

Abstract
We report a catalytic enantioselective electrophilic fluorination of alkenes to form tertiary and quaternary C(sp3)-F bonds and generate β-amino- and β-aryl-allylic fluorides. The reaction takes advantage of the ability of chiral phosphate anions to serve as solid–liquid phase transfer catalysts and hydrogen bond with directing groups on the substrate. A variety of heterocyclic, carbocyclic, and acyclic alkenes react with good to excellent yields and high enantioselectivities. Further, we demonstrate a one-pot, tandem dihalogenation–cyclization reaction, using the same catalytic system twice in series, with an analogous electrophilic brominating reagent in the second step.
Keywords: asymmetric, organocatalysis, hydrogen-bonding
In the history of asymmetric catalysis, directing groups (DGs) have been instrumental for the achievement of selectivity in transformations of great synthetic utility. DGs have likewise been shown to play a central role in transition-metal catalysis, having recently been used for site-selective C-H activation (1–6). The advantage of using a DG lies in the ability to functionalize nonpolar bonds (such as in alkenes) while maintaining beneficial polar interactions between the substrate and chiral source; however, DGs have rarely been used in organocatalysis. In this context, the Miller research group reported the epoxidation reaction of allylic alcohols, using an amino acid catalyst capable of hydrogen bonding with the alcohol DG, and conducted elegant studies delineating the importance of hydrogen-bonding interactions for this and related systems (7–9) (Fig. 1). The Tan research group has elaborated this idea through the concept of induced intramolecularity, in which the catalyst reduces the entropic cost of reaction by bringing together reactants and organizing the transition state through favorable binding interactions (10).
Fig. 1.

(A and B) Modes of reactivity in chiral phosphoric acid/phosphate anion chemistry. (C and D) Substrate–catalyst interactions in directed allylic alkene functionalizations. (E and F) Chiral phosphate anions in directed asymmetric transformations.
The peptide catalysts in Miller’s studies facilitate reaction through organization of the reactants in the transition state (Fig. 1) rather than through formation of covalent adducts or lowest unoccupied molecular orbital-lowering protonation (or hydrogen bonding), which has generally required substrates containing carbonyl or imine moieties (11–13). This generalization applies to chiral phosphoric acids, which have been predominantly used in activation of the electrophile, although hydrogen bonding to the nucleophile has also been implicated as a key component (14). More recently the anionic conjugate bases of chiral phosphoric acids (i.e., chiral phosphate anions) have received attention as counterions for positively charged electrophilic intermediates, wherein ion pairing with the phosphate anion provides a suitable chiral environment for subsequent enantioselective transformations of the electrophilic species (11–13). The generality of the concept of ion pair catalysis has allowed the application of these highly tunable scaffolds to a diverse range of transformations in which carbonyl or imine activation is clearly inapplicable (15–17). As part of both aforementioned modes of reactivity, hydrogen bonds between chiral phosphoric acids or phosphate anions and reacting functionality are often invoked as crucial interactions for achieving high levels of enantioselectivity (11–14). We speculated that hydrogen-bonding interactions with substrate functionality may play a key role, via transition state organization, even when the interacting functionality does not directly participate in the transformation. We saw an opportunity to combine the principle of transition-state organization with our recently reported chiral anion phase-transfer system, with the hypothesis that an allylic hydrogen-bonding DG could effectively direct an ion-paired chiral phosphate species for the enantioselective electrophilic fluorination of alkenes (Fig. 1 E and F).
Our recent enantioselective halocyclization methods (18–20) used chiral anion phase-transfer catalysis, in which metathesis between an insoluble electrophilic salt (such as Selectfluor) and a chiral lipophilic phosphate anion derived from catalysts such as 3,3′-bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate (TRIP) generates a soluble electrophile in a chiral environment (Fig. 2). In addition, we reported the enantioselective fluorinations of enamides, using the same phase-transfer system (21, 22). In the latter examples, hydrogen bonding between the reacting enamide functional group and the phosphate catalyst is hypothesized to facilitate the fluorination reaction.
Fig. 2.

