Co, Carlsbad, CA) and anti-sarcomeric actin (DakoCytomation, Denmark). PMN accumulation in the LV AR area of myocardium was detected with anti-rat granulocytes (Pharmingen, San Diego, CA). The number of PMNs in 10 randomly selected high-power fields (x 400) from the AR regions was counted. PMN accumulation was compared by counting the number of PMNs among 500 cardiomyocytic nuclei and expressed as PMNs as a percentage of myocytic nuclei.
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling (TUNEL)
The cardiomyocytic nuclei were counterstained with Hoechst 33342 (Invitrogen Co, Carlsbad, CA). TUNEL was carried out to detect cells undergoing apoptosis as described previously [22]. To quantify cardiomyocytic apoptosis, 500 cardiomyocytic nuclei were identified in 10 randomly selected 400x high-power fields per section. The results were expressed as the number of TUNEL-positive cardiomyocytic nuclei as a percentage of the total number of cardiomyocytic nuclei.
Data analysis
The data were expressed as mean ± standard deviation (SD). Statistical evaluation of data between groups was performed by analysis of variance, and Student's t test was used when ANOVA followed by a Tukey's test or Kruskal-Wallis tests indicated significance for multiple comparisons. Statistical significance was set at a probability value of less than 0.05.
Results
Effects of ANV-6L15 and hirudin on infarct size and plasma levels of cTnI, TNF-α and sICAM-1
We used a well-characterized rat myocardial I-R model for this study. Regional ischemia was produced by ligation of the left descending coronary artery for 45 min followed by releasing the ligature to allow reperfusion for 4 hours. Figure 1(A) shows that bolus administration of ANV-6L15 at doses of 2.5 μg/kg, 5.0 μg/kg, 10 μg/kg, 50 μg/kg, and 250 μg/kg reduced infarct size by 67–87 % compared with the vehicle control. There was a slight trend toward greater reduction in infarct size when ANV-6L15 was administered 2 minutes post-ligation compared to 1 minute pre-ligation. The difference in infarct size reduction might be due to short circulating half-life (t1/2 ~15 min, unpublished data) of ANV-6L15. For comparison, hirudin was administered to rats by bolus injection (1 mg/kg) 30 min before ischemia followed by continuous infusion at 0.5–1 mg/kg/h. Hirudin treatment reduced infarct size 47% compared with the vehicle control. Thus, hirudin was less potent than ANV-6L15 in protection against I-R induced myocardial infarction.
Figure 1.
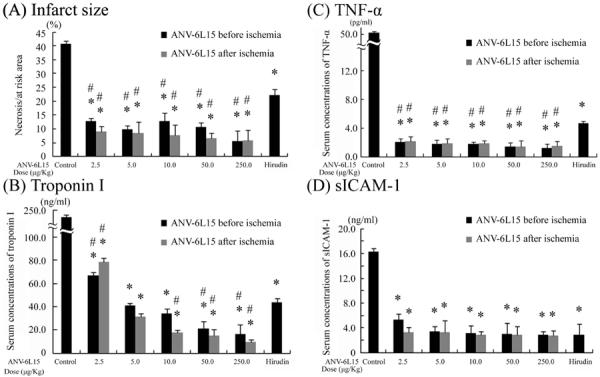
Effects of ANV-6L15 and hirudin on infarct size and plasma levels of Troponin I, TNF-α and sICAM-1. Groups of 10 rats each were subjected to 45 minutes of ischemia followed by 4 hours of reperfusion and treated with the indicated doses of ANV-6L15, hirudin, or vehicle control either 1 minute before or 2 minutes after coronary ligation. As shown in on-line supplements (Fig. S1 and Table S1), the AR/LV% for each group was not significantly different from the control group (p values vs. control were all > 0.05 with an average of 37.1±2.6 % AR/LV across all groups). (A) Infarct size as a percentage of area at risk (AI/AR%); (B) plasma levels of cardiac troponin I; (C) plasma levels of TNF-α; and (D) plasma levels of intercellular adhesion molecule 1 (ICAM-1). Results were normalized to the plasma concentrations of troponin I, TNF-α, and ICAM-1 per mg of total plasma protein (*P < 0.05 vs. control group; #P <0.05 vs. hirudin).
