

Abstract
The ChlR1 DNA helicase is mutated in Warsaw breakage syndrome characterized by developmental anomalies, chromosomal breakage, and sister chromatid cohesion defects. However, the mechanism by which ChlR1 preserves genomic integrity is largely unknown. Here, we describe the roles of ChlR1 in DNA replication recovery. We show that ChlR1 depletion renders human cells highly sensitive to cisplatin; an interstrand-crosslinking agent that causes stalled replication forks. ChlR1 depletion also causes accumulation of DNA damage in response to cisplatin, leading to a significant delay in resolution of DNA damage. We also report that ChlR1-depleted cells display defects in the repair of double-strand breaks induced by the I-PpoI endonuclease and bleomycin. Furthermore, we demonstrate that ChlR1-depeleted cells show significant delays in replication recovery after cisplatin treatment. Taken together, our results indicate that ChlR1 plays an important role in efficient DNA repair during DNA replication, which may facilitate efficient establishment of sister chromatid cohesion.
Keywords: ChlR1 DNA helicase, DNA replication, DNA damage, replication recovery, sister chromatid cohesion, Warsaw breakage syndrome
INTRODUCTION
ChlR1, otherwise known as DDX11, Chl1, Ctf1, or Mcm12, is a DEAH/DEAD box-containing DNA helicase belonging to the FANCJ-like DNA helicase family [1]. The protein was first identified in a yeast genetic screen as CHL1/CTF1, whose deletion resulted in elevated levels of chromosomal loss or missegregation in the budding yeast Saccharomyces cerevisiae [2-4]. Subsequent studies in both budding and fission yeast demonstrated that deletion of chl1 results in premature sister chromatid separation, and that Chl1 genetically interacts with various factors involved in sister chromatid cohesion [5-8]. These factors include Ctf7/Eco1 and RFC-Ctf18, both of which have critical roles in S phase. Ctf7/Eco1 is an acetyltranferase responsible for acetylation of the cohesin subunit Smc3, and is required for cohesion establishment specifically during S phase [9-14]. RFC-Ctf18 is an alternative replication factor C complex involved in cohesion establishment, S-phase checkpoints, and replication fork stabilization [5, 7, 15-22]. These results suggest that ChlR1 plays a critical role during S phase to establish proper sister chromatid cohesion.
The functions of Chl1 appear to be conserved throughout evolution. RNAi-dependent downregulation of ChlR1 causes premature sister separation and a profound delay in mitotic progression in human cells [23-25]. It is also demonstrated that ChlR1 interacts with cohesin subunits, including Scc1, Smc1 and Smc3 [25]. Interestingly, a recent report found that the K879del mutation in ChlR1 is responsible for a cohesinopathy-related disease termed “Warsaw breakage syndrome” (WABS). The patient with WABS displays severe developmental defects, including microcephaly, growth retardation, and facial dysmorphy [26]. On the cellular level, the patient’s lymphocytes show combined phenotypes of Fanconi Anemia and the cohesinopathy Robert’s Syndrome, including abnormal chromosome separation or breakage, and elevated sensitivity to the interstrand-crosslinking (ICL) agent mitomycin C (MMC) and the topoisomerase inhibitor camptothecin [26]. Furthermore, ChlR1 knockout in mice results in embryonic lethality and aneuploidy due to the loss of sister chromatid cohesion [23]. These findings suggest that ChlR1 is required for normal mammalian development and preservation of genomic integrity.
Biochemical studies revealed that ChlR1 possesses a vital ATPase domain, as well as a carboxy-terminal HELICASE domain, both of which are crucial to its enzymatic function [4, 27]. ChlR1 has been shown to preferentially translocate on short single-stranded DNA [27]. Further in-vitro studies showed that ChlR1 interacts preferentially with forked duplex DNA, and efficiently unwinds the 5’ flap structure, a key intermediate of lagging strand processing [28]. Consistently, ChlR1 is able to stimulate the activity of the 5’ flap endonuclease, Fen1 [29]. Importantly, the WABS mutation abrogates ChlR1 helicase activity [28]. These results suggest that ChlR1’s helicase or unwinding activity is crucial to sister-chromatid cohesion and that ChlR1 plays an important role at the replication fork, coordinating lagging strand synthesis with sister chromatid cohesion.
Recent studies have also implicated the role of ChlR1-related proteins in DNA repair. In yeast, chl1 deletion renders cells sensitive to S-phase stressing agents and causes a decrease in the level of DNA damage-induced recombination [5, 30, 31]. In human cells, ChlR1 depletion causes a lower rate of sister chromatid exchange (SCE), which is an indication of a DNA repair process that utilizes sister-chromatids for homologous recombination (HR) [23]. A study in C. elegans showed that the deletion of a FANCJ/ChlR1 homologue affects the ability to resolve secondary structures during replication, a process possibly involving HR [32]. Furthermore, an in-vitro biochemical study showed that ChlR1 is able to unwind a substrate representing an early intermediate of HR, as well as a substrate representing G-quadruplex DNA [28]. Thus, ChlR1’s functions in DNA repair processes may play an important role in establishment of sister chromatid cohesion.
In the course of understanding how DNA replication is coordinated with sister chromatid cohesion, we previously demonstrated that the Timeless protein, which plays a central role in the maintenance of the replication fork [33], interacts with ChlR1 in human cells [24]. We also showed that Timeless depletion leads to cohesion defects, which was alleviated by overexpression of ChlR1 [24]. Furthermore, we also demonstrated in fission yeast that Chl1 overproduction suppresses DNA damage sensitivity of Swi1 (Timeless ortholog) deficient cells [5]. Considering that ChlR1/Chl1 also interacts with replication fork proteins such as PCNA and Fen1 [29, 34], our findings suggested that Timeless and ChlR1 work together at the replication fork to maintain replication fork structures and promote efficient sister chromatid cohesion. In this report, we demonstrate that ChlR1 is required for cellular tolerance to an ICL agent, cisplatin, which is known to cause stalled replication forks. ChlR1-depleted cells accumulate DNA damage and have defects in repair of double stranded breaks. In addition, we show that ChlR1 is required for efficient incorporation of a nucleotide analog after cisplatin treatment. These results indicate that ChlR1 plays an important role in efficient DNA repair during DNA replication.
MATERIALS AND METHODS
Cell culture
Unless indicated otherwise, HeLa and 293T cells were grown as described [24]. For the I-PpoI DNA damage repair assay, HeLa cells were cultured in DMEM supplemented with 10% fetal bovine serum (Gemini Bio-Products, Sacramento, CA), and 100 U/ml of penicillin and 100 μg/ml of streptomycin. After retroviral infection, cells were switched into phenol red-free DMEM supplemented with 10% charcoal-stripped fetal bovine serum (Gemini Bio-Products), 110 mg/ml sodium pyruvate, 2mM L-glutamine, 0.06 mg/ml of penicillin, and 0.1 mg/ml of streptomycin. Where indicated, cells were treated with cis-diammineplatinum(II) dichloride (cisplatin, P4394, Sigma, St Louis, MO) or bleomycin (bleomycin sulfate, 203401, EMD Millipore, Billerica, MA) at the indicated concentrations.
