

Abstract
Endocannabinoids and their attending cannabinoid type 1 receptor (CB1) have been implicated in animal models of posttraumatic stress disorder (PTSD). However, their specific role has not been studied in people with PTSD. Herein, we present an in vivo imaging study using positron emission tomography (PET) and the CB1-selective radioligand [11C]OMAR in individuals with PTSD, and healthy controls with lifetime histories of trauma (trauma controls [TC]) and those without such histories (healthy controls [HC]). Untreated individuals with PTSD (N=25) with non-combat trauma histories, and TC (N=12) and HC (N=23) participated in a magnetic resonance (MR) imaging scan and a resting PET scan with the CB1 receptor antagonist radiotracer [11C]OMAR, which measures volume of distribution (VT) linearly related to CB1 receptor availability. Peripheral levels of anandamide, 2-arachidonoylglycerol (2-AG), oleoylethanolamide (OEA), palmitoylethanolamide (PEA), and cortisol were also assessed. In the PTSD group, relative to the HC and TC groups, we found elevated brain-wide [11C]OMAR VT values (F(2,53)=7.96, p=.001; 19.5% and 14.5% higher, respectively) which were most pronounced in women (F(1,53)=5.52, p=.023). Anandamide concentrations were reduced in the PTSD relative to the TC (53.1% lower) and HC (58.2% lower) groups. Cortisol levels were lower in the PTSD and TC groups relative to the HC group. Three biomarkers examined collectively—OMAR VT, anandamide, and cortisol—correctly classified nearly 85% of PTSD cases. These results suggest that abnormal CB1 receptor-mediated anandamide signaling is implicated in the etiology of PTSD, and provide a promising neurobiological model to develop novel, evidence-based pharmacotherapies for this disorder.
Keywords: PTSD, Cannabinoid receptors, brain imaging, PET, OMAR
INTRODUCTION
Posttraumatic stress disorder (PTSD) is an anxiety disorder that can develop following exposure to traumatic life events1. Central clinical features of PTSD include a persistent, heightened experience of alarm and distress, as well as a failure of extinction processes to diminish the emotional impact of traumatic memories. Investigation of the neural mechanisms that underlie fear acquisition, consolidation, and extinction may thus enhance our understanding of the neurobiological basis of PTSD, and open opportunities for mechanism-based drug discovery and development of the next-generation pharmacotherapies for this disabling disorder.
The process by which emotionally-aversive memories become consolidated is recognized to be an interaction between glucocorticoid hormones and norepinephrine, both of which are released in response to stress2. The primary component of this response appears to be a noradrenergic signal that is necessary for encoding emotionally salient information3. The hyperconsolidation of traumatic memories in PTSD is driven by a glucocorticoid-hormone-facilitated potentiation of norepinephrine inputs to the basolateral amygdala (BLA)4, 5. Recent work has revealed that this glucocorticoid action is mediated by cannabinoid type-1 (CB1) receptors, a mechanism that is critical for the consolidation of aversive memories6, 7 and thus implicates CB1 receptors in the etiology of PTSD. Moreover, there is an emerging body of evidence demonstrating an important role for CB1 receptor-mediated endocannabinoid signaling in the extinction of aversive memories8. Augmenting levels of anandamide in the amygdala modulates short-term fear extinction9, thereby resulting in long-term reduction in fear10 and highlighting the endocannabinoid system as a candidate system for developing novel pharmacotherapies for PTSD11.
CB1 receptors are the most abundant G-protein-coupled receptors in the central nervous system12, 13, and are found in high concentrations within an amygdala-hippocampal-cortico-striatal circuit responsible for processing and storing fear-related memories and coordinating fear-related behaviors14–16. Animal studies17 have shown that chronic stress is associated with decreased brain levels of the endocannabinoid anandamide and CB1 receptor adaptations17–19, which in turn give rise to an anxious/depressive phenotype20, 21. However, it is not clear whether these animal findings apply to PTSD in humans.
The development of a CB1 receptor selective radiotracer—[11C]OMAR22—now makes it possible for the first time to conduct a quantitative assessment of in vivo CB1 receptor availability using positron emission tomography (PET). In the current study, we hypothesized that, relative to healthy non-trauma-exposed (HC) and trauma-exposed controls (TC), individuals with PTSD would have increased CB1 receptor availability. In light of data from animal studies17–19, 23, we further predicted more pronounced CB1 receptor elevations in women than men with PTSD. A TC group free of lifetime PTSD or other psychiatric illness was recruited in order to assess the relation between trauma exposure alone and CB1 receptor availability. We also assessed peripheral levels of the endocannabinoids anandamide and 2-arachidonoylglycerol (2-AG); levels of the fatty acid ethanolamides oleoylethanolamide (OEA) and palmitoylethanolamide (PEA); and cortisol. We expected to find lower anandamide and cortisol levels in the PTSD group relative to the HC and TC groups24. Finally, psychiatrically relevant biomarkers for PTSD are important yet elusive contributors towards accurate diagnosis and improved clinical care for trauma survivors. We predicted that measures of CB1 receptor availability, anandamide and cortisol would accurately categorize a majority of participants with regard to PTSD diagnostic status relative to healthy and trauma-exposed controls.
