

Abstract
Post-traumatic stress disorder, panic disorder and phobia manifest in ways that are consistent with an uncontrollable state of fear. Their development involves heredity, previous sensitizing experiences, association of aversive events with previous neutral stimuli, and inability to inhibit or extinguish fear after it is chronic and disabling. We highlight recent progress in fear learning and memory, differential susceptibility to disorders of fear, and how these findings are being applied to the understanding, treatment and possible prevention of fear disorders. Promising advances are being translated from basic science to the clinic, including approaches to distinguish risk versus resilience before trauma exposure, methods to interfere with fear development during memory consolidation after a trauma, and techniques to inhibit fear reconsolidation and to enhance extinction of chronic fear. It is hoped that this new knowledge will translate to more successful, neuroscientifically informed and rationally designed approaches to disorders of fear regulation.
The laboratory study of fear learning and memory continues to yield knowledge that holds promise for the understanding and treatment of post-traumatic stress disorder (PTSD) and other fear-related disorders. Here we discuss how these new and exciting observations are being translated from the basic science fields to the clinic. Furthermore, we point areas where basic research using animal models can be improved to better account for the dysregulation of fear seen in many disorders.
Experiencing an extremely traumatic event, such as combat or violent assault, can lead to PTSD. Estimates are that up to 90% of all people will be exposed to a severe traumatic event during their lifetime1. Given the high rates of trauma exposure, the prevalence of PTSD is relatively low, affecting approximately only 5–10% of the general population, with women being twice as likely to develop PTSD as men2. However, the rates of lifetime PTSD are closer to 20–30% in highly exposed trauma populations, such as low-income urban populations1 and repeatedly traumatized soldiers. Recent studies have demonstrated a steep dose-response curve between trauma frequency and PTSD symptom severity, such that the more traumatic events a person experiences, the greater the intensity of PTSD symptoms3, 4. PTSD is the fourth most common psychiatric diagnosis1 and is defined by three primary symptom clusters after an event that elicited fear, helplessness or horror5. The first cluster of symptoms includes re-experiencing the traumatic event through intrusive thoughts, nightmares, flashbacks and related phenomena that are often produced by reminders of the traumatic event. The second cluster is characterized by avoidance symptoms, including loss of interest in social situations and emotional detachment. The third cluster includes psychophysiological reactivity in response to trauma-related stimuli, including exaggerated startle, hypervigilance, elevated perspiration and shortness of breath.
Several other anxiety disorders are also characterized primarily by a dysregulated fear response. These include simple phobia; social phobia (also called social anxiety disorder), which involves fear and avoidance of social situations; and panic disorder. What is particularly interesting about this collection of disorders is that they all share a similar set of fear or panic symptoms that now have a clearly understood neurological basis (Fig. 1). Anxiety disorders affect around 18% percent of adults in the United States in a given year. Moreover, in 64% of suicide attempts, at least one anxiety disorder is present. Therefore, from a clinical perspective, improving treatment and identifying prevention measures is of critical importance. Furthermore, from a scientific perspective, we would argue that the fear-related anxiety disorders provide among the ‘lowest-hanging fruit’ for understanding the neural circuitry and pathophysiology of psychiatric disorders. This is because (i) the neural substrates of fear have been well worked out through over 50 years of neurobiological studies, (ii) the basic behavioral mechanisms underlying fear have been studied for over 100 years since the time of Pavlov, (iii) these neural and behavioral mechanisms are remarkably well conserved across mammalian species, including humans, and (iv) in many cases of fear-related disorders, particularly PTSD, the traumatic incident that initiates the dysregulated fear response can be identified. As a result of this last component, not only may we improve our understanding of the biological and psychological processes leading to a transformation from a ‘normal’ fear reaction to a pathologically dysregulated fear response, but we also may be able in some cases to prevent the development of inappropriate fear responses through early intervention.
Figure 1.
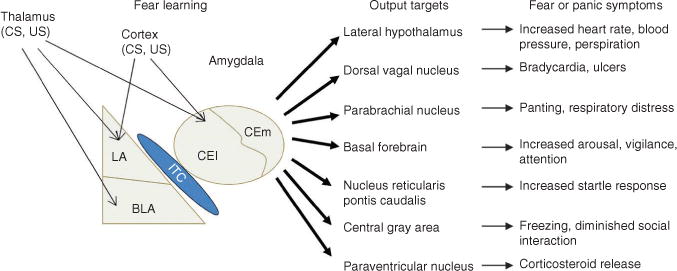
Schematic depicting the amygdala, the brain site most critical for fear learning. Information regarding the conditioned stimulus (CS) and unconditioned stimulus (US) is transmitted to the amygdala by way of sensory areas in the thalamus and cortex. Within the amygdala, the critical plasticity underlying the acquisition of fear conditioning is thought to occur in the lateral amygdala and the lateral portion of the central nucleus (CEl). The medial division of the central nucleus of the amygdala (CEm) projects to various brain areas that produce fear and panic symptoms seen in people with fear-related disorders. LA, lateral nucleus; BLA, basolateral nucleus; ITC, intercalated cells (see also ref. 99).
An important observation in recent years is that there are several different learning components that distinguish normal fear or trauma exposure and recovery from the pathological responses to trauma exposure associated with lack of recovery and/or worsening of symptoms (Fig. 2). Evidence suggests that exposure to trauma in the past, before the ‘index trauma’ associated with the PTSD— particularly childhood trauma exposure—is of substantial importance. Furthermore, it appears that some gene pathways (for example, FKBP5; ref. 3) interact with childhood trauma, but not adult trauma, to predict adult PTSD. One possible reason for this is that developmental critical periods of amygdala function are glucocorticoid dependent6, and FKBP5 regulates glucocorticoid receptor sensitivity. Also, it is known that the level of trauma exposure is of critical import in the later development of post-traumatic symptoms. During the minutes to hours, and possibly days, after trauma exposure, the memory remains in a labile state, called the consolidation period. Some updates on the neurobiology of consolidation are outlined below, and there are exciting areas of inquiry suggesting that new pharmacotherapeutic and psychotherapeutic approaches may be initiated that may inhibit the emotional component of fear memory consolidation, without markedly affecting the explicit memory formation. Such an approach may not cause amnesia per se but could prevent the severe emotional reactions that underlie later development of PTSD. Another aspect of memory modulation that will be addressed below is the idea of reconsolidation, in which reactivation of a memory may cause it to re-enter a labile state after it has become permanent. The extent to which reconsolidation occurs with chronic, long-term memories in humans remains under some debate, but, if robust, it is an extremely exciting potential area of modulation. Finally, there are several further cognitive mechanisms that are associated with pathological reactions, such as generalization and sensitization of reminders of fear or trauma. In contrast, the mechanisms of discrimination and extinction of memory serve to counter these processes. In summary, as outlined below, understanding the different components of fear memory formation and modulation may enable powerful and targeted treatment and intervention approaches.
Figure 2.