RNAi
shRNA mediated depletion of ChlR1 and Timeless was accomplished using the lentiviral vector pLKO.1-PURO encoding shRNA sequences for scrambled control (Addgene plasmid 1684, 5’-CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG-3’) [35], ChlR1/DDX11 (TRC clone ID: TRCN0000151936, 5’-CCCTTACATGATGAGAAAGATCTCGAGATCTTTCTCATCATGTAAGGG-3’), and Timeless (TRC clone ID: TRCN0000157211, 5’-GCCCACACTAACCATTGCATTCTCGAGAATGCAATGGTTAGTGTGGGC-3’) [36] knockdown (Open Biosystems, Huntsville, AL). VSV-G-psuedotyped lentivirus was generated in 293T cells using 10 μg plasmid DNA and packaging vectors as described [37]. HeLa cells were infected with viral particles overnight in medium containing 8 μg/mL polybrene, after which medium was replaced. Two days post-infection, infected cells were selected using 2 μg/mL puromycin for at least 4 days. Cells stably depleted of ChlR1 or Timeless were selected in the presence of puromycin and routinely monitored for ChlR1 levels by Western blotting.
shRNA-mediated ChlR1 depletion was also accomplished using the pGEM-based plasmid expressing an shRNA targeting ChlR1 (ChlR1-5) driven by U6 promoter as described in our previous reports [23, 38].
Transfection of siRNA duplexes or shRNA vectors was performed as described [23, 24, 36]. The sense strands of siRNA oligonucleotides for ChlR1-#1, ChlR1-#2, and ChlR1-#3 are described in our previous report [24].
Antibodies
Purified polyclonal ChlR1 (Hel1) and Timeless antibodies were previously generated [24, 39]. Antibodies to 53BP1 (BP13) and Chk2 (clone 7) were purchased from EMD Millipore (Billerica, MA); phospho-Chk2 (Thr68, #2661) from Cell Signaling (Danvers, MA); Tubulin (alpha-tubulin, B512) from Sigma-Aldrich (St. Louis, MO); and Actin (sc-1616) from Santa Cruz Biotechnology (Santa Cruz, CA).
Clonogenic survival assay
For clonogenic survival assays, HeLa cells permanently depleted of ChlR1 by infecting the lentivirus expressing shRNA were seeded into 6-well plates. After 8 hours, cells were treated with cisplatin at the indicated concentrations. In order for the cells to form colonies, cells were washed once with PBS and grown in drug-free medium for approximately 8 days. Colonies were rinsed once with PBS, fixed with 3.7% formaldehyde, and stained with 0.05% crystal violet (Sigma-Aldrich) for 30 min. Colonies of 50 cells or greater were counted, and the surviving fractions for each RNAi treatment group in triplicate represent the plating efficiency for each treatment divided by the plating efficiency of the corresponding untreated control.
Immunofluorescence
Immunofluorescence was performed as described [40] with minor modifications. Briefly, cells were fixed in 3.7% formaldehyde, permeabilized in 0.5% Triton X-100 (in 1 × PBS), and immunostained with the anti-53BP1 antibody diluted 1:1000 followed by incubation with Alexa-Fluor 568-conjugated anti-mouse IgG (Molecular Probes, Invitrogen, Carlsbad, CA) secondary antibody diluted 1:2000. To visualize DNA, cells were also stained by 4’, 6’-diamidino-2-phenylindole (DAPI, Invitrogen). Microscopic analyses were performed either using an Olympus PROVIS AX70 compound fluorescent microscope equipped with a Retiga EXi camera (QImaging, Surrey, BC, Canada) or using a Nikon E800 microscope equipped with a Nikon DXM1200 camera (Nikon, Tokyo, Japan). Nuclei and foci images were acquired with Ivision software (BioVision Technologies, Exton, PA) or with Nikon N1S-Elements software (Nikon).
EdU fluorescence microscopy
Assessment of cellular incorporation of a thymidine analog was performed as described [36, 41] except that 5-ethynyl-2’-deoxyuridine (EdU) was used in place of 5-bromo-2’-deoxyuridine (BrdU). Briefly, cells were grown on glass coverslips and treated with 1 mM hydroxyurea (HU) for 16 hours to halt replication fork progression. Cells were then washed once with 1 × PBS, and treated with 100 μM cisplatin or vehicle (dH2O) for 1 hour. Cells were washed twice with 1 × PBS, and returned to fresh medium without cisplatin. At the indicated times, cells were incubated with medium containing 15 μM EdU for 20 minutes. After EdU treatment, cells were fixed and prepared for detection of EdU using “Click-iT” Azide-488 EdU detection reaction cocktail as recommended by manufacturer’s instruction (Invitrogen). Microscopic analyses were performed as described above.
I-PpoI DNA damage repair assay
I-PpoI DNA damage repair assay was performed as described [42-44]. Cells depleted of ChlR1, Timeless, or scramble (control) were infected with retrovirus expressing HA-ER-I-PpoI [43, 45], which results in expression of estrogen receptor-I-PpoI fusion protein. Twelve hours post-infection, cells were switched into phenol red-free DMEM. Seventy-two hours post infection, cells were treated with 8 μM tamoxifen (Invitrogen) to induce ER-I-PpoI nuclear localization and double strand break generation at I-PpoI genomic DNA target sites on chromosome 1 (single site) and within the rDNA repeat genes [43, 44]. At the indicated times, cells were lysed in 300 μl of “Genomic DNA Isolation Buffer” (50 mM Tris-HCl pH 8.0, 50 mM EDTA, 10 mM NaCl, and 1% SDS). Lysates were sonicated at 20% duty for 20 seconds with 1-second pulse intervals, digested with 1.25 μl RNase (10 mg/ml) for 30 minutes at 65° C, and incubated with 1 μl Proteinase K (20 mg/ml) overnight at 37° C. DNA was precipitated with 3 volumes of 100% ethanol for 30 minutes and resuspended in 10% Chelex resin (BioRad) and boiled (at 100° C) for 10 minutes. Samples were spun briefly, and supernatants containing DNA were stored. DNA concentrations were measured using a NanoDrop spectrophotometer (ND 1000, Thermo Fisher Scientific) with accompanying software, and samples were equalized to the same concentration.
Real-time (RT) PCR reactions were run in triplicates using iQ SYBR Green Supermix (Quanta Biosciences, Gaithersburg, MD) and I-PpoI chromosome 1 target-site flanking primers (chrm1-flank-fwd: 5’-TCACTGAAGACTTGGTGGGA-3’ and chrm1-flank-rev: 5’-AAACCATACGTGGCAGAGTG-3’) [43]. As a control, RT-PCR was also performed using primers specific to a region adjacent to the I-PpoI target site (280 bp away from the cut site), but unaffected by the enzyme itself (chrom1ADJ-fwd: 5’-TGCTGCTTTTTCTTCTTCTCC-3’ and chrom1ADJ-rev: 5’-CTTCTTTCCCACCAAGTCTTC-3’) [43]. A threshold was set to obtain CT values from the cycle runs, ΔΔCT values were determined as previously described [43], and represented as “Percent damaged DNA”.