METHODS
Participants
Participants were recruited via public advertisements seeking individuals with non-combat trauma histories and healthy control participants with and without lifetime histories of trauma. None of the participants had ever been treated with psychotropic medications. In addition, none were receiving psychotherapy at the time of scanning. The protocol was approved by the New York University Institutional Review Board, the Yale University School of Medicine Human Investigation Committee, the Yale University Magnetic Resonance Research Center, and the Yale New Haven Hospital Radiation Safety Committee. After providing written informed consent, participants underwent a thorough medical and psychiatric evaluation that included physical examination, electrocardiogram, standard blood chemistry and hematology laboratory tests, urine analysis and toxicology, followed by a magnetic resonance (MR) imaging scan and a resting PET scan with the CB1 receptor antagonist radiotracer [11C]OMAR. Psychiatric diagnoses were made using Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition – Text Revision (DSM-IV-TR) criteria and the Structured Clinical Interview for DSM-IV (SCID), which was administered by an experienced psychiatric clinician25, 26. PTSD symptom severity was assessed using the Clinician-Administered PTSD Scale for DSM-IV (CAPS)27 and trauma history was assessed using the Traumatic Life Events Questionnaire (TLEQ)28. Only traumatic events meeting DSM-IV-TR PTSD criterion A1 for severe trauma exposure, as well as criterion A2, which confirms the emotional response to the trauma (i.e., response involved intense fear, horror, or helplessness), were counted towards participants' trauma history in this study. Additional assessments included the Hamilton Rating Scale for Anxiety (HAM-A)29, the Montgomery-Åsberg Depression Rating Scale (MADRS)30, the Alcohol Module of the Addiction Severity Index31 and the Fagerström Test for Nicotine Dependence (FTND)32. To meet TC inclusion criteria, individuals must have been exposed to at least one potentially traumatic event that met DSM-IV-TR PTSD Criteria A1 and A2, but have no lifetime PTSD or other Axis I diagnosis. Participants with significant medical or neurological conditions, substance abuse within 12 months of the PET scan, a lifetime history of substance dependence (including cannabis), or history of head injury with loss of consciousness were excluded from the study. The absence of substance use (including cannabis) was determined by self-report and confirmed by urine toxicology and breathalyzer test at screening, and on the days of MR and PET imaging. Participants were asked to abstain from food, nicotine, and caffeinated beverages after midnight on the day prior to the imaging study until after completion of the scan. Blood samples were collected at the time of tracer injection and processed immediately after collection in the laboratory, which is adjacent to the scan room and frozen at −80°Celsius until analyzed, as previously described33.
PET imaging
[11C]OMAR was prepared in high specific activity (109±74 MBq/nmol at end of synthesis). The radiotracer (injected dose: 589±122 MBq, injected mass: 0.05±0.03 μg/kg) was infused over 1 minute through the antecubital vein. The radioactivity concentration in blood from the radial artery was measured continuously using an automated system (PBS101, Veenstra Instruments, Joure, The Netherlands) for the first 7 min after radiotracer administration and manually drawn and counted thereafter. Discrete samples were acquired at selected times and measured on a gamma counter (Wizard 1480, Perkin-Elmer, Waltham, Massachusetts, United States) to determine radioactivity concentration in whole blood and plasma. Five discrete blood samples (5, 15, 30, 60, 90 minutes) were analyzed for the fraction of unchanged [11C]OMAR and its radiometabolites using a column-switching high pressure liquid chromatography method34. The fraction of tracer unbound to plasma proteins was determined in triplicate by ultrafiltration35. Listmode emission data were collected for 120 minutes after radiotracer administration using the High Resolution Research Tomograph (HRRT; Siemens Medical Systems, Knoxville, Tennessee, United States), a dedicated brain PET scanner with spatial resolution better than 3 mm36. Head motion was measured using the Polaris Vicra optical tracking system (Northern Digital Inc., Waterloo, Ontario, Canada) and incorporated into PET image reconstruction with all corrections.37. The PET images were registered to subject-specific T1-weighted magnetic resonance images (256 × 256 × 176 grid of 1 mm isotropic voxels) acquired on a 3 Tesla Trio imaging system (Siemens Medical Systems, Erlangen, Germany). Anatomical MR images were in turn nonlinearly registered to an MR template where regions of interest (ROIs) were defined38. Regional time activity curves (TACs) were extracted from the dynamic PET data and analyzed using the multilinear analysis method39 with metabolite-corrected arterial input functions and cutoff time t*=30 minutes. The kinetic analysis yielded regional estimates of total volume of distribution (VT), the equilibrium ratio of radioligand concentration in tissue relative to arterial plasma40, which is directly proportional to CB1 receptor availability.
Data Analysis
Shapiro-Wilk tests were conducted to assess data distributions of all study variables for normality. Non-normally distributed variables (e.g., [11C]OMAR VT values, cortisol levels) were transformed using logarithmic-base-10 prior to analysis. Analyses of variance (ANOVA) were then used to compare continuously-distributed demographic and clinical variables of the HC, TC, and PTSD groups; χ2 tests were used to compare categorical variables. Because [11C]OMAR VT values across brain regions were highly correlated (r values=.73 to .96), mean composite [11C]OMAR VT values were computed by averaging [11C]OMAR VT values across all brain regions for each individual. A series of analyses of covariance (ANCOVA) were then conducted to test for group differences in mean composite [11C]OMAR VT values, as well as in regions that comprise the amygdala-hippocampal-cortico-striatal circuit implicated in PTSD41. In these analyses, group (HC, TC, and PTSD) and sex were entered as independent variables, age as a covariate, and [11C]OMAR VT values as the dependent variable. Pairwise comparisons—least-squares difference tests—were computed to compare [11C]OMAR VT values in each of the three groups, with p<.01 used to indicate significant group differences. Effect sizes of differences in [11C]OMAR VT values in the TC and PTSD groups relative to the HC group were expressed using percent difference and Cohen's d ((Mgroup1-Mgroup2)/SDpooled). Similar ANCOVAs were conducted for anandamide, 2-AG, OEA, and PEA and cortisol values. To examine the relation between [11C]OMAR VT values, anandamide, and cortisol biomarkers, and PTSD group membership, a series of binary logistic regression analyses were conducted, with main effects and all combinations of these variables entered as explanatory variables in separate analyses, and PTSD (coded “1”) vs. TC+HC group (coded “0”) entered as the dependent variable.
RESULTS
Seventy-two participants were recruited into the study and 60 completed the protocol. Reasons for exclusion were previous medication exposure (n=9) and medical reasons that would interfere with correct interpretation of the collected data (n=4). Table 1 shows demographic, trauma, and clinical characteristics of the HC, TC, and PTSD groups whose data were used for analyses. [11C]OMAR injection parameters, age, sex, education, nature of trauma histories, and body mass index did not differ among the groups; there was a greater proportion of white individuals in the HC than TC and PTSD groups. The PTSD group was significantly more likely than the HC and TC groups to currently smoke cigarettes, and to have a lifetime history of mood or anxiety disorder, and alcohol or drug abuse, but the groups did not differ with respect to lifetime and current alcohol use and nicotine dependence. The PTSD group scored higher on the MADRS and HAM-A relative to both control groups, and on the CAPS relative to the TC group.
Table 1.
