

Abstract
Background
Perineuronal nets (PNNs) are extracellular matrix structures that enwrap many neurons in the brain. They regulate the postnatal experience-dependent maturation of brain circuits and maintain their functional integrity in the mature brain by stabilizing their synaptic architecture.
Methods
Eighty-six postmortem human brains were included in this study. We used Wisteria Floribunda agglutinin histochemistry to visualize PNNs to investigate whether the densities of PNNs in the prefrontal cortex (PFC) and primary visual cortex were altered in subjects with schizophrenia or bipolar disorder. In addition, we quantified the normal postnatal development of PNNs in the human PFC.
Results
Compared to the normal control subjects, the densities of PNNs were decreased by 70–76% in layers 3 and 5 of the PFC in schizophrenia but not in bipolar disorder. This finding was replicated in a separate group of schizophrenia and normal control subjects. In addition, PNN densities in the primary visual cortex were unaltered in either condition. Finally, the number of PNNs in the PFC increased during postnatal development through the peripubertal period until late adolescence and early adulthood.
Conclusions
These findings suggest that PNN deficit contributes to PFC dysfunction in schizophrenia. The fact that the timing of PNN development overlaps with the period when schizophrenia symptomatology gradually emerges raises the possibility that aberrant PNN formation may contribute to the onset of illness. Thus, characterization of the molecular mechanisms underlying PNN deficit may have important implications for the conceptualization of novel strategies for the diagnosis, treatment, early intervention and prevention of schizophrenia.
Keywords: cerebral cortex, schizophrenia, bipolar disorder, development, GABA, synaptic pruning
INTRODUCTION
Chondroitin sulfate proteoglycans (CSPGs) are the main lectican component of the extracellular matrix (ECM) in the brain (1, 2). CSPGs, together with other ECM components, including hyaluronic acid, link proteins and the glycoprotein tenascin-R, form perineuronal nets (PNNs), which are mesh-like lattice structures that enwrap the cell body and dendrites of neurons, including the inhibitory neurons that contain the calcium-binding protein parvalbumin (PV) and many pyramidal neurons (3–8). PNNs are thought to serve as a buffer for cations in the ECM and, as such, in the case of PV neurons, may facilitate their fast-spiking firing (9). Therefore, deficit of PNNs can lead to the dysfunction of PV neurons, which is thought to be a core pathophysiological mechanism of schizophrenia (10–12). In addition, because PNNs may also play an important role in maintaining the integrity of the connectional architecture of pyramidal cell network by regulating synaptic plasticity (13–15), PNN deficit can destabilize synaptic connectivities and thereby contribute to cortical circuitry dysfunction in schizophrenia (16).
Studies in animals have revealed that PNNs in the cerebral cortex are developmentally regulated. For instance, in the visual cortex, the number of PNNs gradually increases during postnatal development, which temporally parallels the critical period of developmental synaptic plasticity (17–20). In fact, it has been suggested that the maturation of PNNs may play a prominent role in the closure of critical period (21–24), whereas experimental dissolution of PNNs in the adult cortex by tissue plasminogen activator (tPA) has been found to reactivate the molecular machinery of synaptic plasticity (15, 18, 19, 24). Interestingly, during postnatal development, tPA level first rises, and then declines, which signals critical period closure (18, 19, 25). Taken together, it appears that developmental increase in PNNs, together with the gradual reduction in the availability of tPA, stabilizes synaptic connectional architecture by anatomically constraining synaptic plasticity. At present, the postnatal development of PNNs in the cerebral cortex in humans has not been explored.
PNNs have recently been implicated in the pathophysiology of schizophrenia. Specifically, it has been found that the density of PNNs in limbic brain structures, such as the amygdala and the entorhinal cortex, was decreased by as much as 10-fold in subjects with schizophrenia, but it was unchanged in those with bipolar disorder (26). In this context, a goal of the present study is to determine whether PNN deficit in schizophrenia may also occur in the neocortex. In addition, we characterized PNNs in subjects with bipolar disorder in order to establish any possible disease specificity of PNN deficit. In fact, we found that the density of PNNs was decreased by 70–76 % in layers 3 and 5 of the PFC in subjects with schizophrenia, but it was unchanged in any of the layers in the primary visual cortex. Furthermore, PNN density in either the PFC or the primary visual cortex was unaltered in the subjects with bipolar disorder. Finally, we quantified the postnatal development of PNNs in the human PFC and found that their number increased through the peripubertal period until late adolescence and early adulthood, which happens to be the period of time when schizophrenia symptomatology typically begins to gradually emerge. Taken together, these findings suggest that PNN deficit contributes to PFC dysfunction in schizophrenia and raise the possibility that PNN deficit may be a consequence of aberrant peripubertal PNN formation, which may contribute to the onset of illness by disturbing developmental synaptic reorganization.
METHODS AND MATERIALS
Human Subjects
A total of 86 postmortem human brains were included in this study (Table 1). Sixty-seven of them were adult human brains obtained from the Harvard Brain Tissue Resource Center (HBTRC) at McLean Hospital in Belmont, MA. In 47 of these brains, we compared PNN densities in the PFC (Brodmann’s area 9) in subjects with schizophrenia (N=16) or bipolar disorder (N=15) with that of demographically matched normal control subjects (N=16). None of these subjects had a history of substance dependence based on review of medical records and report by the next-of-kin; this was further corroborated by negative toxicology reports. The fact that most of the subjects were free of substance abuse or dependence history is typical for brains that come to the HBTRC, which receives exclusively community-based donations. After it was determined that PNN densities were decreased in the PFC in subjects with schizophrenia, we then attempted to replicate these findings in a separate cohort of brains from 5 schizophrenia and 5 normal control subjects (Table 1). Furthermore, using these 10 brains, together with 5 additional brains for each of the diagnostic groups (Table 1), we quantified PNN densities in the primary visual cortex (Brodmann’s area 17) in schizophrenia and demographically matched normal control subjects in order to determine any region specificity of PNN deficit. Finally, we obtained 19 brains from healthy control human subjects at different postnatal ages from the National Institute of Child and Human Development (NICHD) Brain and Tissue Bank at the University of Maryland in Baltimore, MD in order to survey the trajectory of PNN development in the PFC (Table 2). Brain donation and informed consent procedures at the HBTRC and the NICHD Brank and Tissue Bank have been approved by the Partners Human Research Committee and the Institutional Review Board of the University of Maryland, respectively.