RESULTS
ChlR1-depleted cells are sensitive to cisplatin
WABS patient cells have a lower rate of survival and a higher rate of chromosomal breakage when treated with the interstrand-crosslinking (ICL) agent mitomycin C (MMC) [26]. These results suggest the role of ChlR1 in repair of ICL-induced DNA damage. To understand the function of ChlR1 in DNA repair during replication, we first investigated whether ChlR1 is required for cellular survival to cisplatin, a platinum-containing ICL inducing agent known to inhibit replication fork progression [46]. ChlR1 was stably downregulated in HeLa cells using shRNA-expressing lentivirus. As a control, we also depleted Timeless, via shRNA, since Timeless is required for replication recovery in response to DNA damage [24, 33]. As shown in Fig 1A, ChlR1 and Timeless shRNA-expressing lentiviruses effectively downregulated ChlR1 and Timeless, respectively. When treated with vehicle without cisplatin, ChlR1- or Timeless-depleted cells showed growth rates similar to control cells (Fig 1B). Cisplatin treatment caused a growth delay in control cells probably due to cisplatin-dependent DNA damage, although cell numbers were maintained throughout the experiment (Fig 1C). In contrast, the number of Timeless-depleted cells declined after cisplatin treatment, probably due to cell death (Fig 1C). Interestingly, cisplatin treatment also caused a significant decline of cell numbers in ChlR1-depleted cells (Fig 1C). Cisplatin sensitivity was also assessed by clonogenic survival assays. As shown in Fig 1D, the HeLa cells stably expressing ChlR1 shRNA (Fig 1A) are significantly more sensitive than control HeLa cells. Moreover, HeLa cells transiently transfected with a different ChlR1 shRNA vector (ChlR1 shRNA-5 [23, 38]) also showed considerable cisplatin sensitivity when compared to cells transfected with control shRNA (Fig 1E). These results indicate that ChlR1 is required for cellular tolerance to cisplatin, suggesting that ChlR1 is involved in recovery of cisplatin-induced ICLs.
Figure 1. ChlR1-depleted cells are sensitive to cisplatin.
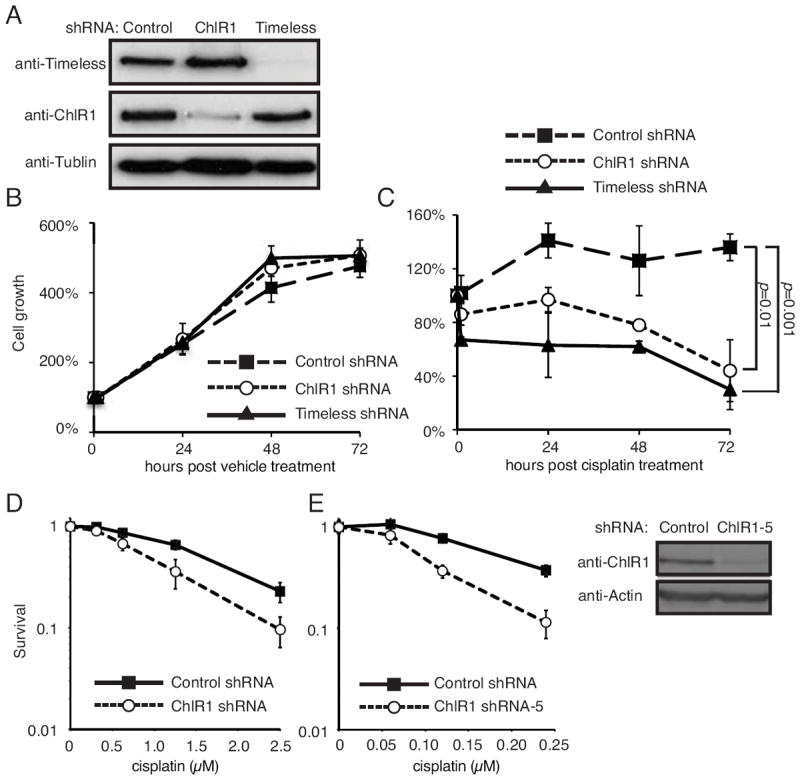
(A) HeLa cells were infected with lentivirus expressing the indicated shRNAs and selected in the presence of puromycin for stable expression of shRNAs. Western blotting results show that Timeless or ChlR1 was efficiently downregulated. (B, C) Cells infected with the indicated shRNA-expressing lentivirus were treated with vehicle (water in B) or 100 μM cisplatin (in C) for one hour and returned to fresh medium. Cells were counted at the indicated time in triplicate, and percentages of cell growth were calculated against the number of cells at the 0 h time point. The error bars represent standard deviation. (D) HeLa cells stably expressing shRNAs used above were incubated with the indicated concentrations of cisplatin for 1 hour. Cells were then washed and released into fresh medium for clonogenic survival assays as described in Material and Methods. (E) HeLa cells were transiently transfected with a plasmid vector expressing ChlR1 shRNA (ChlR1-5) and incubated with the indicated concentrations of cisplatin for 24 hours. Cells were washed and returned to growth medium for clonogenic survival assay as described in D. The error bars represent standard deviation obtained from three independent experiments. Western blotting results show efficient ChlR1 depletion by the vector expressing ChlR1 shRNA.
ChlR1-depleted cells accumulate DNA damage foci in response to cisplatin
The aforementioned results suggest a role for ChlR1 in DNA damage recovery. To address this possibility, we examined whether ChlR1-depleted cells accumulate DNA damage when treated with cisplatin. Levels of DNA damage in these cells were determined by immunofluorescence detection of 53BP1 in HeLa cells (Fig 2). 53BP1 is known to form DNA damage foci and is commonly used as a marker for DNA double-strand breaks (DSBs) [47, 48]. HeLa cells stably expressing ChlR1 shRNA (Fig 2A) were incubated with cisplatin for 1 hour, and then returned to medium without cisplatin. We noticed that ChlR1-depleted cells displayed higher numbers of 53BP1 foci even without cisplatin treatment (Fig 2B and 2C), indicative of spontaneous DSBs in the absence of genotoxic agents. When cells were treated with cisplatin, foci numbers initially increased in both ChlR1-downregulated and control cells (Fig 2D, 1 and 3 hours, Fig S1). In control cells, foci levels peaked around 3-5 hours past treatment and receded by 7 hours (Fig 2D, 5 and 7 hours, Fig S1). However, in ChlR1 downregulated cells, foci numbers continued to increase throughout the time-course. At 5 and 7 hours post-treatment, ChlR1-downregulated cells display a significantly higher level of 53BP1 foci than control cells in response to cisplatin (Fig 2B bottom panels and Fig 2D, 7 hours). These results suggest that ChlR1-depleted cells are less capable of repairing DNA damage caused by cisplatin. To confirm the specificity of ChlR1 downregulation, we transiently transfected HeLa cells with siRNA oligonucleotides targeting control (luciferase) or ChlR1 (Fig 2E). Cells were then treated with cisplatin for 1 hour and returned to fresh medium for 7 hours without cisplatin. Importantly, all three ChlR1 siRNAs significantly increased the number of 53BP1 foci in HeLa cells in response to an ICL-inducing agent, cisplatin. Considering that ICLs induce a replication block leading to DNA damage, our findings may suggest that ChlR1 has an important role in preventing or resolving DNA damage at the replication fork.
Figure 2. ChlR1 depletion leads to accumulation of 53BP1 foci.