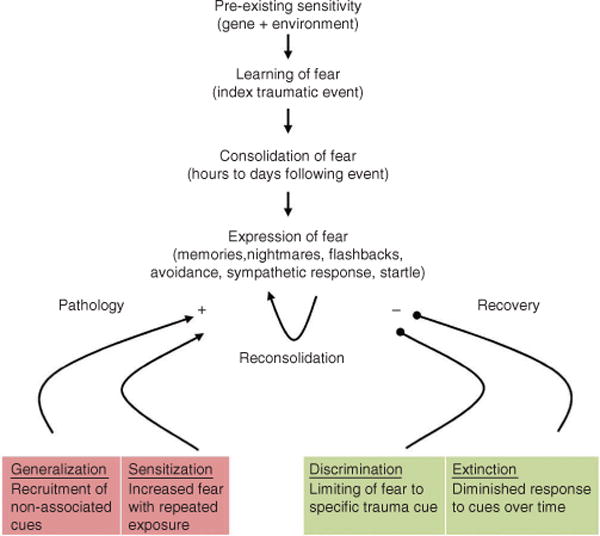
A model for the development of fear-related disorders. Certain individuals are predisposed to the development of fear-related disorders on the basis of early life experience and genetic background, among other risk factors. When a traumatic event occurs, people learn to fear the cues that are associated with the traumatic event, and this memory consolidates over the course of the subsequent hours and days. The expression of fear comes in several different forms, including flashbacks of the traumatic event, nightmares, avoidance of situations that trigger memory for the traumatic event and altered sympathetic responses such as increased startle. The expression of the fear triggered by memory for the traumatic event may serve to sensitize those who develop psychopathology, resulting in increased fear. Additionally, fear may generalize to cues not associated with the traumatic event in those people who go on to develop a fear-related disorder. In contrast, with resilience, fear responses to cues related to the traumatic event extinguish over time, and discrimination occurs between cues that are associated with the traumatic event and those that are not.
Although there are several existing pharmacological and psychotherapeutic treatments for fear-related disorders7, 8, all of these rely on empirically derived approaches. The first-line medication approach for all anxiety disorders includes the antidepressant and anxiolytic classes of selective and nonselective serotonin and other monoamine reuptake inhibitors (for example, fluoxetine, sertraline or venlafaxine). Although our understanding of the monoaminergic regulation of fear circuitry is improving, it is clear that these are not specific in their actions, they can have difficult side effects and they are only effective in some cases. The second-most-common class of agents used to treat these disorders are the benzodiazepines (for example, clonazepam, alprazolam or lorazepam), which act through enhancement of GABAA activity. Enhancing inhibitory transmission in the amygdala and bed nucleus of the stria terminalis (BNST) has been shown to diminish fear responses, but these agents have all of the same limitations as the monoaminergic anxiolytics, in addition to having abuse and tolerance potential.
A particularly promising area of inquiry arises from the burgeoning understanding of the neurobiological mechanisms of learning and memory. Although there are many ways to model the disorders of fear regulation, among the most robust approaches results from functionally dissecting the differential cognitive, learning and memory components that regulate fear learning, consolidation, modulation, generalization, sensitization, discrimination and extinction. Below we will review some of the differential learning and memory aspects underlying fear processing and illustrate how breakthroughs in these areas are leading to new approaches to the modulation of memory, and thus new treatment and intervention approaches targeting disorders of fear regulation.
The essential neural circuit supporting fear conditioning
The progress made in developing strategies to treat fear-related disorders has been greatly aided by the knowledge gained in the past several decades regarding the brain circuitry involved in Pavlovian fear conditioning and the cellular and molecular mechanisms in this network that support this form of learning. Fear conditioning involves learning an association between a neutral conditioned stimulus, such as a light or tone, and an aversive unconditioned stimulus, typically a foot shock (Fig. 3a). Memory for fear conditioning is inferred by presenting the cue that signaled the shock (Fig. 3b), and several conditioned responses consistent with a state of fear can be assessed. Some commonly measured fear responses in rats and mice include freezing behavior and potentiated startle; in humans, potentiated startle and skin conductance responses are often measured.
Figure 3.
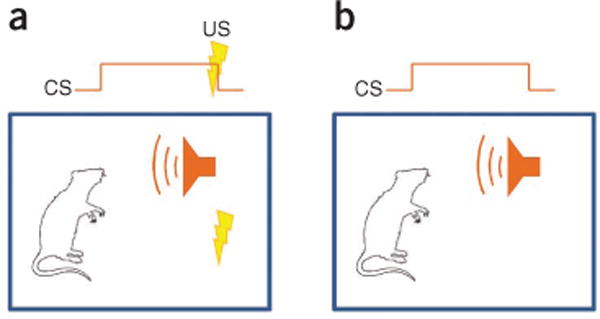
Basic fear conditioning and testing procedures. (a) Fear conditioning involves training an animal to fear a neutral conditioned stimulus (CS) such as an auditory cue by having it signal an aversive unconditioned stimulus (US) such as an electrical shock. (b) Memory for fear conditioning is tested by presenting the conditioned stimulus alone and measuring fear responses.
At the heart of the brain circuitry mediating fear learning and fear responses is a group of subcortical nuclei referred to collectively as the amygdala (Fig. 1). The lateral nucleus of the amygdala receives multimodal sensory information regarding the conditioned stimulus from thalamic and sensory cortical areas9, 10. This converges with input regarding the unconditioned stimulus, believed to arrive from somatosensory thalamic and cortical areas11, 12 and the periaqueductal gray13. This convergence of the conditioned stimulus and unconditioned stimulus, along with other types of data, indicates that the lateral nucleus is a critical site for plasticity underlying fear learning14. Because the central nucleus of the amygdala sends projections to several brain areas responsible for generating fear responses15, it typically has been thought of as an output structure. However, the central nucleus also receives direct thalamic and cortical inputs, and work has shown that preventing the activity of NMDA-type glutamate receptors and preventing the synthesis of protein in the central nucleus blocks the acquisition and consolidation of fear conditioning, respectively16, 17. Recent studies18, 19 showed that the lateral (CEl) and medial (CEm) divisions of the central nucleus make distinct contributions to fear conditioning, with the CEl being necessary for the acquisition of fear and the CEm responsible for the production of fear responses.
Learning of environmental contextual cues also occurs during standard fear conditioning. Although contextual fear conditioning also depends on the amygdala20, it requires the dorsal hippocampus, which is not normally involved in fear conditioning to discrete cues. Lesions of the dorsal hippocampus shortly after fear conditioning were found to block the formation of contextual fear21, and subsequent work showed that pharmacological disruptions in the hippocampus around the time of learning have similar effects22. The neural interactions of the fear circuit external to and within the amygdala are more complex than is being presented here, and we direct the reader to a recent review for a more detailed description14.
Data from studies of fear conditioning in humans largely mirror findings from rodents with respect to the brain areas engaged during acquisition. People with damage to the amygdala show a disruption in fear learning, as measured by changes in skin conductance responses23. Functional brain imaging studies have shown increased amygdala activation during acquisition of fear conditioning24 and during the production of fear responses25. Human brain imaging studies have also demonstrated that the hippocampus and related areas are active during contextual fear learning26, which parallels findings from rodent research.