Demographic and clinical characteristics of study groups
| Healthy controls | Trauma controls | PTSD | Test of difference | Pairwise comparisons | |
|---|---|---|---|---|---|
| N | 23 | 12 | 25 | ||
| M (SD) or n (%) | M (SD) or n (%) | M (SD) or n (%) | |||
| Age | 32.1 (8.5) | 29.7 (7.9) | 32.2 (9.9) | F(2,57)=.36, p=.70 | - |
| Male sex | 11 (47.8%) | 7 (58.3%) | 11 (44.0%) | χ2(2)=.67, p=.71 | - |
| White race/ethnicity | 14 (60.9%) | 2 (16.7%) | 8 (32.0%) | χ2(2)=7.56, p=.023 | HC>TC,PTSD |
| Education (years) | 16.0 (2.1) | 14.1 (1.8) | 14.6 (3.1) | F(2,57)=2.86, p=.066 | - |
| Body mass index | 25.0 (3.4) | 26.6 (4.5) | 25.3 (4.7) | F(2,57)=.56, p=.57 | - |
| Ever drank alcohol in lifetime | 11 (73.3%) | 3 (42.9%) | 12 (57.1%) | χ2(2)=2.04, p=.36 | - |
| Number of years used alcohol | 5.7 (6.3) | 8.0 (11.5) | 5.0 (6.0) | F(2,38)=.44, p=.65 | - |
| Drank alcohol in past 30 days | 9 (56.3%) | 6 (54.5%) | 10 (47.6%) | χ2(2)=.31, p=.86 | - |
| Number of days drank alcohol in past 30 days | 2.4 (3.3) | 3.7 (5.0) | 2.5 (3.4) | F(2,45)=.48, p=.62 | - |
| Current smoker | 0 (0%) | 0 (0%) | 9 (36.0%) | χ2(2)=14.82, p=.001 | PTSD>HC,TC |
| Nicotine dependence | 0 (0%) | 0 (0%) | 3 (12.0%) | χ2(2)=4.42, p=.11 | - |
| Indices of lifetime trauma | |||||
| Age at first trauma | - | 13.0 (4.8) | 13.9 (10.3) | F(1,35)=.09, p=.77 | - |
| Age at presenting trauma | - | 16.5 (8.7) | 18.2 (11.1) | F(1,35)=.22, p=.64 | - |
| Number of traumas | - | 3.0 (2.0) | 3.5 (3.7) | F(1,35)=.18, p=.68 | - |
| Index traumatic event | Fisher's exact test=6.38, p<.001 | ||||
| Physical assault | - | 9 (75.0%) | 22 (88.0%) | ||
| Motor vehicle accident | - | 3 (25.0%) | 0 (0%) | ||
| Witnessed suicide | - | 0 (0%) | 3 (12.0%) | ||
| Lifetime mood or anxiety disorder | 0 (0%) | 0 (0%) | 13 (52.0%) | χ2(2)=23.23, p<.001 | PTSD>HC, TC |
| Lifetime alcohol or drug abuse | 0 (0%) | 0 (0%) | 9 (36.0%) | χ2(2)=14.82, p=.001 | PTSD>HC, TC |
| CAPS score | - | 5.6 (7.6) | 75.5 (17.4) | F(1,35)=175.99, p<.001 | PTSD>TC |
| MADRS score | 2.2 (3.0) | 3.5 (6.5) | 22.7 (10.0) | F(2,57)=54.89, p<.001 | PTSD>HC, TC |
| HAM-A score | 1.6 (2.5) | 2.3 (3.3) | 21.4 (10.4) | F(2,57)=55.77, p<.001 | PTSD>HC, TC |
Note. HC=Healthy controls; TC=Trauma Controls; SD=standard deviation; PTSD=Posttraumatic stress disorder; SD=standard deviation; CAPS=Clinician-Administered PTSD Scale; MADRS= Montgomery-Åsberg Depression Rating Scale; HAM-A=Hamilton Anxiety Rating Scale. Physical assault includes sexual abuse, domestic violence, and other non-combat related physical violence. Some frequencies and denominator degrees of freedom do not sum to total n due to missing data.
Bivariate correlations revealed that composite [11C]OMAR VT values correlated negatively with age (r= −.34, p=.007) and positively with female sex (r= .39, p=.002). Composite [11C]OMAR VT values also correlated negatively with anandamide levels (r= −.27, p=.038), but not cortisol levels (r= −.06, p=.67); and the correlation between anandamide and cortisol levels was also not significant (r= .02, p=.88). Other demographic variables, BMI, lifetime and current alcohol use, current cigarette smoking status and nicotine dependence, and trauma-related variables and lifetime history of mood or anxiety disorders, and alcohol or drug abuse were not associated with [11C]OMAR VT values (all r values<|.22|, all p values>.10).
An ANCOVA examining mean composite [11C]OMAR VT values in the HC, TC, and PTSD groups revealed significant main effects of group (F(2,53)=7.96, p=.001), sex (F(1,53)=5.52, p=.023), and age (F(1,53=11.95, p=.001); there was a trend towards a significant group-by-sex interaction (F(2,53)=2.75, p=.073). Women in the full sample had higher mean composite [11C]OMAR VT values than men (M=1.233, SE=.040 vs. M=1.369, SE=.042, Cohen's d=.61). As shown in Table 2, mean composite [11C]OMAR VT values differed by group, such that PTSD group VT was higher than the TC and HC groups, which did not differ. This same general pattern was also observed in brain regions that comprise the amygdala-hippocampal-cortico-striatal neural circuit implicated in PTSD. As shown in Figure 1, effect sizes for the differences between the PTSD group and HC and TC groups were consistently large in magnitude. Differences between the TC and HC groups were generally small in magnitude. Analyses of [11C]OMAR VT values in brain regions outside of the amygdala-hippocampal-cortico-striatal neural circuit implicated in PTSD revealed this same pattern of results, with the PTSD group having significantly greater [11C]OMAR VT values than both the HC and TC groups (all F's for group effect>4.91, all p's<.01; all p's for pairwise comparisons<.01).
Table 2.