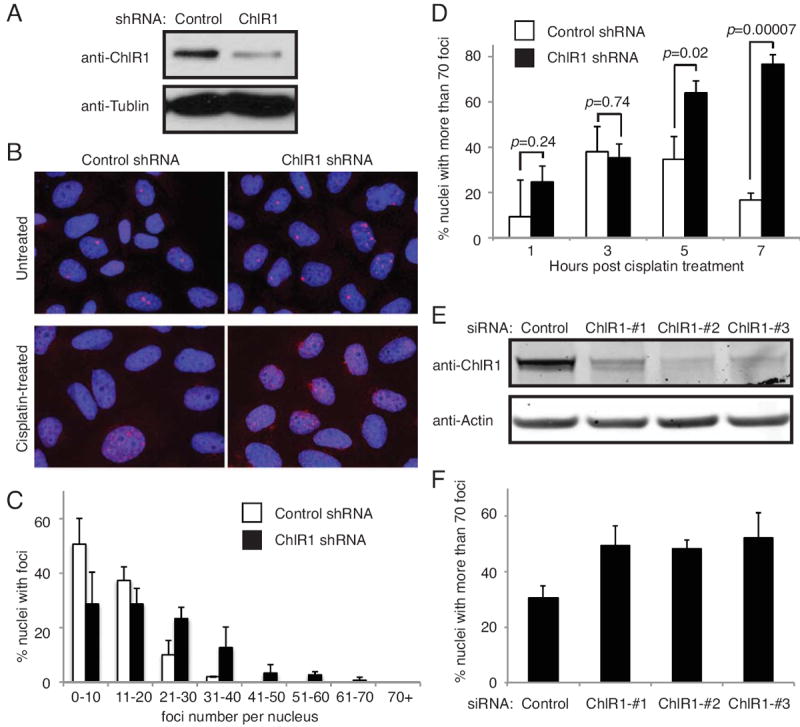
(A) Western blot analysis of HeLa cells stably expressing the indicated shRNAs. (B) Representative images of 53BP1 foci in control and ChlR1-depleted cell nuclei. In the top panel, cells infected with the indicated shRNAs were grown on coverslips and stained with the 53BP1 antibody (red). In the bottom panel, cells were treated with cisplatin for 1 hour and returned to fresh medium for 7 hours. Cells were then stained with the 53BP1 antibody (red). DNA was costained with DAPI (blue). The merged images of DNA and 53BP1 are shown. (C) Quantification of 53BP foci in untreated cells. 53BP1 foci were counted in at least 50 nuclei in each experiment. The number of 53BP1 foci in each nucleus was counted and scored within a category of foci number per nucleus. For each category, an average percentage of nuclei and standard deviation were determined. (D) Quantification of 53BP1 foci in cisplatin treated cells was performed as in C. The number of 53BP1 foci in each nucleus was counted at the indicated time after 1 hour of cisplatin treatment. Percent nuclei with more than 70 foci is shown. (E) HeLa cells were transiently transfected with the indicated siRNA oligonucleotides. Cells were collected 4 days after transfection, and levels of ChlR1 were monitored by Western blotting, using antibody against ChlR1. (F) siRNA-treated cells used in E were incubated with cisplatin for 1 hour and returned to fresh medium without cisplatin for 7 hours. Cells were then stained with the 53BP1 antibody, and cells with more than 70 53BP foci were scored in at least 100 nuclei. Data were obtained from four independent experiments and error bars represent standard deviation.
Inefficient double-strand break repair in ChlR1-depleted cells
Thus far, our data suggests that ChlR1 plays a role in repair of DNA damage. DNA ICL repair involves removal of the crosslinked DNA, which is thought to result in the generation of a double-strand break (DSB) followed by homologous recombination (HR) [49]. To assess ChlR1’s role in DSB repair, ChlR1 was stably downregulated in HeLa cells via shRNA-expressing lentivirus (Fig 3A, Fig S2B). As a control, Timeless was also stably downregulated in HeLa cells (Fig 3A, Fig S2A). These cells were then infected with retrovirus containing HA-ER-I-PpoI, which results in expression of estrogen receptor (ER) fused to the eukaryotic homing endonuclease I-PpoI [43, 45] [44]. Seventy-two hours post-infection, cells were treated with tamoxifen, which induces ER-I-PpoI relocalization from the cytosol to the nucleus. I-PpoI then introduces DSBs at defined genomic sites, promoting DSB repair. I-PpoI induction is reported to cause activation of the ATM-dependent checkpoint pathway [43]. Consistently, we observed increased phosphorylation of an ATM target, Chk2, in response to tamoxifen (Fig 3A, Fig S2C). In control cells, we observed a sharp increase of I-PpoI-dependent DNA damage in 2 h, which decreased rapidly at 4 h. There was then a fluctuation in the level of damaged DNA at 6, 8 and 10 h time point, although DSBs never reached as high as the initial peak (Fig 3B). This fluctuation is similar to those seen by Berkovich and colleagues [43]. In Timeless-depleted cells, DNA damage increased at a similar rate as control cells. However, DNA damage continued to increase until 4 hours after tamoxifen addition, and gradually decreased during the course of the experiment (Fig 3B). In addition, levels of DNA damage in Timeless-depleted cells are consistently higher over the time course, when compared to control cells (Fig 3B). In ChlR1-depleted cells, damage levels peaked 2 hours after induction, similar to control cells. Interestingly, although damage levels subsided, the level of DNA damage in ChlR1-depleted cells remained consistently higher than in control cells throughout the experiment (Fig 3B). These results suggest that Timeless- and ChlR1-depleted cells have defects in efficient repair of DSBs, leading to accumulation of DNA damage. Consistently, pChk2 levels are elevated in Timeless and ChlR1-depleted cells when compared to control cells (Fig 3A, Fig S2C).
Figure 3. ChlR1 is required for efficient DSB repair.