Figure 1(B) shows the effects of ANV-6L15 and hirudin on the plasma levels of cTnI. ANV-6L15 dose-dependently decreased plasma cTnI level by 67.2% to 95.8 % compared with vehicle-treated control. Hirudin decreased cTnI levels 81.6% relative to the control. At 10 μg/kg dose or greater, ANV-6L15 was more effective than hirudin in reducing plasma level of cTnI. Administration of ANV-6L15 before or after coronary ligation resulted in slightly different plasma levels of cTnI, however, the differences were not statistically significant. Figure 1(C) shows that ANV-6L15 at all doses (2.5–250 μg/kg) reduced plasma TNF-α levels approximately 95.8%–97.6 % while hirudin reduced plasma TNF-α 90.8% compared with the control. The plasma levels of sICAM-1 was reduced approximately 67.5%–83.3 % by all doses of ANV-6L15 and hirudin compared with the control [Figue 1(D)].
Effects of ANV-6L15 and hirudin on hemodynamics
Hmodynamics (mean blood pressure, maximum and minimum dP/dT, and heart rate) recorded at 2-minute after coronary ligation, during reperfusion, and after 4-hour reperfusion are shown in Figure 2. Mean pressure decreased progressively with time during I-R from 83% to 74 % of the baseline value in the control group. ANV-6L15 treated groups showed smaller decrease of mean pressure below baseline during I-R at lower doses (2.5–5 μg/kg), and could maintain the mean pressure close to baseline at higher doses (50–250 μg/kg). In comparison, the mean pressure in the hirudin-treated group was 95 % that of baseline at 2-minute after coronary ligation, but dropped to 80% of baseline after 4-hour reperfusion. Thus, ANV-6L15 dose-dependently protected against the decrease of mean pressure during I-R and this protection was greater than that conferred by hirudin. The maximum and minimum dP/dT decreased progressively with time during I-R to 68 % and 64 % of baselines, respectively, during I-R. ANV-6L15 treatment dose-dependently decreased the drop in maximum and minimum dP/dT and restored them towards baseline values. Hirudin treatment only partially reduced the changes of maximum and minimum dP/dT during IR. The heart rate decreased to 67% of baseline during I-R in the control group. ANV-6L15 treatment dose-dependently decreased the change in heart rate and restored the heart rate to near baseline during I-R. Hirudin treatment was also effective in preventing decreases in heart rate during I-R.
Figure 2.
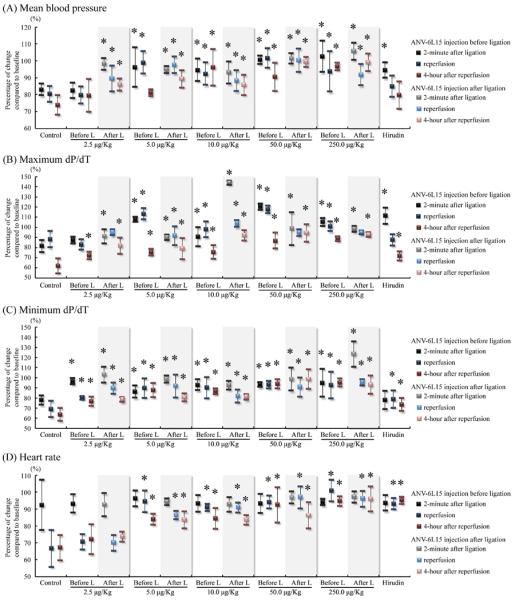
Percentage of changes of (A) mean blood pressure, (B) maximum dP/dT, (C) minimum dP/dT, and (D) heart rate, compared with baseline of various groups through 45-minute ischemia and 4-hour reperfusion period. (*P < 0.05 vs. control group)
PT and aPTT
Plasma samples were collected at four time points (baseline, 2-minute after ligation, 45-minute after ligation and 4-hour reperfusion) during I-R for the measurement of PT and aPTT. Figure 3 (A) shows PT of different groups treated with hirudin and different doses of ANV-6L15 before coronary ligation. ANV-6L15 did not elevate PT over baseline significantly except at the highest dose (250 μg/kg). Injection of 250 μg/kg ANV-6L15 before coronary ligation elevated PT less than 2-fold at 2-minutes after ligation, but the PT returned to baseline when measured at 45-minutes after ligation and 4-hour reperfusion. In contrast, hirudin treatment did not elevate PT during ischemia, but the PT increased approximately 4-fold over baseline at 4-hour reperfusion. Figure 3(B) shows similar PT results when ANV-6L15 was administered 2-minutes after coronary ligation. Figure 3 (C) and (D) show that hirudin treatment prolonged the aPTT up to 4-fold of baseline; none of the ANV-6L15 treated groups showed detectable change in aPTT.