Many studies spanning the preclinical to clinical in humans have demonstrated that the brain areas implicated in rodent models are also robustly involved in human fear learning and modulation. Furthermore, these areas appear to be notably dysregulated in fear-related disorders such as panic disorder, specific and social phobia, and PTSD. Perhaps the most replicated and robust finding is the activation of amygdala nuclei in the presence of fearful cues, most notably fearful faces27(Fig. 4a). Several studies have demonstrated hyperactive amygdala response in people with PTSD and other fear disorders relative to healthy subjects28. In addition to the action of the amygdala in directly mediating the fear response reflex, many areas are involved in the inhibition and modulation of amygdala activity, most notably the hippocampus29–31 and medial prefrontal cortex32–37. These areas have also been demonstrated to have abnormal responses to fearful cues and fear inhibition in human functional magnetic resonance imaging studies38–41(Fig. 4b–f). These data provide face and construct validity for the power of understanding the learning and modulation mechanisms of fear memories in rodent models to provide new therapeutic approaches to amygdala, hippocampal and ventromedial prefrontal cortex (vmPFC) regulation of fear in human disorders.
Figure 4.
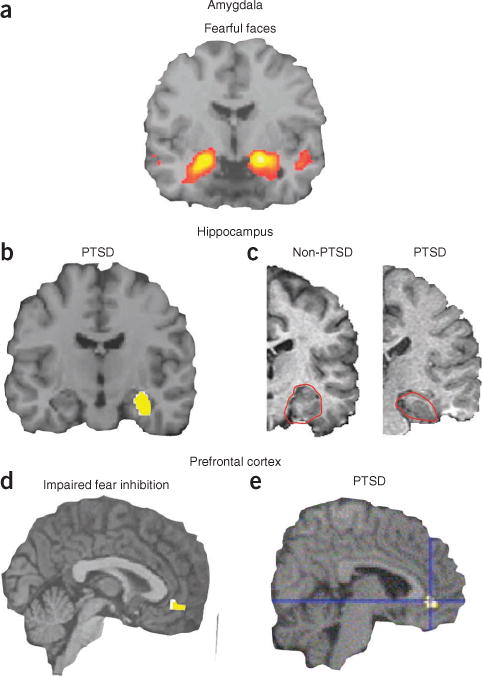
Human neural circuitry involved in fear-related disorders and PTSD. (a) Viewing of fearful or angry faces (compared to shapes) robustly activates human amygdala across protocols and cohorts (reproduced with permission from ref. 27). Often this amygdala activation is increased in fear-related disorders. (b) Right hippocampal activity is lower in youths with post-traumatic stress symptoms than in healthy controls (reproduced with permission from ref. 38). (c) Reduced hippocampal volume in a patient with PTSD (right) compared to that in a subject without PTSD (left). Hippocampus outlined in red (adapted with permission from ref. 39). (d) Reduced neural activation of vmPFC during an inhibition task is associated with impaired fear inhibition (reproduced with permission from ref. 41). (e) Subjects with PTSD show lower regional cerebral blood flow activity in the rostral anterior cingulate during exposure to traumatic or stressful script-driven imagery (reproduced with permission from ref. 40).
Blocking fear memory formation to prevent fear disorders
To appreciate how the study of fear conditioning can help develop strategies to treat fear disorders, it is critical to understand the different phases of learning and how they are typically studied in the laboratory. Acquisition of fear conditioning (Fig. 5a) refers to the process by which the organism learns that the conditioned stimulus predicts the unconditioned stimulus. Treatments that block the acquisition of fear conditioning are applied before conditioned-unconditioned stimulus pairings and prevent the development of short-term memory (STM) memory, tested within a few hours, and consequently the formation of long-term memory (LTM), tested many hours or days later. There are several cellular and molecular processes known to underlie the acquisition of fear conditioning. For example, NMDA antagonists applied just before training prevent the acquisition of fear conditioning, resulting in disrupted STM and LTM42.
Figure 5.
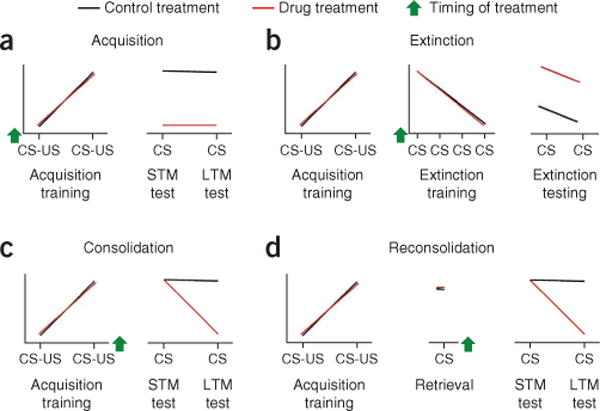
Different components of fear learning and modulation as they are studied in the laboratory. (a) Acquisition training involves pairings of a conditioned stimulus (CS) with an aversive unconditioned stimulus (US). A treatment is said to prevent acquisition of fear if it is applied before training and blocks both STM and LTM from forming. (b) Fear memories can also undergo extinction by repeated presentation of the CS without the US during extinction training. Treatments that block the formation of extinction memory are typically given before extinction training and result in more fear during extinction testing in comparison to control treatments. (c) The consolidation of fear memory refers to time-dependent stabilization of memory after acquisition. Treatments that block memory consolidation are usually given shortly after training and result in disrupted LTM but intact STM. (d) When a memory is retrieved it may undergo reconsolidation, which results in a period of time during which the memory is labile. Reconsolidation of memory is considered to be disrupted when a drug is applied shortly after retrieval and leaves STM intact yet disrupts LTM. Green arrows indicates the timing of a given treatment or manipulation for each of the different learning phases; x axes represent time; y axes, fear behavior.
The consolidation of fear conditioning refers to the transformation of memory from a labile state immediately after acquisition to a more permanent state with the passage of time. Treatments that disrupt the consolidation of memory are usually applied a few minutes to a few hours after conditioned-unconditioned pairings, leaving STM intact but resulting in disrupted LTM (Fig. 5b). The time window for memory consolidation is defined by the period of time after acquisition during which memory can be disrupted by amnesic treatments. For example, protein synthesis inhibitors applied after acquisition of fear conditioning do not affect STM and are only effective in disrupting LTM if they are delivered within a few hours after conditioned–unconditioned stimulus pairings43. The acquisition and consolidation of fear conditioning require many cellular and molecular changes in addition to the two examples given here, a full explanation of which is beyond the scope of this review. We point the reader to some excellent recent reviews that describe these in depth44, 45.