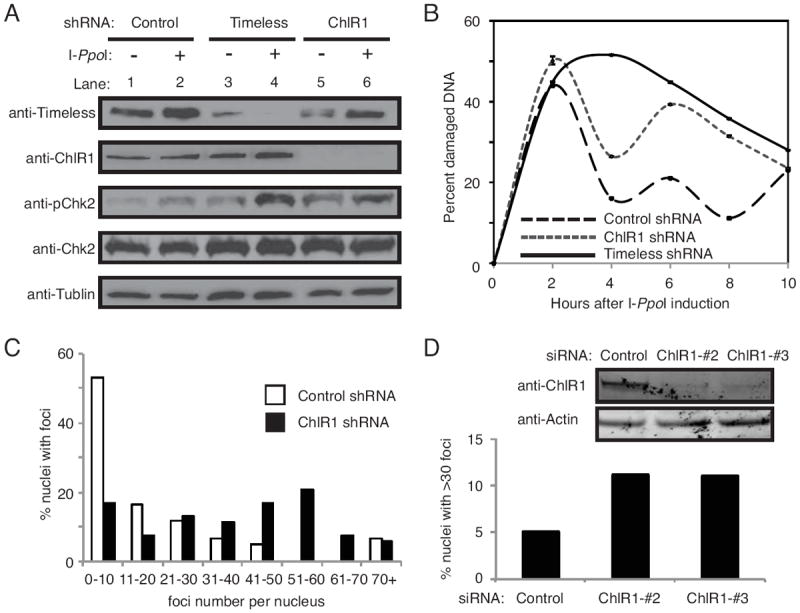
(A) Western blot analysis of cell extracts prepared from HeLa cells stably expressing the indicated shRNAs. Timeless or ChlR1 was efficiently downregulated. Cell extracts were collected before tamoxifen treatment (-), and 8 hours after tamoxifen treatment (+) to assess activation of DNA damage response by I-PpoI nuclear relocalization. Tamoxifen-dependent induction of I-PpoI induced phosphorylation of Chk2 (pChk2). (B) Cells were collected at the indicated times after I-PpoI induction, and genomic DNA was prepared. Triplicate real-time PCR reactions were run by amplifying the I-PpoI target site on chromosome 1, along with a site adjacent to the I-PpoI target site for normalization. ΔΔCT values were determined, and represented as the percentage of damaged DNA at the I-PpoI target. Error bars represent standard deviations. (C) HeLa cells stably expressing the indicated shRNA were treated with 20 μg/ml of bleomycin for 4 hours. Cells were washed, returned to fresh medium without bleomycin for 18 hours, and stained with the 53BP1 antibody. Quantification of 53BP1 foci in these cells were performed as described in Fig 2C. (D) HeLa cells were transiently transfected with the indicated siRNA oligonucleotides and treated with 3 μg/ml bleomycin for 1 h. Cells were then washed, returned to fresh medium for 7 hours, and stained with the 53BP1 antibody. Percent nuclei with more than thirty 53BP1 foci is shown. Representative results of repeat experiments are shown.
To further confirm the role of ChlR1 in DNA repair, HeLa cells stably expressing ChlR1 shRNA were treated with bleomycin, a radiomimetic drug that causes DSBs. After bleomycin treatment, cells were washed and released into fresh medium without bleomycin. As shown in Fig 3C, ChlR1-depleted cells displayed more 53BP1 foci when compared to control cells. Moreover, we transiently transfected HeLa cells with ChlR1-tageting siRNA oligonucleotides and found that ChlR1 downregulation causes nuclear accumulation of 53BP1 foci (Fig 3D), further supporting an idea that ChlR1 is involved in repair of DSBs.
ChlR1 is involved in recovery of DNA replication in response to cisplatin
ChlR1 interacts with replication fork proteins [24, 29, 34] and appears to play a role in DNA damage recovery (Fig 1 - 3). Therefore, we sought to determine whether the loss of ChlR1 affects replication recovery after DNA damage. Cells were incubated with hydroxyurea (HU) to induce replication fork arrest. Cells were then treated with cisplatin for 1 hour to induce DNA damage at the replication fork, and then returned to fresh medium. To monitor replication recovery, cells were treated with the thymidine analog EdU, at various time points after cisplatin treatment, to allow cells to incorporate EdU into replicated DNA. After EdU treatment, cells were fixed and prepared for EdU detection by fluorescence microscopy. When cells were treated with vehicle, ChlR1-depleted cells incorporated EdU at a slightly slower rate than their control counterparts over the time-course (Fig 4B, 4C). More importantly, when treated with cisplatin, the rate of EdU incorporation in ChlR1-depleted cells was significantly slower (Fig 4B, 4C). Considering that cisplatin introduces ICLs on the template DNA, our results suggest that ChlR1 may be needed to repair the damaged DNA before the replication fork can move forward. Taken together with our results on damage recovery (Fig 2 - 4), our findings indicate an important role for ChlR1 at the replication fork to maintain genomic integrity.
Figure 4. ChlR1 depletion negatively affects resumption of replication forks.
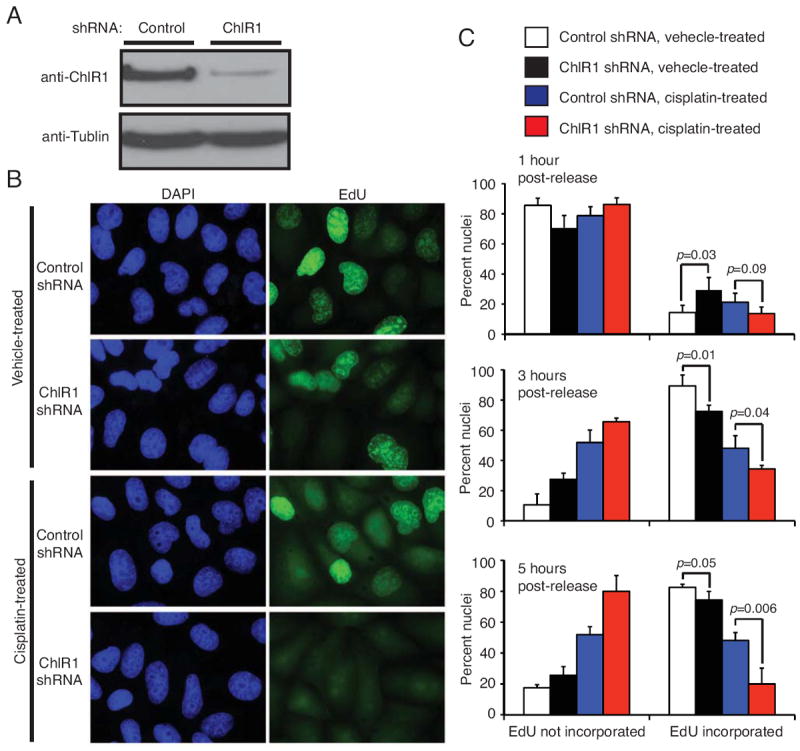
(A) HeLa cells were infected by lentivirus expressing the indicated shRNA. Western analysis shows that ChlR1 was efficiently downregulated. (B) Representative images of EdU incorporation. Cells were treated with 1 mM HU for 16 hours to arrest replication forks. Cells were then released into media containing vehicle (water) or 100 μM cisplatin for 1 hour. At the indicated times after vehicle or cisplatin treatment, cells were incubated with 15 μM EdU for 15 minutes and fixed. Representative images of control or ChlR1-depleted cells incubated with EdU five hours after vehicle or cisplatin treatment are shown. (C) Quantification of levels of nuclear EdU incorporation at 1, 3, and 5 hours after vehicle or cisplatin treatment in control vs. ChlR1-depleted cells. The nuclei were scored into 2 different categories: EdU not incorporated and EdU incorporated. Forty nuclei were counted for each experiment. For each category, an average percentage of nuclei and standard deviation were calculated. Error bars represent standard deviations.
DISCUSSION
In this report, we have described the role of the human ChlR1 DNA helicase in DNA damage recovery. We showed that ChlR1-depleted cells display elevated sensitivity to cisplatin and accumulation of DNA damage represented by 53BP1 foci (Fig 1 and 2). Importantly, ChlR1 depletion led to a significant delay in the resolution of 53BP1 foci in response to cisplatin, an ICL agent that causes stalled replication forks (Fig 2). It is thought that repair of ICL involves the generation of DSBs, which leads to homologous recombination (HR)-driven repair at the replication fork [49]. Therefore, our results suggest that ChlR1 plays an important role in DSB repair at the replication fork. Consistently, we demonstrated that ChlR1-depleted cells have defects in efficient repair of I-PpoI and bleomycin induced DSBs (Fig 3), further supporting the role of ChlR1 in DSB repair processes. Interestingly, it has been reported that SCE rate is reduced in ChlR1-depleted cells in response to a replication fork-damaging agent [23]. Since SCE is the outcome of HR, it is highly possible that ChlR1 is involved in HR. This idea is supported by the fact that ChlR1 enhances unwinding of D-loop DNA structures (HR intermediates) to facilitate the process of HR [28]. Considering that ChlR1 interacts with several replication fork proteins including PCNA, Fen1 and Timeless [24, 29, 34], it is straightforward to suggest that ChlR1’s functions are required at the replication fork during S-phase. Consistently, cells deficient for ChlR1/Chl1 are also sensitive to MMC, MMS, and CPT, all of which cause DNA breaks during replication [5, 26, 30, 31], further supporting a notion that ChlR1 is involved in DNA repair at the replication fork.