Figure 3.
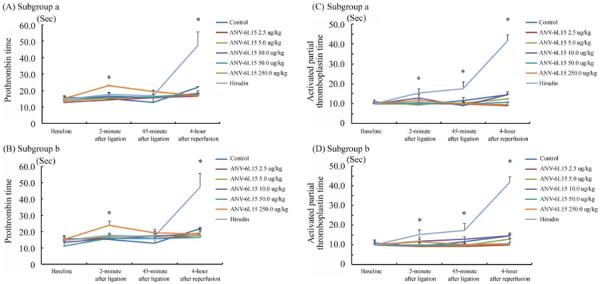
Prothrombin time (PT) and activated partial thromboplastin time (aPTT) over the time course of I-R. Blood samples were taken at the indicated time points. (A) PT after ANV-6L15 injection at 1 minute before coronary ligation; (B) PT after ANV-6L15 injection at 2 minute after coronary ligation; (C) aPTT after ANV-6L15 injection at 1 minute before coronary ligation; and (D) aPTT after ANV-6L15 injection at 2 minute after coronary ligation. Data represented mean ± SD. Asterisks indicate a statistically significant difference (P < 0.05) compared to baseline.
PMN accumulation
Leukocyte infiltration into the AR area in the reperfused hearts was assessed after 4-hours of reperfusion as described in Methods. Figure 4(A) shows representative images of PMN infiltration. The number of PMNs infiltrating the AR area was scored. Figure 4(B) shows that ANV-6L15 injection at various doses reduced PMN infiltration 54.7%–82.3 % after 4 hours of reperfusion compared with the vehicle-treated group. Hirudin treatment reduced the PMN infiltration approximately 50% compared with the vehicle-treated control.
Figure 4.

ANV-6L15 treatment reduced PMN accumulation after myocardial I-R in vivo. ANV-6L15 was injected 1 minute before or 2 minutes after coronary ligation as indicated. (A) Representative images of PMN infiltration after 4-hours reperfusion; and (B) Number of infiltrated PMNs as a percentage of myocytic nuclei in the AR area. The number of PMNs among 500 myocytic nuclei in 10 high-power fields (400x) were counted and expressed as % of myocytic nuclei. Data were expressed as mean ± SD (n=10 per group). *P<0.05 in comparison to control group; #P<0.05 in comparison to hirudin group.
TUNEL detection of apoptosis in myocardium
Figure 5(A) shows representative examples of TUNEL-positive cardiomyocytic nuclei with nicked DNA in the vehicle control and groups treated with ANV-6L15 and hirudin. Figure 5(B) shows that apoptotic cell counts, expressed as a percentage of the total number of cardiomyocytic nuclei counted. ANV-6L15 at lower doses (2.5 and 5.0 μg/kg) did not protect against cardiomyocyte apoptosis compared with control group; however, apoptosis protective effect increased as the ANV-6L15 dosage increased from 10 μg/kg to 250 μg/kg. The result was in sharp contrast with that of the infarct size estimation, in which even the lowest dose caused a large decrease in infarct size (Fig. 1A). This discrepancy might be related to the multiple possible mechanisms of cell death (e.g. necrosis vs. apoptosis) after I-R injury reported previously. Hirudin treatment also decreased cardiomyocyte apoptosis. Doses of ANV-6L15 over 50.0 μg/kg had a significantly greater protective effect against cardiomyocyte apoptosis than hirudin.
Figure 5.
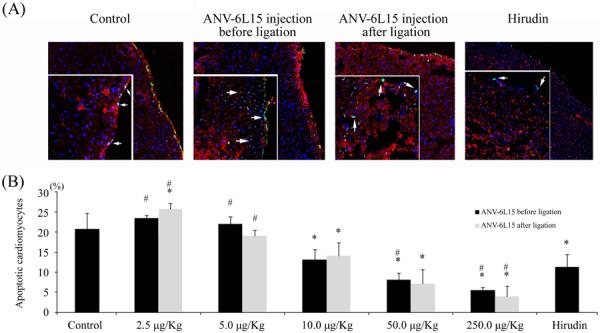
Effects of ANV-6L15 and hirudin on I-R induced cardiomyocyte apoptosis detected by TUNEL method. (A) Representative TUNEL staining of apoptotic nuclei (green) in tissues treated as indicated. Magnifications: large frame, 20x; small insert, 100x fluorescence microscopy. (B) The percentage of TUNEL(+) apoptotic cardiomyocytic nuclei in tissues treated as indicated (*P < 0.05 compared with control group; #P<0.05 compared with hirudin group).