A source of electrophilic fluorine soluble in hydrophobic nonpolar solvents, via solid–liquid phase transfer. Both solubility and chirality are imparted by the organophosphate catalyst ion paired with the positively charged 1,4-diazabicyclo[2.2.2]octane-fluorine moiety.
On the basis of our previous success with amides as pendant nucleophiles, we posited that allylic amides could be used as DGs for direct alkene fluorination to provide access to allylic fluoride products (23). The synthesis of enantioenriched allylic fluorides is a synthetic challenge for which only a handful of approaches have been disclosed. Recently reported general methods for the enantioselective synthesis of allylic fluorides include the reaction of allylic chlorides with palladium catalysts (24, 25) as well as the fluorination of allylsilanes using cinchona alkaloid catalysis (26, 27). Furthermore, we envisioned that, upon removal of the amide directing group, this method would provide access to enantioenriched β-fluoroamines bearing a fluorinated quaternary center, a privileged bioactive motif and longstanding synthetic challenge (28–33).
In initial experiments, enantioselective fluorination of substrates containing no hydrogen-bond donor functionality was attempted (23). As expected, in the absence of a suitable DG, enantioselectivities no higher than 10% enantiomeric excess (ee) were obtained. In stark contrast, under typical chiral anion phase transfer conditions, allylic amide 1a underwent fluorination to provide allylic fluoride 1b in 82% yield and 85% ee, along with cyclized side product 1c (Table 1, Entry 1). Switching the solvent to toluene increased selectivity for the desired allylic fluoride over 1c, as well as enantioselectivity (Table 1, Entry 2). Finally, use of the recently developed 12-hydroxy-1,10-bis(2,4,6-triisopropyphenyl)-4,5,6,7-tetrahydrodiindeno[7,1-de:1′,7′-fg][1,3,2]dioxaphosphocine 12-oxide (STRIP) (3, see Table 1) in place of TRIP (2) as catalyst further increased enantioselectivity and chemoselectivity in favor of the desired allylic fluoride (Table 1, Entry 3) (34). The crystal structure of STRIP confirms earlier suggestions that it may have a tighter binding pocket than TRIP, which may in turn contribute to the enhanced enantioselectivities observed for some systems. Interestingly, under homogeneous conditions (MeCN), cyclization product 1c predominates, regardless of the presence of catalyst 3, illustrating the unique reactivity achievable under anionic phase transfer conditions (Table 1, Entries 4 and 5).
Table 1.
Optimization of product distribution and enantioselectivity
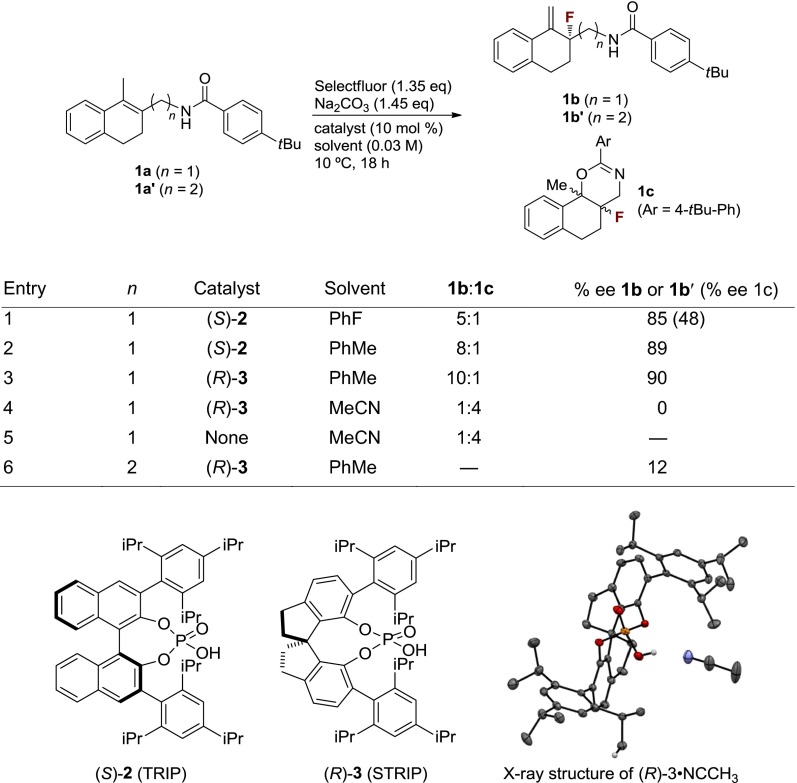 |
A few experiments and observations allowed us to gain some insight into the mechanism of this transformation. In accord with our hypothesis that a hydrogen bond donor is required to direct the fluorination, the N-methylated analog of 1a was unreactive (<10% conversion) under otherwise identical conditions. Furthermore, homoallylic amide 1a′ reacted to deliver allylic fluoride 1b′ in only 12% ee, suggesting that there is a distance requirement for the directing group (Table 1, Entry 6). We considered several possible pathways in which 1b could form from 1a (Fig. 3). We first considered the possibility that allylic fluoride 1b formed from E2 elimination of initially formed cyclization product 1c (Fig. 3A). However, subjecting 1c to standard reaction conditions did not result in the formation of 1b.