Previous studies reported that ChlR1 enhances activity of Fen1, a critical enzyme for Okazaki fragment processing during lagging strand DNA synthesis [29]. This is consistent with the notion that ChlR1 works in S phase. Fen1 is known for its role in RNA primer removal during Okazaki fragment processing and long-patch base excision repair (BER) [50, 51], but it is also reported to interact with WRN for efficient fork cleavage for resolution of stalled replication fork [52]. Consistently, it has been shown that Fen1 is important for fork re-initiation [41]. We also showed that ChlR1 is important for recovery of fork after cisplatin treatment (Fig 4). These results suggest that ChlR1 might assist Fen1 to promote efficient fork recovery after ICL-induced DNA damage.
How might ChlR1’s function in DNA repair affect sister chromatid cohesion? A number of studies demonstrated that ChlR1-related proteins are involved in sister chromatid cohesion, which is known to be established during S phase near the replication fork [5-8, 23-25]. How DNA replication is coordinated with sister chromatid cohesion is unknown. Considering that Fen1 activity is stimulated by ChlR1 in vitro [29], it is possible that lagging-strand processing may affect the efficiency of cohesion. Consistently, Fen1 depletion has been shown to cause sister chromatid cohesion defects [29]. In addition, the length of duplex DNA displaced by ChlR1 helicase was shown to be increased by RFC-Ctf18, an alternative replication factor C involved in sister chromatid cohesion [29]. These findings suggest that ChlR1 supports sister chromatid cohesion by promoting efficient lagging strand synthesis. This is consistent with the idea that delayed lagging strand processing would cause a long stretch of single stranded DNA, which may impede migration of the replication fork through the cohesin ring. It is possible that a large loop structure generated at the fork causes disruption of the ring [16, 29, 53]. Interestingly, we have previously shown that Timeless, which interacts with ChlR1, is required for stable association of cohesin subunits with chromatin [24]. Timeless is also required for maintaining the level of chromatin-bound ChlR1[24], and Timeless downregulation causes accumulation of ssDNA [54]. Importantly, yeast genetic studies show that Swi1 (Timeless homolog) is required for coordinating leading- and lagging-strand DNA synthesis to prevent ssDNA accumulation [55-57]. Thus, we suggest that efficient lagging-strand processing may have a critical role in establishment of sister chromatid cohesion. However, we cannot rule out the possibility that DNA repair defects in ChlR1-depleted cells are downstream consequences of the loss of cohesion establishment. Indeed, it has been suggested that cohesin rings form at sites of DNA damage, assisting in the repair process [58, 59]. In addition, ChlR1 has been shown to interact with cohesin subunits [25]. Therefore, in this scenario, ChlR1-dependent cohesion establishment might mediate accurate DNA repair at the replication forks. Further studies addressing the role ChlR1 in cohesin recruitment would clarify how, mechanistically, ChlR1 promotes sister chromatid cohesion.
In conclusion, we have shown that ChlR1 depletion causes cellular sensitivity to cisplatin, an ICL agent that causes replication fork stalling. Consistently, ChlR1-depleted cells accumulate DNA damage and show defects in efficient DSB repair. In addition, we have shown that ChlR1 is involved in recovery of stalled or damage replication forks in response to cisplatin. Thus, our findings suggest that ChlR1 is required for efficient repair of DSB at the replication fork. It is important to note that ChlR1 is thought to be involved in lagging-strand processing. Such a function of ChlR1 may contribute to the establishment of sister chromatid cohesion at the replication fork. Further investigation warrants a better understanding of the mechanistic link between DNA replication and cohesion processes.
Supplementary Material
Highlights.
ChlR1 depletion causes cellular sensitivity to cisplatin, an ICL agent.
ChlR1 depletion causes accumulation of DNA damage in response to cisplatin.
ChlR1-depleted cells display defects in DSB repair.
ChlR1 is involved in replication recovery after cisplatin treatment.
ChlR1 is involved in efficient DNA repair during DNA replication.
Acknowledgments
We thank Drs. Jane Clifford, Naoaki Fujii, Bradford Jameson and Mauricio Reginato for reagents, comments, advice, and continuous support. We also thank Dr. Michael Kastan for providing reagents for the I-PpoI assay. Members of the Noguchi laboratory are thanked for their support and encouragement. This work was supported by National Institutes of Health (R01GM077604 to E.N) and American Cancer Society (#RSG CDD-120969 to A.I).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
LIST OF REFERENCES
- 1.Wu Y, Suhasini AN, Brosh RM., Jr Welcome the family of FANCJ-like helicases to the block of genome stability maintenance proteins. Cell Mol Life Sci. 2009;66:1209–1222. doi: 10.1007/s00018-008-8580-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerring SL, Spencer F, Hieter P. The CHL 1 (CTF 1) gene product of Saccharomyces cerevisiae is important for chromosome transmission and normal cell cycle progression in G2/M. Embo J. 1990;9:4347–4358. doi: 10.1002/j.1460-2075.1990.tb07884.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haber JE. Bisexual mating behavior in a diploid of Saccharomyces cerevisiae: evidence for genetically controlled non-random chromosome loss during vegetative growth. Genetics. 1974;78:843–858. doi: 10.1093/genetics/78.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holloway SL. CHL1 is a nuclear protein with an essential ATP binding site that exhibits a size-dependent effect on chromosome segregation. Nucleic Acids Res. 2000;28:3056–3064. doi: 10.1093/nar/28.16.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ansbach AB, Noguchi C, Klansek IW, Heidlebaugh M, Nakamura TM, Noguchi E. RFCCtf18 and the Swi1-Swi3 complex function in separate and redundant pathways required for the stabilization of replication forks to facilitate sister chromatid cohesion in Schizosaccharomyces pombe. Mol Biol Cell. 2008;19:595–607. doi: 10.1091/mbc.E07-06-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mayer ML, Pot I, Chang M, Xu H, Aneliunas V, Kwok T, Newitt R, Aebersold R, Boone C, Brown GW, Hieter P. Identification of protein complexes required for efficient sister chromatid cohesion. Molecular biology of the cell. 2004;15:1736–1745. doi: 10.1091/mbc.E03-08-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petronczki M, Chwalla B, Siomos MF, Yokobayashi S, Helmhart W, Deutschbauer AM, Davis RW, Watanabe Y, Nasmyth K. Sister-chromatid cohesion mediated by the alternative RF-CCtf18/Dcc1/Ctf8, the helicase Chl1 and the polymerase-alpha-associated protein Ctf4 is essential for chromatid disjunction during meiosis II. J Cell Sci. 2004;117:3547–3559. doi: 10.1242/jcs.01231. [DOI] [PubMed] [Google Scholar]