[11C]OMAR volume of distribution (VT) values, anandamide, 2-AG, OEA,PEA, and cortisol levels by group
| Healthy controls | Trauma controls | PTSD | Test of difference | Pairwise comparisons | Mean % difference PTSD vs. HC | Mean % difference PTSD vs. TC | Mean % difference TC vs. HC | |
|---|---|---|---|---|---|---|---|---|
| N | 23 | 12 | 25 | |||||
| M (SE) | M (SE) | M (SE) | ||||||
| [11C]OMAR VT values | ||||||||
| Mean composite | 1.205 (.044) | 1.258 (.062) | 1.440 (.042) | F(2,53)=7.96, p=.001 | PTSD>HC,TC | +19.5% | +14.5% | +4.4% |
| Anterior cingulate | 1.400 (.049) | 1.432 (.069) | 1.648 (.047) | F(2,53)=7.54, p=.001 | PTSD>HC,TC | +17.7% | +15.1% | +2.3% |
| Amygdala | 1.322 (.056) | 1.444 (.079) | 1.594 (.054) | F(2,53)=6.13, p=.004 | PTSD>HC | +20.6% | +10.4% | +9.2% |
| Caudate | 1.080 (.047) | 1.104 (.067) | 1.287 (.046) | F(2,53)=5.62, p=.006 | PTSD>HC,TC | +19.2% | +16.6% | +2.2% |
| Hippocampus | 1.214 (.052) | 1.257 (.073) | 1.440 (.050) | F(2,53)=5.36, p=.008 | PTSD>HC,TC | +18.6% | +14.6% | +3.5% |
| Pallidum | 1.638 (.085) | 1.666 (.121) | 2.008 (.082) | F(2,53)=5.60, p=.006 | PTSD>HC,TC | +22.6% | +20.5% | +1.7% |
| Orbitofrontal cortex | 1.265 (.043) | 1.321 (.061) | 1.490 (.042) | F(2,53)=7.31, p=.002 | PTSD>HC,TC | +17.8% | +12.8% | +4.4% |
| Anandamide | 2.43 (.25) | 2.73 (.35) | 1.14 (.24) | F(2,53)=9.75, p<.001 | HC,TC>PTSD | −53.1% | −58.2% | +12.3% |
| 2-AG | 7.16 (2.41) | 10.01 (3.71) | 13.31 (2.16) | F(2,41)=1.81, p=.18 | - | - | - | - |
| OEA | 156.41 (23.20) | 25.01 (38.78) | 35.12 (22.16) | F(2,45)=8.45, p=.001 | HC>TC,PTSD | −77.5% | +40.4% | −84.0% |
| PEA | 8.87 (1.54) | 10.22 (2.58) | 9.68 (1.48) | F(2,45)=13, p=.88 | - | - | - | - |
| Cortisol | 12.47 (.71) | 7.18 (1.00) | 8.33 (.70) | F(2,52)=12.69, p<.001 | HC>TC,PTSD | −33.2% | +16.0% | −42.4% |
Note. SE=standard error of the mean. VT=volume of distribution. 2-AG=2-Arachidonoylglycerol; OEA=Oleoylethanolamine; PEA=Palmitoylethanolamide. All models are adjusted for age and sex. Denominator degrees of freedom are lower for F tests examining group differences in 2-AG, PEA, OEA, and cortisol due to missing data.
Figure 1.

Cohen's d and 95% confidence intervals of effect size differences in [11C]OMAR volume of distribution (VT) values in PTSD and TC groups relative to HC group
Figure 2 shows mean [11C]OMAR VT values for the PTSD, and HC and TC groups by sex. Pairwise comparisons revealed that mean [11C]OMAR VT values were significantly higher among women in the HC (p=.009; d=1.26) and PTSD (p=.011; d=1.16) groups, but not in the TC group (p=.65; d=.32).
Figure 2.
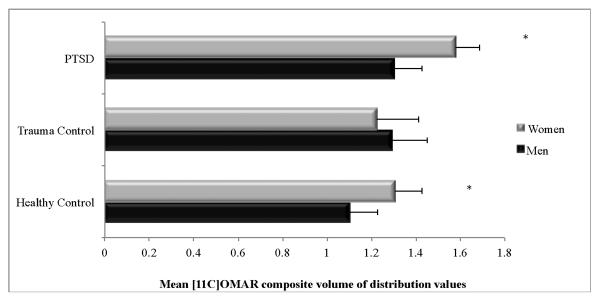
Cohen's d and 95% confidence intervals of effect size differences in [11C]OMAR volume of distribution (VT) values in PTSD and TC groups relative to HC group by sex
An ANCOVA examining anandamide levels in the HC, TC, and PTSD groups revealed significant main effects of group (F(2,53)=9.75, p<.001) and sex (F(1,53)=4.23, p=.045), but age (F(1,53)=1.00, p=.32) and the interaction of group-by-sex (F(2,53)=1.47, p=.24) were not significant. Pairwise comparisons revealed that the PTSD group had lower anandamide levels than both the HC (p=.001; Cohen's d=1.10) and TC (p=.001; Cohen's d=1.36) groups; anandamide levels did not differ between the HC and TC groups (p=.51; Cohen's d=.26). Women had lower levels than men (M=2.44, SE=.24 vs. M=1.76, SE=.23; Cohen's d=.54). OEA levels also differed by group, with the HC group having significantly higher OEA levels than the TC and PTSD groups; age (F(1,45)=2.91, p=.095), sex (F(1,45)=.55, p=.46), and the interaction of group × sex (F(2,45)=.02, p=.98) were not significant. 2-AG and PEA levels did not differ by group.
An ANCOVA examining cortisol levels in the HC, TC, and PTSD groups revealed a significant main effect of group (F(2,52)=12.69, p<.001), but sex (F(1,52)=.05, p=.82), age (F(1,52)=1.84, p=.18), and the interaction of group-by-sex were not significant (F(2,52)=.33, p=.72). Pairwise comparisons revealed that the HC group had higher cortisol levels than both the TC (p<.001; Cohen's d=1.59) and PTSD (p<.001; Cohen's d=1.22) groups; cortisol levels did not differ between the TC and PTSD groups (p=.35; Cohen's d=.34).
Table 3 shows results of binary logistic regression analyses that examined how each of the biomarkers ([11C]OMAR VT values, anandamide, and cortisol) independently and in various combinations related to PTSD vs. HC/TC group membership. Results revealed that the classification accuracy increased as additional biomarkers were added to the model, with the highest classification accuracy observed when [11C]OMAR VT values, anandamide, and cortisol were entered simultaneously. None of the interaction terms were significant (all p values>.21).
Table 3.