The point at which a traumatic event occurs represents the first opportunity to use treatments designed to disrupt the acquisition and/or consolidation of the memory. There are several recent studies that suggest that molecular mediators of fear consolidation may be impaired by specific treatments targeting this memory process. Among the most robust are data suggesting that modulation of the endogenous opioid system may inhibit fear consolidation. Rodent studies have suggested that μ-opioid pathway activation opposes fear consolidation and enhances extinction46, 47. Moreover, κ-opioid antagonists have similar effects on fear learning48 and mediate stress effects on attention49. Morphine treatment after the experience of traumatic burns may decrease later PTSD symptoms in children50. More recent studies in civilians and soldiers suggest that acute morphine administration during the immediate aftermath of traumatic injury may prevent the subsequent development of PTSD51. It has not been fully clarified, however, whether opioid treatment is acting at the level of pain control and, by thus decreasing the pain—the unconditioned stimulus—decreasing the initial fear learning. Alternatively, given the animal results, the opioid pathways may be directly acting in amygdala and brainstem areas involved in fear consolidation and thus may have a direct neural mechanism for decreasing the fear memory, independent of pain regulation.
Another pathway that has been associated with fear memory consolidation is activation of β-adrenergic receptors in amygdala52. As propranolol has been used in humans safely for decades for blockade of cardiovascular sympathetic activity, as well as for inhibiting social anxiety responses, it is a safe medication to use potentially to intervene in fear and trauma memory consolidation. Although preliminary studies were promising53 and propranolol appears to decrease amygdala activation in humans54, more recent, larger studies have not found a significant effect of propranolol administration after trauma55–57.
There have also been exciting recent approaches focused on non-medication-based psychotherapeutic approaches. Specifically, it has previously been shown in animal models that re-exposure to a conditioned cue in the absence of reinforcement can impair the initial consolidation of that fear memory process58 (but see ref. 59). Translating this to humans, Rothbaum and colleagues recently performed a proof-of-concept trial with 137 traumatized civilians, with full exposure-based psychotherapy in the emergency department in the hours after the trauma60. Exposure therapy is thought to rely on extinction mechanisms (see below) and to be well modeled by extinction in rodents. It was found that this early intervention may have a significant protective effect on development of PTSD and depression symptoms assessed 4 and 12 weeks later. Larger, randomized trials are needed, but this suggests the possibility that exposure to appropriate therapy after trauma may lead to more rapid recovery or even prevention of PTSD formation.
There are many questions that have been raised regarding the wisdom and ethics of potential prevention of memory formation in the aftermath of trauma exposure. In particular are issues related to the ethics of induced amnesia and the potential complexity of complete forgetting of an event that may be important to remember for reasons related to future safety or possible legal ramifications. One potential solution to this issue is the recognition of multiple memory systems—that a given traumatic experience is encoded in parallel across declarative, emotional and motor pathways, which all have different underlying neurocircuitry. If the field of neuroscience is able to identify ways to target the overlearning of the emotional component of the memory while leaving the declarative trace intact, it may be possible to convert an overly strong, indelible, overwhelming emotional experience—one that becomes a ‘black hole’ of memories for many with PTSD—to simply a bad memory, which can then be managed, modulated and overcome in appropriate ways, leading to recovery.
Enhancing fear extinction to treat fear–related disorders
The extinction of fear conditioning refers to the decrease in fear responses during repeated presentations of the conditioned stimulus without unconditioned stimulus reinforcement. Extinction can refer to the within-session decrement in fear responses while animals are receiving presentations of the conditioned stimulus alone during extinction training. It can also refer to the retention of extinction learning when animals are presented with the conditioned stimulus at later time points (Fig. 5c). Extinction is thought to involve new learning rather than erasure or unlearning of the association. Evidence for this assertion comes from the observation that fear responses spontaneously recover with passage of time61, that fear responses show renewed responding when the conditioned stimulus is presented in a different environmental context from that in which extinction training occurred62, and that presentation of the unconditioned stimulus alone reinstates fear to a cue that has undergone extinction training63. The extinction of fear conditioning relies on some of the same brain circuitry necessary for acquiring fear memories, including the amygdala64 and hippocampus29. There is good evidence that extinction also requires activity of the vmPFC, which is not normally involved in the acquisition of fear conditioning. In rats, the infralimbic portion of the vmPFC appears to be critical for the extinction of fear conditioning. Lesions of this area have been shown to disrupt the retention of extinction32, and neurons in the infralimbic cortex show increased firing during the recall of extinction memory33. Neurons in the infralimbic cortex are thought to decrease fear responses by means of projections to GABAergic intercalated neurons positioned between the lateral or basal and the central nuclei of the amygdala, which inhibit the output of the central nucleus. Studies of extinction learning in humans largely parallel studies rats, demonstrating that the vmPFC36, 38, amygdala24 and hippocampus31 are all engaged during extinction learning or the recall of extinction.
Pharmacological approaches that enhance fear extinction are being evaluated for treatment efficacy in PTSD. The use of D-cycloserine (DCS), a partial NMDA receptor agonist, as a potential treatment for PTSD arose as a result of many preclinical studies implicating NMDA receptor activity in learning and memory processes65, 66. DCS was first tried in humans for anxiety disorders in combination with virtual reality exposure (VRE) therapy for the fear of heights67. After treatment, those patients that received DCS in combination with VRE showed greater improvement than those who received placebo and VRE. Since that study, DCS has been shown to be an effective therapeutic compound for increasing the rate of recovery with exposure-based psychotherapy several fear- and anxiety-related disorders, including panic disorder68, social anxiety disorder69, obsessive-compulsive disorder70 and PTSD71. Although there have been some negative trials, most of these can be explained retrospectively as the mechanism of DCS is further understood, and two recent meta-analyses support the conclusion that it is an effective augmentation strategy to enhance the rate of emotional learning underlying exposure-based psychotherapy72, 73. Other methods of augmenting NMDA receptor activity in conjunction with extinction are also now being explored.
More recent work has identified brain-derived neurotrophic factor (BDNF) as a molecular target for facilitating extinction learning and a potential treatment for fear disorders74. Studies have shown that blocking the activity of BDNF in the amygdala75 or hippocampus30 disrupts the retention of extinction. Other studies indicate that memory for extinction can be facilitated by infusion of recombinant BDNF in the infralimbic cortex or dorsal hippocampus35 or by systemic injection of an agonist for its receptor TrkB76. Further very interesting work involves the Val66Met variant of BDNF in humans. Carriers of the methionine-encoding allele release less BDNF peptide77. Recently humans with this allele have been shown to have been found to have diminished extinction of conditioned fear78, which may serve as a partial explanation for the increased prevalence of anxiety-related disorders in people with this genotype79. Most intriguingly, in the same study78, it was shown in ‘humanized’ mouse models using knock-ins of each of the human alleles to the mouse Bdnf gene locus that these alleles lead to phenotypes in mice similar to those in human: decreased extinction of fear in the methionine allele carriers relative to that in the valine allele carriers. Some meta-analyses have failed to find increased incidence of anxiety disorders in methionine allele carriers80; however, this might be the result of low samples sizes. Together these data extend our understanding and appreciation of the role of BDNF in extinction and recovery from fear and fear-related disorders. They also provide further evidence for the face validity of the usefulness of the extinction-of-fear model in mice for extinction of fear in humans.