Table 1.
| Case1 | Group | Sex | Age (years) | PMI2 (hours) | Freezer Storage Time (days) | Cause of Death | Antipsychotics | Anticonvulsants/Mood Stabilizers | Area 93 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Control | M | 69 | 15.3 | 5258 | Chronic obstructive pulmonary disease | None | None | + |
| 2 | Control | F | 78 | 14.1 | 5088 | Myocardial infarction | None | None | + |
| 3 | Control | M | 36 | 24.5 | 4684 | Myocardial infarction | None | None | + |
| 4# | Control | M | 37 | 18.7 | 4426 | Electrocution | None | None | + |
| 5 | Control | M | 49 | 24.6 | 4410 | Myocardial infarction | None | None | + |
| 6 | Control | M | 40 | 16.6 | 4246 | Cardiac arrest | None | None | + |
| 7 | Control | F | 74 | 12.5 | 4244 | Renal failure | None | None | + |
| 8 | Control | M | 67 | 22.3 | 4028 | Cardiac arrest | None | None | + |
| 9 | Control | M | 79 | 20.9 | 3874 | Pancreatic cancer | None | None | + |
| 10# | Control | F | 78 | 23.9 | 3865 | Breast cancer | None | None | + |
| 11 | Control | F | 65 | 24.2 | 3831 | Lung cancer | None | None | + |
| 12 | Control | F | 66 | 7.4 | 3844 | Lung cancer | None | None | + |
| 13 | Control | M | 89 | 7.4 | 3724 | Cancer | None | None | + |
| 14 | Control | M | 50 | 24.1 | 3422 | Myocardial infarction | None | None | + |
| 15 | Control | M | 73 | 20.5 | 3334 | Cardiac arrest | None | None | + |
| 16 | Control | M | 62 | 26.1 | 4957 | Myocardial infarction | None | None | + |
| Average±SD (cases 1–16) | 63.0±16.0 | 18.9±6.1 | 4152±554 | ||||||
| 17 | Control | M | 76 | 23.9 | 1181 | Pancreatic cancer | None | None | +* |
| 18 | Control | F | 47 | 25.8 | 1142 | Cardiopulmonary arrest | None | None | +* |
| 19 | Control | M | 46 | 28.8 | 1029 | Myocardial infarction | None | None | +* |
| 20 | Control | F | 53 | 34.5 | 656 | Chronic obstructive pulmonary disease | None | None | +* |
| 21 | Control | M | 55 | 23.9 | 773 | Cardiopulmonary arrest | None | None | +* |
| Average±SD (cases 17–21) | 55.0±12.0 | 27.4±4.5 | 1049±232 | ||||||
| 22 | Control | F | 51 | 30.6 | 1171 | Pulmonary embolism | None | None | |
| 23 | Control | M | 65 | 20.9 | 1236 | Myocardial infarction | None | None | |
| 24 | Control | M | 57 | 35.5 | 686 | Myocardial infarction | None | None | |
| 25 | Control | F | 65 | 27.2 | 654 | Unknown | None | None | |
| 26 | Control | M | 62 | 26.1 | 674 | Myocardial infarction | None | None | |
| Average±SD (cases 17–26) | 58.0±9.0 | 27.7±4.7 | 920±252 | ||||||
|
| |||||||||
| 27 | Bipolar | F | 80 | 11.6 | 4921 | Stroke | None | Valproic acid | + |
| 28# | Bipolar | F | 42 | 15.8 | 4613 | Suicide by overdose | Perphenazine | Valproic acid, lithium | + |
| 29 | Bipolar | F | 76 | 22.8 | 4552 | Myocardial infarction | Olanzapine | None | + |
| 30 | Bipolar | M | 50 | 30.5 | 4551 | Cardiopulmonary arrest | None | Lithium | + |
| 31 | Bipolar | M | 74 | 24.8 | 4472 | Pneumonia | Quetiapine | Valproic acid | + |
| 32# | Bipolar | M | 73 | 7.18 | 4032 | Pneumonia | Olanzapine | Gabapentin | + |
| 33# | Bipolar | F | 73 | 20.8 | 3985 | Sepsis | Olanzapine | None | + |
| 34# | Bipolar | M | 74 | 14.3 | 3898 | Pneumonia | Quetiapine | Carbamazepine, valproic acid, lithium | + |
| 35# | Bipolar | F | 73 | 17.0 | 3851 | Cardiopulmonary arrest | Olanzapine | Valproic acid | + |
| 36 | Bipolar | M | 72 | 27.7 | 4793 | Respiratory failure | None | Lithium | + |
| 37 | Bipolar | M | 78 | 30.2 | 3675 | Cardiopulmonary arrest | Fluphenazine, chlorpromazine | Valproic acid, lithium | + |
| 38 | Bipolar | M | 41 | 30.7 | 3613 | Suicide by hanging | Risperidone, ziprasidone | Topiramate, gabapentin | + |
| 39# | Bipolar | M | 83 | 17.5 | 3401 | Unknown | Unknown | Unknown | + |
| 40 | Bipolar | M | 83 | 5.0 | 3376 | Cardiopulmonary arrest | Unknown | Unknown | + |
| 41 | Bipolar | M | 38 | 22.0 | 4921 | Suicide by carbon monoxide poisoning | Olanzapine | Valproic acid | + |
| Average±SD (cases 27–41) | 67.0±16.0 | 19.9±8.2 | 4179±544 | ||||||
|
| |||||||||
| 42 | Schizophrenia | F | 83 | 23.2 | 5086 | Unknown | Haloperidol, fluphenazine | None | + |
| 43 | Schizophrenia | F | 84 | 25.8 | 5054 | Congestive heart failure | Risperidone | Valproic acid | + |
| 44 | Schizophrenia | M | 44 | 19.0 | 4881 | Pneumonia | Clozapine | None | + |
| 45 | Schizophrenia | M | 35 | 28.0 | 4882 | Cardiopulmonary arrest | None | Valproic acid | + |
| 46 | Schizophrenia | M | 42 | 18.1 | 4881 | Suicide by carbon monoxide poisoning | Olanzapine | None | + |
| 47 | Schizophrenia | F | 78 | 13.4 | 4848 | Sick sinus syndrome | Haloperidol | Lithium | + |
| 48 | Schizophrenia | M | 46 | 18.5 | 4840 | Sepsis | Olanzapine | Valproic acid | + |
| 49 | Schizophrenia | F | 72 | 21.7 | 3849 | Cardiac arrest | Risperidone | None | + |
| 50 | Schizophrenia | M | 26 | 16.0 | 4793 | Suicide by hanging | Fluphenazine | None | + |
| 51 | Schizophrenia | F | 42 | 27.1 | 4791 | Cancer | Unknown | Unknown | + |
| 52 | Schizophrenia | F | 47 | 19.2 | 4776 | Cancer | Unknown | Unknown | + |
| 53 | Schizophrenia | M | 31 | 29.0 | 4649 | Unknown | Olanzapine, risperidone | None | + |
| 54 | Schizophrenia | F | 80 | 10.9 | 4547 | Cardiopulmonary arrest | Thioridazine | None | + |
| 55 | Schizophrenia | M | 49 | 19.1 | 4527 | Suicide by hanging | Thiothixene | Valproic acid | + |
| 56 | Schizophrenia | F | 73 | 24.1 | 4062 | Lung cancer | Risperidone | None | + |
| 57 | Schizophrenia | M | 60 | 22.2 | 5086 | Cardiopulmonary arrest | Quetiapine, olanzapine | None | + |
| Average±SD (cases 42–57) | 56.0±20.0 | 20.9±5.2 | 4698±341 | ||||||
| 58 | Schizophrenia | M | 77 | 25.3 | 1246 | Pneumonia | Clozapine | Valproic acid | +* |
| 59 | Schizophrenia | F | 48 | 29.9 | 1177 | Uterine cancer | Olanzapine | None | +* |
| 60# | Schizophrenia | M | 32 | 38.4 | 1130 | Suicide by hanging | None | None | +* |