Classification accuracy statistics for logistic regression model examining relation between [11C]OMAR VT, anandamide, and cortisol bio markers and PTSD
| Biomarker | Overall classification accuracy | % PTSD cases correctly classified | Nagelkerke's R2 |
|---|---|---|---|
| Cortisol | 61.0% | 33.3% | .134 |
| Composite [11C]OMAR VT | 70.0% | 52.0% | .217 |
| Anandamide | 73.3% | 76.0% | .348 |
| Cortisol + AEA | 72.9% | 70.8% | .482 |
| Composite [11C]OMAR + Anandamide | 75.0% | 68.0% | .472 |
| Composite [11C]OMAR + Cortisol | 76.3% | 66.7% | .357 |
| Composite [11C]OMAR + Cortisol + Anandamide | 88.1% | 83.3% | .661 |
|
Final logistic regression model Hosmer and Lemeshow test=9.17, p=.33 |
|||
| Wald χ2, p | OR (95%CI) | ||
| Composite [11C]OMAR VT | 6.96, .008 | 3.09 (1.34–7.16) | |
| Cortisol | 7.40, .007 | .24 (.08–.67) | |
| Anandamide | 9.88, .002 | .13 (.03–.46) |
Note. VT=volume of distribution; OR=odds ratio; 95%CI=95% confidence interval.
DISCUSSION
We found that PTSD is associated with a ubiquitously expressed large magnitude elevation (~20%) in [11C]OMAR VT values, which quantitatively reflects CB1 receptor availability. Notably, this elevation was found in an amygdala-hippocampal-cortico-striatal neural circuit implicated in PTSD, as well as in brain regions outside this circuit. These results suggest greater brain-wide CB1 receptor availability in individuals with PTSD relative to control participants with and without histories of trauma exposure. Reduced peripheral anandamide levels in PTSD complemented the brain [11C]OMAR VT results, suggesting that the elevated CB1 receptor availability in PTSD may result from a combination of both receptor up-regulation and low receptor occupancy by anandamide. The lack of displacement of CB1 radioligands by agonists42–44 which has been attributed to a large receptor reserve45 suggests that increased [11C]OMAR VT values are explained to the most part by receptor up-regulation in response to low anandamide levels rather than low receptor occupancy by anandamide. This idea is substantiated by data showing that CB1 receptor up-regulation in response to low stress-induced synaptic anandamide availability was prevented by enhanced anandamide signaling46. OEA levels were higher in HC relative to TC and PTSD participants in the current study, but groups did not differ with respect to 2-AG and PEA levels. Taken together, these data suggest that abnormal CB1 receptor-mediated anandamide signaling is implicated in the etiology of PTSD.
The sex-related results of the current study accord with animal data demonstrating sex differences in CB1 receptor regulation, with stress-related up-regulation of CB1 receptors observed predominantly in female animals18. We also found abnormally low cortisol levels in trauma survivors, corroborating prior work47. Another key contribution of the current study is the finding that collective consideration of all three of the biomarkers examined—OMAR VT, anandamide, and cortisol—was highly accurate in classifying PTSD, with nearly 85% of PTSD cases correctly classified and overall classification accuracy approaching 90%. Results of this study advance the extant literature in three important ways: (1) they contribute to extant knowledge regarding the etiology of PTSD; (2) they identify candidate biomarkers that may be used to support clinical decision-making regarding diagnostic classification of PTSD; and (3) they provide a promising neurobiological rationale to develop novel, evidence-based pharmacotherapies for PTSD.
Our results of reduced peripheral anandamide levels together with a compensatory up-regulation of CB1 receptors in PTSD suggest lower anandamide tone in PTSD. Notably, elevated rates of cannabis abuse/dependence among individuals with PTSD have been reported48, 49. Such findings substantiate, at least in part, emerging evidence that synthetic cannabinoid receptor agonists50 or plant-derived cannabinoids such as marijuana51 may possess some benefits in individuals with PTSD by helping relieve haunting nightmares and other symptoms of PTSD. However, such data do not allow the conclusion that self-medication with cannabis with its primary psychoactive constituent tetrahydrocannabinol should be recommended for the treatment of PTSD, as direct activation of CB1 receptors with plant-derived cannabinoids over an extended period of time leads to down-regulation of CB1 receptors52, 53, which may in turn result in a depression-like phenotype in certain individuals54 and increase risk of addiction55.
Another important finding in this study is the sex differences in anandamide levels and [11C]OMAR VT values in both the HC and PTSD groups. Animal data showing higher CB1 receptor levels in male relative to female animals18, 56, 57 and receptor fluctuations during the estrous cycle together with changes in affinity of agonist binding58 highlight the importance of careful considerations of gender and menstrual cycle phase in assessments of CB1 receptor availability in imaging studies. In addition, we believe, that a conclusive interpretation of the CB1 receptor profile in males and females requires a broad and dynamic perspective rather than a single observation in a cross-sectional study with a single time point. Our results are largely in agreement with a previous study that used the CB1 PET tracer [18F]MK-9470 to investigate the effects of age and gender59. That report found greater plasma parent fraction and higher normalized brain uptake (SUV) in men, which is consistent with our findings. However, because of the nearly irreversible uptake kinetics of the radiotracer and the lack of significant gender differences in the metabolite-corrected input function in the initial cohort that underwent arterial blood sampling, the [18F]MK-9470 study used brain SUV as the final outcome metric of tracer binding. We performed kinetic analysis of [11C]OMAR data using metabolite-corrected arterial input functions in all participants. This methodology provided estimates of VT, which – in contrast to SUV, which was greater in men than women – was reduced in men compared to women60. Thus, our measurements are compatible with those of the previously reported [18F]MK-9470 data and the discrepant interpretations appear to be accounted for by different endpoints centered on our use of arterial input functions in kinetic analyses rather than the simplified outcome of normalized brain concentration. If, as our results suggest, women show higher CB1 receptor availability than men already under basal, non-stress conditions, then they may be at increased risk for PTSD when exposed to trauma. This finding may thus provide a neurobiological explanation for why women are at greater risk for developing PTSD following exposure to various types of trauma than men even when sexual trauma—which is more common in women—is excluded61.