Disrupting traumatic memories after retrieval
Recently there has been renewed interest in the notion that LTM becomes susceptible to disruption after a consolidated memory is retrieved. In fear conditioning studies, memory is retrieved by presenting the animal with a single presentation of the conditioned stimulus used to signal shock during acquisition (Fig. 5d). The seminal finding was that when a protein synthesis inhibitor is given after retrieval, LTM is impaired on subsequent tests81. This result generated wide interest, and this phenomenon, termed reconsolidation, has now been observed in organisms ranging from invertebrates to humans82. Somewhat less is known about memory reconsolidation than about initial consolidation, but the available evidence suggests that the molecular and cellular mechanisms supporting reconsolidation are similar to those necessary for consolidation, although they do not overlap completely83.
The observation that fear memories can be disrupted by combining retrieval of memory with drug treatment opens up the possibility of using this strategy to treat fear-related disorders. Theoretically, patients could be brought into a clinical setting, presented with a stimulus that retrieves the fearful memory and given a drug, and the fear memory would be weakened. Recent laboratory studies have used this basic approach to determine whether fear memories can be disrupted by combining retrieval with a memory-impairing drug. In one study84, human subjects were fear conditioned, given a retrieval trial the next day in conjunction with oral administration of the β-adrenergic blocker propranolol, and tested the day after. The results showed that those given the drug while the memory was reactivated showed significantly less fear-potentiated startle during testing the next day than those given placebo. At least one study85 has shown that a similar approach can be taken to disrupt traumatic memories in humans. In this study, PTSD patients were asked to describe a traumatic experience and were given a single dose of propranolol or a placebo. Patients given propranolol showed reduced physiological signs of fear when they were asked to once again describe the traumatic experience a week later.
Although there are some differences, there is also evidence that disruption of reconsolidation and extinction may share some interesting properties86. Of note, in vivo and ex vivo physiological studies have suggested that fear learning leads to LTP-like potentiation of synapses with fear learning. Extinction of fear then appears to be associated with depotentiation and LTD-like mechanisms in some models87, 88. Thus, diminished representation of synaptic strength may be achieved, in part, both through strengthened extinction and through inhibited reconsolidation.
Although this strategy is promising, laboratory studies of reconsolidation indicate that there may be limitations to using a reconsolidation-disruption approach as a way to treat fear-related disorders. Several studies have indicated that retrieval does not always trigger reconsolidation, including the observation that both older and stronger memories are less susceptible to disruption after retrieval89, 90. If this pattern of data extends to humans with fear-related disorders, it may prove difficult to disrupt traumatic memories after retrieval because these memories are most certainly strong and in many cases have persisted for some time. In fact, many PTSD patients may take years to seek treatment, and chronic PTSD is often the most difficult to treat. Another consideration is that memory retrieval happens outside of the clinical context, often in the form of re-experiencing of the traumatic event. Replaying the traumatic event over and over again can sensitize patients with fear-related disorders and lead to worsening of the disorder. As in sensitization in humans with fear-related disorders, animal studies have also shown that repeated retrieval can strengthen fear memory and make it impervious to disruption with treatments that normally disrupt memory reconsolidation91. Thus, even if a drug is given each time a patient re-experiences a traumatic event, it may not be sensitive to disruption.
Future directions
Further areas of interest that are less well developed include studies of generalization versus discrimination, avoidance behavior and combined extinction-reconsolidation processes. The use of more sophisticated behavioral techniques in the laboratory to understand how fear generalizes to stimuli not originally associated with the traumatic event, which is a hallmark of PTSD and panic disorder, may provide powerful insight. An approach to studying generalization is to use differential fear conditioning whereby, in addition to a cue that signals shock, there is also a cue that is not followed by shock. Studies have shown that in rats92 some animals show good discrimination, whereas others generalize fear to the safe cue, similarly to what is seen in patients with fear-related disorders. Another approach is to use conditioned inhibition training to identify animals that do not inhibit fear in the presence of a safety signal93. Both of these strategies can address a potential limitation of animal studies: that the variability of responses is often not factored into the analyses, even though in people who experience a traumatic event there is great variability in responses, with some developing a pathological disorder and others being resilient94. In a similar vein, early life stress and previous trauma experience factor into the development PTSD (Fig. 2), yet there are relatively few laboratory studies determining the effects of previous trauma and early life stress on fear learning and fear extinction. More refined protocols are needed to model this important aspect of susceptibility to developing PTSD.
Another line of research that could potentially be relevant for the treatment of fear-related disorders is based on recent behavioral studies95–97 demonstrating that, if extinction training occurs shortly after a single retrieval trial, fear memories are diminished and show no evidence of recovery. Although this finding is not always consistent98, the ability to diminish fear memories in this manner opens another potential avenue by which traumatic memories can be targeted in patients with fear-related disorders.
Conclusions
Our goal is to describe how knowledge of basic learning and memory processes has translated into potential treatments for PTSD and other fear-related disorders. We wish to point to recent areas that have potential to drive clinical treatments in the future. If animal models are modified to better account for fear dysregulation in these disorders, we may improve the impact of preclinical research on prevention and treatment. Recent advances in molecular and cellular approaches to cognitive function are rapidly advancing our understanding of fear-related disorders. Progress in this area is exciting, not only in its potential to affect and improve treatment but also in the hope that it provides to biological psychiatry in general. Success in this arena suggests that if the neural circuitry underlying functional pathophysiology can be defined, then powerful behavioral neuroscience approaches can be effectively translated to the clinic, even in debilitating and previously mysterious psychiatric disorders.
Acknowledgments
Support was provided by the US National Institutes of Health (F32MH090700, R01MH071537, R01MH094757 and R01MH096764), the Burroughs Wellcome Fund and a US National Institutes of Health National Center for Research Resources base grant (P51RR000165) to Yerkes National Primate Research Center.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details accompany the online version of the paper.