| 61 | Schizophrenia | F | 56 | 26.5 | 1081 | Colon cancer | Olanzapine, Chlorpromazine, | None | +* |
| 62 | Schizophrenia | M | 58 | 25.3 | 947 | Coronary artery disease | risperidone | None | +* |
| Average±SD (cases 58–62) | 54.0±16.0 | 29.1±5.5 | 1077±193 | ||||||
| 63 | Schizophrenia | F | 47 | 31.8 | 492 | Dilated cardiomyopathy | Risperidone | None | |
| 64 | Schizophrenia | M | 65 | 21.1 | 750 | Congestive heart failure | Thioridazine | Valproic acid | |
| 65 | Schizophrenia | M | 59 | 29.7 | 939 | Colon cancer | Clozapine | Valproic acid | |
| 66# | Schizophrenia | F | 69 | 23.1 | 325 | Cardiopulmonary arrest | Olanzapine | None | |
| 67# | Schizophrenia | M | 60 | 22.2 | 454 | Cardiopulmonary arrest | Quetiapine, olanzapine | None | |
| Average±SD (cases 58–67) | 57.0±13.0 | 27.3±5.2 | 854±331 | ||||||
Table 2.
Human subjects included for PNN quantification during PFC development.
| Age1 | Race2 | Sex | PMI (hours) | Freezer Storage Time (days) |
|---|---|---|---|---|
| 2 days | B | M | 6 | 1731 |
| 42 days | B | M | 2 | 5238 |
| 2 | C | M | 16 | 1098 |
| 4 | B | F | 15 | 1599 |
| 11 | C | M | 20 | 4853 |
| 12 | B | M | 15 | 1046 |
| 12 | B | M | 15 | 2189 |
| 13 | C | F | 17 | 3253 |
| 15 | C | M | 9 | 1216 |
| 16 | C | M | 16 | 2494 |
| 16 | C | F | 13 | 1688 |
| 16 | C | F | 13 | 1744 |
| 18 | C | M | 17 | 2231 |
| 18 | B | M | 14 | 1300 |
| 19 | B | M | 5 | 1924 |
| 20 | B | M | 5 | 2349 |
| 22 | B | M | 13 | 2186 |
| 23 | C | M | 18 | 2830 |
| 24 | C | M | 14 | 1945 |
Age in years except for the first two subjects.
B=black, C=Caucasian; M=male;
PMI=postmortem interval.
Tissue Preparation
For the initial quantification of PNN densities in the PFC in the 47 normal control, bipolar disorder and schizophrenia subjects (cases 1–16, 27–41 and 42–57, respectively; see Table 1), tissue blocks approximately 3 mm-thick containing Brodmann’s area 9 of the PFC were post-fixed in 0.1M phosphate buffer (PB) containing 4% paraformaldehyde and 0.1 M Na azide at 4 °C for 3 weeks, cryoprotected at 4 °C for another 3 weeks (30% glycerol, 30% ethylene glycol and 0.1% Na azide in 0.1M PB), embedded in agar, and then sectioned at 40 μm using a freezing microtome, as previously described (26). Sections were then stored in cryoprotectant at −20 °C until use. Due to limited availability of additional paraformaldehyde-fixed tissue, tissue blocks snap-frozen in liquid nitrogen vapor (LNV) were used for the PFC replication experiment (cases 17–21 and 58–62 for the normal control and schizophrenia groups, respectively; see Table 1) and for the quantification of PNNs in Brodmann’s area 17 (cases 17–26 and 58–67 for the normal control and schizophrenia groups, respectively; see Table 1). LNV blocks were sectioned to a thickness of 20 μm using a cryostat, mounted on slides and then post-fixed in 4% paraformaldehyde for 20 minutes at room temperature.
Wisteria Floribunda Agglutinin Histochemistry
CSPGs were visualized histochemically using biotinylated Wisteria Floribunda agglutinin (WFA), which selectively binds to N-acetyl-galactosamine (6, 27–29), a core component of the long chondroitin sulfate glycosaminoglycan chains characteristic of CSPGs. For each of the subjects, two sections were processed for WFA histochemistry. For the paraformaldehyde-fixed tissue, antigen retrieval was performed prior to WFA histochemical procedures, as previously described (26). Briefly, free-floating sections were incubated in citric acid buffer (0.1 M citric acid, 0.2 M Na2HPO4) overnight. They were then placed in the same buffer heated to 80 °C for 30 minutes, followed by incubation in WFA (1:1,000; Vector Laboratories, CA) in 1% bovine serum albumin for 24 hours at 4 °C, in horseradish peroxidase- conjugated streptavidin for 2 hours (1:5,000, Zymed, CA), and, finally, in nickel-enhanced diaminobenzidine/peroxidase reaction (0.02% diaminobenzidine, 0.08% nickel-sulphate, 0.006% hydrogen peroxide in PB). Sections were then washed and counterstained. For the LNV tissue, sections were incubated in WFA (1:3,000) overnight at room temperature, followed by a 2-hour incubation in horseradish peroxidase conjugated streptavidin (1:5,000, Zymed, CA) at room temperature and subsequent WFA visualization, washing and counterstaining as described above. All of the sections derived from the paraformaldehyde-fixed PFC blocks were processed at the same time and so were those derived from LNV blocks of the PFC and primary visual cortex to ensure that they were subjected to identical experimental conditions.
Quantification of PNN Densities
For each of the subjects, two sections were included for quantification. For each of the two sections, we quantified the density of WFA-labeled PNNs for each of the layers in two 500 μm-wide cortical traverses extending from the pial surface to the white matter border. Thus, for each of the subjects, PNN density for each cortical layer was determined by averaging the density measures derived from the four traverses (i.e. two traverses per section and two sections per case). Cortical layer determination was made in reference to percentages of depth relative to the pial surface measured using neighboring Nissl-stained sections. To avoid the introduction of any systemic biases, all of the cases for each of the experiments (with the exception of the 5 schizophrenia and 5 normal control cases used for the replication study due to the small sample size) were quantified following a stratified random sampling procedure in which cases were stratified into three blocks of equal proportion of schizophrenia, bipolar and normal control cases per block, and cases within each block were quantified in a random sequence. In addition, the investigator performing the quantification was blind to the diagnostic status of the cases.