Fig. 3.

(A–C) Possible mechanisms for the formation of allylic fluoride 1b from substrate 1a.
We next considered the possibility that a carbocationic intermediate could be formed upon initial fluorination, followed by loss of a proton to generate 1b (Fig. 3B). Indeed, previous studies of the kinetics of Selectfluor-mediated fluorofunctionalization of alkenes in polar solvents supported a stepwise mechanism involving the rate-determining formation of a fluorocarbocation, followed by rapid nucleophilic trapping (35). On the basis of these studies, if a stepwise mechanism were operative in the allylic fluorination reaction, the formation of the fluorocarbocation intermediate would be expected to be rate determining as well, especially considering the low polarity of the reaction medium (toluene). Under this scenario the fluorination step would also be enantiodetermining, and products 1b and 1c should be formed with the same level of enantioselectivity. However, the observed enantioselectivity of the cyclization product (1c) is considerably lower (48% ee) than that of the allylic fluoride (1b, 85% ee; Table 1, Entry 1). This observation suggests that fluorination to give 1b is unlikely to take place through a stepwise fluorination–deprotonation mechanism. Therefore, we propose that this transformation most likely occurs through a concerted process in which fluorination and loss of proton, potentially assisted by the phosphate catalyst or substrate amide, occur in the same step (Fig. 3C).
We anticipated that better understanding of the properties required for an effective DG would aid in identification of a readily cleaved amide DG. Thus, investigations of the impact of steric and electronic changes to amide DGs on enantioselectivity were performed (Table 2). Halogenated acetyl derivatives provided insight into the influence of changing electronegativity, whereas alkyl derivatives revealed the effect of electronically negligible changes in steric bulk. Increased enantioselectivity was observed with increased steric bulk (t-Bu > i-Pr > Me; Table 2, Entries 1–3). Identical enantioselectivities were achieved with CH3 and CHF2 substrates (Table 2, Entries 1 and 5). It was, however, observed that one additional fluorine increased enantioselectivity dramatically (Table 2, Entry 5 vs. Entry 6). This may be due to a conformation attained with rotation of the CHF2 group to minimize steric interactions that is not possible with the larger CF3 group. In line with this proposal, an increase in size to CCl3, constituting a trichloroacetamide (TCA) group, improved enantioselectivity to 93% ee. Thus, both steric and electronic parameters affect enantioselectivity, although steric factors may be more important. More substantial changes such as the use of the more electron-withdrawing p-toluenesulfonamide as the directing group provided product with 67% conversion by NMR analysis and only 18% ee. In addition to being highly efficient for the desired transformation, the TCA group was readily hydrolyzed with potassium hydroxide to provide free amine in 90% yield with no racemization of the fluorinated stereocenter, thus allowing access to primary β-fluoroamines and potential derivatives not necessarily containing the DGs presented here (see Fig. 6A).
Table 2.