References
- 1.Breslau N, et al. Trauma and posttraumatic stress disorder in the community: the 1996 Detroit Area Survey of Trauma. Arch Gen Psychiatry. 1998;55:626–632. doi: 10.1001/archpsyc.55.7.626. [DOI] [PubMed] [Google Scholar]
- 2.Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry. 1995;52:1048–1060. doi: 10.1001/archpsyc.1995.03950240066012. [DOI] [PubMed] [Google Scholar]
- 3.Binder EB, et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. J Am Med Assoc. 2008;299:1291–1305. doi: 10.1001/jama.299.11.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McTeague LM, et al. Aversive imagery in posttraumatic stress disorder: trauma recurrence, comorbidity, and physiological reactivity. Biol Psychiatry. 2010;67:346–356. doi: 10.1016/j.biopsych.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. American Psychiatric Association; Washington, DC: 2000. Text Revision (DSMIV-TR) Ch 7. [Google Scholar]
- 6.Moriceau S, Wilson DA, Levine S, Sullivan RM. Dual circuitry for odor-shock conditioning during infancy: corticosterone switches between fear and attraction via amygdala. J Neurosci. 2006;26:6737–6748. doi: 10.1523/JNEUROSCI.0499-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steckler T, Risbrough V. Pharmacological treatment of PTSD – established and new approaches. Neuropharmacology. 2012;62:617–627. doi: 10.1016/j.neuropharm.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cain CK, Maynard GD, Kehne JH. Targeting memory processes with drugs to prevent or cure PTSD. Expert Opin Investig Drugs. 2012;21:1323–1350. doi: 10.1517/13543784.2012.704020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.LeDoux JE, Cicchetti P, Xagoraris A, Romanski LM. The lateral amygdaloid nucleus: sensory interface of the amygdala in fear conditioning. J Neurosci. 1990;10:1062–1069. doi: 10.1523/JNEUROSCI.10-04-01062.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campeau S, Davis M. Involvement of subcortical and cortical afferents to the lateral nucleus of the amygdala in fear conditioning measured with fear-potentiated startle in rats trained concurrently with auditory and visual conditioned stimuli. J Neurosci. 1995;15:2312–2327. doi: 10.1523/JNEUROSCI.15-03-02312.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi C, Davis M. Pain pathways involved in fear conditioning measured with fear-potentiated startle: lesion studies. J Neurosci. 1999;19:420–430. doi: 10.1523/JNEUROSCI.19-01-00420.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lanuza E, Nader K, LeDoux JE. Unconditioned stimulus pathways to the amygdala: effects of posterior thalamic and cortical lesions on fear conditioning. Neuroscience. 2004;125:305–315. doi: 10.1016/j.neuroscience.2003.12.034. [DOI] [PubMed] [Google Scholar]
- 13.Johansen JP, Tarpley JW, LeDoux JE, Blair HT. Neural substrates for expectation-modulated fear learning in the amygdala and periaqueductal gray. Nat Neurosci. 2010;13:979–986. doi: 10.1038/nn.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pape HC, Pare D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev. 2010;90:419–463. doi: 10.1152/physrev.00037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LeDoux JE, Iwata J, Cicchetti P, Reis DJ. Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci. 1988;8:2517–2529. doi: 10.1523/JNEUROSCI.08-07-02517.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goosens KA, Maren S. Pretraining NMDA receptor blockade in the basolateral complex, but not the central nucleus, of the amygdala prevents savings of conditional fear. Behav Neurosci. 2003;117:738–750. doi: 10.1037/0735-7044.117.4.738. [DOI] [PubMed] [Google Scholar]
- 17.Wilensky AE, Schafe GE, Kristensen MP, LeDoux JE. Rethinking the fear circuit: the central nucleus of the amygdala is required for the acquisition, consolidation, and expression of Pavlovian fear conditioning. J Neurosci. 2006;26:12387–12396. doi: 10.1523/JNEUROSCI.4316-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ciocchi S, et al. Encoding of conditioned fear in central amygdala inhibitory circuits. Nature. 2010;468:277–282. doi: 10.1038/nature09559. [DOI] [PubMed] [Google Scholar]
- 19.Haubensak W, et al. Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature. 2010;468:270–276. doi: 10.1038/nature09553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muller J, Corodimas KP, Fridel Z, LeDoux JE. Functional inactivation of the lateral and basal nuclei of the amygdala by muscimol infusion prevents fear conditioning to an explicit conditioned stimulus and to contextual stimuli. Behav Neurosci. 1997;111:683–691. doi: 10.1037//0735-7044.111.4.683. [DOI] [PubMed] [Google Scholar]
- 21.Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- 22.Sanders MJ, Fanselow MS. Pre-training prevents context fear conditioning deficits produced by hippocampal NMDA receptor blockade. Neurobiol Learn Mem. 2003;80:123–129. doi: 10.1016/s1074-7427(03)00040-6. [DOI] [PubMed] [Google Scholar]
- 23.Bechara A, et al. Double dissociation of conditioning and declarative knowledge relative to the amygdala and hippocampus in humans. Science. 1995;269:1115–1118. doi: 10.1126/science.7652558. [DOI] [PubMed] [Google Scholar]
- 24.LaBar KS, Gatenby JC, Gore JC, LeDoux JE, Phelps EA. Human amygdala activation during conditioned fear acquisition and extinction: a mixed-trial fMRI study. Neuron. 1998;20:937–945. doi: 10.1016/s0896-6273(00)80475-4. [DOI] [PubMed] [Google Scholar]
- 25.Cheng DT, Knight DC, Smith CN, Helmstetter FJ. Human amygdala activity during the expression of fear responses. Behav Neurosci. 2006;120:1187–1195. doi: 10.1037/0735-7044.120.5.1187. [DOI] [PubMed] [Google Scholar]
- 26.Alvarez RP, Biggs A, Chen G, Pine DS, Grillon C. Contextual fear conditioning in humans: cortical-hippocampal and amygdala contributions. J Neurosci. 2008;28:6211–6219. doi: 10.1523/JNEUROSCI.1246-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drabant EM, McRae K, Manuck SB, Hariri AR, Gross JJ. Individual differences in typical reappraisal use predict amygdala and prefrontal responses. Biol Psychiatry. 2009;65:367–373. doi: 10.1016/j.biopsych.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Etkin A, Wager TD. Functional neuroimaging of anxiety: a meta-analysis of emotional processing in PTSD, social anxiety disorder, and specific phobia. Am J Psychiatry. 2007;164:1476–1488. doi: 10.1176/appi.ajp.2007.07030504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corcoran KA, Desmond TJ, Frey KA, Maren S. Hippocampal inactivation disrupts the acquisition and contextual encoding of fear extinction. J Neurosci. 2005;25:8978–8987. doi: 10.1523/JNEUROSCI.2246-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heldt SA, Stanek L, Chhatwal JP, Ressler KJ. Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol Psychiatry. 2007;12:656–670. doi: 10.1038/sj.mp.4001957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knight DC, Smith CN, Cheng DT, Stein EA, Helmstetter FJ. Amygdala and hippocampal activity during acquisition and extinction of human fear conditioning. Cogn Affect Behav Neurosci. 2004;4:317–325. doi: 10.3758/cabn.4.3.317. [DOI] [PubMed] [Google Scholar]