Discussion
Myocardial infarct volume is the most critical determinant for long-term prognosis after acute myocardial infarction (AMI). Thus, development of cardioprotective therapies to reduce the size of the infarct due to myocardial I-R injury has been the main objective of research on ischemic myocardial disease in the past few decades. In this study, we showed that ANV-6L15 reduced myocardial infarct size after I-R by 67–87% at doses (2.5–250 μg/kg) that caused little or no perturbation to coagulation parameters such as aPTT and PT. In comparison, hirudin treatment reduced infarct size by only 47 % in the same model at a supra clinical dosage, but caused greater than four-fold increases in aPTT and PT. The reduction of infarct size by ANV-6L15 was also significantly greater than the 44–61 % infarct size reduction observed in previous studies using bolus doses of 1–2 mg/kg anti-TF antibodies [10], 1 mg/kg bolus dose of FVIIai [11] or 1mg/kg bolus plus 0.5–1 mg/kg continuous infusion of hirudin [10]. These data suggests that treatment with the thrombogenic site-targeted ANV-6L15 could achieve greater reduction of infarct size with less risk for bleeding side effect compared with treatment with conventional anticoagulants that induce a state of systemic anticoagulation. In addition, ANV-6L15 elicited a broad range of cardioprotective effects, including normalization of hemodynamics, reduction of plasma TNF-α and sICAM-1 levels by 96–97 %, and attenuation of PMN infiltration and cardiomyocyte apoptosis. These outstanding efficacy and safety profiles suggest that ANV-6L15 might be useful for cardioprotection against myocardial I-R injury. Limitations remain with this study however, as many therapeutic interventions previously shown to protect against myocardial I-R injury in animal models have failed to translate into human clinical applications. Further verification of the cardioprotective effect of ANV-6L15 in larger animal models might be needed before moving into clinical testing.
Molecular and cellular events underlying myocardial I-R injury are complex. Its pathogenesis reflects the confluence of divergent biological pathways, including ion channels, reactive oxygen species, inflammation, endothelial dysfunction, mitochondria abnormalities, cardiomyocyte apoptosis and necrosis [5,6]. TF-thrombin pathway contributes to myocardial I-R injury, as modulation of this pathway resulted in significant cardioprotection [10–12] similar to interventions of other biological pathways [5–7]. ANV-6L15 effectively blocked the initiation of TF pathway, thereby inhibiting generation of coagulation proteases and PAR signaling. PAR-1 and PAR-2 couples to members of the G-protein families to elicit diverse cardiovascular actions [23–26], including inflammatory responses [23,24,27], modulation of ion channels [28–29], arrhythmogenesis [30], increased microvascular permeability [31] and acute cardiomyocyte death [32]. Thus, TF pathway cascade and PAR-1/PAR-2 signaling are likely critical upstream events that lead to subsequent biochemical and cellular alterations that result in myocardial injury. The cardioprotective effect of ANV-6L15 might be attributed to its efficient blockade of the upstream TF pathway cascade, thereby preventing PAR-1/PAR-2 signaling and myocardial injury. ANV-6L15 bound to PS/PE-exposed cells membranes with increased affinity compared to ANV [33], and could affect membrane-related events [34] and survival of cells undergoing apoptosis [35]. Thus, ANV-6L15 might also exert its cardioprotective effect partly through modulation of membrane functions and cell apoptosis. TF was prominently expressed in myocardium and up-regulated by I-R [10]. PAR-1 and PAR-2 were also abundantly expressed by endothelial cells and cardiomyocytes in the heart [24]. Ischemic insult damages the endothelium, increases endothelial permeability [32] and causes externalization of PS/PE on endothelial cells and cardiomyocytes [14–16]. These changes facilitate the localization of ANV-6L15 to the ischemic endothelium and cardiomyocytes because of high affinity binding of the fusion protein to PS/PE-exposed membranes. The membrane-docking action in turn facilitates the inhibition of the membrane-anchored TF/FVIIa by 6L15 domain, resulting in highly efficient blockade of the initiation of TF pathway cascade. PAR-1/PAR-2 cell signaling has been shown to occur immediately after formation of minute amounts of TF/FVIIa/FXa ternary complex (low pM) [25,26] and thrombin (50 pM) [24] during the initiation of TF pathway cascade. By specific localization on the PS/PE-exposed membranes, ANV-6L15 might inhibit the membrane-anchored TF/VIIa far more efficiently than other anticoagulants that exist in the fluid phase. As a consequence, ANV-6L15 can inhibit TF pathway cascade and PAR-1/PAR-2 cell signaling more effectively than conventional anticoagulants that require high concentrations in circulating blood to suppress TF pathway activation. In this study, bolus injection of 2.5–250 μg/kg of ANV-6L15 conferred highly potent cardioprotection against I-R injury with little or no effect on plasma aPTT and PT. In another animal study, bolus injection of 10–100 μg/kg of ANV-6L15 blocked thrombogenesis after balloon injury for >6 hours despite its short circulating half-life (t1/2 ~15 min). These results suggest that ANV-6L15 could passivate the thrombogenic sites without inducing a state of systemic anticoagulation, and therefore lead to reduced bleeding tendency. Based on these efficacy and safety profiles, we suggest that ANV-6L15 may be a promising drug candidate for cardioprotection against myocardial I-R injury and other antithrombotic applications.