Statistical Analysis
PNN density for each cortical layer of each cortical region (i.e. Brodmann’s areas 9 and 17) was compared separately for subjects with schizophrenia or bipolar disorder relative to the normal control subjects using stepwise regression, as previously described (26). Briefly, we first used correlation analysis to evaluate any potential effect of each of the numerical covariates (i.e. age, PMI, antipsychotic exposure in terms of chlorpromazine equivalent dosage or CED and freezer storage time) on PNN density for each cortical layer. Those variables that were found to have a statistical significant correlation with the density measures were included in the subsequent stepwise linear regression procedure to determine any Bonferroni-corrected differences in PNN densities between diagnostic groups (i.e. between schizophrenia and normal control groups and between bipolar disorder and normal control groups). Finally, linear regression and nonlinear hyperbolic regression were performed to quantitatively survey the course of change in the density of PNNs across development for the entire PFC as a whole and for each of the cortical layers individually. To evaluate the potential influence of PMI or freezer storage on PNN density measures in this developmental cohort of cases, Pearson correlation analysis was performed. We also performed the analysis after removing the female subjects (N=4 per group) to assess if sex might affect the models.
RESULTS
Qualitative Description of PNNs in the Cerebral Cortex
The laminar distribution of PNNs was similar between the PFC and the primary visual cortex. These structures were most prominent in the mid-cortical layers (i.e. layers 3–4), less so in layers 2, 5 and 6, and not present in layer 1 (Figure 1A). Most of the neurons surrounded by PNNs appeared to be interneurons (Figure 1B), although a significant minority of them exhibited clear pyramidal morphology. However, the tissue preparation method utilized in this study does not allow us to accurately and definitively determine the relative proportion of pyramidal versus nonpyramidal cells that were surrounded by PNNs.
Figure 1. Densities of PNNs are decreased in the PFC in subjects with schizophrenia.

A. Representative photomicrographs showing the distribution of PNNs in the PFC in a schizophrenia (right) and a normal control (left) subjects. Scale bar=100μm. B. Photomicrograph showing a WFA-labeled PNN. Scale bar=20μm. C. WFA-labeled PNNs are significantly decreased in layers 3 (70%) and 5 (76%) in subjects with schizophrenia (SZ; N=16). Bar graphs represent the mean and upper 95% confidence interval by cortical layer. Layer 1 is not shown because no PNNs were found in that layer. There are no significant differences in PNN densities between subjects with bipolar disorder (N=15) and normal control (N=16) subjects. p value, F ratio: *(0.016, 6.49); **(0.028, 5.36); ***(0.042, 4.51). D. PNN densities in relation to medications in the schizophrenia subjects. Note that in the two subjects who were on no medications (N=1) at the time of death or on a mood stabilizer only (N=1), PNNs were essentially undetectable in the PFC. C=control, SZ=schizophrenia, BD=bipolar disorder.
Decrease in PNN Density in the Cerebral Cortex in Schizophrenia is Disease-, Layer- and Region-Specific
There was a significant effect of diagnosis on PNN density in layers 3 (F=6.49, p=0.016) and 5 (F=5.36, p=0.028) of the PFC in schizophrenia. Specifically, PNN density was decreased by 70% and 76%, respectively, in layers 3 and 5 in the subjects with schizophrenia (0.31 ± 0.48 cells/mm2, 0.15 ± 0.31 cells/mm2, respectively; N=16), compared to the normal control subjects (1.02 ± 0.97 cells/mm2, 0.60 ± 0.70 cells/mm2, respectively; N=16), with age as a covariate in the model for layer 3 (Figure 1C). When we separated the cases based on medication status, it appears that those who were receiving antipsychotics might have higher PNN densities (Figure 1D); with the caveats that the sample size is very small and we found no statistically significant correlation between CED and PNN densities. In contrast to the finding of decreased PNNs in schizophrenia, no differences in PNN density were observed between subjects with bipolar disorder (N=16) and normal control subjects in any of the cortical layers (Figure 1C).
In an attempt to replicate the finding of PNN deficit in the PFC schizophrenia, we performed PNN quantification in a separate group of schizophrenia (cases 58–62) and normal control (cases 17–21) subjects (see Table 1). As shown in Figure 2, we found that PNN density was significantly decreased by 52% in layer 3 in the subjects with schizophrenia (F=8.87, p=0.018). As discussed above, LNV tissue was used in this replication study whereas paraformaldehyde-fixed tissue was used in the initial experiment. In addition, the LNV tissue had been, on average, stored in the freezer for a significantly shorter duration than the paraformaldehyde-fixed tissue (see Table 1). Interestingly, it appears that the number of PNNs visualized when LNV tissue was used was about an order of magnitude greater than when paraformaldehyde-fixed tissue was examined across diagnostic groups; hence, tissue processing and/or freezer storage may have an impact on the histochemical visualization of PNNs. However, the fact that decreased PNN density was observed in the schizophrenia subjects regardless of the type of tissue used or the duration of freezer storage strongly suggests that PNN deficit in schizophrenia is a disease-specific finding, rather than an epiphenomenon of the effects of fixation or freezer storage. This argument is further strengthened by the fact that decreased PNNs in schizophrenia was found only in the PFC, but not in the primary visual cortex (Figure 2). In summary, our findings indicate that PNN deficit appears to be present only in the subjects with schizophrenia but not in those with bipolar disorder and that it is present primarily in layer 3 and possibly also in layer 5 of the PFC, but not in any of the layers of the primary visual cortex.
Figure 2. Densities of PNNs are unaltered in the primary visual cortex in subjects with schizophrenia.