To date, drug development in PTSD has been opportunistic, building almost entirely on empirical observations with drugs approved for other indications. The data reported herein are the first of which we are aware of to demonstrate the critical role of CB1 receptors and endocannabinoids in the etiology of PTSD in humans. As such, they provide a foundation upon which to develop and validate informative biomarkers of PTSD vulnerability, as well as to guide the rational development of the next generation of evidence-based treatments for PTSD. Blocking anandamide deactivation or re-uptake, both of which will increase synaptic anandamide availability, may lead to a more circumscribed and beneficial spectrum of biological responses than those produced by direct CB1 receptor activation62, 63. This is of particular interest for the development of mechanism-based novel pharmacotherapies for PTSD, as emerging data have revealed that enhanced anandamide signaling can curb the effects of chronic stress, possibly by maintaining normal amygdala function64 via extinction-driven reductions in fear resulting in improved stress-reactivity in humans10.
Acknowledgement
The authors acknowledge the excellent work of the staff of the Yale PET Center and the nursing support from Brenda Breault, R.N., B.S.N., Cynthia D'Amico, R.N., B.S.N., Michelle San Pedro, R.N., C.C.R.C., Jamie Cyr, R.N., C.C.R.N. and Deborah Campbell, R.N. for their contributions with patient care during the PET scans.
Funding Information:
The project described was supported by the National Institutes of Health through the following awards: R21MH096105, R21MH085627, and R01MH096876. This publication was also made possible by CTSA Grant Number UL1 RR024139 from the National Center for Research Resources (NCRR) and the National Center for Advancing Translational Science (NCATS), components of the National Institutes of Health (NIH), and NIH roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH.
Footnotes
Conflict of Interest/Financial Disclosures The authors report no conflicts of interest relevant to this article.
REFERENCES
- 1.Yehuda R. Post-traumatic stress disorder. N Engl J Med. 2002;346(2):108–114. doi: 10.1056/NEJMra012941. [DOI] [PubMed] [Google Scholar]
- 2.Roozendaal B, McGaugh JL. Memory modulation. Behav Neurosci. 2011;125(6):797–824. doi: 10.1037/a0026187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roozendaal B, Okuda S, de Quervain DJ, McGaugh JL. Glucocorticoids interact with emotion-induced noradrenergic activation in influencing different memory functions. Neuroscience. 2006;138(3):901–910. doi: 10.1016/j.neuroscience.2005.07.049. [DOI] [PubMed] [Google Scholar]
- 4.Roozendaal B, Barsegyan A, Lee S. Adrenal stress hormones, amygdala activation, and memory for emotionally arousing experiences. Progress in brain research. 2008;167:79–97. doi: 10.1016/S0079-6123(07)67006-X. [DOI] [PubMed] [Google Scholar]
- 5.Atsak P, Roozendaal B, Campolongo P. Role of the endocannabinoid system in regulating glucocorticoid effects on memory for emotional experiences. Neuroscience. 2012;204:104–116. doi: 10.1016/j.neuroscience.2011.08.047. [DOI] [PubMed] [Google Scholar]
- 6.Campolongo P, Roozendaal B, Trezza V, Hauer D, Schelling G, McGaugh JL, et al. Endocannabinoids in the rat basolateral amygdala enhance memory consolidation and enable glucocorticoid modulation of memory. Proc Natl Acad Sci U S A. 2009;106(12):4888–4893. doi: 10.1073/pnas.0900835106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hill MN, McEwen BS. Endocannabinoids: The silent partner of glucocorticoids in the synapse. Proc Natl Acad Sci U S A. 2009;106(12):4579–4580. doi: 10.1073/pnas.0901519106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418(6897):530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- 9.Pamplona FA, Bitencourt RM, Takahashi RN. Short- and long-term effects of cannabinoids on the extinction of contextual fear memory in rats. Neurobiology of learning and memory. 2008;90(1):290–293. doi: 10.1016/j.nlm.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 10.Gunduz-Cinar O, Macpherson KP, Cinar R, Gamble-George J, Sugden K, Williams B, et al. Convergent translational evidence of a role for anandamide in amygdala-mediated fear extinction, threat processing and stress-reactivity. Mol Psychiatry. 2012 doi: 10.1038/mp.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neumeister A. The endocannabinoid system provides an avenue for evidence-based treatment development for ptsd. Depress Anxiety. 2013;30(2):93–96. doi: 10.1002/da.22031. [DOI] [PubMed] [Google Scholar]
- 12.Glass M, Dragunow M, Faull RL. Cannabinoid receptors in the human brain: a detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience. 1997;77(2):299–318. doi: 10.1016/s0306-4522(96)00428-9. [DOI] [PubMed] [Google Scholar]
- 13.Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, et al. Cannabinoid receptor localization in brain. Proc Natl Acad Sci U S A. 1990;87(5):1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390(6660):604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- 15.LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- 16.Rodrigues SM, Schafe GE, LeDoux JE. Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron. 2004;44(1):75–91. doi: 10.1016/j.neuron.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 17.Hill MN, Patel S, Carrier EJ, Rademacher DJ, Ormerod BK, Hillard CJ, et al. Downregulation of endocannabinoid signaling in the hippocampus following chronic unpredictable stress. Neuropsychopharmacology. 2005;30(3):508–515. doi: 10.1038/sj.npp.1300601. [DOI] [PubMed] [Google Scholar]