Supplementary Material
Acknowledgments
We are grateful to the laboratory animal center and the confocal laser scanning microscope room of Chang Gung Memorial Hospital-Keelung for their excellent technical services.
Sources of Funding Funding for this project was provided by Chang Gung Memorial Hospital, Taiwan (CMRPGs 270261, 270262, 270263, 270281) and National Heart Lung and Blood Institute, NIH, USA (1R43HL77061, 1R43HL093848, 2R44HL093848 to EVAS Therapeutics, LLC).
Footnotes
Addendum Study concept and design: C.H. Yeh, T.P. Chen, C.Y. Wang and T.C. Wun. Acquisition of data: C.H. Yeh and S.W. Fang. Analysis and interpretation of data: C.H. Yeh, T.C. Wun and S.W. Fang. Drafting of the manuscript: C.H. Yeh and T.C. Wun. Critical revision of the manuscript for important intellectual content: C.H. Yeh, T.P. Chen, C.Y. Wang and T.C. Wun. Statistical analysis: C.H. Yeh and S.W. Fang. Administrative, technical and material support: C.H. Yeh, S.W. Fang and T.C. Wun. Study supervision: T.P. Chen, C.Y. Wang and T.C. Wun.
Disclosures T-C W is the proprietor of EVAS Therapeutics LLC, currently developing ANV-KPIs for therapeutic application. Other authors have no conflicts of interest.
References
- 1.Jennings RB, Reimer KA. The cell biology of acute myocardial ischemia. Annu Rev Med. 1991;42:225–246. doi: 10.1146/annurev.me.42.020191.001301. [DOI] [PubMed] [Google Scholar]
- 2.Kloner RA. Does reperfusion injury exist in humans? J Am Coll Cardiol. 1993;21:537–545. doi: 10.1016/0735-1097(93)90700-b. [DOI] [PubMed] [Google Scholar]
- 3.Moens AL, Claeys MJ, Timmermans JP, Vrints CJ. Myocardial ischemia/reperfusion-injury, a clinical view on a complex pathophysiological process. Int J Cardio. 2005;100:179–190. doi: 10.1016/j.ijcard.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 4.Rodríguez-Sinovas A, Abdallah Y, Piper HM, Garcia-Dorado D. Reperfusion injury as a therapeutic challenge in patients with acute myocardial infarction. Heart Fail Rev. 2007;12:207–216. doi: 10.1007/s10741-007-9039-9. [DOI] [PubMed] [Google Scholar]
- 5.Turer AT, Hill JA. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am J Cardiol. 2010;106:360–368. doi: 10.1016/j.amjcard.2010.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 7.Kloner RA, Schwartz Longacre L. State of the science of cardioprotection: Challenges and opportunities--proceedings of the 2010 NHLBI Workshop on Cardioprotection. J Cardiovasc Pharmacol Ther. 2011;16:223–232. doi: 10.1177/1074248411402501. [DOI] [PubMed] [Google Scholar]
- 8.Drake TA, Morrissey JH, Edgington TS. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol. 1989;134:1087–1097. [PMC free article] [PubMed] [Google Scholar]
- 9.Luther T, Dittert DD, Kotzsch M, Erlich J, Albrecht S, Mackman N, Müller M. Functional implications of tissue factor localization to cell-cell contacts in myocardium. J Pathol. 2000;192:121–130. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH667>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 10.Erlich JH, Boyle EM, Labriola J, Kovacich JC, Santucci RA, Fearns C, Morgan EN, Yun W, Luther T, Kojikawa O, Martin TR, Pohlman TH, Verrier ED, Mackman N. Inhibition of the tissue factor-thrombin pathway limits infarct size after myocardial ischemia-reperfusion injury by reducing inflammation. Am J Pathol. 2000;157:1849–1862. doi: 10.1016/S0002-9440(10)64824-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Golino P, Ragni M, Cirillo P, Scognamiglio A, Ravera A, Buono C, Guarino A, Piro O, Lambiase C, Botticella F, Ezban M, Condorelli M, Chiariello M. Recombinant human, active site-blocked factor VIIa reduces infarct size and no-reflow phenomenon in rabbits. Am J Physiol Heart Circ Physiol. 2000;278:H1507–H1516. doi: 10.1152/ajpheart.2000.278.5.H1507. [DOI] [PubMed] [Google Scholar]