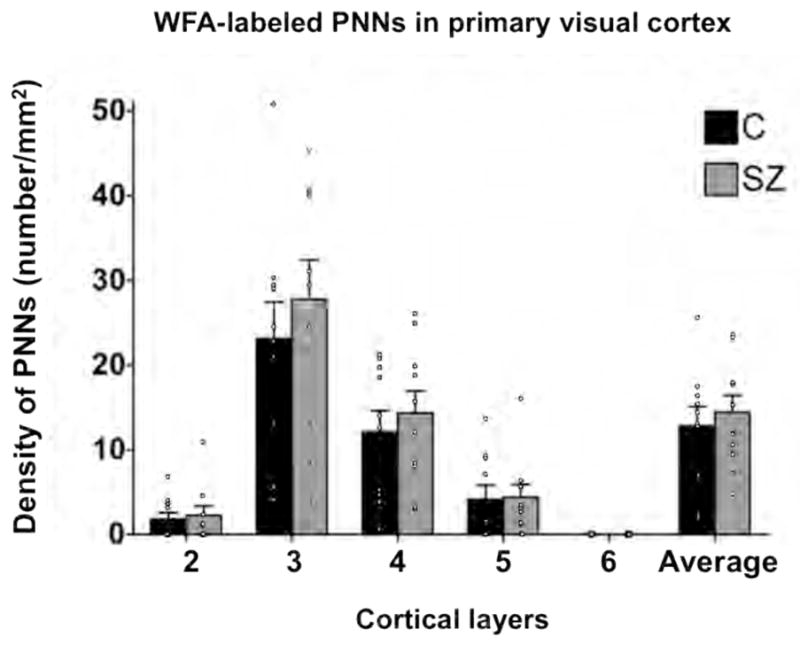
There are no significant differences in PNN densities between subjects with schizophrenia (SZ; N=10) and normal control (C; N=10) subjects.
Density of PNNs in the PFC Undergoes Postnatal Increase
Linear regression analysis indicates that there was a statistically significant effect of age on PNN density in the entire PFC as a whole (R2=0.45, p=0.0017) and specifically in layer 3 (R2=0.49, p=0.0008), suggesting that the density of PNNs in the PFC undergoes a prolonged course of progressive increase during postnatal development through adolescence and early adulthood (Figure 3). However, the nonlinear hyperbolic regression models appear to be a better fit of the data (R2=0.71 and 0.76 for the entire PFC and layer 3, respectively), consistent with the interpretation that PNN density increases during postnatal development with the most pronounced changes occurring around the peripubertal period (Figure 3). Nevertheless, in order to be able to more precisely map the developmental time course of change in PNN density and to distinguish between these two models, it would be necessary to increase the sample size, in particular the number of subjects that are under the age of 12. Finally, it does not appear that our results were influenced by PMI, freezer storage or sex because Pearson analysis revealed that there was no significant correlation between PMI or freezer storage duration and PNN densities, and removing the four female subjects also did not significantly affect the results.
Figure 3. Postnatal development of PNNs in the human PFC.
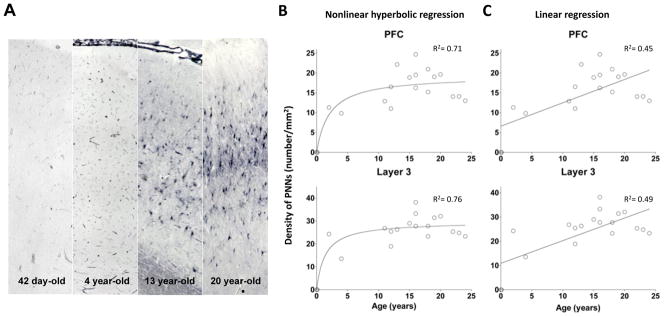
A. Photomicrographs demonstrating the increase in PNNs in the PFC during postnatal development. B. Nonlinear hyperbolic regression model of PNN densities in layer 3 and in the entire PFC. C. Linear regression model of PNN densities in layer 3 and in the entire the PFC.
DISCUSSION
Findings of this study, together with previous observations of decreased PNN densities in the amygdala and entorhinal cortex in subjects with schizophrenia (26), suggest that PNN deficit may be a rather pervasive pathophysiologic feature of this illness, affecting a variety of regions of the brain. In addition, deficit of PNNs appears to be specific for schizophrenia rather than a feature shared by primary psychotic conditions, as decreased PNN density was not found in either of these brain regions in subjects with bipolar disorder. Furthermore, PNN densities appear to be unchanged in the primary visual cortex not only in the subjects with bipolar disorder, but also in the schizophrenia subjects, indicating that, within the neocortex, PNN deficit in schizophrenia is region-specific, although the pathophysiologic significance of such regional dissociation is not immediately clear. Finally, our data suggest that PNNs in the human PFC undergo a protracted course of postnatal maturation, with the density of these structures progressively increasing through the peripubertal period until adolescence and early adulthood, which happens to be the period of time when the symptoms and deficits of schizophrenia typically begin to gradually emerge.
PNN Deficit in the PFC in Schizophrenia
PV-containing inhibitory neurons are thought to play a critical role in the pathophysiology of schizophrenia (10–12). Recent findings suggest that dysfunction of these neurons may contribute to the clinical symptoms and cognitive impairments of the illness by disrupting gamma band synchronized oscillation of pyramidal cell networks in the cerebral cortex (30–35). The etiology of the functional disturbances of PV neurons remains largely unknown, although it has been suggested that oxidative stress and hypofunction of the N-methyl-D-aspartate (NMDA) glutamate receptor, among other factors, may lead to their injury (36–41). Our data, together with findings of a previous study (26), suggest that decreased PNNs, which preferentially (but not selectively) enwrap PV neurons (3, 5, 42, 43), may be an additional pathophysiologic mechanism that contributes to the dysfunction of these neurons. Furthermore, in a recent study, it has been shown that oxidative stress that results from impaired synthesis of glutathione in mice can lead to the disturbances of the functional maturation of PV neurons and the developmental formation of PNNs that normally surround these neurons (44), suggesting that PNN deficit may be a mediator of the pathophysiological effects of oxidative stress. In this context, because PV neurons in the cerebral cortex appear to be relatively intact in bipolar disorder (45, 46), the fact that PNN deficit was not observed in the subjects with bipolar disorder in this study is not unexpected.
Aside from PV neurons, pyramidal neurons in the cerebral cortex, especially those in layer 3, are also known to be disturbed in schizophrenia (47). For instance, the density of dendritic spines on pyramidal neurons in layer 3 of the PFC has been found to be decreased in subjects with schizophrenia (48). Consistent with this observation, the average somal area of these neurons also appears to be decreased (49). PNNs are known to enwrap not only PV neurons, but also the dendritic trees, somata and axon initial segments of many pyramidal neurons and, as such, they are thought to play an important role in the stabilization and maintenance of the synaptic connectivities of pyramidal neurons (3, 5, 42, 43). Hence, PNN deficit may directly contribute to the dysfunction of pyramidal cell networks in schizophrenia by compromising the integrity and stability of their synaptic architecture, thereby leading to aberrant information processing.