- 32.Quirk GJ, Russo GK, Barron JL, Lebron K. The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci. 2000;20:6225–6231. doi: 10.1523/JNEUROSCI.20-16-06225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- 34.Choi DC, et al. Prelimbic cortical BDNF is required for memory of learned fear but not extinction or innate fear. Proc Natl Acad Sci USA. 2010;107:2675–2680. doi: 10.1073/pnas.0909359107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peters J, Dieppa-Perea LM, Melendez LM, Quirk GJ. Induction of fear extinction with hippocampal-infralimbic BDNF. Science. 2010;328:1288–1290. doi: 10.1126/science.1186909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Extinction learning in humans: role of the amygdala and vmPFC. Neuron. 2004;43:897–905. doi: 10.1016/j.neuron.2004.08.042. [DOI] [PubMed] [Google Scholar]
- 37.Milad MR, et al. Recall of fear extinction in humans activates the ventromedial prefrontal cortex and hippocampus in concert. Biol Psychiatry. 2007;62:446–454. doi: 10.1016/j.biopsych.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 38.Carrión VG, Haas BW, Garrett A, Song S, Reiss AL. Reduced hippocampal activity in youth with posttraumatic stress symptoms: an FMRI study. J Pediatr Psychol. 2010;35:559–569. doi: 10.1093/jpepsy/jsp112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bremner JD. Neuroimaging in posttraumatic stress disorder and other stress-related disorders. Neuroimaging Clin N Am. 2007;17:523–538. doi: 10.1016/j.nic.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Britton JC, Phan KL, Taylor SF, Fig LM, Liberzon I. Corticolimbic blood flow in posttraumatic stress disorder during script-driven imagery. Biol Psychiatry. 2005;57:832–840. doi: 10.1016/j.biopsych.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 41.Jovanovic T, et al. Reduced neural activation during an inhibition task is associated with impaired fear inhibition in a traumatized civilian sample. Cortex. 2012 Oct 1; doi: 10.1016/j.cortex.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maren S, Aharonov G, Stote DL, Fanselow MS. N-methyl-D-aspartate receptors in the basolateral amygdala are required for both acquisition and expression of conditional fear in rats. Behav Neurosci. 1996;110:1365–1374. doi: 10.1037//0735-7044.110.6.1365. [DOI] [PubMed] [Google Scholar]
- 43.Schafe GE, LeDoux JE. Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci. 2000;20:RC96. doi: 10.1523/JNEUROSCI.20-18-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johansen JP, Cain CK, Ostroff LE, LeDoux JE. Molecular mechanisms of fear learning and memory. Cell. 2011;147:509–524. doi: 10.1016/j.cell.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Orsini CA, Maren S. Neural and cellular mechanisms of fear and extinction memory formation. Neurosci Biobehav Rev. 2012;36:1773–1802. doi: 10.1016/j.neubiorev.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Good AJ, Westbrook RF. Effects of a microinjection of morphine into the amygdala on the acquisition and expression of conditioned fear and hypoalgesia in rats. Behav Neurosci. 1995;109:631–641. doi: 10.1037//0735-7044.109.4.631. [DOI] [PubMed] [Google Scholar]
- 47.McNally GP, Westbrook RF. Opioid receptors regulate the extinction of Pavlovian fear conditioning. Behav Neurosci. 2003;117:1292–1301. doi: 10.1037/0735-7044.117.6.1292. [DOI] [PubMed] [Google Scholar]
- 48.Knoll AT, et al. Kappa opioid receptor signaling in the basolateral amygdala regulates conditioned fear and anxiety in rats. Biol Psychiatry. 2011;70:425–433. doi: 10.1016/j.biopsych.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van’t Veer A, Yano JM, Carroll FI, Cohen BM, Carlezon WA., Jr Corticotropin-releasing factor (CRF)-induced disruption of attention in rats is blocked by the κ-opioid receptor antagonist JDTic. Neuropsychopharmacology. 2012;37:2809–2816. doi: 10.1038/npp.2012.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saxe G, et al. Relationship between acute morphine and the course of PTSD in children with burns. J Am Acad Child Adolesc Psychiatry. 2001;40:915–921. doi: 10.1097/00004583-200108000-00013. [DOI] [PubMed] [Google Scholar]
- 51.Holbrook TL, Galarneau MR, Dye JL, Quinn K, Dougherty AL. Morphine use after combat injury in Iraq and post-traumatic stress disorder. N Engl J Med. 2010;362:110–117. doi: 10.1056/NEJMoa0903326. [DOI] [PubMed] [Google Scholar]
- 52.LaLumiere RT, Buen TV, McGaugh JL. Post-training intra-basolateral amygdala infusions of norepinephrine enhance consolidation of memory for contextual fear conditioning. J Neurosci. 2003;23:6754–6758. doi: 10.1523/JNEUROSCI.23-17-06754.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pitman RK, et al. Pilot study of secondary prevention of posttraumatic stress disorder with propranolol. Biol Psychiatry. 2002;51:189–192. doi: 10.1016/s0006-3223(01)01279-3. [DOI] [PubMed] [Google Scholar]
- 54.Hurlemann R, et al. Human amygdala reactivity is diminished by the beta-noradrenergic antagonist propranolol. Psychol Med. 2010;40:1839–1848. doi: 10.1017/S0033291709992376. [DOI] [PubMed] [Google Scholar]
- 55.Stein MB, Kerridge C, Dimsdale JE, Hoyt DB. Pharmacotherapy to prevent PTSD: results from a randomized controlled proof-of-concept trial in physically injured patients. J Trauma Stress. 2007;20:923–932. doi: 10.1002/jts.20270. [DOI] [PubMed] [Google Scholar]
- 56.Sharp S, Thomas C, Rosenberg L, Rosenberg M, Meyer W., III Propranolol does not reduce risk for acute stress disorder in pediatric burn trauma. J Trauma. 2010;68:193–197. doi: 10.1097/TA.0b013e3181a8b326. [DOI] [PubMed] [Google Scholar]
- 57.McGhee LL, et al. The effect of propranolol on posttraumatic stress disorder in burned service members. J Burn Care Res. 2009;30:92–97. doi: 10.1097/BCR.0b013e3181921f51. [DOI] [PubMed] [Google Scholar]
- 58.Myers KM, Ressler KJ, Davis M. Different mechanisms of fear extinction dependent on length of time since fear acquisition. Learn Mem. 2006;13:216–223. doi: 10.1101/lm.119806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maren S, Chang CH. Recent fear is resistant to extinction. Proc Natl Acad Sci USA. 2006;103:18020–18025. doi: 10.1073/pnas.0608398103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rothbaum BO, et al. Early intervention may prevent the development of posttraumatic stress disorder: a randomized pilot civilian study with modified prolonged exposure. Biol Psychiatry. 2012;72:957–963. doi: 10.1016/j.biopsych.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quirk GJ. Memory for extinction of conditioned fear is long-lasting and persists following spontaneous recovery. Learn Mem. 2002;9:402–407. doi: 10.1101/lm.49602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bouton ME, King DA. Contextual control of the extinction of conditioned fear: tests for the associative value of the context. J Exp Psychol Anim Behav Process. 1983;9:248–265. [PubMed] [Google Scholar]
- 63.Rescorla RA, Heth CD. Reinstatement of fear to an extinguished conditioned stimulus. J Exp Psychol Anim Behav Process. 1975;1:88–96. [PubMed] [Google Scholar]
- 64.Falls WA, Miserendino MJ, Davis M. Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci. 1992;12:854–863. doi: 10.1523/JNEUROSCI.12-03-00854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci. 2002;22:2343–2351. doi: 10.1523/JNEUROSCI.22-06-02343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Richardson R, Ledgerwood L, Cranney J. Facilitation of fear extinction by D-cycloserine: theoretical and clinical implications. Learn Mem. 2004;11:510–516. doi: 10.1101/lm.78204. [DOI] [PubMed] [Google Scholar]