Studies on electronic and steric factors using amide DGs
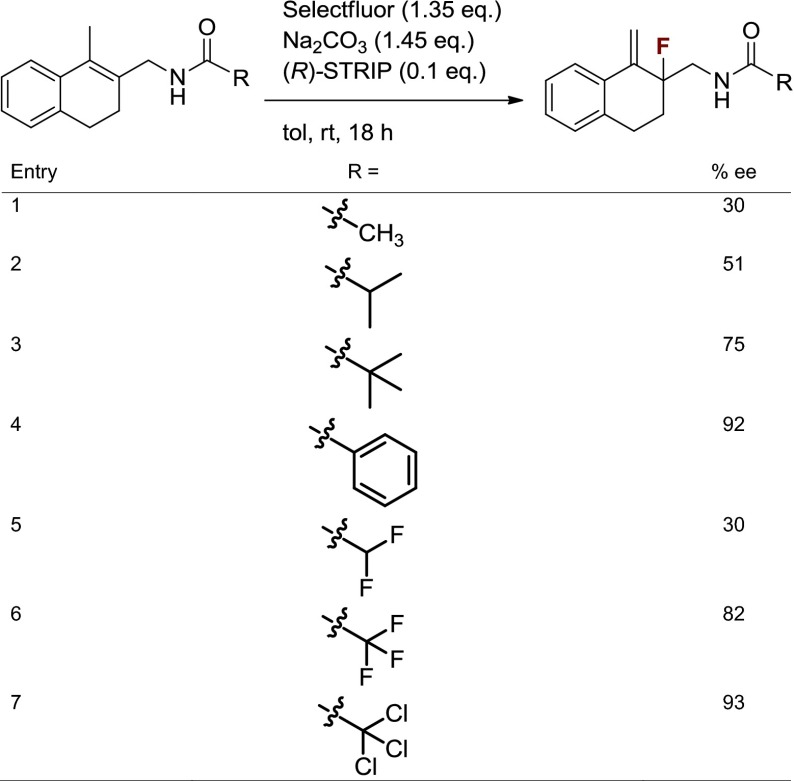 |
Fig. 6.

(A–C) Further reactions with fluorinated products.
In addition to the studies described above, substrates with different ring sizes, benzamide substitution patterns, substitutions on the bicyclic core, and heterocyclic substrates were examined (Fig. 4). With modifications to dihydronaphthalene-derived substrate 5a, we found that both 3,5- and 4-position substitution patterns of strongly electron-withdrawing groups on the benzamide were well tolerated (Fig. 4, Entries 6 and 7). Good yields and high enantioselectivities were maintained with benzosuberene- and indene-derived substrates, allowing access to a variety of ring sizes (Fig. 4, Entries 3 and 4). Neither electron-withdrawing nor electron-donating groups present on the fused benzene ring resulted in diminished enantioselectivity, (Fig. 4, Entries 8 and 10). Fluorination of chromanone-derived heterocyclic substrates 9a and 14a proceeded with good yield and high enantioselectivity (96% ee and 97% ee, respectively).
Fig. 4.

Scope of amide DG substrates. a, reaction run at room temperature; b, product isolated with cyclized by-product in 10:1 ratio; c, reaction run for 36 h; d, all stereochemistry defined in analogy to compound 14b (see SI Appendix for crystallography details).
In addition to amides, 2-hydroxyphenyl was found to be a suitable DG to promote enantioselective fluorination of a variety of alkenes under similar reaction conditions (Fig. 5). We had recently seen phenol act as a competent hydrogen-bond donor in enantioselective fluorinative dearomatization reactions (36). In the examples shown here we observe that increased substitution on the alkene changes the chemoselectivity, and fluorination on the alkene is seen exclusively rather than fluorination of the phenol, as in our previous studies. Both β-phenolic tertiary and quaternary fluorides with alkyl, aryl, or heteroaryl substituents were obtained with good to high enantioselectivity. Further, fluorinated piperidine product 19b can be obtained in moderate yield and enantioselectivity—a facile synthesis of a fluorinated scaffold that has been previously shown to be a privileged core of various biologically active compounds (37). In agreement with previous results, conversion was severely diminished (<10%) when the phenol group was methylated, presumably due to the inability of the methyl ether moiety to effectively participate in hydrogen bonding with the catalysts. We anticipate that the enantioselective construction of both β-amino and β-aryl sp3-C-F bonds will be valuable in drug discovery (38).
Fig. 5.