Because the formation of PNNs is known to be activity-dependent (20, 24, 50–52) and because the dysfunction of both PV and pyramidal neurons in schizophrenia is expected to be associated with altered neuronal activities, PNN deficit in schizophrenia may be a consequence, instead of a cause, of the dysfunction of these neurons. Nevertheless, even in this scenario, PNN deficit triggered by the hypofunction of PV and/or pyramidal neurons can further compromise their functional integrity, which in turn may exacerbate PNN disturbances, hence resulting in a malicious, self-sustaining mechanism of neuronal injury. Our data suggest that PNN deficit occurs predominantly in layer 3, which furnishes long-range connections with widespread cortical regions. This observation is consistent with a large body of literature demonstrating that various pathophysiological features of schizophrenia appear to occur primarily in this layer. In addition, our data suggests that PNN deficit may also occur in layer 5, which furnishes subcortical output and provides feedback regulation of layer 3 circuits. Taken together, it appears that a consequence of PNN deficit in the PFC in schizophrenia may involve large-scale information processing disturbances across multiple cortical and subcortical neural networks.
Many questions remain unanswered. For instance, without colocalizing WFA histochemical signal with specific neuronal markers, the relative proportion of PV versus pyramidal neurons that were associated with decreased PNNs is not known. Furthermore, WFA is only one of the many markers that recognize some but not all PNNs, as various markers recognize chemically distinct populations of these structures (2, 51, 53–57). The relationships, if any, between specific PNN populations recognized by these various markers and the different neuronal types (e.g. PV versus pyramidal neurons) are unknown. Determining such relationships and how these relationships might be altered in schizophrenia is likely to deepen our insight into the molecular abnormalities that underlie cortical circuitry disturbances.
PNNs and Schizophrenia Onset
Studies in the mammalian cerebral cortex suggest that PNNs are developmentally regulated; hence, the number of PNNs gradually increases during postnatal development, which temporally parallels the time course of the critical period of developmental synaptic plasticity (17–20). In fact, it has been shown that the maturation of PNNs may play a crucial role in determining the timing of the closure of critical period, whereas proteolytic treatment in the adult brain (e.g. by tPA) reactivates the molecular machinery of this process (15, 18, 19, 24). Interestingly, during postnatal development, tPA level first rises, and then declines, which appears to signal the endpoint of developmental experience-dependent synaptic remodeling (18, 19, 25). In other words, developmental increase in PNNs, together with the decrease in tPA, stabilizes experience-dependent formation of synapses by preventing them from undergoing further structural modification. If PNNs also regulate the extensive synaptic refinement process that occurs in the human PFC during the periadolescent period (58–61), similar to their important role in determining the time course of critical period in other cortical regions in animals, then it can be expected that deficient developmental PNN formation may compromise the experience-dependent consolidation of synaptic connectivities. As a result, the synaptic architecture of the PFC may remain in an excessively plastic, permanently juvenile state where synapses and thus functional cortical circuits fail to be stabilized, compromising the integrity, stability and fidelity of PFC circuits that are necessary for reliable and predictable information processing and may thereby contribute to the onset of schizophrenia and the persistent symptomatic and cognitive deficits that characterize the course of this chronic debilitating illness. This scenario may, at least in part, explain the previous postmortem findings of decreased dendritic spines and neuropil in subjects with schizophrenia (48, 62, 63). Consistent with this hypothesis, using a novel free-water imaging technique, it has recently been shown that the extracellular space in the cerebral cortex, of which PNNs are a component, was significantly decreased in first-episode schizophrenia patients (64).
Clinical Implications and Future Direction
Given the presumably critical role of PNNs in the normal functioning of PV and pyramidal neurons, the maturation of cortical circuits involving these neurons, and the maintenance of cortical circuit stability, one can speculate that effective therapeutic and preventative strategies may involve restoring the structural and developmental integrity of PNNs. Our findings may also inform the development of novel diagnostic techniques for schizophrenia, using PNNs as a biomarker. For instance, ligands that recognize specific molecular domains that make up PNNs can be developed to detect and quantify these structures in the living human brain, much like imaging amyloid protein in Alzheimer’s disease. In summary, the finding of decreased PNNs in the PFC in schizophrenia suggests that detailed characterization of the molecular and pathogenetic basis of PNN deficit has the potential of leading to breakthroughs in the diagnosis, treatment, early intervention and prevention of this devastating illness.
Acknowledgments
This study was supported by grants MH076060 and MH080272 from the National Institutes of Health.
Footnotes
CONFLICTS OF INTEREST AND FINANCIAL DISCLOSURES
The authors report no biomedical financial interests or potential conflicts of interest.
AUTHORS’ CONTRIBUTIONS
SAM, KMA, NS and EP processed the tissue. SAM, KMA and HP carried out the WFA histochemical staining. SAM and KMA performed PNN density quantification. SAM and TUWW conducted the data analysis. TUWW conceived of the study, participated in its design and data interpretation and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hynes RO, Yamada KM. Extracellular matrix biology. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 2012. [Google Scholar]
- 2.Yamaguchi Y. Lecticans: organizers of the brain extracellular matrix. Cellular and molecular life sciences : CMLS. 2000;57:276–289. doi: 10.1007/PL00000690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Celio MR, Blumcke I. Perineuronal nets--a specialized form of extracellular matrix in the adult nervous system. Brain Res Brain Res Rev. 1994;19:128–145. doi: 10.1016/0165-0173(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 4.Dityatev A, Schachner M. Extracellular matrix molecules and synaptic plasticity. Nat Rev Neurosci. 2003;4:456–468. doi: 10.1038/nrn1115. [DOI] [PubMed] [Google Scholar]