- 8.Skibbens RV. Chl1p, a DNA helicase-like protein in budding yeast, functions in sister-chromatid cohesion. Genetics. 2004;166:33–42. doi: 10.1534/genetics.166.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ivanov D, Schleiffer A, Eisenhaber F, Mechtler K, Haering CH, Nasmyth K. Eco1 is a novel acetyltransferase that can acetylate proteins involved in cohesion. Curr Biol. 2002;12:323–328. doi: 10.1016/s0960-9822(02)00681-4. [DOI] [PubMed] [Google Scholar]
- 10.Rolef Ben-Shahar T, Heeger S, Lehane C, East P, Flynn H, Skehel M, Uhlmann F. Eco1-dependent cohesin acetylation during establishment of sister chromatid cohesion. Science. 2008;321:563–566. doi: 10.1126/science.1157774. [DOI] [PubMed] [Google Scholar]
- 11.Skibbens RV, Corson LB, Koshland D, Hieter P. Ctf7p is essential for sister chromatid cohesion and links mitotic chromosome structure to the DNA replication machinery. Genes Dev. 1999;13:307–319. doi: 10.1101/gad.13.3.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toth A, Ciosk R, Uhlmann F, Galova M, Schleiffer A, Nasmyth K. Yeast cohesin complex requires a conserved protein, Eco1p(Ctf7), to establish cohesion between sister chromatids during DNA replication. Genes Dev. 1999;13:320–333. doi: 10.1101/gad.13.3.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Unal E, Heidinger-Pauli JM, Kim W, Guacci V, Onn I, Gygi SP, Koshland DE. A molecular determinant for the establishment of sister chromatid cohesion. Science. 2008;321:566–569. doi: 10.1126/science.1157880. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J, Shi X, Li Y, Kim BJ, Jia J, Huang Z, Yang T, Fu X, Jung SY, Wang Y, Zhang P, Kim ST, Pan X, Qin J. Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol Cell. 2008;31:143–151. doi: 10.1016/j.molcel.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Bermudez VP, Maniwa Y, Tappin I, Ozato K, Yokomori K, Hurwitz J. The alternative Ctf18-Dcc1-Ctf8-replication factor C complex required for sister chromatid cohesion loads proliferating cell nuclear antigen onto DNA. Proc Natl Acad Sci U S A. 2003;100:10237–10242. doi: 10.1073/pnas.1434308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bylund GO, Burgers PM. Replication protein A-directed unloading of PCNA by the Ctf18 cohesion establishment complex. Mol Cell Biol. 2005;25:5445–5455. doi: 10.1128/MCB.25.13.5445-5455.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanna JS, Kroll ES, Lundblad V, Spencer FA. Saccharomyces cerevisiae CTF18 and CTF4 are required for sister chromatid cohesion. Mol Cell Biol. 2001;21:3144–3158. doi: 10.1128/MCB.21.9.3144-3158.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mayer ML, Gygi SP, Aebersold R, Hieter P. Identification of RFC(Ctf18p, Ctf8p, Dcc1p): an alternative RFC complex required for sister chromatid cohesion in S. cerevisiae. Mol Cell. 2001;7:959–970. doi: 10.1016/s1097-2765(01)00254-4. [DOI] [PubMed] [Google Scholar]
- 19.Merkle CJ, Karnitz LM, Henry-Sanchez JT, Chen J. Cloning and characterization of hCTF18, hCTF8, and hDCC1. Human homologs of a Saccharomyces cerevisiae complex involved in sister chromatid cohesion establishment. J Biol Chem. 2003;278:30051–30056. doi: 10.1074/jbc.M211591200. [DOI] [PubMed] [Google Scholar]
- 20.Naiki T, Kondo T, Nakada D, Matsumoto K, Sugimoto K. Chl12 (Ctf18) forms a novel replication factor C-related complex and functions redundantly with Rad24 in the DNA replication checkpoint pathway. Mol Cell Biol. 2001;21:5838–5845. doi: 10.1128/MCB.21.17.5838-5845.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohta S, Shiomi Y, Sugimoto K, Obuse C, Tsurimoto T. A proteomics approach to identify proliferating cell nuclear antigen (PCNA)-binding proteins in human cell lysates. Identification of the human CHL12/RFCs2-5 complex as a novel PCNA-binding protein. J Biol Chem. 2002;277:40362–40367. doi: 10.1074/jbc.M206194200. [DOI] [PubMed] [Google Scholar]
- 22.Shiomi Y, Shinozaki A, Sugimoto K, Usukura J, Obuse C, Tsurimoto T. The reconstituted human Chl12-RFC complex functions as a second PCNA loader. Genes Cells. 2004;9:279–290. doi: 10.1111/j.1356-9597.2004.00724.x. [DOI] [PubMed] [Google Scholar]
- 23.Inoue A, Li T, Roby SK, Valentine MB, Inoue M, Boyd K, Kidd VJ, Lahti JM. Loss of ChlR1 helicase in mouse causes lethality due to the accumulation of aneuploid cells generated by cohesion defects and placental malformation. Cell Cycle. 2007;6:1646–1654. doi: 10.4161/cc.6.13.4411. [DOI] [PubMed] [Google Scholar]
- 24.Leman AR, Noguchi C, Lee CY, Noguchi E. Human Timeless and Tipin stabilize replication forks and facilitate sister-chromatid cohesion. J Cell Sci. 2010;123:660–670. doi: 10.1242/jcs.057984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parish JL, Rosa J, Wang X, Lahti JM, Doxsey SJ, Androphy EJ. The DNA helicase ChlR1 is required for sister chromatid cohesion in mammalian cells. J Cell Sci. 2006;119:4857–4865. doi: 10.1242/jcs.03262. [DOI] [PubMed] [Google Scholar]
- 26.van der Lelij P, Chrzanowska KH, Godthelp BC, Rooimans MA, Oostra AB, Stumm M, Zdzienicka MZ, Joenje H, de Winter JP. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am J Hum Genet. 2010;86:262–266. doi: 10.1016/j.ajhg.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirota Y, Lahti JM. Characterization of the enzymatic activity of hChlR1, a novel human DNA helicase. Nucleic Acids Res. 2000;28:917–924. doi: 10.1093/nar/28.4.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Y, Sommers JA, Khan I, de Winter JP, Brosh RM., Jr Biochemical characterization of Warsaw breakage syndrome helicase. J Biol Chem. 2012;287:1007–1021. doi: 10.1074/jbc.M111.276022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farina A, Shin JH, Kim DH, Bermudez VP, Kelman Z, Seo YS, Hurwitz J. Studies with the human cohesin establishment factor, ChlR1. Association of ChlR1 with Ctf18-RFC and Fen1. J Biol Chem. 2008;283:20925–20936. doi: 10.1074/jbc.M802696200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laha S, Das SP, Hajra S, Sau S, Sinha P. The budding yeast protein Chl1p is required to preserve genome integrity upon DNA damage in S-phase. Nucleic Acids Res. 2006;34:5880–5891. doi: 10.1093/nar/gkl749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogiwara H, Ui A, Lai MS, Enomoto T, Seki M. Chl1 and Ctf4 are required for damage-induced recombinations. Biochem Biophys Res Commun. 2007;354:222–226. doi: 10.1016/j.bbrc.2006.12.185. [DOI] [PubMed] [Google Scholar]