2-Hydroxyphenyl DG effective for various alkenes. a, Selectfluor (1.35 eq), Na2CO3 (1.45 eq), (R)-STRIP (0.1 eq), tol, rt, 18 h; b, reaction run for 36 h; c, catalyst A (39) used instead of (R)-STRIP.
Our previously reported electrophilic bromination reagent 22 (19) could be used to further functionalize the product from the title reaction via bromocyclization. This reaction shows the capability of chiral anion phase-transfer catalysis to operate sequentially in one pot to facilitate orthogonal reactions through the use of reagents with differing reactivity. Although Selectfluor did not further react with fluorination product 8b, it was found that bromocyclization of 8b took place in the presence of reagent 22 under phase-transfer conditions. With phase-transfer bromination verified stepwise, 8a was subjected to the standard fluorination conditions for 18 h (“F+”), followed by addition of 22 (“Br+”) and base. (This was operationally simple: The cap to the vial was opened and 22 was added without exclusion of air.) Gratifyingly, indeno-1,3-oxazine 20 formed in good yield and high diastereo- and enantioselectivity with the creation of two adjacent stereocenters and two carbon–halogen bonds. Heterocyclic substrate 14a also underwent fluorination with high selectivity, providing highly functionalized tricyclic product 21 (Fig. 6).
A catalytic, enantioselective direct fluorination of alkenes to provide a variety of β-amino and β-phenolic allylic fluorides has been presented. The utilization of solid–liquid chiral anion phase-transfer catalysis and the implementation of DGs were instrumental to achieving enantioselectivity. More generally, the ability of chiral phosphate anions to interact with the cationic electrophiles and with the substrate DGs through hydrogen bonding offers the potential to greatly expand the scope of electrophilic alkene functionalization reactions.
Supplementary Material
Acknowledgments
We thank Natalja Früh and Hunter Shunatona for synthesis of some intermediate materials. We thank William J. Wolf for obtaining X-ray crystallographic data for 3. We thank the University of California for financial support. Y.-M.W. thanks Amgen for a graduate fellowship and R.J.P. thanks the European Commission for a Marie Curie International Outgoing Fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and X-ray structures have been deposited in the Cambridge Structural Database, Cambridge Crystallographic Data Centre, Cambridge CB2 1EZ, United Kingdom (CSD reference nos. 927904, 927905, and 947493).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1304346110/-/DCSupplemental.
References
- 1.Hoveyda AH, Evans DA, Fu GC. Substrate-directable chemical reactions. Chem Rev. 1993;93(4):1307–1370. [Google Scholar]
- 2.Engle KM, Mei T-S, Wasa M, Yu J-Q. Weak coordination as a powerful means for developing broadly useful C-H functionalization reactions. Acc Chem Res. 2012;45(6):788–802. doi: 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colby DA, Tsai AS, Bergman RG, Ellman JA. Rhodium catalyzed chelation-assisted C-H bond functionalization reactions. Acc Chem Res. 2012;45(6):814–825. doi: 10.1021/ar200190g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X-G, Dai H-X, Wasa M, Yu J-Q. Pd(II)-catalyzed ortho trifluoromethylation of arenes and insights into the coordination mode of acidic amide directing groups. J Am Chem Soc. 2012;134(29):11948–11951. doi: 10.1021/ja305259n. [DOI] [PubMed] [Google Scholar]
- 5.Neufeldt SR, Sanford MS. Controlling site selectivity in palladium-catalyzed C-H bond functionalization. Acc Chem Res. 2012;45(6):936–946. doi: 10.1021/ar300014f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu C, Wang R, Falck JR. Amide-directed tandem C-C/C-N bond formation through C-H activation. Chem Asian J. 2012;7(7):1502–1514. doi: 10.1002/asia.201200035. [DOI] [PubMed] [Google Scholar]