- 12.Strande JL, Hsu A, Su J, Fu X, Gross GJ, Baker JE. SCH 79797, a selective PAR1 antagonist, limits myocardial ischemia/reperfusion injury in rat hearts. Basic Res Cardiol. 2007;102:350–358. doi: 10.1007/s00395-007-0653-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antoniak S, Rojas M, Spring D, Bullard TA, Verrier ED, Blaxall BC, Mackman N, Pawlinski R. Protease-activated receptor 2 deficiency reduces cardiac ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol. 2010;30:2136–2142. doi: 10.1161/ATVBAHA.110.213280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Musters RJ, Otten E, Biegelmann E, Bijvelt J, Keijzer JJ, Post JA, Op den Kamp JA, Verkleij AJ. Loss of asymmetric distribution of sarcolemmal phosphatidylethanolamine during simulated ischemia in the isolated neonatal rat cardiomyocyte. Circ Res. 1993;73:514–523. doi: 10.1161/01.res.73.3.514. [DOI] [PubMed] [Google Scholar]
- 15.Maulik N, Kagan VE, Tyurin VA, Das DK. Redistribution of phosphatidylethanolamine and phosphatidylserine precedes reperfusion-induced apoptosis. Am J Physiol. 1998;274(1 Pt 2):H242–H248. doi: 10.1152/ajpheart.1998.274.1.H242. [DOI] [PubMed] [Google Scholar]
- 16.Kenis H, Zandbergen HR, Hofstra L, Petrov AD, Dumont EA, Blankenberg FD, Haider N, Bitsch N, Gijbels M, Verjans JW, Narula N, Narula J, Reutelingsperger CP. Annexin A5 uptake in ischemic myocardium: demonstration of reversible phosphatidylserine externalization and feasibility of radionuclide imaging. J Nucl Med. 2010;51:259–267. doi: 10.2967/jnumed.109.068429. [DOI] [PubMed] [Google Scholar]
- 17.Chen HH, Vicente CP, He L, Tollefsen DM, Wun TC. Fusion proteins comprising annexin V and Kunitz protease inhibitors are highly potent thrombogenic site-directed anticoagulants. Blood. 2005;105:3902–3909. doi: 10.1182/blood-2004-11-4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang YP, Maeta H, Mizoguchi K, Suzuki T, Yamashita Y, Oe M. Intestinal ischemia preconditions myocardium: role of protein kinase C and mitochondrial K(ATP) channel. Cardiovasc Res. 2002;55:576–582. doi: 10.1016/s0008-6363(02)00245-6. [DOI] [PubMed] [Google Scholar]
- 19.Gertz SD, Fallon JT, Gallo R, Taubman MB, Banai S, Barry WL, Gimple LW, Nemerson Y, Thiruvikraman S, Naidu SS, Chesebro JH, Fuster V, Sarembock IJ, Badimon JJ. Hirudin reduces tissue factor expression in neointima after balloon injury in rabbit femoral and porcine coronary arteries. Circulation. 1998;98:580–587. doi: 10.1161/01.cir.98.6.580. [DOI] [PubMed] [Google Scholar]
- 20.Lubenow N, Eichler P, Lietz T, Greinacher A. Lepirudin in patients with heparin-induced thrombocytopenia - results of the third prospective study (HAT-3) and a combined analysis of HAT-1, HAT-2, and HAT-3. J Thromb Haemost. 2005;3:2428–2436. doi: 10.1111/j.1538-7836.2005.01623.x. [DOI] [PubMed] [Google Scholar]
- 21.Askari Arman T., Lincoff A. Michael. Antithrombotic Drug Therapy in Cardiovascular Disease. Springer; 2009. pp. 440–441. ISBN 978-1-60327-234-6. [Google Scholar]