- 32.Chung G, O’Neil NJ, Rose AM. CHL-1 provides an essential function affecting cell proliferation and chromosome stability in Caenorhabditis elegans. DNA Repair (Amst) 2011;10:1174–1182. doi: 10.1016/j.dnarep.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 33.Leman AR, Noguchi E. Local and global functions of Timeless and Tipin in replication fork protection. Cell Cycle. 2012;11:3945–3955. doi: 10.4161/cc.21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moldovan GL, Pfander B, Jentsch S. PCNA controls establishment of sister chromatid cohesion during S phase. Mol Cell. 2006;23:723–732. doi: 10.1016/j.molcel.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 35.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 36.Leman AR, Dheekollu J, Deng Z, Lee SW, Das MM, Lieberman PM, Noguchi E. Timeless preserves telomere length by promoting efficient DNA replication through human telomeres. Cell Cycle. 2012;11:2337–2347. doi: 10.4161/cc.20810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Zhang M, Ihrig MM, McManus MT, Gertler FB, Scott ML, Van Parijs L. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- 38.Inoue A, Hyle J, Lechner MS, Lahti JM. Mammalian ChlR1 has a role in heterochromatin organization. Exp Cell Res. 2011;317:2522–2535. doi: 10.1016/j.yexcr.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amann J, Kidd VJ, Lahti JM. Characterization of putative human homologues of the yeast chromosome transmission fidelity gene, CHL1. J Biol Chem. 1997;272:3823–3832. doi: 10.1074/jbc.272.6.3823. [DOI] [PubMed] [Google Scholar]
- 40.Douglas P, Zhong J, Ye R, Moorhead GB, Xu X, Lees-Miller SP. Protein phosphatase 6 interacts with the DNA-dependent protein kinase catalytic subunit and dephosphorylates gamma-H2AX. Mol Cell Biol. 2010;30:1368–1381. doi: 10.1128/MCB.00741-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saharia A, Teasley DC, Duxin JP, Dao B, Chiappinelli KB, Stewart SA. FEN1 ensures telomere stability by facilitating replication fork re-initiation. J Biol Chem. 2010;285:27057–27066. doi: 10.1074/jbc.M110.112276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beishline K, Kelly CM, Olofsson BA, Koduri S, Emrich J, Greenberg RA, Azizkhan-Clifford J. Sp1 facilitates DNA double-strand break repair through a nontranscriptional mechanism. Mol Cell Biol. 2012;32:3790–3799. doi: 10.1128/MCB.00049-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berkovich E, Monnat RJ, Jr, Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007;9:683–690. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- 44.Berkovich E, Monnat RJ, Jr, Kastan MB. Assessment of protein dynamics and DNA repair following generation of DNA double-strand breaks at defined genomic sites. Nat Protoc. 2008;3:915–922. doi: 10.1038/nprot.2008.54. [DOI] [PubMed] [Google Scholar]
- 45.Monnat RJ, Jr, Hackmann AF, Cantrell MA. Generation of highly site-specific DNA double-strand breaks in human cells by the homing endonucleases I-PpoI and I-CreI. Biochem Biophys Res Commun. 1999;255:88–93. doi: 10.1006/bbrc.1999.0152. [DOI] [PubMed] [Google Scholar]
- 46.Henry-Mowatt J, Jackson D, Masson JY, Johnson PA, Clements PM, Benson FE, Thompson LH, Takeda S, West SC, Caldecott KW. XRCC3 and Rad51 modulate replication fork progression on damaged vertebrate chromosomes. Mol Cell. 2003;11:1109–1117. doi: 10.1016/s1097-2765(03)00132-1. [DOI] [PubMed] [Google Scholar]
- 47.Anderson L, Henderson C, Adachi Y. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol. 2001;21:1719–1729. doi: 10.1128/MCB.21.5.1719-1729.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 53BP1, a mediator of the DNA damage checkpoint. Science. 2002;298:1435–1438. doi: 10.1126/science.1076182. [DOI] [PubMed] [Google Scholar]
- 49.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shen B, Singh P, Liu R, Qiu J, Zheng L, Finger LD, Alas S. Multiple but dissectible functions of FEN-1 nucleases in nucleic acid processing, genome stability and diseases. Bioessays. 2005;27:717–729. doi: 10.1002/bies.20255. [DOI] [PubMed] [Google Scholar]
- 51.Liu Y, Kao HI, Bambara RA. Flap endonuclease 1: a central component of DNA metabolism. Annu Rev Biochem. 2004;73:589–615. doi: 10.1146/annurev.biochem.73.012803.092453. [DOI] [PubMed] [Google Scholar]
- 52.Sharma S, Otterlei M, Sommers JA, Driscoll HC, Dianov GL, Kao HI, Bambara RA, Brosh RM., Jr WRN helicase and FEN-1 form a complex upon replication arrest and together process branchmigrating DNA structures associated with the replication fork. Mol Biol Cell. 2004;15:734–750. doi: 10.1091/mbc.E03-08-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lengronne A, McIntyre J, Katou Y, Kanoh Y, Hopfner KP, Shirahige K, Uhlmann F. Establishment of sister chromatid cohesion at the S. cerevisiae replication fork. Mol Cell. 2006;23:787–799. doi: 10.1016/j.molcel.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 54.Smith KD, Fu MA, Brown EJ. Tim-Tipin dysfunction creates an indispensible reliance on the ATR-Chk1 pathway for continued DNA synthesis. J Cell Biol. 2009;187:15–23. doi: 10.1083/jcb.200905006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dalgaard JZ, Klar AJ. swi1 and swi3 perform imprinting, pausing, and termination of DNA replication in S. pombe. Cell. 2000;102:745–751. doi: 10.1016/s0092-8674(00)00063-5. [DOI] [PubMed] [Google Scholar]
- 56.Noguchi E, Noguchi C, McDonald WH, Yates JR, 3rd, Russell P. Swi1 and Swi3 are components of a replication fork protection complex in fission yeast. Mol Cell Biol. 2004;24:8342–8355. doi: 10.1128/MCB.24.19.8342-8355.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sommariva E, Pellny TK, Karahan N, Kumar S, Huberman JA, Dalgaard JZ. Schizosaccharomyces pombe Swi1, Swi3, and Hsk1 are components of a novel S-phase response pathway to alkylation damage. Mol Cell Biol. 2005;25:2770–2784. doi: 10.1128/MCB.25.7.2770-2784.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Strom L, Karlsson C, Lindroos HB, Wedahl S, Katou Y, Shirahige K, Sjogren C. Postreplicative formation of cohesion is required for repair and induced by a single DNA break. Science. 2007;317:242–245. doi: 10.1126/science.1140649. [DOI] [PubMed] [Google Scholar]
- 59.Unal E, Heidinger-Pauli JM, Koshland D. DNA double-strand breaks trigger genome-wide sister-chromatid cohesion through Eco1 (Ctf7) Science. 2007;317:245–248. doi: 10.1126/science.1140637. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.