- 7.Vasbinder MM, Jarvo ER, Miller SJ. Incorporation of peptide isosteres into enantioselective peptide-based catalysts as mechanistic probes. Angew Chem Int Ed Engl. 2001;40(15):2824–2827. doi: 10.1002/1521-3773(20010803)40:15<2824::AID-ANIE2824>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 8.Peris G, Jakobsche CE, Miller SJ. Aspartate-catalyzed asymmetric epoxidation reactions. J Am Chem Soc. 2007;129(28):8710–8711. doi: 10.1021/ja073055a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jakobsche CE, Peris G, Miller SJ. Functional analysis of an aspartate-based epoxidation catalyst with amide-to-alkene peptidomimetic catalyst analogues. Angew Chem Int Ed Engl. 2008;47(35):6707–6711. doi: 10.1002/anie.200802223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan KL. Induced intramolecularity: An effective strategy in catalysis. ACS Catalysis. 2011;1(8):877–886. [Google Scholar]
- 11.Phipps RJ, Hamilton GL, Toste FD. The progression of chiral anions from concepts to applications in asymmetric catalysis. Nature Chem. 2012;4(8):603–614. doi: 10.1038/nchem.1405. [DOI] [PubMed] [Google Scholar]
- 12.Mahlau M, List B. Asymmetric counteranion-directed catalysis: Concept, definition, and applications. Angew Chem Int Ed Engl. 2013;52(2):518–533. doi: 10.1002/anie.201205343. [DOI] [PubMed] [Google Scholar]
- 13.Brak K, Jacobsen EN. Asymmetric ion-pairing catalysis. Angew Chem Int Ed Engl. 2013;52(2):534–561. doi: 10.1002/anie.201205449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simón L, Goodman JM. A model for the enantioselectivity of imine reactions catalyzed by BINOL-phosphoric acid catalysts. J Org Chem. 2011;76(6):1775–1788. doi: 10.1021/jo102410r. [DOI] [PubMed] [Google Scholar]
- 15.Hamilton GL, Kang EJ, Mba M, Toste FD. A powerful chiral counterion strategy for asymmetric transition metal catalysis. Science. 2007;317(5837):496–499. doi: 10.1126/science.1145229. [DOI] [PubMed] [Google Scholar]
- 16.Hamilton GL, Kanai T, Toste FD. Chiral anion-mediated asymmetric ring opening of meso-aziridinium and episulfonium ions. J Am Chem Soc. 2008;130(45):14984–14986. doi: 10.1021/ja806431d. [DOI] [PubMed] [Google Scholar]
- 17.Liao S, List B. Asymmetric counterion-directed transition-metal catalysis: Enantioselective epoxidation of alkenes with manganese(III) salen phosphate complexes. Angew Chem Int Ed. 2010;49(3):628–631. doi: 10.1002/anie.200905332. [DOI] [PubMed] [Google Scholar]
- 18.Rauniyar V, Lackner AD, Hamilton GL, Toste FD. Asymmetric electrophilic fluorination using an anionic chiral phase-transfer catalyst. Science. 2011;334(6063):1681–1684. doi: 10.1126/science.1213918. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y-M, Wu J, Hoong C, Rauniyar V, Toste FD. Enantioselective halocyclization using reagents tailored for chiral anion phase-transfer catalysis. J Am Chem Soc. 2012;134(31):12928–12931. doi: 10.1021/ja305795x. [DOI] [PubMed] [Google Scholar]
- 20.Shunatona HP, Früh N, Wang Y-M, Rauniyar V, Toste FD. 2013 doi: 10.1002/anie.201302002. Enantioselective fluoroamination: 1,4-Addition to conjugated dienes using anionic phase-transfer catalysis. Angew Chem Int Ed Engl 52(30):7724–7727. [DOI] [PubMed] [Google Scholar]
- 21.Honjo T, Phipps RJ, Rauniyar V, Toste FD. A doubly axially chiral phosphoric acid catalyst for the asymmetric tandem oxyfluorination of enamides. Angew Chem Int Ed Engl. 2012;51(38):9684–9688. doi: 10.1002/anie.201205383. [DOI] [PubMed] [Google Scholar]