- 18.Reich CG, Taylor ME, McCarthy MM. Differential effects of chronic unpredictable stress on hippocampal CB1 receptors in male and female rats. Behav Brain Res. 2009;203(2):264–269. doi: 10.1016/j.bbr.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hill MN, Carrier EJ, McLaughlin RJ, Morrish AC, Meier SE, Hillard CJ, et al. Regional alterations in the endocannabinoid system in an animal model of depression: effects of concurrent antidepressant treatment. J Neurochem. 2008;106(6):2322–2336. doi: 10.1111/j.1471-4159.2008.05567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haller J, Bakos N, Szirmay M, Ledent C, Freund TF. The effects of genetic and pharmacological blockade of the CB1 cannabinoid receptor on anxiety. Eur J Neurosci. 2002;16(7):1395–1398. doi: 10.1046/j.1460-9568.2002.02192.x. [DOI] [PubMed] [Google Scholar]
- 21.Haller J, Varga B, Ledent C, Freund TF. CB1 cannabinoid receptors mediate anxiolytic effects: convergent genetic and pharmacological evidence with CB1-specific agents. Behav Pharmacol. 2004;15(4):299–304. doi: 10.1097/01.fbp.0000135704.56422.40. [DOI] [PubMed] [Google Scholar]
- 22.Horti AG, Fan H, Kuwabara H, Hilton J, Ravert HT, Holt DP, et al. 11C-JHU75528: a radiotracer for PET imaging of CB1 cannabinoid receptors. J Nucl Med. 2006;47(10):1689–1696. [PubMed] [Google Scholar]
- 23.Viveros MP, Llorente R, Lopez-Gallardo M, Suarez J, Bermudez-Silva F, De la Fuente M, et al. Sex-dependent alterations in response to maternal deprivation in rats. Psychoneuroendocrinology. 2009;34(Suppl 1):S217–226. doi: 10.1016/j.psyneuen.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 24.Hill MN, Patel S, Campolongo P, Tasker JG, Wotjak CT, Bains JS. Functional interactions between stress and the endocannabinoid system: from synaptic signaling to behavioral output. J Neurosci. 2010;30(45):14980–14986. doi: 10.1523/JNEUROSCI.4283-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.First MB, Spitzer RL, Gibbons M, Williams JBW. Structured Clinical Interview for DSM-IV Axis I Disorders. New York State Psychiatric Institute, Biometrics Research; New York: 1995. [Google Scholar]
- 26.DSM-IV APATFo . Diagnostic and Statisical Manual of Mental Disorders: DSM-IV-TR. 4th, text revision edn American Psychiatric Association; Washington, DC: 2000. [Google Scholar]
- 27.Blake DD, Weathers FW, Nagy LM, Kaloupek DG, Gusman FD, Charney DS, et al. The development of a Clinician-Administered PTSD Scale. J Trauma Stress. 1995;8(1):75–90. doi: 10.1007/BF02105408. [DOI] [PubMed] [Google Scholar]
- 28.Kubany ES, Haynes SN, Leisen MB, Owens JA, Kaplan AS, Watson SB, et al. Development and preliminary validation of a brief broad-spectrum measure of trauma exposure: the Traumatic Life Events Questionnaire. Psychol Assess. 2000;12(2):210–224. doi: 10.1037//1040-3590.12.2.210. [DOI] [PubMed] [Google Scholar]
- 29.Hamilton M. The assessment of anxiety states by rating. British Journal of Medical Psychology. 1959;(32):50–55. doi: 10.1111/j.2044-8341.1959.tb00467.x. [DOI] [PubMed] [Google Scholar]
- 30.Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–389. doi: 10.1192/bjp.134.4.382. [DOI] [PubMed] [Google Scholar]
- 31.McLellan AT, Luborsky L, Woody GE, O'Brien CP. An improved diagnostic evaluation instrument for substance abuse patients. The Addiction Severity Index. J Nerv Ment Dis. 1980;168(1):26–33. doi: 10.1097/00005053-198001000-00006. [DOI] [PubMed] [Google Scholar]
- 32.Heatherton TF, Kozlowski LT, Frecker RC, Fagerstrom KO. The Fagerstrom Test for Nicotine Dependence: a revision of the Fagerstrom Tolerance Questionnaire. Br J Addict. 1991;86(9):1119–1127. doi: 10.1111/j.1360-0443.1991.tb01879.x. [DOI] [PubMed] [Google Scholar]
- 33.Mangieri RA, Hong KI, Piomelli D, Sinha R. An endocannabinoid signal associated with desire for alcohol is suppressed in recently abstinent alcoholics. Psychopharmacology (Berl) 2009;205(1):63–72. doi: 10.1007/s00213-009-1518-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hilton J, Yokoi F, Dannals RF, Ravert HT, Szabo Z, Wong DF. Column-switching HPLC for the analysis of plasma in PET imaging studies. Nucl Med Biol. 2000;27(6):627–630. doi: 10.1016/s0969-8051(00)00125-6. [DOI] [PubMed] [Google Scholar]
- 35.Gandelman MS, Baldwin RM, Zoghbi SS, Zea-Ponce Y, Innis RB. Evaluation of ultrafiltration for the free-fraction determination of single photon emission computed tomography (SPECT) radiotracers: beta-CIT, IBF, and iomazenil. J Pharm Sci. 1994;83(7):1014–1019. doi: 10.1002/jps.2600830718. [DOI] [PubMed] [Google Scholar]
- 36.de Jong HW, van Velden FH, Kloet RW, Buijs FL, Boellaard R, Lammertsma AA. Performance evaluation of the ECAT HRRT: an LSO-LYSO double layer high resolution, high sensitivity scanner. Physics in medicine and biology. 2007;52(5):1505–1526. doi: 10.1088/0031-9155/52/5/019. [DOI] [PubMed] [Google Scholar]
- 37.Proceedings of the Conf Record IEEE Nuclear Science Symposium and Medical Imaging. Portland, OR: 2003. Design of a motion-compensation OSEM List-mode Algorithm for Resolution-Recovery Reconstruction of the HRRT. [Google Scholar]
- 38.Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage. 2002;15(1):273–289. doi: 10.1006/nimg.2001.0978. [DOI] [PubMed] [Google Scholar]
- 39.Ichise M, Toyama H, Innis RB, Carson RE. Strategies to improve neuroreceptor parameter estimation by linear regression analysis. J Cereb Blood Flow Metab. 2002;22(10):1271–1281. doi: 10.1097/01.WCB.0000038000.34930.4E. [DOI] [PubMed] [Google Scholar]
- 40.Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, et al. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab. 2007;27(9):1533–1539. doi: 10.1038/sj.jcbfm.9600493. [DOI] [PubMed] [Google Scholar]
- 41.Murrough JW, Czermak C, Henry S, Nabulsi N, Gallezot JD, Gueorguieva R, et al. The effect of early trauma exposure on serotonin type 1B receptor expression revealed by reduced selective radioligand binding. Arch Gen Psychiatry. 2011;68(9):892–900. doi: 10.1001/archgenpsychiatry.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terry G, Liow JS, Chernet E, Zoghbi SS, Phebus L, Felder CC, et al. Positron emission tomography imaging using an inverse agonist radioligand to assess cannabinoid CB1 receptors in rodents. Neuroimage. 2008;41(3):690–698. doi: 10.1016/j.neuroimage.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gatley SJ, Gifford AN, Volkow ND, Lan R, Makriyannis A. 123I-labeled AM251: a radioiodinated ligand which binds in vivo to mouse brain cannabinoid CB1 receptors. Eur J Pharmacol. 1996;307(3):331–338. doi: 10.1016/0014-2999(96)00279-8. [DOI] [PubMed] [Google Scholar]