- 5.Celio MR, Chiquet-Ehrismann R. ‘Perineuronal nets’ around cortical interneurons expressing parvalbumin are rich in tenascin. Neurosci Lett. 1993;162:137–140. doi: 10.1016/0304-3940(93)90579-a. [DOI] [PubMed] [Google Scholar]
- 6.Bruckner G, Brauer K, Hartig W, Wolff JR, Rickmann MJ, Derouiche A, et al. Perineuronal nets provide a polyanionic, glia-associated form of microenvironment around certain neurons in many parts of the rat brain. Glia. 1993;8:183–200. doi: 10.1002/glia.440080306. [DOI] [PubMed] [Google Scholar]
- 7.Giamanco KA, Matthews RT. Deconstructing the perineuronal net: Cellular contributions and molecular composition of the neuronal extracellular matrix. Neuroscience. 2012;218:367–384. doi: 10.1016/j.neuroscience.2012.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carulli D, Rhodes KE, Brown DJ, Bonnert TP, Pollack SJ, Oliver K, et al. Composition of perineuronal nets in the adult rat cerebellum and the cellular origin of their components. J Comp Neurol. 2006;494:559–577. doi: 10.1002/cne.20822. [DOI] [PubMed] [Google Scholar]
- 9.Hartig W, Derouiche A, Welt K, Brauer K, Grosche J, Mader M, et al. Cortical neurons immunoreactive for the potassium channel Kv3.1b subunit are predominantly surrounded by perineuronal nets presumed as a buffering system for cations. Brain Res. 1999;842:15–29. doi: 10.1016/s0006-8993(99)01784-9. [DOI] [PubMed] [Google Scholar]
- 10.Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- 11.Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25:1–27. doi: 10.1016/S0893-133X(01)00225-1. [DOI] [PubMed] [Google Scholar]
- 12.Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2011 doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dityatev A, Schachner M, Sonderegger P. The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat Rev Neurosci. 2010;11:735–746. doi: 10.1038/nrn2898. [DOI] [PubMed] [Google Scholar]
- 14.Wlodarczyk J, Mukhina I, Kaczmarek L, Dityatev A. Extracellular matrix molecules, their receptors, and secreted proteases in synaptic plasticity. Dev Neurobiol. 2011;71:1040–1053. doi: 10.1002/dneu.20958. [DOI] [PubMed] [Google Scholar]
- 15.Galtrey CM, Fawcett JW. The role of chondroitin sulfate proteoglycans in regeneration and plasticity in the central nervous system. Brain Res Rev. 2007;54:1–18. doi: 10.1016/j.brainresrev.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 16.Berretta S. Extracellular matrix abnormalities in schizophrenia. Neuropharmacology. 2012;62:1584–1597. doi: 10.1016/j.neuropharm.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maeda N, Fukazawa N, Ishii M. Chondroitin sulfate proteoglycans in neural development and plasticity. Front Biosci. 2010;15:626–644. doi: 10.2741/3637. [DOI] [PubMed] [Google Scholar]
- 18.Hensch TK. Critical period mechanisms in developing visual cortex. Curr Top Dev Biol. 2005;69:215–237. doi: 10.1016/S0070-2153(05)69008-4. [DOI] [PubMed] [Google Scholar]
- 19.Mataga N, Nagai N, Hensch TK. Permissive proteolytic activity for visual cortical plasticity. Proc Natl Acad Sci U S A. 2002;99:7717–7721. doi: 10.1073/pnas.102088899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hockfield S, Kalb RG, Zaremba S, Fryer H. Expression of neural proteoglycans correlates with the acquisition of mature neuronal properties in the mammalian brain. Cold Spring Harb Symp Quant Biol. 1990;55:505–514. doi: 10.1101/sqb.1990.055.01.049. [DOI] [PubMed] [Google Scholar]
- 21.Frischknecht R, Gundelfinger ED. The brain’s extracellular matrix and its role in synaptic plasticity. Advances in experimental medicine and biology. 2012;970:153–171. doi: 10.1007/978-3-7091-0932-8_7. [DOI] [PubMed] [Google Scholar]
- 22.Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–888. doi: 10.1038/nrn1787. [DOI] [PubMed] [Google Scholar]
- 23.Miyata S, Komatsu Y, Yoshimura Y, Taya C, Kitagawa H. Persistent cortical plasticity by upregulation of chondroitin 6-sulfation. Nat Neurosci. 2012;15:414–422. S411–412. doi: 10.1038/nn.3023. [DOI] [PubMed] [Google Scholar]
- 24.Pizzorusso T, Medini P, Berardi N, Chierzi S, Fawcett JW, Maffei L. Reactivation of ocular dominance plasticity in the adult visual cortex. Science. 2002;298:1248–1251. doi: 10.1126/science.1072699. [DOI] [PubMed] [Google Scholar]
- 25.Mataga N, Mizuguchi Y, Hensch TK. Experience-dependent pruning of dendritic spines in visual cortex by tissue plasminogen activator. Neuron. 2004;44:1031–1041. doi: 10.1016/j.neuron.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 26.Pantazopoulos H, Woo T-UW, Lim MP, Lange N, Berretta S. Extracellular matrix-glial abnormalities in the amygdala and entorhinal cortex of subjects diagnosed with schizophrenia. Arch Gen Psychiatry. 2010;67:155–166. doi: 10.1001/archgenpsychiatry.2009.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartig W, Brauer K, Bruckner G. Wisteria floribunda agglutinin-labelled nets surround parvalbumin-containing neurons. Neuroreport. 1992;3:869–872. doi: 10.1097/00001756-199210000-00012. [DOI] [PubMed] [Google Scholar]
- 28.Nakagawa F, Schulte BA, Spicer SS. Selective cytochemical demonstration of glycoconjugate-containing terminal N-acetylgalactosamine on some brain neurons. J Comp Neurol. 1986;243:280–290. doi: 10.1002/cne.902430210. [DOI] [PubMed] [Google Scholar]
- 29.Celio MR, Spreafico R, De Biasi S, Vitellaro-Zuccarello L. Perineuronal nets: past and present. Trends Neurosci. 1998;21:510–515. doi: 10.1016/s0166-2236(98)01298-3. [DOI] [PubMed] [Google Scholar]
- 30.Woo TUW, Spencer K, McCarley RW. Gamma oscillation deficits and the onset and early progression of schizophrenia. Harv Rev Psychiatry. 2010;18:173–189. doi: 10.3109/10673221003747609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez-Burgos G, Hashimoto T, Lewis DA. Alterations of Cortical GABA Neurons and Network Oscillations in Schizophrenia. Curr Psychiatry Rep. 2010 doi: 10.1007/s11920-010-0124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uhlhaas PJ, Haenschel C, Nikolic D, Singer W. The role of oscillations and synchrony in cortical networks and their putative relevance for the pathophysiology of schizophrenia. Schizophr Bull. 2008;34:927–943. doi: 10.1093/schbul/sbn062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uhlhaas PJ, Singer W. Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron. 2006;52:155–168. doi: 10.1016/j.neuron.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 34.Spencer KM, Nestor PG, Perlmutter R, Niznikiewicz MA, Klump MC, Frumin M, et al. Neural synchrony indexes disordered perception and cognition in schizophrenia. Proc Natl Acad Sci U S A. 2004;101:17288–17293. doi: 10.1073/pnas.0406074101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cho RY, Konecky RO, Carter CS. Impairments in frontal cortical {gamma} synchrony and cognitive control in schizophrenia. Proc Natl Acad Sci U S A. 2006;103:19878–19883. doi: 10.1073/pnas.0609440103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bitanihirwe BK, Woo T-UW. Oxidative stress in schizophrenia: an integrated approach. Neurosci Biobehav Rev. 2011;35:878–893. doi: 10.1016/j.neubiorev.