- 22.Phipps RJ, Hiramatsu K, Toste FD. Asymmetric fluorination of enamides: Access to α-fluoroimines using an anionic chiral phase-transfer catalyst. J Am Chem Soc. 2012;134(20):8376–8379. doi: 10.1021/ja303959p. [DOI] [PubMed] [Google Scholar]
- 23.Luo H-Q, Loh T-P. Synthesis of aryl allylic fluorides by direct electrophilic fluorination of alkenes. Tet Lett. 2009;50(14):1554–1556. [Google Scholar]
- 24.Katcher MH, Sha A, Doyle AG. Palladium-catalyzed regio- and enantioselective fluorination of acyclic allylic halides. J Am Chem Soc. 2011;133(40):15902–15905. doi: 10.1021/ja206960k. [DOI] [PubMed] [Google Scholar]
- 25.Katcher MH, Doyle AG. Palladium-catalyzed asymmetric synthesis of allylic fluorides. J Am Chem Soc. 2010;132(49):17402–17404. doi: 10.1021/ja109120n. [DOI] [PubMed] [Google Scholar]
- 26.Ishimaru T, et al. Cinchona alkaloid catalyzed enantioselective fluorination of allyl silanes, silyl enol ethers, and oxindoles. Angew Chem Int Ed Engl. 2008;47(22):4157–4161. doi: 10.1002/anie.200800717. [DOI] [PubMed] [Google Scholar]
- 27.Tredwell M, Tenza K, Pacheco MC, Gouverneur V. Synthesis of novel enantiopure fluorinated building blocks from acyclic chiral allylsilanes. Org Lett. 2005;7(20):4495–4497. doi: 10.1021/ol0518535. [DOI] [PubMed] [Google Scholar]
- 28. Pacheco MH, Purser S, Gouverneur V (2008) The chemistry of propargylic and allylic fluorides. Chem Rev 108(6):1943–1981. [DOI] [PubMed]
- 29.Brunet VA, O’Hagan D. Catalytic asymmetric fluorination comes of age. Angew Chem Int Ed Engl. 2008;47(7):1179–1182. doi: 10.1002/anie.200704700. [DOI] [PubMed] [Google Scholar]
- 30.Lectard S, Hamashima Y, Sodeoka M. Recent advances in catalytic enantiose-lective fluorination reactions. Adv Synth Catal. 2010;352(16):2708–2732. [Google Scholar]
- 31.Duthion B, Gomez Pardo D, Cossy J. Enantioselective synthesis of β-fluoroamines from β-amino alcohols: Application to the synthesis of LY503430. Org Lett. 2010;12(20):4620–4623. doi: 10.1021/ol1019579. [DOI] [PubMed] [Google Scholar]
- 32.Kalow JA, Schmitt DE, Doyle AG. Synthesis of β-fluoroamines by Lewis base catalyzed hydrofluorination of aziridines. J Org Chem. 2012;77(8):4177–4183. doi: 10.1021/jo300433a. [DOI] [PubMed] [Google Scholar]
- 33.Fadeyi OO, Lindsley CW. Rapid, general access to chiral β-fluoroamines and β,β-difluoroamines via organocatalysis. Org Lett. 2009;11(4):943–946. doi: 10.1021/ol802930q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Čorić I, Müller S, List B. Kinetic resolution of homoaldols via catalytic asymmetric transacetalization. J Am Chem Soc. 2010;132(49):17370–17373. doi: 10.1021/ja108642s. [DOI] [PubMed] [Google Scholar]
- 35.Papež-Iskra M, Zupan M, Stavber S. The effect of reaction conditions on activation parameters for the mild introduction of fluorine into phenyl substituted alkenes with Selectfluor F-TEDA-BF4. Acta Chim Slov. 2005;52(3):200–206. [Google Scholar]
- 36.Phipps RJ, Toste FD. Chiral anion phase-transfer catalysis applied to the direct enantioselective fluorinative dearomatization of phenols. J Am Chem Soc. 2013;135(4):1268–1271. doi: 10.1021/ja311798q. [DOI] [PubMed] [Google Scholar]
- 37.Chang M-Y, Lee N-C, Lee M-F, Huang Y-P, Lin C-H. Selectfluor-promoted fluorination of piperidinyl olefins. Tet Lett. 2010;51(45):5900–5903. [Google Scholar]
- 38.Hagmann WK. The many roles for fluorine in medicinal chemistry. J Med Chem. 2008;51(15):4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 39.Guo Q-S, Du D-M, Xu J. The development of double axially chiral phosphoric acids and their catalytic transfer hydrogenation of quinolines. Angew Chem Int Ed Engl. 2008;47(4):759–762. doi: 10.1002/anie.200703925. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.