- 44.Berding G, Muller-Vahl K, Schneider U, Gielow P, Fitschen J, Stuhrmann M, et al. [123I]AM281 single-photon emission computed tomography imaging of central cannabinoid CB1 receptors before and after Delta9-tetrahydrocannabinol therapy and whole-body scanning for assessment of radiation dose in tourette patients. Biol Psychiatry. 2004;55(9):904–915. doi: 10.1016/j.biopsych.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 45.Gifford AN, Bruneus M, Gatley SJ, Lan R, Makriyannis A, Volkow ND. Large receptor reserve for cannabinoid actions in the central nervous system. J Pharmacol Exp Ther. 1999;288(2):478–483. [PubMed] [Google Scholar]
- 46.Bortolato M, Mangieri RA, Fu J, Kim JH, Arguello O, Duranti A, et al. Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol Psychiatry. 2007;62(10):1103–1110. doi: 10.1016/j.biopsych.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 47.Morris MC, Compas BE, Garber J. Relations among posttraumatic stress disorder, comorbid major depression, and HPA function: a systematic review and meta-analysis. Clin Psychol Rev. 2012;32(4):301–315. doi: 10.1016/j.cpr.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vetter S, Rossegger A, Rossler W, Bisson JI, Endrass J. Exposure to the tsunami disaster, PTSD symptoms and increased substance use - an Internet based survey of male and female residents of Switzerland. BMC Public Health. 2008;8:92. doi: 10.1186/1471-2458-8-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cornelius JR, Kirisci L, Reynolds M, Clark DB, Hayes J, Tarter R. PTSD contributes to teen and young adult cannabis use disorders. Addict Behav. 35(2):91–94. doi: 10.1016/j.addbeh.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fraser GA. The use of a synthetic cannabinoid in the management of treatment-resistant nightmares in posttraumatic stress disorder (PTSD) CNS Neurosci Ther. 2009;15(1):84–88. doi: 10.1111/j.1755-5949.2008.00071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Passie T, Emrich HM, Karst M, Brandt SD, Halpern JH. Mitigation of post-traumatic stress symptoms by Cannabis resin: a review of the clinical and neurobiological evidence. Drug testing and analysis. 2012;4(7–8):649–659. doi: 10.1002/dta.1377. [DOI] [PubMed] [Google Scholar]
- 52.Hirvonen J, Goodwin RS, Li CT, Terry GE, Zoghbi SS, Morse C, et al. Reversible and regionally selective downregulation of brain cannabinoid CB(1) receptors in chronic daily cannabis smokers. Mol Psychiatry. doi: 10.1038/mp.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leweke FM, Koethe D. Cannabis and psychiatric disorders: it is not only addiction. Addict Biol. 2008;13(2):264–275. doi: 10.1111/j.1369-1600.2008.00106.x. [DOI] [PubMed] [Google Scholar]
- 54.Beyer CE, Dwyer JM, Piesla MJ, Platt BJ, Shen R, Rahman Z, et al. Depression-like phenotype following chronic CB1 receptor antagonism. Neurobiol Dis. 2010;39(2):148–155. doi: 10.1016/j.nbd.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 55.Klugmann M, Klippenstein V, Leweke FM, Spanagel R, Schneider M. Cannabinoid exposure in pubertal rats increases spontaneous ethanol consumption and NMDA receptor associated protein levels. Int J Neuropsychopharmacol. 14(4):505–517. doi: 10.1017/S1461145710001562. [DOI] [PubMed] [Google Scholar]
- 56.Riebe CJ, Hill MN, Lee TT, Hillard CJ, Gorzalka BB. Estrogenic regulation of limbic cannabinoid receptor binding. Psychoneuroendocrinology. 2010;35(8):1265–1269. doi: 10.1016/j.psyneuen.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rubino T, Vigano D, Realini N, Guidali C, Braida D, Capurro V, et al. Chronic delta 9-tetrahydrocannabinol during adolescence provokes sex-dependent changes in the emotional profile in adult rats: behavioral and biochemical correlates. Neuropsychopharmacology. 2008;33(11):2760–2771. doi: 10.1038/sj.npp.1301664. [DOI] [PubMed] [Google Scholar]
- 58.Rodriguez de Fonseca F, Cebeira M, Ramos JA, Martin M, Fernandez-Ruiz JJ. Cannabinoid receptors in rat brain areas: sexual differences, fluctuations during estrous cycle and changes after gonadectomy and sex steroid replacement. Life Sci. 1994;54(3):159–170. doi: 10.1016/0024-3205(94)00585-0. [DOI] [PubMed] [Google Scholar]
- 59.Van Laere K, Goffin K, Casteels C, Dupont P, Mortelmans L, de Hoon J, et al. Gender-dependent increases with healthy aging of the human cerebral cannabinoid-type 1 receptor binding using [(18)F]MK-9470 PET. Neuroimage. 2008;39(4):1533–1541. doi: 10.1016/j.neuroimage.2007.10.053. [DOI] [PubMed] [Google Scholar]
- 60.Normandin MD, Zheng M-Q, Ropchan J, Najafzadeh S, Hull R, Qian W, et al. Test-retest reproducibility and gender differences in binding of CB1 PET tracer [11C]OMAR in humans. 57th Annual Meeting of SNM; Salt Lake City, Utah. 2010. [Google Scholar]
- 61.Stein MB, Walker JR, Forde DR. Gender differences in susceptibility to posttraumatic stress disorder. Behav Res Ther. 2000;38(6):619–628. doi: 10.1016/s0005-7967(99)00098-4. [DOI] [PubMed] [Google Scholar]
- 62.Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, et al. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3'-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J Pharmacol Exp Ther. 2005;313(1):352–358. doi: 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- 63.Piomelli D. The endocannabinoid system: a drug discovery perspective. Curr Opin Investig Drugs. 2005;6(7):672–679. [PubMed] [Google Scholar]
- 64.Hill MN, Kumar SA, Filipski SB, Iverson M, Stuhr KL, Keith JM, et al. Disruption of fatty acid amide hydrolase activity prevents the effects of chronic stress on anxiety and amygdalar microstructure. Mol Psychiatry. 2012 doi: 10.1038/mp.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]