- 22.Yeh CH, Chen TP, Lee CH, Wu YC, Lin YM, Lin PJ. Cardiomyocytic apoptosis following global cardiac ischemia and reperfusion can be attenuated by peroxisome proliferator-activated receptor-alpha but not gamma activators. Shock. 2006;26:262–270. doi: 10.1097/01.shk.0000225863.56714.96. [DOI] [PubMed] [Google Scholar]
- 23.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 24.Steinberg SF. The cardiovascular actions of protease-activated receptors. Mol Pharmacol. 2005;67:2–11. doi: 10.1124/mol.104.003103. [DOI] [PubMed] [Google Scholar]
- 25.Camerer E, Huang W, Coughlin SR. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc Natl Acad Sci U S A. 2000;97:5255–5260. doi: 10.1073/pnas.97.10.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riewald M, Ruf W. Mechanistic coupling of protease signaling and initiation of coagulation by tissue factor. Proc Natl Acad Sci U S A. 2001;98:7742–7747. doi: 10.1073/pnas.141126698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cirino G, Cicala C, Bucci M, Sorrentino L, Maraganore J, Stone S. Thrombin functions as an inflammatory mediator through activation of its receptor. J Exp Med. 1996;183:821–827. doi: 10.1084/jem.183.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Long M, Yang L, Huang G, Liu L, Dong Y, Du Z, Tang A, Hu C, Gu R, Gao X, Tang L. Thrombin and its receptor enhance ST-segment elevation in acute myocardial infarction by activating the KATP channel. Mol Med. 2010;16:322–332. doi: 10.2119/molmed.2010.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yasutake M, Haworth RS, King A, Avkiran M. Thrombin activates the sarcolemmal Na(+)-H+ exchanger. Evidence for a receptor-mediated mechanism involving protein kinase C. Circ Res. 1996;79:705–715. doi: 10.1161/01.res.79.4.705. [DOI] [PubMed] [Google Scholar]
- 30.Jacobsen AN, Du XJ, Lambert KA, Dart AM, Woodcock EA. Arrhythmogenic action of thrombin during myocardial reperfusion via release of inositol 1,4,5-triphosphate. Circulation. 1996;93:23–26. doi: 10.1161/01.cir.93.1.23. [DOI] [PubMed] [Google Scholar]
- 31.Dauber IM, VanBenthuysen KM, McMurtry IF, Wheeler GS, Lesnefsky EJ, Horwitz LD, Weil JV. Functional coronary microvascular injury evident as increased permeability due to brief ischemia and reperfusion. Circ Res. 1990;66:986–998. doi: 10.1161/01.res.66.4.986. [DOI] [PubMed] [Google Scholar]
- 32.Mirabet M, Garcia-Dorado D, Ruiz-Meana M, Barrabés JA, Soler-Soler J. Thrombin increases cardiomyocyte acute cell death after ischemia and reperfusion. J Mol Cell Cardiol. 2005;39:277–283. doi: 10.1016/j.yjmcc.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 33.Yen T-C, Wey S-P, Liao C-H, Yeh C-H, Shen D-W, Achilef S, Wun T-C. Measurement of the binding parameters of annexin derivatives-erythrocyte membrane interactions. Anal Biochem. 2010;406:70–79. doi: 10.1016/j.ab.2010.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gerke V, Moss SE. Annexins: from structure to function. Physiol Rev. 2002;82:331–71. doi: 10.1152/physrev.00030.2001. [DOI] [PubMed] [Google Scholar]
- 35.Gidon-Jeangirard C, Hugel B, Holl V, Toti F, Laplanche JL, Meyer D, Freyssinet JM. Annexin V delays apoptosis while exerting an external constraint preventing the release of CD4+ and PrPc+ membrane particles in a human T lymphocyte model. J Immunol. 1999;162:5712–5718. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.