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M. Redox dysregulation, neurodevelopment, and schizophrenia. Curr Opin Neurobiol. 2009 doi: 10.1016/j.conb.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 38.Behrens MM, Sejnowski TJ. Does schizophrenia arise from oxidative dysregulation of parvalbumin-interneurons in the developing cortex? Neuropharmacology. 2009 doi: 10.1016/j.neuropharm.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, et al. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008;31:234–242. doi: 10.1016/j.tins.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM. A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci. 2006;26:1604–1615. doi: 10.1523/JNEUROSCI.4722-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. Journal of Psychiatric Research. 1999;33:523–533. doi: 10.1016/s0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- 42.Alpar A, Gartner U, Hartig W, Bruckner G. Distribution of pyramidal cells associated with perineuronal nets in the neocortex of rat. Brain Res. 2006;1120:13–22. doi: 10.1016/j.brainres.2006.08.069. [DOI] [PubMed] [Google Scholar]
- 43.Bruckner G, Szeoke S, Pavlica S, Grosche J, Kacza J. Axon initial segment ensheathed by extracellular matrix in perineuronal nets. Neuroscience. 2006;138:365–375. doi: 10.1016/j.neuroscience.2005.11.068. [DOI] [PubMed] [Google Scholar]
- 44.Cabungcal JH, Steullet P, Kraftsik R, Cuenod M, Do KQ. Early-Life Insults Impair Parvalbumin Interneurons via Oxidative Stress: Reversal by N-Acetylcysteine. Biol Psychiatry. 2013;73:574–582. doi: 10.1016/j.biopsych.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 45.Bitanihirwe BK, Lim MP, Kelley JF, Kaneko T, Woo T-UW. Glutamatergic deficits and parvalbumin-containing inhibitory neurons in the prefrontal cortex in schizophrenia. BMC Psychiatry. 2009;9:71. doi: 10.1186/1471-244X-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bitanihirwe BK, Lim MP, Woo T-UW. N-methyl-D-aspartate receptor expression in parvalbumin-containing inhibitory neurons in the prefrontal cortex in bipolar disorder. Bipolar Disord. 2010;12:95–101. doi: 10.1111/j.1399-5618.2009.00785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lewis DA, Gonzalez-Burgos G. Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology. 2008;33:141–165. doi: 10.1038/sj.npp.1301563. [DOI] [PubMed] [Google Scholar]
- 48.Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Archives of General Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- 49.Pierri JN, Volk CL, Auh S, Sampson A, Lewis DA. Decreased somal size of deep layer 3 pyramidal neurons in the prefrontal cortex of subjects with schizophrenia. Archives of General Psychiatry. 2001;58:466–473. doi: 10.1001/archpsyc.58.5.466. [DOI] [PubMed] [Google Scholar]
- 50.Nakamura M, Nakano K, Morita S, Nakashima T, Oohira A, Miyata S. Expression of chondroitin sulfate proteoglycans in barrel field of mouse and rat somatosensory cortex. Brain Res. 2009;1252:117–129. doi: 10.1016/j.brainres.2008.11.022. [DOI] [PubMed] [Google Scholar]
- 51.Lander C, Kind P, Maleski M, Hockfield S. A family of activity-dependent neuronal cell-surface chondroitin sulfate proteoglycans in cat visual cortex. J Neurosci. 1997;17:1928–1939. doi: 10.1523/JNEUROSCI.17-06-01928.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foscarin S, Ponchione D, Pajaj E, Leto K, Gawlak M, Wilczynski GM, et al. Experience-dependent plasticity and modulation of growth regulatory molecules at central synapses. PLoS ONE. 2011;6:e16666. doi: 10.1371/journal.pone.0016666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pizzorusso T, Medini P, Landi S, Baldini S, Berardi N, Maffei L. Structural and functional recovery from early monocular deprivation in adult rats. Proc Natl Acad Sci U S A. 2006;103:8517–8522. doi: 10.1073/pnas.0602657103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lander C, Zhang H, Hockfield S. Neurons produce a neuronal cell surface-associated chondroitin sulfate proteoglycan. J Neurosci. 1998;18:174–183. doi: 10.1523/JNEUROSCI.18-01-00174.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Virgintino D, Perissinotto D, Girolamo F, Mucignat MT, Montanini L, Errede M, et al. Differential distribution of aggrecan isoforms in perineuronal nets of the human cerebral cortex. Journal of cellular and molecular medicine. 2009;13:3151–3173. doi: 10.1111/j.1582-4934.2009.00694.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ajmo JM, Eakin AK, Hamel MG, Gottschall PE. Discordant localization of WFA reactivity and brevican/ADAMTS-derived fragment in rodent brain. BMC Neurosci. 2008;9:14. doi: 10.1186/1471-2202-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bruckner G, Grosche J, Hartlage-Rubsamen M, Schmidt S, Schachner M. Region and lamina-specific distribution of extracellular matrix proteoglycans, hyaluronan and tenascin-R in the mouse hippocampal formation. J Chem Neuroanat. 2003;26:37–50. doi: 10.1016/s0891-0618(03)00036-x. [DOI] [PubMed] [Google Scholar]
- 58.Woo TUW, Pucak ML, Kye CH, Matus CV, Lewis DA. Peripubertal refinement of the intrinsic and associational circuitry in monkey prefrontal cortex. Neuroscience. 1997;80:1149–1158. doi: 10.1016/s0306-4522(97)00059-6. [DOI] [PubMed] [Google Scholar]
- 59.Huttenlocher PR. Neural Plasticity. Cambridge: Harvard University Press; 2002. [Google Scholar]
- 60.Bourgeois JP, Goldman-Rakic PS, Rakic P. Synaptogenesis in the prefrontal cortex of rhesus monkeys. Cerebral Cortex. 1994;4:78–96. doi: 10.1093/cercor/4.1.78. [DOI] [PubMed] [Google Scholar]
- 61.Goldman-Rakic PS, Bourgeois J-P, Rakic P. Synaptic substrate of cognitive development: Life-span analysis of synaptogenesis in the prefrontal cortex of the nonhuman primate. In: Krasnegor NA, Lyon GR, Goldman-Rakic PS, editors. Development of the prefontal cortex. Baltimore: Paul H. Brookes Publishing; 1997. pp. 27–48. [Google Scholar]
- 62.Selemon LD, Goldman-Rakic PS. The reduced neuropil hypothesis: a circuit based model of schizophrenia. Biological Psychiatry. 1999;45:17–25. doi: 10.1016/s0006-3223(98)00281-9. [DOI] [PubMed] [Google Scholar]
- 63.Glantz LA, Lewis DA. Reduction of synaptophysin immunoreactivity in the prefrontal cortex of subjects with schizophrenia. Regional and diagnostic specificity. Archives of General Psychiatry. 1997;54:660–669. doi: 10.1001/archpsyc.1997.01830190088009. [DOI] [PubMed] [Google Scholar]
- 64.Pasternak O, Westin CF, Bouix S, Seidman LJ, Goldstein JM, Woo TU, et al. Excessive Extracellular Volume Reveals a Neurodegenerative Pattern in Schizophrenia Onset. J Neurosci. 2012;32:17365–17372. doi: 10.1523/JNEUROSCI.2904-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]