- 67.Ressler KJ, et al. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61:1136–1144. doi: 10.1001/archpsyc.61.11.1136. [DOI] [PubMed] [Google Scholar]
- 68.Otto MW, et al. Efficacy of D-cycloserine for enhancing response to cognitive-behavior therapy for panic disorder. Biol Psychiatry. 2010;67:365–370. doi: 10.1016/j.biopsych.2009.07.036. [DOI] [PubMed] [Google Scholar]
- 69.Hofmann SG, et al. Augmentation of exposure therapy with D-cycloserine for social anxiety disorder. Arch Gen Psychiatry. 2006;63:298–304. doi: 10.1001/archpsyc.63.3.298. [DOI] [PubMed] [Google Scholar]
- 70.Kushner MG, et al. Cycloserine augmented exposure therapy for obsessive-compulsive disorder. Biol Psychiatry. 2007;62:835–838. doi: 10.1016/j.biopsych.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 71.de Kleine RA, Hendriks GJ, Kusters WJ, Broekman TG, van Minnen A. A randomized placebo-controlled trial of D-cycloserine to enhance exposure therapy for posttraumatic stress disorder. Biol Psychiatry. 2012;71:962–968. doi: 10.1016/j.biopsych.2012.02.033. [DOI] [PubMed] [Google Scholar]
- 72.Norberg MM, Krystal JH, Tolin DF. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63:1118–1126. doi: 10.1016/j.biopsych.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 73.Bontempo A, Panza KE, Bloch MH. Cycloserine augmentation of behavioral therapy for the treatment of anxiety disorders: a meta-analysis. J Clin Psychiatry. 2012;73:533–537. doi: 10.4088/JCP.11r07356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Andero R, Ressler KJ. Fear extinction and BDNF: translating animal models of PTSD to the clinic. Genes Brain Behav. 2012;11:503–512. doi: 10.1111/j.1601-183X.2012.00801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chhatwal JP, Stanek-Rattiner L, Davis M, Ressler KJ. Amygdala BDNF signaling is required for consolidation but not encoding of extinction. Nat Neurosci. 2006;9:870–872. doi: 10.1038/nn1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Andero R, et al. Effect of 7,8-dihydroxyfavone, a small-molecule TrkB agonist, on emotional learning. Am J Psychiatry. 2011;168:163–172. doi: 10.1176/appi.ajp.2010.10030326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Egan MF, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:257–269. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- 78.Soliman F, et al. A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science. 2010;327:863–866. doi: 10.1126/science.1181886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rakofsky JJ, Ressler KJ, Dunlop BW. BDNF function as a potential mediator of bipolar disorder and post-traumatic stress disorder comorbidity. Mol Psychiatry. 2012;17:22–35. doi: 10.1038/mp.2011.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Frustaci A, Pozzi G, Gianfagna F, Manzoli L, Boccia S. Meta-analysis of the brain-derived neurotrophic factor gene (BDNF) Val66Met polymorphism in anxiety disorders and anxiety-related personality traits. Neuropsychobiology. 2008;58:163–170. doi: 10.1159/000182892. [DOI] [PubMed] [Google Scholar]
- 81.Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- 82.Nader K, Hardt O. A single standard for memory: the case for reconsolidation. Nat Rev Neurosci. 2009;10:224–234. doi: 10.1038/nrn2590. [DOI] [PubMed] [Google Scholar]
- 83.Tronson NC, Taylor JR. Molecular mechanisms of memory reconsolidation. Nat Rev Neurosci. 2007;8:262–275. doi: 10.1038/nrn2090. [DOI] [PubMed] [Google Scholar]
- 84.Kindt M, Soeter M, Vervliet B. Beyond extinction: erasing human fear responses and preventing the return of fear. Nat Neurosci. 2009;12:256–258. doi: 10.1038/nn.2271. [DOI] [PubMed] [Google Scholar]
- 85.Brunet A, et al. Effect of post-retrieval propranolol on psychophysiologic responding during subsequent script-driven traumatic imagery in post-traumatic stress disorder. J Psychiatr Res. 2008;42:503–506. doi: 10.1016/j.jpsychires.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 86.Fiorenza NG, Sartor D, Myskiw JC, Izquierdo I. Treatment of fear memories: interactions between extinction and reconsolidation. An Acad Bras Cienc. 2011;83:1363–1372. doi: 10.1590/s0001-37652011000400023. [DOI] [PubMed] [Google Scholar]
- 87.Lin CH, Lee CC, Gean PW. Involvement of a calcineurin cascade in amygdala depotentiation and quenching of fear memory. Mol Pharmacol. 2003;63:44–52. doi: 10.1124/mol.63.1.44. [DOI] [PubMed] [Google Scholar]
- 88.Mao SC, Hsiao YH, Gean PW. Extinction training in conjunction with a partial agonist of the glycine site on the NMDA receptor erases memory trace. J Neurosci. 2006;26:8892–8899. doi: 10.1523/JNEUROSCI.0365-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Milekic MH, Alberini CM. Temporally graded requirement for protein synthesis following memory reactivation. Neuron. 2002;36:521–525. doi: 10.1016/s0896-6273(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 90.Suzuki A, et al. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci. 2004;24:4787–4795. doi: 10.1523/JNEUROSCI.5491-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Inda MC, Muravieva EV, Alberini CM. Memory retrieval and the passage of time: from reconsolidation and strengthening to extinction. J Neurosci. 2011;31:1635–1643. doi: 10.1523/JNEUROSCI.4736-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Duvarci S, Bauer EP, Pare D. The bed nucleus of the stria terminalis mediates inter-individual variations in anxiety and fear. J Neurosci. 2009;29:10357–10361. doi: 10.1523/JNEUROSCI.2119-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jovanovic T, Ressler KJ. How the neurocircuitry and genetics of fear inhibition may inform our understanding of PTSD. Am J Psychiatry. 2010;167:648–662. doi: 10.1176/appi.ajp.2009.09071074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yehuda R, LeDoux J. Response variation following trauma: a translational neuroscience approach to understanding PTSD. Neuron. 2007;56:19–32. doi: 10.1016/j.neuron.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 95.Monfils MH, Cowansage KK, Klann E, LeDoux JE. Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science. 2009;324:951–955. doi: 10.1126/science.1167975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Clem RL, Huganir RL. Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science. 2010;330:1108–1112. doi: 10.1126/science.1195298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schiller D, et al. Preventing the return of fear in humans using reconsolidation update mechanisms. Nature. 2010;463:49–53. doi: 10.1038/nature08637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chan WY, Leung HT, Westbrook RF, McNally GP. Effects of recent exposure to a conditioned stimulus on extinction of Pavlovian fear conditioning. Learn Mem. 2010;17:512–521. doi: 10.1101/lm.1912510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Davis M. The role of the amygdala in fear and anxiety. Annu Rev Neurosci. 1992;15:353–375. doi: 10.1146/annurev.ne.15.030192.002033. [DOI] [PubMed] [Google Scholar]